
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructured anode materials for lithium-ion batteries: principle, recent progress and future perspectives
Wen
Qi
a,
Joseph G.
Shapter
 b,
Qian
Wu
a,
Ting
Yin
a,
Guo
Gao
*a and
Daxiang
Cui
*a
b,
Qian
Wu
a,
Ting
Yin
a,
Guo
Gao
*a and
Daxiang
Cui
*a
aInstitute of Nano Biomedicine and Engineering, Shanghai Engineering Research Center for Intelligent Diagnosis and Treatment Instrument, Department of Instrument Science and Technology, School of Electronic Information and Electrical Engineering, Shanghai Jiao Tong University, Shanghai, China. E-mail: guogao@sjtu.edu.cn; dxcui@sjtu.edu.cn; Fax: +86-21-34206886; Tel: +86-21-34206886
bSchool of Chemical and Physical Sciences, Flinders University, Bedford Park, Adelaide 5042, Australia
First published on 8th September 2017
Abstract
As the most commonly used potential energy conversion and storage devices, lithium-ion batteries (LIBs) have been extensively investigated for a wide range of fields including information technology, electric and hybrid vehicles, aerospace, etc. Endowed with attractive properties such as high energy density, long cycle life, small size, low weight, few memory effects and low pollution, LIBs have been recognized as the most likely approach to be used to store electrical power in the future. This review will start with a brief introduction to charge–discharge principles and performance assessment indices. The advantages and disadvantages of several commonly studied anode materials including carbon, alloys, transition metal oxides and silicon along with lithium intercalation will be reviewed. The mechanism and synthesis methods, followed by strategies to enhance battery performance by virtue of interesting structural designs will be examined. Finally, a few issues needing further exploration will be discussed followed by a brief outline of the prospects and outlook for the LIB field.
 Wen Qi | Wen Qi was born in Sichuan, China in 1994. She received her BE degree from the Harbin Institute of Technology in 2016. Now, she is pursuing her ME under the guidance of Prof. Guo Gao at Shanghai Jiao Tong University. Her current research directions include nanomaterial-based energy storage and conversion and rare-earth luminescent nanomaterials. |
 Joseph G. Shapter | Joe Shapter obtained his PhD from the University of Toronto in 1990 and from 1990 to 1996, he worked at the University of Western Ontario. In 1996 he moved to Flinders and is now Professor of Nanotechnology. He is currently the Director of the South Australian node of the Australian Microscopy and Microanalysis Facility (AMMRF). His group is working in Nanotechnology using various techniques to examine materials on the micron or nanometer scale. Work with carbon nanomaterials is an active area of research in his group. These materials have been used for applications in areas such as sensing and solar cells. |
 Qian Wu | Qian Wu received his BE degree in Industrial Design from the Harbin Institute of Technology, China in 2016. Currently, he is a graduate student in Industrial Design under the guidance of Prof. Jianguang Li in the Lab. of Digital Manufacturing Technology at the Harbin Institute of Technology, China. His current research directions are engineering material evaluation and industrial design theory in digital manufacturing technology. |
 Ting Yin | Ting Yin was born in Shanxi, China in 1984. She obtained her ME degree from Chongqing University in 2011. At present, she is a PhD candidate focusing on the design and preparation of multifunctional nanoprobes under the guidance of Prof. Guo Gao and Prof. Daxiang Cui at Shanghai Jiao Tong University. Her current research specialties include the immunomagnetic bead cell sorting technology and early diagnosis of gastric cancer therapy. |
 Guo Gao | Guo Gao obtained his PhD from the Dalian University of Technology in 2007. After a postdoctoral fellowship at Tsinghua University from 2007 to 2009, he was appointed as a lecturer in Shanghai Jiao Tong University at the end of 2009, and was appointed as an associate professor at Shanghai Jiao Tong University in 2011. His research interests are focused on inorganic functional nanomaterials, biological imaging and cancer therapy, and energy conversion and storage. |
 Daxiang Cui | Daxiang Cui obtained his PhD in biochemistry and molecular biology from the Fourth Military Medical University in China in 1998, was appointed as an associate professor at the Fourth Military Medical University in 2000, and professor at Shanghai Jiao Tong University in 2004. He was a post-doc staff at the Max Planck Institute for Metals Research, and a visiting professor at Waseda University. His interests focus on the preparation of multifunctional nanomaterials and their application in biomedical engineering and energy. |
1. Introduction
Increasingly more serious environmental pollution problems have seen expanding research activity concerning new clean energy sources and efficient energy conversion and storage techniques. Lead-acid batteries have a relatively stable charge/discharge state but suffer from high weight and large volume, far from the requirements of portable, light electric devices.1 One of the earliest types of batteries used in mobile phones and other portable electronic devices was the Ni–Cd battery, which had a high capacity under large current, but also had severe memory effects leading to shortened battery life coupled with significant toxicity issues given its components.2,3 Another battery type, Ni-MH, is environmentally friendly and has longer lifetime but has leakage problems.4 Therefore, the lithium-ion battery was pioneered with great potential. The solid polymer electrolyte in LIBs solves the liquid leakage issues in conventional secondary batteries but the volume contracts to a small size in a lightweight architecture satisfying the demands of portable facilities. At present, a large majority of laptops and mobile phones use LIBs as the power storage technology. However, the relatively low power available for some applications leading to short on times for devices, short battery lifetimes and slow charging speed still impedes the further development of these indispensable electronic products. Moreover, the advent of electric vehicles and hybrid electric vehicles (EVs and HEVs) has broadened the application of LIBs. In an EV or HEV, the rechargeable battery provides power and the electrical energy will be converted into mechanical energy by a motor. Furthermore, worries about the exhaustion of fossil fuels can be relieved because electric power could be obtained from a variety of sources such as nuclear, hydro, wind, solar, etc. In a word, the popularity of EVs and HEVs is a core development to the construction of an environmentally friendly society.5,6 Hence, studies about improving the properties of LIBs are necessary and critical.The anode plays a crucial role in the lithium-ion battery as the characteristics of the anode directly influence the battery's electrochemical performance. The physical and chemical properties of the anode's active materials determine battery behavior and thus must be considered and controlled appropriately. Some unprecedented characteristics of raw materials admittedly provide convenience for their application as active anode materials but inherent defects in materials hinder their optimized utilization. As a result, in addition to material choice and preparation, appropriate architectural modification and design is essential for better battery performance. This review will analyze a variety of anode materials pointing out advantages and limitations, summarizing original and novel synthesis routes and exhibiting advanced performance improvement through smart and suitable structures (Scheme 1). Recently, the most frequently used anode materials have been one-dimensional (1D), 2D, 3D carbon-based materials as well as porous and core–shell structures. The distinct hierarchical graphite structure accommodates lithium ions and electrons with little shape deformation. Silicon anodes with ten times the capacity of carbon anodes have attracted considerable research attention to solve the severe volumetric change issue, which is also a common drawback in alloy anodes. Low coulombic efficiency limits the application of transition metal oxide anodes in LIBs and composites with carbon may be a good choice to improve the performance of these systems. Meanwhile, combining the strengths of different materials such as alloys, transition metal oxides or silicon with carbon complexes to prepare composite anodes is an effective and promising way to improve electrochemical behavior.
 | ||
| Scheme 1 Schematic diagram of the recent anode materials for lithium ion batteries. | ||
2. Principle of lithium-ion batteries
Just as its name implies, the work of a lithium-ion battery mainly relies on repeated transfer of lithium ions between the anode and the cathode. The anode, also called the negative electrode, releases lithium ions into the electrolyte leading to their transport to the cathode, also called the positive electrode which absorbs ions in a discharge process. Not surprisingly, during the charging process, lithium ions transfer from the cathode to the anode through the electrolyte.7–9 The structure of the secondary battery is illustrated in Fig. 1. As a crucial component of the rechargeable battery, the anode dramatically influences the performance of the whole battery. At present, most of the commercially available anodes are made of graphite due to its special hierarchical structure. When lithium ions insert into the anode material, the relatively wide interspace between two adjacent carbon layers provides insertion locations thus avoiding the structure, shape and size variations of the electrode material during the charge–discharge process.10–13 Besides this conventional mode, there are also other novel mechanisms for lithium interactions such as the displacement reaction in alloy anodes and the redox reaction in transition metal oxide anodes.14,15 In order to assess the performance of batteries, some parameters including reversible/irreversible capacity, coulombic efficiency and rate capacity are important. The reversible recycling of lithium ions was thought to be the reason for the excellent rechargeable properties of LIBs, but the reality is that various losses occur unavoidably in the real interaction/de-interaction process. One huge loss comes from the SEI (solid electrolyte interface) layer, which forms in the first charging–discharging process as a result of the reaction between the electrode material and electrolyte and helps to guarantee the freedom of lithium movement while preventing solvent intercalation into the electrode. Since the formation process consumes some lithium and SEI layers inevitably bring irreversible capacity to batteries, actions need to be taken to control the loss of the reversible capacity of LIBs.13 Another vital index is coulombic efficiency which is the ratio of lithium removal capacity to lithium insertion capacity in the same cycle and equates to discharge capacity to charge capacity for anode material and the opposite for cathode material. The decomposition of the electrolyte and physical or chemical variations of electrode active materials can lower the value of coulombic efficiency. LIB capacity varies for different currents, high capacity tends to be obtained under a low current, and hence the discharge capacity under high currents, which is called the rate capacity, becomes a significant factor to be considered. It should be noted that excessive charging and discharging of lithium ion batteries will cause permanent damage to the anode and cathode. In the practical HEV application, hard carbon anodes and manganese-based cathodes are the most common types, and besides specific capacity and volumetric capacity, power density is also a crucial element to be considered. Reports pointed out that manufacturing electrodes with as thin as possible may be the most effective way to increase the battery power density for the decrease of both current density and resistance. Moreover, the improvement of manufacture technology and production efficiency is of vital importance to meet the increasing market demand for portable, small, lightweight electric products.16 | ||
| Fig. 1 Schematic diagram of the lithium intercalation–de-intercalation reaction mechanism in a rechargeable lithium-ion battery containing solid electrodes and a liquid electrolyte. Reproduced with permission from ref. 7. Copyright 2015, J. Mater. Chem. A. | ||
3. Anode material
As is obvious in the previous section, the anode plays a key role in the whole battery performance. The battery behavior not only hinges on the inherent properties of the anode material including the physical and/or chemical properties and energy storage capacity, but also depends on the crystallinity or amorphous structure of the anode material as well as the shape, size and component state. Although some inherent properties of the material make it stand out for lithium storage, crude material without architectural arrangement may present issues during charge/discharge process (Table 1). Herein, the appropriate structural design is of greater importance than the selected material. In the remaining sections, we will introduce several common anode materials by category where the impact of the diverse structure of each material will be demonstrated in detail separately. Effective improvement techniques are reviewed for advanced rechargeable batteries.| Material | Advantages | Disadvantages |
|---|---|---|
| Carbon | (1) High electronic conductivity | (1) Low specific capacity |
| (2) Nice hierarchical structure | (2) Low rate capacity | |
| (3) Abundant and low-cost resources | (3) Safety issues | |
| Alloys | (1) High specific capacity (400–2300 mA h g−1) | (1) Low electronic conductivity |
| (2) Good security | (2) Large volume change (100%) | |
| Transition metal oxides | (1) High specific capacity (600–1000 mA h g−1) | (1) Low coulombic efficiency |
| (2) Nice stability | (2) Large potential hysteresis | |
| Silicon | (1) Highest specific capacity (3579 mA h g−1) | (1) Large volume change (300%) |
| (2) Rich, low-cost, clean resources |
Nanotechnology has been applied in a wide range of fields and energy storage is no exception. Nanoscale electrode materials have almost completely replaced the traditional materials in energy storage. Researchers have confirmed that not only the small size of particles, but also the small grains inside the particles contribute to the outstanding performances of nanomaterial electrodes. When the size of particles inside materials decreases to match with the wavelength of electrons, phonons and magnons, some small size effects occur, including the improvement of electrical conductivity, increase in magnetic coercivity, improvement in usual mechanical and optical properties and even appearance of superparamagnetic behavior,17due to the fact that the nano-size increases the surface area. The interface of the nanomaterial has a stronger adsorption of Li ions in the electrolyte and contributes to a higher battery capacity as a result of the modification of the phase transition boundary and the augmentation of the surface and interfacial area.18–20 Furthermore, the contraction of the material size helps to accelerate lithium ion diffusion since the rate of Li+ ion diffusion relies on the diffusion coefficient and diffusion length in host materials, which is represented as follows:7
| τ = Lion2/DLi |
3.1 Carbon-based materials
Commercial lithium-ion battery anodes now mainly depend on carbon due to its favorable features, such as the excellent electronic conductivity, beneficial hierarchical structure for lithium ion intercalation, ready availability and low cost. However, quite a few shortcomings of graphite anodes such as low specific capacity and rate capacity, and safety issues have led to more research on the performance enhancement of carbon-based material anodes.29 The low working potential of carbon and continuous deposition of lithium ions lead to the formation of dendrites. These structures are the sources of some of the safety issues—one of the biggest challenges for the prospect of carbon-based anodes in large-scale applications.Carbonaceous materials can be roughly divided into two types: graphitic carbon and non-graphitic carbon. The former has better crystallinity and the latter is amorphous. We usually call graphitic carbon “soft carbon” and the non-graphitic carbon “hard carbon” because the latter is relatively mechanically harder. In a graphite structure, carbon atoms exist in the form of sp2 or sp3 hybridization and the whole structure of the carbon material consists of stacked graphene planes composed of carbon double bonds. At most six graphitized carbon atoms can capture one lithium ion—the formation of LiC6 appears in electrodes most frequently and thus leads to a theoretical capacity of 372 mA h g−1, while amorphous carbon structures adsorb less Li. The lithium insertion/extraction mechanism of the carbon material anode can be described as:
| Li+ + e− + xC ⇌ LiCx | (3-1) |
The form of CNTs in the anode can be divided into an entangled random network (ECNT) or an array structure (CNTA). As shown in Fig. 2, carbon nanotubes are interconnected with each other in the ECNT arrangement. The “bridge” morphology favors the conductive integrity of the whole electrode (a), while the parallel CNTs standing vertically on the substrate shorten the lithium ion diffusion and charge transfer path lengths (b). The conventional method of preparing ECNT-based electrodes is scattering CNTs uniformly on the current collector via blending the CNT powder into a PVDF (polyvinylidene fluoride, a common binder in electrodes) suspension followed by coating on a metal planar or foam disk (such as Cu, Ni or Pt sheets). Nevertheless, the conductive contact between the active material and the substrate is very poor in the conventional electrode. Binder-free CNT anodes can address this issue and can be realized through electrophoretic deposition (EPD) or layer by layer (LBL) deposition techniques. There are many reports of high lithium storage capacity using EPD techniques37–39 and the LBL route can achieve accurate control of thickness and morphology by virtue of the interaction of cations and anions with functional groups attached on the outer walls of CNTs (as shown in Fig. 3).40–42 In fact, carbon nanotubes could act as the active material, binder and conductive substrate simultaneously to constitute a so-called free-standing electrode. As the weight of anodes could be greatly reduced by the lack of additives, free-standing electrodes have been regarded as an effective way to improve the energy density of secondary batteries.29,43 Moreover, production costs can be lowered significantly because the preparation process for free-standing CNT paper anodes is much simpler than that of the traditional CNT slurry coated substrate anode structures. The most common process is a filtration method to filter CNTs and a carbon black mixture suspension through certain size pores contained in a PVDF membrane under positive pressure followed by peeling off the CNT mat from the PVDF membrane after vacuum drying.36,44,45 The CNT array (CNTA) anode can be prepared by transfer or direct growth methods. Generally, silicon and quartz wafers are more suitable for CNT growth, and thus as-grown CNTs have to be peeled off and transferred to the current collector. However the as-obtained CNTs are usually short.46,47 The synthesized CNTs can be metallic or semiconducting depending on their chirality. Harutyunyan et al.48 initiated a synthesis technique capable of tuning the conductivity of CNTs. The authors successfully improved the conductivity of CNTs through variation of the inert gas atmosphere where the Fe catalyst (may be pre-built or directly CVD on the substrate) was heated. On the contrary, a direct growth method is appealing due to the absence of intricate preparation steps.49 Hiraoka and co-workers clarified that Ni alloys doped with Cr or Fe enable CNTA growth under the catalytic action of Al2O3 thin films.50 When Xiong et al.51 planted CNTs on the MgO substrate with the Fe catalyst via the CVD method, high-purity CNTAs with a length of 2.2 mm are obtained. Catalysts on the substrate in both methods, however, play a key role in the final growth morphology of CNTs. After promoting the decomposition of hydrocarbon molecules into carbon atoms, the metal catalyst nanoparticles (Fe, Co, Ni, Au, etc.) on the substrate (such as SiO2 and Al2O3) then act as an orientation guider for the formation of CNTs.46,47,49–52 It is necessary to manipulate the content of the catalyst and other reaction conditions. Z. J. Han and his partners proved that a reduction in the thickness of the Fe catalyst brings impurities into SWCNTs and MWCNTs and also leads to the formation of amorphous carbon once the thickness is over 3 nm. At the same time, they claimed that too low a temperature or pressure will prohibit the formation of SWCNTs.52 Limited by the ability of carbon itself to capture lithium ions, an increasing number of studies are focused on CNT-based composite anodes in which a small proportion of the CNTs serves as an efficient conductor, linker and current collector by blending with a large part of active substances such as iron oxides, alloys and silicon (detailed illustration is provided in a later section).
 | ||
| Fig. 2 Schematic of (a) ECNTs (entangled CNTs). Reproduced with permission from ref. 41. Copyright 2004, Langmuir, and (b) CNTAs (CNT arrays). Reproduced with permission from ref. 45. Copyright 2007, Nanotechnology. | ||
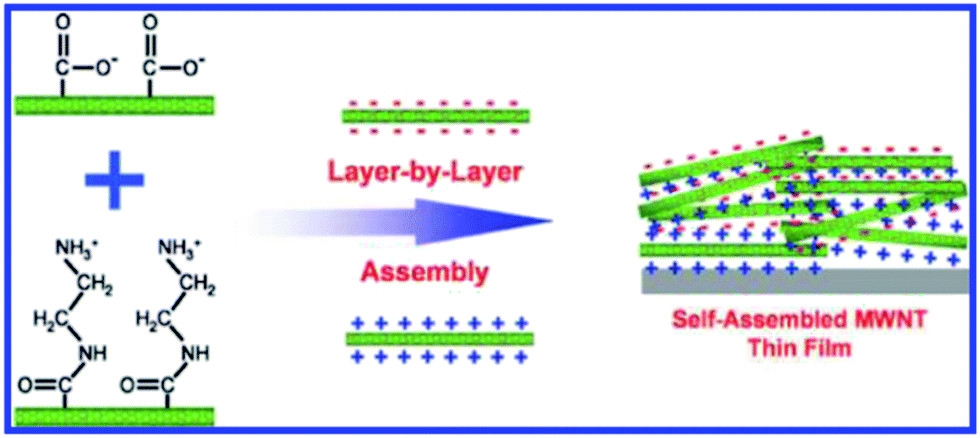 | ||
| Fig. 3 Layer-by-layer assembled MWNT thin film with positively and negatively charged MWNTs. Reproduced with permission from ref. 40. Copyright 2009, J. Am. Chem. Soc. | ||
Interestingly, simple variations of several reaction conditions can lead to the generation of carbon nanorods or CNWs (carbon nanowires) instead of CNTs. The morphology of carbon differs considerably between these carbon nanostructures: CNTs consist of ordered graphitic carbon with sp2 bonding and amorphous carbon exists in CNWs with sp2 and sp3 mixed hybridization. As shown in Fig. 4, the addition of H2 into CH4 gas generates CNWs with the catalyst nanoparticles on the top of the wires while SWCNTs grow attached to Fe particles in the absence of H2.52 There is evidence that has shown that the catalyst metal for CNWs tends to be larger than the counterpart for CNTs.52,53 Long and his group synthesized N-doped porous carbon nanofiber webs (CNFWs) with an extraordinary lithium-ion battery performance—a high reversible capacity of 943 mA h g−1 at 2 A g−1 even after 600 cycles, an excellent first-cycle coulombic efficiency of 48.4% at 0.1 A g−1 and 35.5% at 2 A g−1 with an increase to 98% after 30 cycles. The CNFWs were prepared through chemical activation of polypyrrole as the raw material (containing ∼16 wt% N content) and KOH as the activating agent (to form a porous structure). The outstanding ability could be ascribed to two important structural advantages: one is that the porous structures enlarge the electrode/electrolyte interfacial area, facilitating lithium ion diffusion and charge transfer (indeed, nanopores tend to accommodate more ions than interlayer structures); the other is the doping of heteroatoms improves the electrochemical activity and conductivity.54
 | ||
| Fig. 4 The schematic illustration of the growth mechanism of the single-walled carbon nanotubes and the carbon nanowires at different temperatures and hydrogen conditions. Reproduced with permission from ref. 51. Copyright 2011, Nanoscale. | ||
Despite these excellent properties, there are still some drawbacks of graphene hindering its further research and development: the low density of graphene sheets reduces their volumetric capacity; re-stacking of graphene sheets may cause loss of their unique strengths; synthesizing graphene with both large surface area and high conductivity is difficult and the growth control of defects/impurities on graphene sheets is challenging.
In general, synthesis methods of graphene can be divided into “top-down” and “bottom-up” categories. The former includes mechanical exfoliation, synthesis based on SiC, reducing graphene oxide and liquid phase exfoliation which usually give relatively low yields. Compared with other methods, mechanical exfoliation is low-cost, simple to operate but harder to control. The same goes for the liquid phase exfoliation except for scalability, and electrochemical exfoliation (being regarded as a type of liquid phase exfoliation) is a promising route for mass production because electro-force enables electrolyte ion intercalation into graphene sheets which helps to realize a more complete exfoliation. The reduction of GO has obvious advantages in yield at the cost of purity. Structural defects, multiple stacked graphene sheets and oxidized functional groups may appear in the as-obtained material unavoidably and favor graphene application in energy storage since they offer additional active sites. Graphene-based composites may be needed in some cases and thus reducing graphene oxide facilitates synthesis of composites due to the superior activity of oxidized functional groups in contrast to the drastically inert graphene surface. The “bottom-up” strategies include chemical vapor deposition (CVD) and surface segregation processes. In addition to high production, the graphene films fabricated through the CVD technique and surface segregation method also have an excellent scalability and large-area. Generally, the CVD route to prepare few-layer graphene involves utilizing C-containing gases to release C on several kinds of metal substrates including Ni, Fe, Pt, Pd, Cu and Co,15,55–62 while the latter one involves the dissolution of carbon in the bulk of such metals. Actually, the combination of these two methods is common in application. For example, CH4 gas decomposes C on the surface of Ni and carbon atoms diffuse into metal with a concentration gradient between the surface and the bulk.63 To manipulate a uniform thickness of the graphene layer, annealing the substrate before the formation of the graphene film is an effective way.61
Researchers have devised more feasible structural patterns for lithium storage and conversion (Fig. 5). On the one hand, lithium ions tend to aggregate between pristine graphene sheets and rarely diffuse through platelets. On the other hand, structural defects usually bring electronic discontinuity and architectural instability into the graphene framework and affect lithium storage capacity. Therefore, doping heteroatoms (N, B, and P) or halogen ions onto graphene sheets provides a compromise. A higher electronegativity than that of carbon atoms and a similar diameter size to that of carbon atoms are essential elements for doped heteroatoms, whether it is for single or multi-doped heteroatoms. N and S doped in graphene simultaneously act well in the battery anode, resulting in a reversible capacity of 1090 mA h g−1 after 500 cycles (over twice that of rGO).64 Halogen ions mingled in graphene nanoplatelets (GnPs) favor lithiation and de-lithiation for they have a lower electrostatic repulsion than O2−. Among different halogen ions, IGnP demonstrated the highest capacity and longest cycling life because of its high electronegativity and large diameter size which facilitates electrolyte ion diffusion.65
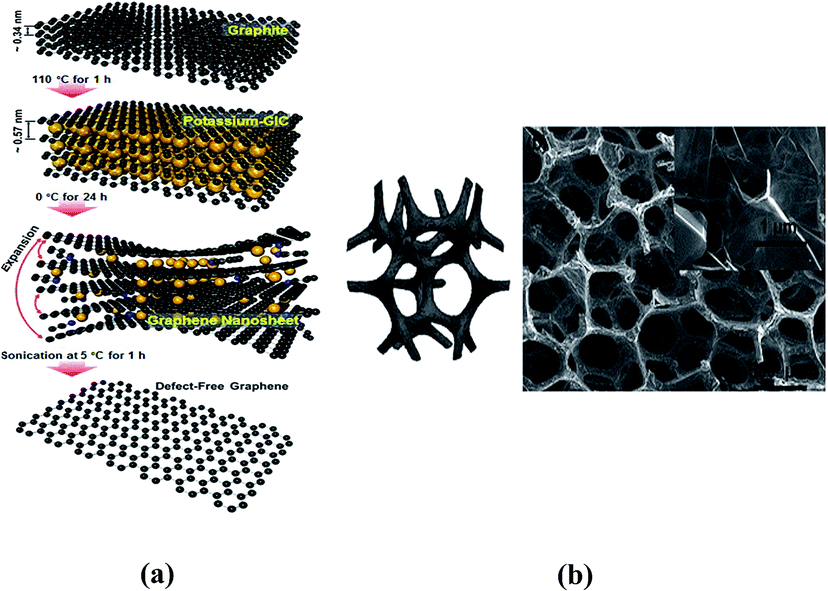 | ||
| Fig. 5 (a) Schematic illustration of the steps to defect-free graphene. Reproduced with permission from ref. 65. Copyright 2014, Nano Lett. (b) 3D graphene sponge composite. Reproduced with permission from ref. 68. Copyright 2013, Chem. Soc. Rev. | ||
Graphene-based composites are obtained by mixing electro-active materials with graphene to form composites for lithium-ion batteries. For instance, Co3O4 anchored on defect-free graphene sheets presented a high capacity of 1050 mA h g−1 at 500 mA g−1 and 900 mA h g−1 at 1000 mA g−1 over 200 cycles.66 In addition to 2D structures, researchers have focused on the 3D patterns of graphene-based composites. For example, an MoS2-wrapped 3D graphene network (3DGN) manifested reversible capacities as high as 877 and 665 mA h g−1 after 50 cycles at 100 and 500 mA g−1, respectively which were attributed to the easier access for lithium ions provided by the macroporous construction. Reports showed that sealing 2DGnP into a 3D construction yields extraordinary performance of LIBs. Fe2O3/graphene sheet aerogels (Fe2O3 uniformly scattered on GnPs which was designed into a three-dimensional hydrogel model), as a kind of metal-oxide-coated 3D graphene composite, were prepared by using metal–organic frameworks (MOFs) as the precursors.67 Benefitting from its macroporous structure, large surface area, high electrical conductivity, and good electrode homogeneity, the hybrid electrode exhibited a specific capacity of 930, 660 and 520 mA h g−1 at 500, 2000 and 4000 mA g−1, respectively and excellent cycling stability (733 mA h g−1 even after 1000 cycles at 2000 mA g−1).68
 | ||
| Fig. 6 (1) Schematic illustration of the fabrication steps for various carbon spheres (HCSs). Reproduced with permission from ref. 73. Copyright 2006, J. Mater. Chem. (2) Schematics of the possible formation process of dewatering sugar spherules through the emulsion polymerization mechanism. Reproduced with permission from ref. 76. Copyright 2002, Solid State Ionics. (3) Hollow mesoporous carbon spheres. Reproduced with permission from ref. 74. Copyright 2011, J. Phys. Chem. C. | ||
Limited by low theoretical capacity, carbonaceous electrode materials alone cannot satisfy the increasing requirements of modern power. New promising anodes consist of composites with other powerful Li storage materials in a carbon matrix and thus have been the subject of a lot of studies. Core–shell structures (carbonaceous material as the shell) have shown remarkable performances in high power/energy batteries. Before a detailed introduction of various core–shell structures, we first give descriptions of several common materials that could potentially be used for anodes.
3.2 Alloy material
Graphite anodes suffer from poor capacity as well as safety risks. Alloy anodes (such as Sn, Sb, Al, Mg, Ag and their alloys) can compensate for these defects. It has been reported that alloy anodes have theoretical specific capacity 2–10 times higher than carbon anodes. The danger of lithium deposition often observed in graphite anodes can also be avoided by the higher onset voltage over Li/Li+ in Sn containing alloy anodes.79 Furthermore, the additional benefits like good processing performance and fast charge–discharge capacity make alloy anodes an interesting path forward.Alloy negative materials may refer to pure metals or multicomponent alloys, which have a high Li storage competence. The conducting principles of metal anodes are generally based on either the insertion or chemical reaction. The active metal reacts with lithium ions and forms a kind of LixM (M represents metal). The value of x is over one, which explains why alloys have a much higher theoretical lithium capacity than carbon.80 However, it has been reported that the huge volumetric expansion because of the formation of a new phase dramatically undermines LIB properties. Other metals which are inactive toward lithium can store lithium by providing layered, porous or interval structures for Li intercalation/de-intercalation. These structures are suitable for Li accommodation with little structural transformation and retain their electronic and architectural integrity.81
Alloy anodes have their own problems like inferior electronic conductivity and large volume changes during the lithium ion intercalation/de-intercalation process, which brings about several issues like high irreversible capacity loss and rapid loss of capacity in use.14 To address these performance weaknesses, it is necessary to have a comprehensive understanding of the electrochemical mechanism causing these issues. Five reasons have been summarized as follows:14,82–85 (1) cracking of nanoparticles and the matrix supporting them leads to electric disconnection between the active material and the current collector as a result of drastic volume changes during cycling (especially when Li ions are extracted from the electrode). (2) Unlike the graphite anodes, the solid–electrolyte interface (SEI) films forming on the surface of alloy anodes go through a rupture and regeneration process due to the volume expansion and compression. The SEI layer consists of Li2CO3, ROCO2Li, lithium oxalates, and/or the ROLi mixture and the chemical composition is altered with increasing Li2CO3 content during the cycling process.86 The SEI films become thicker and salt-degradation products accumulate with increasing cycle number thus reducing both the first-cycle efficiency and later capacity. (3) Even though lithiation/de-lithiation are recognized as reversible actions generally, Li ions may be trapped in alloy anodes as a consequence of low lithium ion diffusion rates, formation of lithium compounds or adsorption to uncoordinated atoms at metal structure defects and obviously cause irreversible capacity loss. (4) Large amounts of Li can react with even a little oxygen in a thin oxide layer on the surface of metal or alloy particles because of the low atomic weight of oxygen. Li loss leads to irreversible loss of capacity. (5) Electrochemical aggregation of nanosized alloy particles occurs commonly for several reasons including the reaction of Li ions with these components to form a new alloy, volumetric enlargement of a single particle contributing to the contact of separated particles, active material particle agglomeration after Li extraction, etc.
Among these five factors, SEI film formation, surface oxidation and particle aggregation occur in the interfacial region and these surface factors can be controlled by surface modification to alleviate capacity loss. For example, coating a protective layer on the surface of nanoalloy particles is a good method to hinder oxidation and aggregation. Li conductors like ROCO2Li and Li3PO4 can be applied as the protective layers since they can also act as a kind of SEI film.
In addition, the superiority of carbon as mentioned can just compensate for the inferiority of alloy electrodes. Specifically, growing high conductivity and layered structure graphite as a dispersant or a substrate for alloy anodes can eliminate the loss of capacity to some degree. Studies on carbon-supported tin anodes have proven that the carbon matrix can prolong the battery life and narrow the potential over Li/Li+ in contrast to the pure tin anode. Various configurations of carbon have been used including carbon coating layers, hollow carbon spheres, carbon fibers and carbon nanotubes each of which presents its own strengths as well as weaknesses.79,87–90 Take a hollow shell structure as an example. Zhang et al. prepared TNHCs (tin nanoparticles with less than 100 nm diameter encapsulated into a hollow carbon sphere shell with near 20 nm thickness) for use as an anode. The structure shows an exceedingly high specific capacity and cycling retention of more than 800 mA h g−1 and maintained over 550 mA h g−1 after 100 cycles as well as a good working potential. Researchers pointed out that the larger the amount of graphite, the longer the cycle life of the secondary battery but a decrease in specific capacity and an increase in irreversible capacity loss were also observed.91
Apart from the graphite matrix, a great deal of other active/inactive matrices including SnSb, SbAl, Mg2Si, etc., and inactive matrices including metals like Fe, Cu, Nb and alloys like FeSi2 and oxides like Al2O3 and Li2O are appealing for researchers. The active or inactive here refers to whether it reacts with lithium ions—active composite (both the component and the matrix are active).
Research on the mechanism of varied metal composite anodes appeared early before large scale study of nanoscale electrode materials.87,92–94 Take the compounds of Sn-based alloys as an example. Tamura et al.95 demonstrated that Sn–Cu alloy plating on the Cu foil current collector can minimize volumetric changes by virtue of the formation of Cu6Sn5 and Cu3Sn-like phases, and the rough surface of the Cu foil contributes to the stress relief among alloy micro-particles. The team later studied a Sn–Co alloy active composite attached on a Cu current collector which showed more desirable battery behavior.96 The hardness of the active material is considered to be one reason for the performance enhancement. Much higher material hardness brings a mechanical balance to inhibit the cracking of micro-scale alloy particles.
In recent years, alloy electrodes have exhibited a higher level of electrochemical performance for advanced LIBs. As for the inactive matrix, an amorphous Sn–Ca alloy has been prepared via a solution technique utilizing the reduction effect of NaBH4, where the alloy appears in the form of the Li5Sn2 crystallite with the Ca matrix during the charge/discharge process. Sixty percent of the capacity of 400 mA h g−1 remains over 70 cycles.97 M. Anji Reddy and his group prepared a NbSb2 alloy anode where Sb reacts with Li to form Li3Sb and Nb serves as the buffer substrate. The battery performance is enhanced due to the release of stress and decrease of volume variation.98 An Ag–Sn alloy has been synthesized by Yin et al.99 for use as an anode where Ag acts as an inert matrix with buffer action. The two kinds of composites, Ag52Sn48 and Ag46Sn54, both show a commendable reversible capacity of 800 mA h g−1 maintaining 350 mA h g−1 over 50 cycles. A Sn–Fe–C alloy powder has been achieved by Mao et al.100via mechanical alloying methods to constitute the negative electrode. Here, Sn2Fe is an active phase, namely, it reacts with Li, while SnFe3C serves as the inactive matrix. They suggested that when Sn2Fe accounts for 25% and SnFe3C accounts for 75% of the composite, the battery demonstrates high reversible capacities close to 1600 mA h cm−3 and brilliant cyclability. Silver serves as an inert additive that can improve the electrochemical behavior of TiO2. He et al.101 deposited Ag NPs onto TiO2 NTs through a hydrothermal method by virtue of the conventional silver mirror reaction. The addition of Ag enormously enhanced the conductivity, reversible capacity and cyclability in contrast to single TiO2 nanotubes.
The inactive and carbon containing matrices have also been studied. Guo et al.102 applied Ni and carbon matrices to constitute a kind of spherical Sn–Ni–C alloy anode by carbon thermal reduction at 900 °C and a stable reversible capacity is obtained. The latest achievement by Liu and his group is a negative electrode consisting of Sn–SnO2 hollow spheres coated with a uniformly dispersed carbon layer which exhibits excellent reversible capacity, cyclability and rate capacity. The initial charge and discharge capacities were 878.7 and 1061.6 mA h g−1 and the discharge capacities remained at 401 mA h g−1 after 50 cycles at 0.1C, 1156.8 mA h g−1 at 0.1C, 752.2 mA h g−1 at 0.2C, 481.4 mA h g−1 at 0.5C, 289.5 mA h g−1 at 1C, and 120.6 mA h g−1 at 2C.103 Tian and his partners prepared nanospheres composed of SnO2, Sn and carbon in a 3D nanowire network with a reversible capacity of 678.6 mA h g−1 at 800 mA g−1 after 500 cycles.104
For active composites, the more active component reacts with lithium while the other component acts as a buffer to release the stress during charge/discharge on the basis of different onset potentials.105 For instance, Xie et al.106 exploited a solvothermal method to synthesize a CoSb2 alloy which exhibits better battery behavior when acting as an anode material for LIBs. In particular, the cyclability enhancement due to the morphology was very good. (CoSb2 is centrally crystallized at 900 °C and exhibits a nearly single phase). Yang and his group adopted a Sn–SnSb nanopowder composite (prepared by chemical reduction) as the negative electrode. Both Sn and SnSb react with Li and the successive reactions during the charge/discharge process lead to remarkable cycling behavior.107 The main drawback of the active-matrix anode is the reduction of reversible capacity with increasing cycle number. By contrast, the inert component in the inactive-matrix composite enhances the cyclability to a large extent although the specific capacity drops.
Active materials mixed with carbon substrates have been studied extensively. One example is that nanosized electrode materials of Co–Sn alloys coated with a carbon layer present outstanding battery performance due to the combined effect of successive active reactions, the porous framework, improved electrical conductivity and volumetric change accommodation.108 Chen et al.109 devised a new LIB negative electrode material through adding graphene nanosheets into the Sn–Sb@C core–shell structure. Extremely high rate capacities from the composite network of 978, 850 and 668 mA h g−1 respectively at 0.1C, 2C and 5C with excellent cycle stability were observed.
Porous structures can also act as a matrix to accommodate volumetric changes. The cycling properties of the electrode material dispersed on porous current collectors are better than those of the one grown on the planar collector.14 Ni–Sn macroporous alloy anodes have been prepared via an electroplating method by Nishikawa et al.110 However, the cycling stability is not good despite the increase of capacity. Kim et al.111 synthesized a nanosized Cu6Sn5 intermetallic compound powder via chemical reduction for use as an anode material. They proved that lithium was inserted into the Cu6Sn5 lattice first as LixCu6Sn5 and then as (Li4.4Sn + Cu) which are both shown to have better reversible capacity than the pure Sn metal anode. The improvement of cyclability was attributed to the nanoscale morphology as opposed to the bulk electrodes and the chemical reduction synthesis method used rather than sintering and mechanical alloying. Xue et al.112 further clarified the superiority of the 3D porous Cu6Sn5 nanostructured compound by plating it on a Cu foam current collector as opposed to other Cu–Sb-based anodes like Sn–Cu6Sb5 and Cu3Sn–Cu10Sn3–Cu6Sn5 alloy composites. A wonderful reversible capacity and cycling stability of 404 mA h g−1 after 100 cycles were observed. Similarly, Fan and his group applied a 3D porous nanoalloy anode framework into a Sn–Co alloy that was shown as a CoSn3 compound and attached on a porous Cu collector, which showed superb performance with 726 and 563 mA h g−1 initial discharge and charge capacities as well as over 70% retention after 50 cycles.113 Nevertheless, the increase of the surface area and volume brought by holes in matrices becomes a critical issue which needs further exploration (Fig. 7).110
 | ||
| Fig. 7 Several typical structural designs of alloy anode materials: (a) tin-encapsulated hollow carbon spheres. Reproduced with permission from ref. 7. Copyright 2015, J. Mater. Chem. A. (b) Microsized alloy ball composed of many sub-micro particles. Reproduced with permission from ref. 101. Copyright 2007, Electrochem. Commun. (c) Sn/SnO2@carbon nanospheres and nanowire networks. Reproduced with permission from ref. 103. Copyright 2014, J. Mater. Chem. A. (d) 3D porous Cu6Sn5 plated on the Cu foam current collector. Reproduced with permission from ref. 103. (e) Graphene supported Sn–Sb@carbon nanoparticles. Reproduced with permission from ref. 103. (f) Sn–Co alloy anode using porous Cu as the current collector (left) and after 50 charge/discharge cycles (right). Reproduced with permission from ref. 112. Copyright 2009, J. Alloys Compd. | ||
3.3 Transition metal oxides
In our effort to solve the problem of the cyclability of the electrode material, the excellent stability of transition metal oxides (TMO) undoubtedly brings about a new pathway for the development of electrode materials. Transition metal oxides not only possess relatively high capacity and good cyclability but also are cheap and easily available. Nonetheless, the commercial application of transition metal oxide anodes is far from mature. Quite a few problems including high working potential, low coulombic efficiency and large potential hysteresis between discharge and charge are tough to handle. Before presenting feasible solutions to their weakness, lithiation mechanisms are discussed and different kinds of TMOs are illustrated in the next section.According to previous research, the reaction mechanism of transition metal oxide anodes can be divided into two groups: one is the insertion reaction and the other is the conversion reaction. Several transition metal oxides such as MoOx and WOx rely on the first lithiation mechanism that Li ions insert into the interlayer place among the lattice of the electrode material free of structural transformation. In spite of their relatively high potential and low capacity, MOx-based (M represents transition metal) anodes have unique strengths including remarkable cycle stability attributed to nearly no stress release during lithium intercalation/de-intercalation as well as large rate capability free of electrolyte decomposition. Here, nanosizing also plays a crucial role in that the smaller the electrode material, the larger the quantity of lithium that can combine with the matrix. The other Li storage mechanism, conversion reaction, is also applied massively to TMOs accompanied by the reaction between Li+ and MOx, which means that the number of Li ions captured by the matrix is much more than for the insertion mechanism. The reaction between TMO and Li can be described as follows:114
| MO + 2Li+ + 2e− ↔ Li2O + M (M represents Fe, Ni, Co, etc.) |
It is clear from the equation that the reversible transition of Li2O plays an important role in the battery's reversible capacity. Though we generally regard lithiation/de-lithiation as a reversible process, various TMO components may have various impacts. Take cobalt oxide as an example. Co3O4 has twice the capacity (700 mA h g−1) compared to a graphite anode and retains 93.4% of its initial capacity after 100 cycles. Here Co is not only the product of the above reaction, but also acts as a catalyst for Li2O decomposition and formation, which contributes to the reversible transition of Li.115 The electrochemical behavior of the cobalt oxide anode can be further enhanced by virtue of other effective active materials. Xu et al.116 employed functional CNTs combined with Co3O4 spheres via a hydrothermal method. The framework looks like a necklace with beads and the battery behavior including rate capacity and cycling stability improved considerably. Kang and his group prepared a composite architecture of porous CuCo2O4 nanocubes wrapped by rGO nanopaper which exhibited outstanding battery performance at high current densities particularly.117 Several kinds of CoO–NiO–C composites have been synthesized via a high-yield, easy-to-operate method (co-precipitation and subsequent pyrolytic carbon reduction) and their optical morphology has been explored.118
More popular in the TMO group is iron oxide with tremendously high theoretical capacity (up to 1007 mA h g−1 (ref. 119)), environmentally friendly properties and enhanced safety. It has attracted a great deal of research as a high energy density and superior stability electrode. Iron oxides of diverse types (Fe3O4, Fe2O3, etc.) and structures (nanoparticles, nanowires, nanospheres, etc.) require various synthesis methods. Hematite (α-Fe2O3) nanocapsules with a length of 65 nm, a diameter of 15 nm and a thickness of 5 nm have been synthesized through wrapping–baking–peeling method (silicon coating, annealing and removal of the silicon coating) by Kim et al.120 The structure demonstrated an initial capacity of 888 mA h g−1, maintaining 740 mA h g−1 after 30 cycles as a result of the thin shell and large surface area with interior space to allow for volume variation. Liu and his group prepared α-Fe2O3 nanowires via a facile hydrothermal method. The initial charge/discharge capacities are 947/1303 mA h g−1 at the rate of 0.1C, and the reversible capacity was maintained at 456 mA h g−1 after 100 cycles. The high capacity and good capacity retention may be attributed to the short diffusion length and large contact area between electrodes and the electrolyte.121 In fact, the hydrothermal method has been recognized as an optimal synthesis route so far, which possesses a wide range of applications for various iron oxide preparation.
With iron oxide as an example, we explore the primary drawbacks of the TMO anode, the introduction of other components to make up for the weakness of TMO as well as methods to create a new synergistic action between different constituents in composite anodes. Previous studies illustrated that the electrochemical behavior of TMO electrodes with a conductive carbon matrix is much better than those of any other nanocomposite anodes with ceramic matrices (Al2O3 and MgO),122 which was attributed to the conductivity and buffer effect of the amorphous carbon matrix. For example, Zhang et al.123 synthesized a free-standing Fe3O4/rebar graphene (FRG) composite film (as shown in Fig. 8(d)), exhibiting an extraordinary rate capability with an excellent cycling performance as a result of the advantages of graphene and the absence of binders and additives. Zou et al.124 devised an anode composite of Ag-incorporated carbon nanofibers (CNFs) confined with Fe2O3 nanoparticles (Ag–Fe2O3/CNFs) which delivers great LIB performance of high rate capability and cycling stability even at low temperature. Gao and his group prepared diverse Fe2O3/CNT models via hydrothermal reactions which gave LIBs with outstanding specific and rate capability as well as cycling stability.125,126 A composite framework, porous Fe2O3 nanorods distributed on nitrogen-doped graphene sheets (NGr), has been synthesized in the same way (on the basis of a hydrothermal method). Extraordinary battery performances were observed. A lithium storage capacity of 508 mA h g−1 can be obtained at 2 A g−1 for 200 cycles and a remarkable capacity of 249 mA h g−1 at 20 A g−1 can be maintained without capacity loss up to 2000 cycles.127 There are also synthesis paths containing MOF (metal organic framework) precursors, e.g., a nanocomposite with Fe3O4 NPs (about 30 nm diameter) anchored on the amorphous carbon layer, which showed a high initial discharge capacity of 1586 mA h g−1.128
 | ||
| Fig. 8 (a) Different kinds of Co3O4-CNT heterostuctures. Reproduced with permission from ref. 115. Copyright 2013, Nanoscale. (b) Schematic diagram of the formation of a porous CuCo2O4 nanocube/rGO composite. Reproduced with permission from ref. 116. Copyright 2014, Nanoscale. (c) Ti2Nb10O29/rGO composite structure. Reproduced with permission from ref. 129. Copyright 2015, J. Power Sources. (d) Free-standing Fe3O4/rebar graphene composite film. Reproduced with permission from ref. 122. Copyright 2016, J. Alloys Compd. (e) Synthetic procedure of uniform hematite nanocapsules by the wrapping–baking–peeling process. Reproduced with permission from ref. 119. Copyright 2010, Electrochem. Commun. (f) α-Fe2O3-CNT architectures. Reproduced with permission from ref. 125. Copyright 2015, ACS Appl. Mater. Interfaces. (g) Synthesis procedure of a peanut-like α-MnO2 sphere@C composite. Reproduced with permission from ref. 131. Copyright 2014, Nanoscale. | ||
Titanium oxide is another typical transition metal oxide that has various structures including rutile, anatase, TiO2 (B) and so on. TiO2 nanosheets are usually anisotropic in terms of charge transfer (both lithium ion and electron transfer). Therefore, appropriate control of the direction of conductive structures is able to provide good conductivity for electrodes and the electrolyte and the entire battery. The ultrathin TiO2 nanosheets (NSs) with a thickness of only a few nanometers along the [001] direction and exposed (001) facets have proven to be a suitable candidate for fast speed lithiation processes in LIBs.129 Utilizing the unique strength of a carbon substrate in the same way, Wang et al.130 adopted rGO (reduced graphene oxide) as the matrix for a kind of bimetal oxide (Ti2Nb10O29) to collectively constitute the anode whose rate capability and cycle performance are both enhanced compared to those of the bare transition metal oxide.
There has been considerable work examining MnOx due to its high density (5.43 g cm−3) and the fact that it is abundant and easily available. The theoretical lithium storage capacity of manganese oxide is relatively low. Apart from size contraction, TMO composite and substrate studies are of vital importance to make high power manganese-based anode batteries. Wang and his group anchored Mn3O4 NPs on RGO nanosheets through a simple two-step solution-phase method. The strong attachment between the active particles and the substrate dramatically improved the specific capacity of LIBs.131 Carbon layer coating and carbon shell addition contribute to kinetic performance improvement similarly. Wang et al.132 creatively devised a peanut-like MnO@C core–shell structure relying on an in situ synchronous graphitization and reduction process. The performance boost was ascribed to the buffer space and conductive network inside the graphite-like carbon shell.
These performance enhancement approaches can be extended to other TMO material negative electrodes such as MoOx and NiO. Different structures containing a great deal of transition metal oxide materials has been studied and developed for advanced LIB application, such as carbon-coated MoO3 nanobelts, MoO2 NPs plated on carbon layers, porous structural NiO powder and fibers and NiO nanopaper on graphene, etc.133–137
Various metal active materials are blended together to make superior negative electrodes for battery performance improvement. For instance, Yang et al.138 designed a new nanocomposite thin film composed of NiO and NiSe, for which the first cycle discharge capacity is 577.7 mA h g−1 and holds the reversible capacity of 495 mA h g−1 after 50 cycles. The authors attributed the boost in kinetic performance compared to a single NiO electrode to the synergistic action of two components—the high capacity from NiO and the cyclability from NiSe. Bresser et al.139 synthesized transition-metal-doped zinc oxide nanoparticles for use in the secondary battery anode material with a remarkable reversible capacity of over 900 mA h g−1 and the addition of a carbon coating on the nanoparticles further improved the battery electrochemical properties. Wang and his group fabricated a composite structure of anatase and TiO2 to form a high power LIB anode. The interfacial charge storage in allomorphs (ICSA) plays a key role in higher capacity, better integrity and more stable cyclability for the combined action of TiO2(A) and TiO2(B). The interfacial (001) plane of TiO2(A) is core to electron capture while the (100) plane of TiO2(B) provides accommodation for lithium diffusion.140
3.4 Silicon
Silicon is another extremely promising anode material since it has the highest gravimetric and volumetric capacity among all the elements known and also is low cost. At the same time, the low working potential of Si-based anodes can avoid risks compared with graphite electrodes. Nevertheless, huge volume variation is a significant obstacle for Si anodes. For Si-based electrodes, the lithiation/de-lithiation process is through a conversion reaction where each Si atom is able to capture around 4 Li atoms. The most rich phase Li22Si5(Li4.4Si) at 415 °C, along with a high lithium storage capacity of 4200 mA h g−1, lead to a large volume expansion close to 310%.141,142 Another Li15Si4 phase exists at room temperature and has a lithium capacity of 3579 mA h g−1 giving a lower expansion ratio of 280%.143–145 As for alloy anodes, the huge dimension variations bring numerous issues including particle cracking, intermittent electrical contact, ineffective electron transfer and repeated dynamic formation of SEI layers. Hence, the actual capacity is far lower than theoretical values and cycling properties are poor for commercial applications. The lack of electrical conductivity also plagues the development of new-age secondary batteries.During the initial charge/discharge process, silicon usually goes through a phase transition from a crystal to an amorphous state. Amorphous silicon can bear force evenly from each direction and thus alleviate volumetric variation. So preventing the reverse transition (from amorphous Si to crystalline) in the later cycles favors the architectural and electronic integrity of LIB electrodes.145,146 Previous reports have demonstrated that crystalline bulk Si can be cycled smoothly by careful control of external voltage conditions. If the lithiation voltage of amorphous silicon can be limited above 50 mV, the amorphous state of silicon can be maintained instead of converting into crystalline Si.143 At the same time, the capacity drops dramatically in the first discharge process in comparison to the charge process from 5149 mA h g−1 to 4032 mA h g−1 (take a kind of Si-based composite anode with 80 wt% silicon, 12 wt% carbon black and 8 wt% sodium carboxymethyl cellulose as an example).146 Besides the above mentioned drawbacks arising from large volume changes, the solid-state phase transition may be another key factor causing huge irreversible capacity loss. The low initial coulombic efficiency has become a big problem hindering the commercial application of silicon serving as an electrode material for LIBs.146–148 Moreover, the depth of discharge and charge impacts internal bulk changes to some degree.
Based on the above analysis, scientific and engineering researchers have realized that the necessity of the rational design of the nanostructure of the anode is especially necessary to overcome the shortcomings of Si electrodes. Some studies have indicated that Si nanowires perform better due to their specific conductive direction and lower pulverization than nanoparticles. Experiments revealed that the Si NWs became mostly amorphous with some crystalline Si regions embedded inside the core at 50 mV and all of the Si turned to the amorphous state at 10 mV.25 To accommodate the huge volume variation of silicon, inactive matrices are introduced into silicon-based anodes to constitute composite electrodes. Compared with polymer matrices, inorganic materials are more advantageous to buffer the expansion and contraction of silicon during the charge and discharge process as well as provide good electrical contact to the active material, and carbon materials, in particular, are more lightweight than other inorganic matrix materials such as TiN and TiB2 and therefore they are suitable as additive materials for Si-based composite electrodes.149 Actually, Si/C composite anodes have been studied extensively in different frame structures (detailed design will be described in the later paragraph). Silicon owns extremely high specific and volumetric capacity but suffers from large volume expansion and contraction, while the carbon matrix could accommodate the volumetric changes, hold electrical stability and maintain structural integrity but at the same time reduce battery capacity. Therefore, the content of silicon in the Si/C composite plays a key role in rechargeable battery properties and the amount of silicon determines two development directions of silicon-based composite anodes—low capacity good cyclability one and high capacity poor cyclability one.150–154 Through maximizing the amount of carbon to relax strain produced during the lithiation/de-lithiation process, the battery capacity of the former type would be largely reduced though stable cycles could be guaranteed via cautious selection of the surface coating material and heat treatment with high security. The latter one aims at elevating the specific and volumetric battery capacity to the greatest extent in spite of great irreversible capacity losses, poor cyclability and low power efficiency. The maximum of silicon content in the Si/C composite for high capacity brings about many problems including poor processability, low economic efficiency and weak compatibility with the current battery system at the same time. Ren et al.154 demonstrated that composite electrodes have the highest initial coulombic efficiency when the C/Si ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 compared with 1:0.1 and 1:0.3 in a porous silicon/carbon microsphere structure, due to the fact that a moderate increase of the amount of Si can elevate the porosity of the Si/C microspheres while a part of silicon particles will agglomerate as the ratio of C/Si further drops. Moreover, the compatibility of the high capacity Si anode with existing battery systems is usually poor because of its precise constitution. Current research studies on the high capacity Si anode are based on a self-designed assembly system, which impedes the development of its commercial application. In a word, current and future studies on silicon/carbon anode materials are mostly focused on a better cycle performance, high energy density, high rate charge and discharge performance, stable safety performance and large-scale preparation with low cost and stable properties. The main synthesis methods of silicon/carbon composite anode materials contain CVD, sol–gel, pyrolysis, mechanical milling, hydrothermal and electrospinning methods. Unfortunately, most of the conventional synthesis routes are low-yield and non-eco-friendly, and thus the innovation of scalable processes enabling to achieve high productivity is essential for the practical application of Si-based composite anodes.155,156 Ren et al.154 have reported a low-cost and pollution-free preparation process of porous silicon/carbon microspheres (GPSCMs). They utilized graphitized needle coke produced from industrial waste as the carbon source and sucrose to bind silicon nanoparticles with porous carbon microspheres, and the optimal Si/C ratio, reaction time, treatment temperature, pre-mixing methods and carbon-coating have been discussed.
:0.2 compared with 1:0.1 and 1:0.3 in a porous silicon/carbon microsphere structure, due to the fact that a moderate increase of the amount of Si can elevate the porosity of the Si/C microspheres while a part of silicon particles will agglomerate as the ratio of C/Si further drops. Moreover, the compatibility of the high capacity Si anode with existing battery systems is usually poor because of its precise constitution. Current research studies on the high capacity Si anode are based on a self-designed assembly system, which impedes the development of its commercial application. In a word, current and future studies on silicon/carbon anode materials are mostly focused on a better cycle performance, high energy density, high rate charge and discharge performance, stable safety performance and large-scale preparation with low cost and stable properties. The main synthesis methods of silicon/carbon composite anode materials contain CVD, sol–gel, pyrolysis, mechanical milling, hydrothermal and electrospinning methods. Unfortunately, most of the conventional synthesis routes are low-yield and non-eco-friendly, and thus the innovation of scalable processes enabling to achieve high productivity is essential for the practical application of Si-based composite anodes.155,156 Ren et al.154 have reported a low-cost and pollution-free preparation process of porous silicon/carbon microspheres (GPSCMs). They utilized graphitized needle coke produced from industrial waste as the carbon source and sucrose to bind silicon nanoparticles with porous carbon microspheres, and the optimal Si/C ratio, reaction time, treatment temperature, pre-mixing methods and carbon-coating have been discussed.
The first structural design concept to tackle the inherent problems of the Si anode is to provide extra empty space for silicon expansion which can be done using a conductive carbon matrix. Hertzberg et al.157 devised a kind of Si@C composite (named Si-in-C tubes) with a Si nanotube inside a carbon nanotube. The Si nanotube expands inward due to the restriction of the outer CNT during lithiation and contracts freely inside the CNT during delithiation. The pore walls of CNTs provide sufficient room for silicon volume variation and ensure whole structural integrity. The battery performance test with the Si-in-C structure as the electrode showed excellent electrochemical performance. They tested frameworks of different silicon wall thicknesses and found that there is a delamination phenomenon for the 33 and 46 wt% Si samples that Si nanotubes delaminated from CNTs leading to the reduction of electrical contact and the 9 and 46 wt% Si samples have the highest capacity which is close to theoretical capacity. Although the coulombic efficiency after 2 and more cycles is as high as 99.5%, the initial coulombic capacity can only reach 75%. Besides the improvement of the thickness of Si and C walls, other more effective modification ways are in urgent need. Wu et al.158 proposed a model of SiNPs@CNTs that had Si nanoparticles scattered on the pore wall of carbon tubes. The volume change and the stability of the SEI layer can be controlled separately by allowing free expansion of Si nanoparticles using empty space between the particles and inhibiting Si nanoparticles from touching the electrolyte using the carbon tubes. Forney led his team to deposit silicon on CNTs via low-pressure and plasma-enhanced chemical vapor deposition methods. They proposed that the latter method is better for the free-standing Si-CNT anode to get a high extraction capacity at a low Si consumption, which makes the battery competitive in price.159 Another ingenious self-supported anode was designed by Ji et al.160 They prepared a Si/graphene/UGF composite in which Si NPs encapsulated in graphene cover a UGF (ultrathin-graphite foam) surface using aqueous suspensions. In addition to the strengths of graphene, the foam graphite surface not only guarantees good electronic contact in the electrode, but also does not require massive Si loading. The chemical treatment here is essential to architectural stability and further determines the capacity and cyclability of the whole cell. Liu et al.161 prepared a pomegranate-like Si@C composite in which each Si particle is sealed inside a carbon sphere with well-confined free space left for volume enlargement/reduction during the Li insertion/de-insertion process (the carbon shell will be fully occupied while lithiation). The initial coulombic efficiency can reach 82% for 9% carbon and the average later CE (500th to 1000th cycles) can even reach 99.87%. The root of relatively low ICE may be the amorphous morphology of the carbon shell and searching for other alternative shells may be the entrance for further improvement of ICE to surpass 85%. It is worth mentioning that the synthesis route is free of complex procedures and this Si pomegranate anode can be well compatible with the current battery system. Wu et al. invented double-wall Si nanotubes via electrospinning methods, where the inner wall is active Si with SiOx covering as the outermost layer. The free-standing carbon nanofibers are applied in the early step and removed to save inner space for DWSiNTs. The DWSiNT based anode exhibits an extraordinary rate capacity of 1780 mA h g−1 at 0.2C and 600 mA h g−1 at 12C with a cycling stability of 88% retained after 6000 cycles.162
Surface modification of silicon-based anodes has become a common method. Depositing a carbon layer on the surface of silicon will undoubtedly improve the conductivity of the electrode and inhibit the formation of the unstable SEI layer.163,164 Moreover, the working voltage of LIBs can be lower when using carbon-coated Si as the anode material – about 0.3 V lower than those of graphite electrodes. Yoshio et al.165 fabricated carbon-coated Si electrodes through a thermal vapor decomposition reaction. A high reversible capacity over 800 mA h g−1, high coulombic efficiency, good cyclability, and better thermal stability in contrast to a graphite anode were achieved. Lee and his partners exploited a mechanochemical reduction means to manufacture carbon-coated nano-Si uniformly distributed oxide/graphite anodes using ball milling of SiO2 and Al. Each component of the composite jointly contributes to the whole performance improvement.166 Chen and his group devised an atmospheric pressure plasma technology to make large area Si/SiC anodes (N-APPJ). They utilized a nitrogen atmosphere to modify the silicon surface and a Li–N matrix was formed during the charge/discharge process. The Li–N matrix offers a layer of nitrogen to impede the formation and spreading of SEI films when Li inserts and is extracted. The N-APPJ altered the surface properties of the active material and brought impressive progress in the cycle retention and coulombic efficiency of LIBs.167 To overcome the complex procedures and conditions of CVD, TVD, and atmospheric pressure plasma technology, researchers have also developed some effective, green, simple and high-compatible synthesis methods for Si/C composite anodes. Hu adopted a one-step procedure to synthesize a Si@SiOx/C nanocomposite with a thin layer of SiOx and carbon by the hydrothermal carbonization of glucose. A remarkable reversible capacity of almost 1100 mA h g−1 stands out beyond high coulombic efficiency and cyclability.168 Bin and his partners devised a one step and scalable high energy mechanical milling (HEMM) method to obtain a carbon-coated silicon electrode from low grade raw sources (metallurgical silicon (M–Si, ∼99 wt% Si) and ferrosilicon (F–Si, ∼83 wt% Si, ∼13 wt% Fe)). The full cell with the as-obtained anode has the initial coulombic efficiency as high as 86% which is much higher than those of traditional Si/C composite electrodes as the well-defined carbon coating hinders the dynamic formation of the SEI layer and withstands the volumetric variation during lithiation and de-lithiation.153 Apart from carbon coating, atomic layer deposition (ALD) of other materials such as aluminum can be employed to modify the surface of the silicon anode. He et al.169 illustrated that the thickness of the alumina layer can be controlled and 2 nm or so may be the optimal thickness to help raise the coulombic efficiency and avert great capacity losses. They also clarified the great advantages of patterned Si anodes in contrast to unetched Si electrodes, and the relative integrity of Si electrodes after cycles has guaranteed an extremely high initial coulombic efficiency of 83.2% and considerable capacity remaining after several cycling numbers.
Utilizing the excellent characteristics of graphene to use it as a support material to form Si–graphene hybrid electrodes has become a novel developing trend to design high capacity batteries, since graphene can help boost electrical conductivity and accommodate the volumetric variation of silicon. Lee et al.170 made a structure using flexible graphene sheets with Si nanoparticles sandwiched between other sectional reconstituted phase graphene sheets to maintain rigidity. In this case, the latter is the key conductive section as well as the robust framework bracing the former bending sheets trapping Si NPs. Therefore, appropriate reconstituted graphene phase content is a guarantee for high capacity and long cycle life. Another point of the Si–graphene sandwich structure is its self-supporting property free of binders, additives and even current collectors. Zhao et al.171 fabricated a similar composition structure adding in-plane defects on graphene sheets, which provide easy access for lithium ions to diffuse throughout the graphene sheets and thus ensure the high specific capacity and rate capacity as well as outstanding cyclability. Luo et al.172 designed a composite anode with silicon nanoparticles wrapped by crumpled graphene shells by virtue of capillary force in aerosol droplets. The internal void space in the crumpled morphology provides great buffer for silicon nanoparticle expansion and extraction, but on the other hand lacks the whole electrical contact in the composite since some part of silicon was not made use of. Therefore this structure cannot avoid great irreversible capacity loss though both the capacity and cyclability of the LIB using these electrodes far exceed those of the LIB using bare Si nanoparticles. To disperse silicon nanoparticles homogeneously between graphene paper layers, Zhou et al.173 adopted an electrostatic attraction self-assembly preparation method to built a new structure of the silicon nanoparticle/graphene composite anode. The negatively charged PDDA has been appended on the surface of Si nanoparticles to combine with the functional group with positive charges of two graphene sheets, and nanospaces have been confirmed to exist between Si nanoparticles and graphene layers, which ensured excellent cycle performance and high rate capacity. Kim and his partners constructed a fresh Si/C nanocomposite internally wired with a graphene network (Si/C-IWGN), obtaining super specific and volumetric capacity and the cycle stability can also be ensured. They established a series of Si/C-IWGN composites under differed preparation conditions and corresponding control groups and clarified that the initial coulombic efficiency improves as the R/C molar ratio increases. R represents resorcinol which was the carbon precursor and C represents the gelation catalyst (Na2CO3 or NH4OH). With a higher R/C molar ratio, the surface oxidized layer (SiOx layer) is thinner, and thus more reversible capacity can be maintained, especially in the first cycle.174
Another improvement method is to fabricate silicon porous structures to buffer the stress from volume expansion and contraction. Several studies have obtained pioneering results of high capacity, cycle stability and fast lithium ion diffusion.175–178 Guo and his partners designed a 3D porous carbon scaffold structured silicon by carbonization of porous Si-PVdF precursors. The synthesis procedure simultaneously anchored the electrode immediately on the current collector without binders or additives. A high rate capacity and good cycling property were shown in this novel-structured silicon anode because of the buffer action and conductive transmission of the pores inside the Si framework. The authors suggested that the control of pores, from size to distribution, will promote new research.179 A kind of nano-porous Si/C composite has been synthesized by Zheng et al.180via two-step ball-milling and the etching process. The composite electrodes show a reversible capacity of ca. 700 mA h g−1 with almost no capacity loss above 120 cycles (at 0.2 mA cm−2). Ge and his group deduced that the large pores size and porosity inside Si nanowires facilitate structural stability. By etching boron-doped silicon wafers, the porous SiNWs employed in battery anodes gave a drastic performance improvement. The capacity can reach respectively more than 2000, 1600, and 1100 mA h g−1 at current rates of 2, 4, and 18 A g−1 beyond 250 cycles.181 Zhang and his partners successfully built a porous Ni/Si hybrid anode through hydrogen bubble template electrodeposition. The rechargeable battery displayed a superb rate capability of 1420 and 1273 mA h g−1 at 4.2 A g−1 and 8.4 A g−1 current densities respectively for cycle numbers above 100. The lithium storage capacity reached 2444 mA h g−1 at 0.84 A g−1 with 83% capacity retention for 100 cycles.182 Mesoporous structured Si nanorods, nanofibers and other shapes gradually became the focus of recent research.183–186 For example, a lotus-root-like mesoporous Si framework was conceived by Jia and his team. The uniformly distributed mesopores contribute to a stable capacity of 1500 mA h g−1 after 100 cycles at 1C and high rate capability up to 15C.187 As for mass production, conventional physical methods cannot satisfy the demands of the porous Si nanostructure output. Zhao et al.188 proposed a wet-chemical etching method to fulfill the large scale, efficient production of porous-Si anodes in secondary batteries. They have demonstrated that the amorphous carbon coating on porous silicon electrodes can hinder the formation of SEI layers and improve the initial coulombic efficiency.
Beside the design of the Si-based anode itself, other parts of the full cell including the cathode, binder and membrane should also be taken into consideration and the coordination between them may bring an unexpected outcome. The latest achievement reported by Choi, Sunghun and his group in accommodating volume changes of silicon is to manufacture highly elastic binders with polyrotaxanes by the pulley principle. They added a small amount of polyurethane into PAA to form a “molecular pulley” (PR-PAA). The structure of this “molecular pulley” is unique in that it is covalently bound by the linear PAA polymer framework and cyclic polysaccharide and then the PEG chain penetrates through the ring. Some of the rings in the molecular pulley have strong adhesive properties, and the others have unique free sliding properties. The elasticity of the binder has been greatly improved with the combination of two properties and can withstand the expansion and contraction of volume changes of the silicon anode and release the stress produced in the process of charge and discharge without rupture, thereby enhancing the stability (Fig. 9).189
 | ||
| Fig. 9 (a) Schematic of a Si-in-C tube after Li extraction. Reproduced with permission from ref. 157. Copyright 2010, J. Am. Chem. Soc. (b)Three-dimensional and two-dimensional cross-sectional views of Si@C pomegranate microparticles. Reproduced with permission from ref. 161. Copyright 2014, Nat. Nanotechnol. (c) 3D network of graphite (red) anchoring regions of the graphene–Si composite. Blue circles: Si nanoparticles, black lines: graphene sheets. Reproduced with permission from ref. 170. Copyright 2010 Chem. Commun. (Camb). (d) 3D porous nano-Ni film supported silicon anode architecture. Reproduced with permission from ref. 182. Copyright 2012, J. Power Sources. (e) 3D mesoporous Si particles. Reproduced with permission from ref. 187. Copyright 2011, Adv. Energy Mater. (f) Si nanoparticles in a graphene scaffold with in-plane carbon vacancy defects. Reproduced with permission from ref. 171. Copyright 2011, Adv. Energy Mater. (g) Fabrication process of Si nanoparticles homogeneously dispersed between graphene layers. Reproduced with permission from ref. 173. Copyright 2012, Adv. Energy Mater. | ||
4. Summary and future perspectives
In recent years, there have been a steady stream of research studies examining superior energy conversion and storage devices due to the pressing demand for energy in modern life. Because of their high capacity, high power density and low-pollution, lithium-ion batteries are a promising candidate for future energy strategies. Ranging from low capacity for smart phones, panel computers and laptops to large capacity for EVs and HEVs, common applications in daily life put forward higher requirements for maximized energy storage with minimized weight and volume for LIBs. Accordingly, the unique role of the nanostructured anode makes it worth researching. In this review, we summarized the common anode materials examining the conducting mechanism, morphologies, synthesis methods and novel structural modifications and designs. Given the excellent unique hierarchical structures, good electrical conductivity and plentiful resources, quality electrodes can be made of carbon materials from various frameworks, morphologies and assemblies. The exceptional conductivity of one-dimensional carbon structures is better than those of other structures and contributes to superb electric stability and capacity for secondary batteries. ECNTs set up a conductive network upon current collectors or in free-standing architectures while CNTAs guide current directly to substrates. Ultrathin 2D nanomaterials like graphene possess compelling physical and chemical properties due to electron confinement in-plane. Three-dimensional architectures, especially porous and core–shell spheres can accommodate large volumetric variations during the charge/discharge process. Carbon in CNTs and graphene offers layered structures for lithiation and de-lithiation free of shape deformations, and the amorphous carbon present in CNWs, carbon nanorods and CSs provides better cyclability for LIBs due to its stable nature. Limited by poor specific capacity and potential safety risks, carbon anodes have limitations for further development.Due to much larger specific capacity than graphite anodes, alloy electrodes have attracted enormous attention. However, they suffer from huge volume changes during charge and discharge. Active and inactive matrices are both good approaches to overcome the large volume change issues where one matrix component reacts with Li and the other serves as a buffer. Surface modification and coating could avoid exterior oxidation and cracking of nanoparticles. Heteroatom, halogen atom or other atom doping is an effective way to increase electric connection inside electrode materials. A porous active material or porous current collector equally accommodates volume expansion and contraction during lithium insertion and extraction.
Some transition metal oxides such as cobalt oxide and iron oxide could capture more lithium ions through conversion reactions than others like molybdenum oxide which use an insertion mechanism. Sufficient efforts toward blending metal and metal oxide to make anodes have been reported and give impressive battery behavior. Appropriate preparation methods for TMO have become a research focus to accurately control electrode behavior.
Nowadays, silicon electrodes stand out because they potentially have nearly ten times the capacity compared to a graphite anode. Smart structural designs aim to address instability due to huge volumetric changes. Low specific capacity as compared to theoretical values has been reported from nanowires, porous and core–shell structures, and surface modified composites formed with CNTs and graphene.
There is a trend to combine alloys, TMOs or silicon with carbon to constitute composite anodes. Though various related structures have been created and considerable progress has been made in energy storage, there are still many challenges that need further exploration. First, cleverly designed structures optimize certain properties of LIBs, at the same time these anodes present other issues. For example, porous or hollow structures are able to accommodate the volume changes but reduce the volumetric capacity of the whole cell. For the prevalent application of portable, light, small electric devices, volumetric capacity as well as specific capacity is highly valued. The comprehensive assessment of anode materials and structures is thus in urgent need, aiming to produce designs of electrode architectures endowed with better overall performance. Even though composites of multi-material blending and combination of different structures do help, more theoretical research including modelling and computing should be applied to maximize the feature which leads to improved performance while also minimizing shortcomings among the different materials and structures. The experimental research is far from the requirements of commercial production. On one hand, the experimental conditions in research labs may be straightforward and produce small amounts of relatively pure material while realizing the production requirements in the real commercial environment can be very tough and require sophisticated procedures. On the other hand, the consideration of the matching among the anode, cathode, electrolyte and membranes cannot be ignored since there may be repulsive actions between different parts of the battery which could offset each advantage and even produce new drawbacks. We hope the present review will help a little to provide a global view of the latest developments in anode materials for LIBs and give insight into promising perspectives in the energy conversion and storage field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 91634108, 81671737) and the National Key Program (2017FYA0205300).Notes and references
- J. F. Manwell and J. G. McGowan, Sol. Energy, 1993, 50, 399–405 CrossRef CAS.
- R. Sebastián and R. P. Alzola, Renewable Energy, 2010, 35, 952–965 CrossRef.
- A. Hammouche, E. Karden and R. W. De Doncker, J. Power Sources, 2004, 127, 105–111 CrossRef CAS.
- F. Feng, M. Geng and D. O. Northwood, Int. J. Hydrogen Energy, 2001, 26, 725–734 CrossRef CAS.
- J. Van Mierlo, P. Van den Bossche and G. Maggetto, J. Power Sources, 2004, 128, 76–89 CrossRef CAS.
- J. Speirs, M. Contestabile, Y. Houari and R. Gross, Renewable Sustainable Energy Rev., 2014, 35, 183–193 CrossRef.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 CAS.
- Y. Zhong, M. Yang, X. Zhou and Z. Zhou, Mater. Horiz., 2015, 2, 553–566 RSC.
- X. Li and C. Wang, J. Mater. Chem. A, 2013, 1, 165–182 CAS.
- M. Osiak, H. Geaney, E. Armstrong and C. O'Dwyer, J. Mater. Chem. A, 2014, 2, 9433 CAS.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- M.-H. Sun, S.-Z. Huang, L.-H. Chen, Y. Li, X.-Y. Yang, Z.-Y. Yuan and B.-L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294–1305 RSC.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- Y. Kim, W. Song, S. Y. Lee, C. Jeon, W. Jung, M. Kim and C. Y. Park, Appl. Phys. Lett., 2011, 98, 263106–2631063 CrossRef CAS PubMed.
- R. B. M. Yoshio and A. Kozawa, Lithium-ion Batteries, Springer, 2009 Search PubMed.
- J. Schoonman, Solid State Ionics, 2000, 135, 5–19 CrossRef CAS.
- D. Liu and G. Cao, Energy Environ. Sci., 2010, 3, 1218 CAS.
- J. Maier, Nat. Mater., 2005, 4, 805–815 CrossRef CAS PubMed.
- P. G. Bruce, Solid State Ionics, 2008, 179, 752–760 CrossRef CAS.
- S. J. L. Billinge and I. Levin, Science, 2007, 316, 561–565 CrossRef CAS PubMed.
- V. Petkov, B. Prasai, Y. Ren, S. Shan, J. Luo, P. Joseph and C. J. Zhong, Nanoscale, 2014, 6, 10048–10061 RSC.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- M.-S. Balogun, Y. Luo, W. Qiu, P. Liu and Y. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- X. Chen, Y. Du, N. Q. Zhang and K. N. Sun, J. Nanomater., 2012, 2012, 1–19 Search PubMed.
- Y. Dong, S. Liu, Y. Liu, Y. Tang, T. Yang, X. Wang, Z. Wang, Z. Zhao and J. Qiu, J. Mater. Chem. A, 2016, 4, 17718–17725 CAS.
- J. K. Lee, C. Oh, N. Kim, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 5366–5384 CAS.
- Z. Xiong, Y. Yun and H.-J. Jin, Materials, 2013, 6, 1138–1158 CrossRef CAS PubMed.
- R. E. W. Bradley and A. Johnson, J. Power Sources, 1998, 70, 48–54 CrossRef.
- J. Zhao, A. Buldum, J. Han and a. J. P. Lu, Phys. Rev. Lett., 1999, 1706–1709 Search PubMed.
- M. Senami, Y. Ikeda, A. Fukushima and A. Tachibana, AIP Adv., 2011, 1, 042106 CrossRef.
- M. Zhao, Y. Xia, X. Liu, Z. Tan, B. Huang, F. Li, Y. Ji and C. Song, Phys. Lett. A, 2005, 340, 434–439 CrossRef CAS.
- N. Pierard, A. Fonseca, Z. Konya, I. Willems, G. Van Tendeloo and J. B. Nagy, Chem. Phys. Lett., 2001, 335, 1–8 CrossRef CAS.
- K. Nishidate and M. Hasegawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 245418 CrossRef.
- B. J. Landi, M. J. Ganter, C. M. Schauerman, C. D. Cress and A. R. P. Raffaelle, J. Phys. Chem. C, 2008, 7509–7515 CAS.
- C. S. Du and N. Pan, J. Power Sources, 2006, 160, 1487–1494 CrossRef CAS.
- J. Li and I. Zhitomirsky, J. Mater. Process. Technol., 2009, 209, 3452–3459 CrossRef CAS.
- I.-S. Hwang, J.-C. Kim, S.-D. Seo, S. Lee, J.-H. Lee and D.-W. Kim, Chem. Commun., 2012, 48, 7061–7063 RSC.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B.-S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS PubMed.
- S. W. Lee, B.-S. Kim, S. Chen, Y. Shao-Horn and P. T. Hammond, J. Am. Chem. Soc., 2009, 131, 671–679 CrossRef CAS PubMed.
- M. Zhang, Y. Yan, K. Gong, L. Mao, Z. Guo and Y. Chen, Langmuir, 2004, 20, 8781–8785 CrossRef CAS PubMed.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638 CAS.
- S. H. Ng, J. Wang, Z. P. Guo, J. Chen, G. X. Wang and H. K. Liu, Electrochim. Acta, 2005, 51, 23–28 CrossRef CAS.
- S. Y. Chew, S. H. Ng, J. Wang, P. Novák, F. Krumeich, S. L. Chou, J. Chen and H. K. Liu, Carbon, 2009, 47, 2976–2983 CrossRef CAS.
- H. Zhang, G. P. Cao and Y. S. Yang, Nanotechnology, 2007, 18, 195607 CrossRef.
- K. Hata, D. N. Futaba, K. Mizuno, T. Namai, M. Yumura and S. Iijima, Science, 2004, 306, 1362–1364 CrossRef CAS PubMed.
- A. R. Harutyunyan, G. Chen, T. M. Paronyan, E. M. Pigos, O. A. Kuznetsov, K. Hewaparakrama, S. M. Kim, D. Zakharov, E. A. Stach and G. U. Sumanasekera, Science, 2009, 326, 116–120 CrossRef CAS PubMed.
- S. Talapatra., S. Kar., S. K. Pal, R. Vajtai., L. Ci., P. Victor., M. M. Shaijumon, S. Kaur., O. Nalamasu. and P. M. Ajayan, Nat. Nanotechnol., 2006, 1, 112–116 CrossRef CAS PubMed.
- T. Hiraoka, T. Yamada, K. Hata, D. N. Futaba, H. Kurachi, S. Uemura, M. Yumura and S. Iijima, J. Am. Chem. Soc., 2006, 128, 13338–13339 CrossRef CAS PubMed.
- G.-Y. Xiong, D. Z. Wang and Z. F. Ren, Carbon, 2006, 44, 969–973 CrossRef CAS.
- Z. J. Han, S. Yick, I. Levchenko, E. Tam, M. M. A. Yajadda, S. Kumar, P. J. Martin, S. Furman and K. Ostrikov, Nanoscale, 2011, 3, 3214–3220 RSC.
- D. Takagi, Y. Kobayashi, H. Hibino, S. Suzuki and Y. Homma, Nano Lett., 2008, 8, 832–835 CrossRef CAS PubMed.
- L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- P. Avouris and C. Dimitrakopoulos, Mater. Today, 2012, 15, 86–97 CrossRef CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- S. Bhaviripudi, X. Jia, M. S. Dresselhaus and J. Kong, Nano Lett., 2010, 10, 4128–4133 CrossRef CAS PubMed.
- Z.-Y. Juang, C.-Y. Wu, A.-Y. Lu, C.-Y. Su, K.-C. Leou, F.-R. Chen and C.-H. Tsai, Carbon, 2010, 48, 3169–3174 CrossRef CAS.
- V. Y. Aristov, G. Urbanik, K. Kummer, D. V. Vyalikh, O. V. Molodtsova, A. B. Preobrajenski, A. A. Zakharov, C. Hess, T. Hanke, B. Buchner, I. Vobornik, J. Fujii, G. Panaccione, Y. A. Ossipyan and M. Knupfer, Nano Lett., 2010, 10, 992–995 CrossRef CAS PubMed.
- L. G. D. Arco, Y. Zhang, A. Kumar and C. Zhou, IEEE Trans. Nanotechnol., 2009, 8, 135–138 CrossRef.
- Q. Yu, J. Lian, S. Siriponglert, H. Li, Y. P. Chen and S.-S. Pei, Appl. Phys. Lett., 2008, 93, 113103 CrossRef.
- S. Garaj, W. Hubbard and J. A. Golovchenko, Appl. Phys. Lett., 2010, 97, 183103 CrossRef PubMed.
- H. H. Angermann and G. Hörz, Appl. Surf. Sci., 1993, 70, 163–168 CrossRef.
- W. Ai, Z. Luo, J. Jiang, J. Zhu, Z. Du, Z. Fan, L. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186–6192 CrossRef CAS PubMed.
- J. Xu, I.-Y. Jeon, J.-M. Seo, S. Dou, L. Dai and J.-B. Baek, Adv. Mater., 2014, 26, 7317–7323 CrossRef CAS PubMed.
- K. H. Park, D. Lee, J. Kim, J. Song, Y. M. Lee, H.-T. Kim and J.-K. Park, Nano Lett., 2014, 14, 4306–4313 CrossRef CAS PubMed.
- X. Cao, B. Zheng, X. Rui, W. Shi, Q. Yan and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1404–1409 CrossRef CAS PubMed.
- R. Wang, C. Xu, M. Du, J. Sun, L. Gao, P. Zhang, H. Yao and C. Lin, Small, 2014, 10, 2260–2269 CrossRef CAS PubMed.
- S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794–830 RSC.
- H.-s. Qian, F.-m. Han, B. Zhang, Y.-c. Guo, J. Yue and B.-x. Peng, Carbon, 2004, 42, 761–766 CrossRef CAS.
- Y. Z. Jin, C. Gao, W. K. Hsu, Y. Zhu, A. Huczko, M. Bystrzejewski, M. Roe, C. Y. Lee, S. Acquah, H. Kroto and D. R. M. Walton, Carbon, 2005, 43, 1944–1953 CrossRef CAS.
- A. Nieto-Marquez, R. Romero, A. Romero and J. L. Valverde, J. Mater. Chem., 2011, 21, 1664–1672 RSC.
- Y. Wang, F. Su, C. D. Wood, J. Y. Lee and X. S. Zhao, Ind. Eng. Chem. Res., 2008, 47, 2294–2300 CrossRef CAS.
- F. Su, X. S. Zhao, Y. Wang, L. Wang and J. Y. Lee, J. Mater. Chem., 2006, 16, 4413–4419 RSC.
- X. Chen, K. Kierzek, Z. Jiang, H. Chen, T. Tang, M. Wojtoniszak, R. J. Kalenczuk, P. K. Chu and E. Borowiak-Palen, J. Phys. Chem. C, 2011, 115, 17717–17724 CAS.
- Q. Wang, H. Li, L. Chen and X. Huang, Carbon, 2001, 39, 2211–2214 CrossRef CAS.
- Q. Wang, H. Li, L. Chen and X. Huang, Solid State Ionics, 2002, 152–153, 43–50 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- S. Menkin, Z. Barkay, D. Golodnitsky and E. Peled, J. Power Sources, 2014, 245, 345–351 CrossRef CAS.
- Y. Hu and X. Sun, J. Mater. Chem. A, 2014, 2, 10712 CAS.
- L. Su, Y. Jing and Z. Zhou, Nanoscale, 2011, 3, 3967–3983 RSC.
- L. Y. Beaulieu, K. W. Eberman, R. L. Turner, L. J. Krause and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A137 CrossRef CAS.
- H. Li, L. Shi, W. Lu, X. Huang and L. Chen, J. Electrochem. Soc., 2001, 148, A915 CrossRef CAS.
- P. Nithyadharseni, M. V. Reddy, B. Nalini, M. Kalpana and B. V. R. Chowdari, Electrochim. Acta, 2015, 161, 261–268 CrossRef CAS.
- H. Li, L. Shi, Q. Wang, L. Chen and X. Huang, Solid State Ionics, 2002, 148, 247–258 CrossRef CAS.
- J.-T. Li, J. Swiatowska, A. Seyeux, L. Huang, V. Maurice, S.-G. Sun and P. Marcus, J. Power Sources, 2010, 195, 8251–8257 CrossRef CAS.
- M. R. Wagner, P. R. Raimann, A. Trifonova, K. C. Moeller, J. O. Besenhard and M. Winter, Electrochem. Solid-State Lett., 2004, 7, A201 CrossRef CAS.
- M. Noh, Y. Kwon, H. Lee, J. Cho, Y. Kim and M. G. Kim, Chem. Mater., 2005, 1926–1929 CrossRef CAS.
- K. T. Lee, Y. S. Jung and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652–5653 CrossRef CAS PubMed.
- W.-M. Zhang, J.-S. Hu, Y.-G. Guo, S.-F. Zheng, L.-S. Zhong, W.-G. Song and L.-J. Wan, Adv. Mater., 2008, 20, 1160–1165 CrossRef CAS.
- G. K. Simon and T. Goswami, Metall. Mater. Trans. A, 2011, 42, 231–238 CrossRef CAS.
- K. D. Kepler, J. T. Vaughey and M. M. Thackeray, J. Power Sources, 1999, 383–387 CrossRef CAS.
- A. M. Wilson and J. R. Dahn, J. Electrochem. Soc., 1995, 142, 326–332 CrossRef CAS.
- Y. P. Wu, E. Rahm and R. Holze, J. Power Sources, 2003, 114, 228–236 CrossRef CAS.
- N. Tamura, R. Ohshita, M. Fujimoto, M. Kamino and S. Fujitani, J. Electrochem. Soc., 2003, 150, A679 CrossRef CAS.
- N. Tamura, M. Fujimoto, M. Kamino and S. Fujitani, Electrochim. Acta, 2004, 49, 1949–1956 CrossRef CAS.
- L. Fang and B. V. R. Chowdari, J. Power Sources, 2001, 97–98, 181–184 CrossRef CAS.
- M. A. Reddy and U. V. Varadaraju, J. Power Sources, 2006, 159, 336–339 CrossRef CAS.
- J. Yin, M. Wada, S. Yoshida, K. Ishihara, S. Tanase and T. Sakai, J. Electrochem. Soc., 2003, 150, A1129 CrossRef CAS.
- O. Mao and J. R. Dahn, J. Electrochem. Soc., 1999, 423–427 CrossRef CAS.
- B.-L. He, B. Dong and H.-L. Li, Electrochem. Commun., 2007, 9, 425–430 CrossRef CAS.
- H. Guo, H. Zhao and X. Jia, Electrochem. Commun., 2007, 9, 2207–2211 CrossRef CAS.
- R. P. Liu, W. M. Su, P. He, C. Shen, C. Zhang, F. B. Su and C. A. Wang, J. Alloys Compd., 2016, 688, 908–913 CrossRef CAS.
- Q. Tian, Z. Zhang, L. Yang and S.-i. Hirano, J. Mater. Chem. A, 2014, 2, 12881–12887 CAS.
- A. Trifonova, M. Wachtler, M. Winter and J. O. Besenhard, Ionics, 2002, 8, 321–328 CrossRef CAS.
- J. Xie, X. B. Zhao, G. S. Cao, Y. D. Zhong, M. J. Zhao and J. P. Tu, Electrochim. Acta, 2005, 50, 1903–1907 CrossRef CAS.
- J. Yang, M. Wachtler, M. Winter and J. O. Besenhard, Electrochem. Solid-State Lett., 1999, 2, 161–163 CrossRef CAS.
- W. Guo, F. Li, X. C. Duan and J. M. Ma, J. Alloys Compd., 2016, 685, 720–723 CrossRef CAS.
- S. Chen, P. Chen, M. Wu, D. Pan and Y. Wang, Electrochem. Commun., 2010, 12, 1302–1306 CrossRef CAS.
- K. Nishikawa, K. Dokko, K. Kinoshita, S.-W. Woo and K. Kanamura, J. Power Sources, 2009, 189, 726–729 CrossRef CAS.
- D. G. Kim, H. Kim, H. J. Sohn and T. Kang, J. Power Sources, 2002, 104, 221–225 CrossRef CAS.
- L. Xue, Z. Fu, Y. Yao, T. Huang and A. Yu, Electrochim. Acta, 2010, 55, 7310–7314 CrossRef CAS.
- X.-Y. Fan, F.-S. Ke, G.-Z. Wei, L. Huang and S.-G. Sun, J. Alloys Compd., 2009, 476, 70–73 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- Y.-M. Kang, M.-S. Song, J.-H. Kim, H.-S. Kim, M.-S. Park, J.-Y. Lee, H. K. Liu and S. X. Dou, Electrochim. Acta, 2005, 50, 3667–3673 CrossRef CAS.
- M. Xu, F. Wang, Y. Zhang, S. Yang, M. Zhao and X. Song, Nanoscale, 2013, 5, 8067–8072 RSC.
- W. Kang, Y. Tang, W. Li, Z. Li, X. Yang, J. Xu and C. S. Lee, Nanoscale, 2014, 6, 6551–6556 RSC.
- Y. F. Wang and L. J. Zhang, J. Power Sources, 2012, 209, 20–29 CrossRef CAS.
- Z. Sun, E. Madej, C. Wiktor, I. Sinev, R. A. Fischer, G. van Tendeloo, M. Muhler, W. Schuhmann and E. Ventosa, Chemistry, 2015, 21, 16154–16161 CrossRef CAS PubMed.
- H. S. Kim, Y. Piao, S. H. Kang, T. Hyeon and Y.-E. Sung, Electrochem. Commun., 2010, 12, 382–385 CrossRef CAS.
- H. Liu, D. Wexler and G. Wang, J. Alloys Compd., 2009, 487, L24–L27 CrossRef CAS.
- B.-C. Yu, J.-O. Lee, J. H. Song, C.-M. Park, C. K. Lee and H.-J. Sohn, J. Solid State Electrochem., 2012, 16, 2631–2638 CrossRef CAS.
- G. Zhang, J. Li, J. Sha, C. He, E. Liu, N. Zhao and C. Shi, J. Alloys Compd., 2016, 661, 448–454 CrossRef CAS.
- M. Zou, J. Li, W. Wen, L. Chen, L. Guan, H. Lai and Z. Huang, J. Power Sources, 2014, 270, 468–474 CrossRef CAS.
- G. Gao, Q. Zhang, K. Wang, H. Song, P. Qiu and D. Cui, Nano Energy, 2013, 2, 1010–1018 CrossRef CAS.
- G. Gao, Q. Zhang, X. B. Cheng, P. Qiu, R. Sun, T. Yin and D. Cui, ACS Appl. Mater. Interfaces, 2015, 7, 340–350 CAS.
- T. Hu, M. Xie, J. Zhong, H.-t. Sun, X. Sun, S. Scott, S. M. George, C.-s. Liu and J. Lian, Carbon, 2014, 76, 141–147 CrossRef CAS.
- S.-Z. Kang, L. Bo, X. Li and A. J. Mu, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 46, 647–652 CrossRef.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- W. L. Wang, B.-Y. Oh, J.-Y. Park, H. Ki, J. Jang, G.-Y. Lee, H.-B. Gu and M.-H. Ham, J. Power Sources, 2015, 300, 272–278 CrossRef CAS.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- S. Wang, Y. Ren, G. Liu, Y. Xing and S. Zhang, Nanoscale, 2014, 6, 3508–3512 RSC.
- L. C. Yang, Q. S. Gao, Y. Tang, Y. P. Wu and R. Holze, J. Power Sources, 2008, 179, 357–360 CrossRef CAS.
- M. F. Hassan, Z. P. Guo, Z. Chen and H. K. Liu, J. Power Sources, 2010, 195, 2372–2376 CrossRef CAS.
- C. Wang, D. Wang, Q. Wang and H. Chen, J. Power Sources, 2010, 195, 7432–7437 CrossRef CAS.
- B. Wang, J. L. Cheng, Y. P. Wu, D. Wang and D. N. He, Electrochem. Commun., 2012, 23, 5–8 CrossRef CAS.
- Y. Zou and Y. Wang, Nanoscale, 2011, 3, 2615–2620 RSC.
- Y. Yang, J. Liu, H.-Q. Dai, Y. Cui, J. Liu, X. Liu and Z.-W. Fu, J. Alloys Compd., 2016, 661, 190–195 CrossRef CAS.
- D. Bresser, F. Mueller, M. Fiedler, S. Krueger, R. Kloepsch, D. Baither, M. Winter, E. Paillard and S. Passerini, Chem. Mater., 2013, 25, 4977–4985 CrossRef CAS.
- R. Wang, X. Xue, W. Lu, H. Liu, C. Lai, K. Xi, Y. Che, J. Liu, S. Guo and D. Yang, Nanoscale, 2015, 7, 12833–12838 RSC.
- J. Wang, I. D. Raistrick and R. A. Huggins, J. Electrochem. Soc., 1986, 133, 457–460 CrossRef CAS.
- C. J. Wen and R. A. Huggins, J. Electrochem. Soc., 1981, 128, 1636–1641 CrossRef CAS.
- M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103–A108 CrossRef CAS.
- M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93–A96 CrossRef CAS.
- T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A838–A842 CrossRef CAS.
- J. Li and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A156 CrossRef CAS.
- P. Limthongkul, Y.-I. Jang, N. J. Dudney and Y.-M. Chiang, J. Power Sources, 2003, 119, 604–609 CrossRef.
- G. X. Wang, J. Yao and H. K. Liu, Electrochem. Solid-State Lett., 2004, 7, A250 CrossRef CAS.
- I.-s. Kim, G. E. Blomgren and P. N. Kumta, J. Power Sources, 2004, 130, 275–280 CrossRef CAS.
- K. Kierzek, J. Machnikowski and F. Béguin, J. Appl. Electrochem., 2015, 45, 1–10 CrossRef CAS.
- F. Luo, B. Liu, J. Zheng, G. Chu, K. Zhong, H. Li, X. Huang and L. Chen, J. Electrochem. Soc., 2015, 162, A2509–A2528 CrossRef CAS.
- J. S. Bridel, T. Azaïs, M. Morcrette, J. M. Tarascon and D. Larcher, Chem. Mater., 2010, 22, 1229–1241 CrossRef CAS.
- B. Zhu, Y. Jin, Y. Tan, L. Zong, Y. Hu, L. Chen, Y. Chen, Q. Zhang and J. Zhu, Nano Lett., 2015, 15, 5750–5754 CrossRef CAS PubMed.
- W. Ren, Z. Zhang, Y. Wang, Q. Tan, Z. Zhong and F. Su, J. Mater. Chem. A, 2015, 3, 5859–5865 CAS.
- W. Ren, Z. Zhang, Y. Wang, G. Kan, Q. Tan, Z. Zhong and F. Su, RSC Adv., 2015, 5, 11115–11123 RSC.
- W. Ren, Y. Wang, Z. Zhang, Q. Tan, Z. Zhong and F. Su, Appl. Surf. Sci., 2016, 360, 192–197 CrossRef CAS.
- B. Hertzberg, A. Alexeev and A. G. Yushin, J. Am. Chem. Soc., 2010, 8548–8549 CrossRef CAS PubMed.
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904–909 CrossRef CAS PubMed.
- M. W. Forney, R. A. DiLeo, A. Raisanen, M. J. Ganter, J. W. Staub, R. E. Rogers, R. D. Ridgley and B. J. Landi, J. Power Sources, 2013, 228, 270–280 CrossRef CAS.
- J. Ji, H. Ji, L. L. Zhang, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, Adv. Mater., 2013, 25, 4673–4677 CrossRef CAS PubMed.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed.
- N. Dimov, S. Kugino and M. Yoshio, Electrochim. Acta, 2003, 48, 1579–1587 CrossRef CAS.
- S.-H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z.-P. Guo and H.-K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896–6899 CrossRef CAS PubMed.
- M. Yoshio, H. Wang, K. Fukuda, T. Umeno, N. Dimov and Z. Ogumi, J. Electrochem. Soc., 2002, 149, A1598 CrossRef CAS.
- H.-Y. Lee and S.-M. Lee, Electrochem. Commun., 2004, 6, 465–469 CrossRef CAS.
- B. H. Chen, S. I. Chuang, W. R. Liu and J. G. Duh, ACS Appl. Mater. Interfaces, 2015, 7, 28166–28176 CAS.
- Y.-S. Hu, R. Demir-Cakan, M.-M. Titirici, J.-O. Müller, R. Schlögl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 1645–1649 CrossRef CAS PubMed.
- Y. He, X. Yu, Y. Wang, H. Li and X. Huang, Adv. Mater., 2011, 23, 4938–4941 CrossRef CAS PubMed.
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Chem. Commun., 2010, 46, 2025–2027 RSC.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, Adv. Energy Mater., 2011, 1, 1079–1084 CrossRef CAS.
- J. Luo, X. Zhao, J. Wu, H. D. Jang, H. H. Kung and J. Huang, J. Phys. Chem. Lett., 2012, 3, 1824–1829 CrossRef CAS PubMed.
- X. Zhou, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Adv. Energy Mater., 2012, 2, 1086–1090 CrossRef CAS.
- J. Kim, C. Oh, C. Chae, D.-H. Yeom, J. Choi, N. Kim, E.-S. Oh and J. K. Lee, J. Mater. Chem. A, 2015, 3, 18684–18695 CAS.
- X. Xin, X. Zhou, F. Wang, X. Yao, X. Xu, Y. Zhu and Z. Liu, J. Mater. Chem., 2012, 22, 7724 RSC.
- J. Cho, J. Mater. Chem., 2010, 20, 4009 RSC.
- F.-H. Du, B. Li, W. Fu, Y.-J. Xiong, K.-X. Wang and J.-S. Chen, Adv. Mater., 2014, 26, 6145–6150 CrossRef CAS PubMed.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., 2008, 120, 10305–10308 CrossRef.
- J. Guo, X. Chen and C. Wang, J. Mater. Chem., 2010, 20, 5035 RSC.
- Y. Zheng, J. Yang, J. Wang and Y. NuLi, Electrochim. Acta, 2007, 52, 5863–5867 CrossRef CAS.
- M. Ge, J. Rong, X. Fang and C. Zhou, Nano Lett., 2012, 12, 2318–2323 CrossRef CAS PubMed.
- Y. Q. Zhang, X. H. Xia, X. L. Wang, Y. J. Mai, S. J. Shi, Y. Y. Tang, C. G. Gu and J. P. Tu, J. Power Sources, 2012, 213, 106–111 CrossRef CAS.
- Y. Zhou, X. Jiang, L. Chen, J. Yue, H. Xu, J. Yang and Y. Qian, Electrochim. Acta, 2014, 127, 252–258 CrossRef CAS.
- D. J. Lee, H. Lee, M.-H. Ryou, G.-B. Han, J.-N. Lee, J. Song, J. Choi, K. Y. Cho, Y. M. Lee and J.-K. Park, ACS Appl. Mater. Interfaces, 2013, 5, 12005–12010 CAS.
- W. Chen, Z. Fan, A. Dhanabalan, C. Chen and C. Wang, J. Electrochem. Soc., 2011, 158, A1055–A1059 CrossRef CAS.
- X. Li, M. Gu, S. Hu, R. Kennard, P. Yan, X. Chen, C. Wang, M. J. Sailor, J.-G. Zhang and J. Liu, Nat. Commun., 2014, 5, 4105 CAS.
- H. Jia, P. Gao, J. Yang, J. Wang, Y. Nuli and Z. Yang, Adv. Energy Mater., 2011, 1, 1036–1039 CrossRef CAS.
- Y. Zhao, X. Liu, H. Li, T. Zhai and H. Zhou, Chem. Commun., 2012, 48, 5079–5081 RSC.
- S. Choi, T.-w. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |