
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing the relation between the structure, Li-ion conductivity and solid-state battery performance of the argyrodite Li6PS5Br solid electrolyte†
Chuang
Yu
a,
Swapna
Ganapathy
a,
Ernst R. H.
van Eck
b,
Lambert
van Eijck
a,
Shibabrata
Basak
c,
Yanyan
Liu
d,
Long
Zhang
d,
Henny W.
Zandbergen
c and
Marnix
Wagemaker
 *a
*a
aDepartment of Radiation Science and Technology, Delft University of Technology, Mekelweg 15, 2629JB Delft, The Netherlands. E-mail: M.Wagemaker@tudelft.nl
bInstitute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
cKavli Institute of Nanoscience Delft, Department of Quantum Nanoscience, Delft University of Technology, Lorentzweg 1, 2628 CJ Delft, The Netherlands
dState Key Laboratory of Metastable, Materials Science and Technology, Yanshan University, Qinhuangdao, Hebei 066004, China
First published on 14th September 2017
Abstract
Based on its high Li-ion conductivity, argyrodite Li6PS5Br is a promising solid electrolyte for all-solid-state batteries. However, more understanding is required on the relation between the solid electrolyte conductivity and the solid-state battery performance with the argyrodite structure, crystallinity and particle size that depend on the synthesis conditions. In the present study, this relationship is investigated using neutron and X-ray diffraction to determine the detailed structure and impedance as well as 7Li solid state NMR spectroscopy to study the Li-ion kinetics. It is found that depending on the synthesis conditions the distribution of the Br dopant over the crystallographic sites in Li6PS5Br is inhomogeneous, and that this may be responsible for a larger mobile Li-ion fraction in the interfacial regions in the annealed argyrodite materials. Comparing the bulk and interfacial properties of the differently prepared Li6PS5Br materials, it is proposed that optimal solid-state battery performance requires a different particle size for the solid electrolyte only region and the solid electrolyte in the cathode mixture. In the electrolyte region, the grain boundary resistance is minimized by annealing the argyrodite Li6PS5Br resulting in relatively large crystallites. In the cathode mixture however, additional particle size reduction of the Li6PS5Br is required to provide abundant Li6PS5Br–Li2S interfaces that reduce the resistance of this rate limiting step in Li-ion transport. Thereby the results give insight into how to improve solid-state battery performance by controlling the solid electrolyte structure.
Introduction
Inorganic solid electrolytes for all-solid-state Li-ion batteries are attracting significant attention because of the improved safety they provide in comparison with the conventional liquid electrolyte based Li-ion batteries.1,2 Other potential advantages include improved chemical and electrochemical stability and more efficient cell packaging, allowing higher practical energy densities, using the same anode and cathode chemistries. One of the crucial parameters for solid electrolytes is their high room temperature Li-ion conductivity that for practical applications should be in the order of 10−4 S cm−1.3,4Intensive research efforts have led to the discovery of several new solid electrolytes with high Li-ion conductivities including sulphides (Li2S–P2S5, Li2S–SiS2, and Li2S–GeS2),5–7 oxides (Li7La3Zr2O12 and Li3xLa2/3−3xTiO3)8 and phosphates (LiPON, Li1+xAlxGe2−x(PO4)3, and Li1+xTi2−xAlx(PO4)3).1,2,9,10 Among them, the chemically stable oxides and phosphates display relatively low ionic conductivities and high grain boundary resistances.1 Sulphides generally offer higher conductivities11 and their relatively low annealing temperatures, compared to the oxides and phosphates, apparently lead to a relatively low grain boundary resistance,2,12,13 which makes them promising solid electrolytes for solid-state cells. An important family of sulphide based solid electrolytes are the Li-argyrodites Li6PS5X (X = Cl, Br and I) providing Li-ion conductivities in the range of 10−2 to 10−3 S cm−1 at room temperature.7 The high conductivity and the low costs of starting materials make the Li-argyrodites attractive for application in all-solid-state batteries. Previous work has demonstrated that the Li6PS5Cl argyrodite, with high Li-ion conductivity, can be synthesized by direct ball-milling followed by annealing, resulting in a good solid state Li–S battery performance with both S and Li2S cathodes.14–17 Although for argyrodite solid electrolytes large electrochemical stability windows have been reported,18,19 calculations indicate a much smaller window for sulphides20 with an upper limit just exceeding the potential of Li2S, motivating the use of Li2S as a cathode material in combination with sulphide electrolytes.
To improve the all-solid-state battery performance, it is essential to develop an understanding of the influence of synthesis parameters and cycling conditions on the electrochemical performance of solid electrolytes. For argyrodite solid electrolytes, the annealing temperature appears to be very important for the Li-ion conductivity and electrochemical performance, as illustrated by the role of the halogen dopant position in the lattice,14 recently studied by DFT.21 The energy barriers for short range jumps and long range jumps were investigated by MD simulations and valence calculations indicating the detailed influence of the halogen on the diffusion mechanism.15,21 However, little is known about how annealing and ball milling strategies influence the bulk mobility as well as the macroscopic Li-ion conductivity, including across grain boundaries, and how this affects the solid-state battery performance. Additionally, little is known about the impact of the electrochemical (dis)charge window on the cycle life, which is in particular relevant in the context of the predicted limited electrochemical stability window for these sulfide electrolytes.
In the present work, a detailed study of the effect of annealing on the Li-ion dynamics in the argyrodite Li6PS5Br solid electrolyte has been performed. By employing both impedance spectroscopy and spin–lattice relaxation (SLR) NMR measurements, differences in the local Li-ion mobility and bulk diffusion as well as the impact of grain boundaries are revealed between differently prepared Li6PS5Br solid electrolyte materials. Combined Rietveld refinement of neutron and X-ray powder diffraction data were performed to probe structural differences between the prepared Li6PS5Br materials. Transmission electron microscopy (TEM) measurements were performed to investigate the crystallinity and the distribution of the Br dopant. Finally, cyclic voltammetry (CV) was performed to determine the optimum (dis)charge voltage window for the nano-Li2S/Li6PS5Br/In solid-state cells. By employing the optimized electrochemical window for (dis)charging, a significant difference in the performance is observed for the differently prepared solid electrolytes, where only the solid-state cell using annealed nano-sized Li6PS5Br as the solid electrolyte maintained capacities above 500 mA h g−1 (per gram of active material) for 25 cycles.
Experimental
Reagent-grade Li2S (99.98%, Sigma-Aldrich), P2S5 (99%, Sigma-Aldrich), and LiBr (99.0%, Sigma-Aldrich) crystalline powders were used as starting materials. The required amount of the starting materials was ball milled together in a tungsten carbide (WC) coated (inner) stainless steel jar with 10 WC balls (8 g per ball) in an argon filled glovebox (H2O, O2 < 0.3 ppm) because of the reactivity of the sample with oxygen and moisture. The total weight of the mixture was almost 2.0 grams, and the ball milling speed was fixed at 500 rpm. The milling duration was varied to find the optimal milling time. After subsequent milling times, a small amount of powder was collected to perform powder XRD measurements. After the ball milling process, half of the sample was set aside as the ball-milled only Li6PS5Br (Li6PS5Br), and the remaining material was sealed in a quartz tube and annealed at 300 °C for 5 hours to obtain the annealed Li6PS5Br (AN-Li6PS5Br) solid electrolyte. The AN-Li6PS5Br was further milled with a rotation speed of 450 rpm for 4 h to obtain the AN-BM-Li6PS5Br sample.Powder XRD patterns were collected over a 2θ range of 10–80° to identify the crystalline phases of the different samples using CuKα X-rays (1.5406 Å at 45 kV and 40 mA) on an X'Pert Pro X-ray diffractometer (PANalytical). To prevent the reaction with moisture and oxygen, the powders were sealed in an airtight XRD sample holder in an argon filled glove box. Neutron diffraction data were collected on a new neutron powder diffractometer PEARL of the TU Delft.22 Data were collected at room temperature using the (533) reflection of the germanium monochromator (λ = 1.665 Å). The sample was loaded under argon in a 6 mm diameter air-tight vanadium sample can. The sample was measured for 18 hours from 10.4–160 degrees 2θ. The sample can is under vacuum during the data collection. The data treatment consisted of a relative correction for the detection efficiency of (each of) the 1408 detector pixels and a subtraction of the background, caused by the instrument and the sample can. The X-ray and neutron diffraction data were refined simultaneously using the Rietveld method implemented in GSAS.23 Besides the atomic and lattice parameters, the absorption and line-broadening parameters, the crystal phase fractions, and the background were fitted. The S and Br occupancies on both the 4a and 4c crystallographic sites were restricted by setting the sum to 1 on each site.
For the TEM investigation, a suspension of each Li6PS5Br sample in hexane was prepared which was dropcast onto a standard gold grid with a holey carbon film, inside an argon-filled glove box. To prevent any contact with air TEM grids with the sample were loaded into a custom-made vacuum transfer TEM holder. TEM measurements were carried out on a FEI-Tecnai operating at 200 kV. The morphology of the materials and the corresponding distribution of S, P, and Br were investigated by SEM and EDS.
The ionic conductivities of the Li6PS5Br electrolyte materials were determined by AC impedance. Stainless-steel disks were attached on both faces of the 10 mm diameter powder pellets.16,17 AC impedance measurements were performed using an Autolab (PGSTAT302N) in the frequency range of 0.1 Hz to 1 MHz with an applied voltage of 0.05 V.
Solid-state NMR measurements were performed on a Chemagnetics 400 Infinity spectrometer (B0 = 9.4T), operating at the 155.506 MHz 7Li resonance frequency. The π/2 pulse length was determined to be 3.1 μs with an RF field strength of 85 kHz. Chemical shifts were referenced with respect to a 0.1 M LiCl solution. The air sensitive Li6PS5Br materials were sealed in custom-made Teflon tubes in an argon filled glove box (H2O, O2 < 0.3 ppm). Variable temperature measurements were performed using a 5 mm static goniometer probe. For all three Li6PS5Br samples spectra were acquired in the temperature range of −100 °C to +180 °C. T1 relaxation times were determined at various temperatures using a saturation recovery experiment. Corresponding T1ρ measurements were performed using the spin-lock method at a lock frequency of 20 kHz. Additional single pulse experiments were performed at different temperatures to determine the evolution of the line-width as a function of temperature employing a spectral width of 250 kHz. In each case a recycle delay of 3T1 was used.
Laboratory-scale solid-state Li2S/Li6PS5Br/In batteries were prepared. Commercial Li2S was first milled with a rotation speed of 500 rpm for 4 h, and then milled with Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br separately and super P (TIMCAL) with a weight ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2 using a rotation speed of 450 rpm for 2 h to obtain the final cathode mixture. A two-layer pellet, 10 mm in diameter, consisting of 12 mg of the described cathode mixture and 88 mg Li6PS5Br, AN-Li6PS5Br, or AN-BM-Li6PS5Br solid electrolyte, was obtained by pressing the electrode and electrolyte powders by applying 6 tons of pressure. A piece of In foil was subsequently attached to the other side. Finally, the full solid-state battery pellet was pressed with 2 tons of pressure for 30 s. Cyclic voltammetry (CV) measurements of the solid-state battery with the AN-Li6PS5Br solid electrolyte were performed at different voltage windows applying a sweep rate of 0.5 mV s−1 to determine the optimum (dis)charge voltage window. The assembled cells, either with Li6PS5Br, AN-Li6PS5Br or AN-BM-Li6PS5Br, were charged and discharged applying a current density of 0.064 mA cm−2 between 0 and 3.5 V vs. In order to evaluate the electrochemical performance. The obtained capacity was normalized by the weight of Li2S in the cathode.
:4:2 using a rotation speed of 450 rpm for 2 h to obtain the final cathode mixture. A two-layer pellet, 10 mm in diameter, consisting of 12 mg of the described cathode mixture and 88 mg Li6PS5Br, AN-Li6PS5Br, or AN-BM-Li6PS5Br solid electrolyte, was obtained by pressing the electrode and electrolyte powders by applying 6 tons of pressure. A piece of In foil was subsequently attached to the other side. Finally, the full solid-state battery pellet was pressed with 2 tons of pressure for 30 s. Cyclic voltammetry (CV) measurements of the solid-state battery with the AN-Li6PS5Br solid electrolyte were performed at different voltage windows applying a sweep rate of 0.5 mV s−1 to determine the optimum (dis)charge voltage window. The assembled cells, either with Li6PS5Br, AN-Li6PS5Br or AN-BM-Li6PS5Br, were charged and discharged applying a current density of 0.064 mA cm−2 between 0 and 3.5 V vs. In order to evaluate the electrochemical performance. The obtained capacity was normalized by the weight of Li2S in the cathode.
Results and discussion
Impedance, diffraction and TEM characterization of Li6PS5Br
The XRD patterns of the Li6PS5Br samples synthesized by mechanical milling at 500 rpm for different durations are shown in Fig. 1a. As illustrated, the diffraction peaks of the sample milled at 110 rpm for 1 hour could be indexed to the starting materials Li2S, LiBr and P2S5. This step appears to be necessary in order to mix the raw materials homogeneously. After that, the milling speed was increased to 500 rpm to provide the mechanical energy necessary to obtain the Li6PS5Br phase. After 1 h of milling, the initially sharp diffraction peaks originating from the Li2S phase broadened, indicating a decrease in particle size. Moreover, a halo pattern located in the low 2θ region appeared, indicating the formation of an amorphous phase.24 As the milling duration increases, diffraction peaks at 25.2, 29.7, 31.1 and 51.9° appear which can be attributed to the Li6PS5Br phase. Compared to the pattern measured after 1 h of milling, the intensity of the Li2S diffraction peaks decreases and after a milling time of 10 h, the diffraction peak due to Li2S located at 53.3° disappears completely. | ||
| Fig. 1 (a) XRD patterns of the Li2S, P2S5 and LiBr mixtures ball-milled for different milling durations. (b) Complex impedance plots for the samples ball-milled for different durations. The dotted lines represent fits using an equivalent circuit (RbulkQbulk)(RgbQgb)Q. (c) The AC impedance results of Li6PS5Br (ball milled for 8 hours at 500 rpm) (blue), AN-Li6PS5Br (Li6PS5Br subsequently annealed at 300 °C for 5 hours) (red) and AN-BM-Li6PS5Br (AN-Li6PS5Br subsequently ball milled for 4 hours at 450 rpm) (pink). The dotted lines represent fits using an equivalent circuit (RbulkQbulk)(RgbQgb)Q. | ||
The influence of the milling time on the Li-ion conductivity is investigated by impedance spectroscopy. The resistance of the mixture decreases sharply with increasing milling duration until a constant value is obtained after 8 h of milling, as shown in Fig. 1b. The increase in Li+ conductivity during the milling process is associated with the concurrent decrease of the poorly conducting Li2S phase and the increase of the Li-ion conducting Li6PS5Br phase in the mixture. Because the material obtained after 8 hours reaches the maximum conductivity, this material is used for further investigations and referred to as Li6PS5Br. Subsequently, this material is annealed at 300 °C for 5 h and the resulting material is referred to as AN-Li6PS5Br. Finally, the AN-Li6PS5Br is ball-milled at a rotation speed of 450 rpm for 4 hours, the resulting material of which is referred to as AN-BM-Li6PS5Br. The AC impedance and fitting results of the Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br samples are shown in Fig. 1c. These result in bulk and grain boundary resistances of (924.8 ± 16.58) and (3.02 ± 0.64) × 106 Ω for Li6PS5Br, (516.8 ± 32.28) and (619.9 ± 22.26) Ω for AN-Li6PS5Br and (381.7 ± 10.81) and (686.8 ± 35.88) Ω for AN-BM-Li6PS5Br. The corresponding room temperature bulk conductivities are 6.20 × 10−4 S cm−1, 1.11 × 10−3 S cm−1 and 1.38 × 10−3 S cm−1 for Li6PS5Br AN-Li6PS5Br and AN-BM-Li6PS5Br respectively. The annealing process increases the conductivity significantly and the subsequent ball milling step appears to further increase the bulk conductivity. Annealing dramatically lowers the grain boundary resistance as observed by impedance spectroscopy, whereas the subsequent ball milling process results in a small increase of the grain boundary resistance. Perhaps the most surprising result is that the bulk conductivity of AN-BM-Li6PS5Br is larger than that of the AN-Li6PS5Br, suggesting that decreasing the particle size of AN-Li6PS5Br enhances the Li-ion bulk conductivity. However, it should be realized that for these geometrically complex systems it is difficult to distinguish the bulk conductivity from the grain boundary resistance using impedance spectroscopy.
Both powder neutron and X-ray diffraction were performed, the data of which were simultaneously refined in the cubic F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group using the Rietveld method as implemented in GSAS.23 The neutron powder patterns and the X-ray patterns of the three Li6PS5Br materials are shown in Fig. 2 and the crystallographic parameters resulting from the refinements are provided in Table S1 of the ESI.† The smaller line width of the reflections of the AN-Li6PS5Br material in Fig. 2(c and d) compared to that of Li6PS5Br in Fig. 2(a and b) indicates an increase in crystallinity and crystallite size upon annealing. From the refinement of the patterns, assuming only size broadening, the average crystallite size of the Li6PS5Br material is 15 nm and of the AN-Li6PS5Br material is 74 nm. As shown in Fig. 2(e and f), subsequent ball milling of AN-Li6PS5Br resulting in the AN-BM-Li6PS5Br material increases the width of the reflections again, leading to an average crystallite size of 22 nm from the refinement. The substantial background observed in the diffraction pattern of Li6PS5Br, especially visible at low angles in Fig. 2a, indicates the presence of a significant amorphous fraction. The much lower background in Fig. 2c indicates that, as expected based on the glass temperature of 265 °C, annealing the sample at 300 °C converts most of the amorphous fraction to the crystalline Li6PS5Br phase. For the AN-BM-Li6PS5Br material, the background increases again, suggesting that the final ball milling procedure increases the amorphous fraction again. However, the larger lattice parameters of the AN-BM-Li6PS5Br material compared to that of the Li6PS5Br material and the smaller background suggest that the crystalline fraction of the AN-BM-Li6PS5Br material is larger than that of the Li6PS5Br material, which is also confirmed by the TEM experiments discussed in the next paragraph.
3m space group using the Rietveld method as implemented in GSAS.23 The neutron powder patterns and the X-ray patterns of the three Li6PS5Br materials are shown in Fig. 2 and the crystallographic parameters resulting from the refinements are provided in Table S1 of the ESI.† The smaller line width of the reflections of the AN-Li6PS5Br material in Fig. 2(c and d) compared to that of Li6PS5Br in Fig. 2(a and b) indicates an increase in crystallinity and crystallite size upon annealing. From the refinement of the patterns, assuming only size broadening, the average crystallite size of the Li6PS5Br material is 15 nm and of the AN-Li6PS5Br material is 74 nm. As shown in Fig. 2(e and f), subsequent ball milling of AN-Li6PS5Br resulting in the AN-BM-Li6PS5Br material increases the width of the reflections again, leading to an average crystallite size of 22 nm from the refinement. The substantial background observed in the diffraction pattern of Li6PS5Br, especially visible at low angles in Fig. 2a, indicates the presence of a significant amorphous fraction. The much lower background in Fig. 2c indicates that, as expected based on the glass temperature of 265 °C, annealing the sample at 300 °C converts most of the amorphous fraction to the crystalline Li6PS5Br phase. For the AN-BM-Li6PS5Br material, the background increases again, suggesting that the final ball milling procedure increases the amorphous fraction again. However, the larger lattice parameters of the AN-BM-Li6PS5Br material compared to that of the Li6PS5Br material and the smaller background suggest that the crystalline fraction of the AN-BM-Li6PS5Br material is larger than that of the Li6PS5Br material, which is also confirmed by the TEM experiments discussed in the next paragraph.
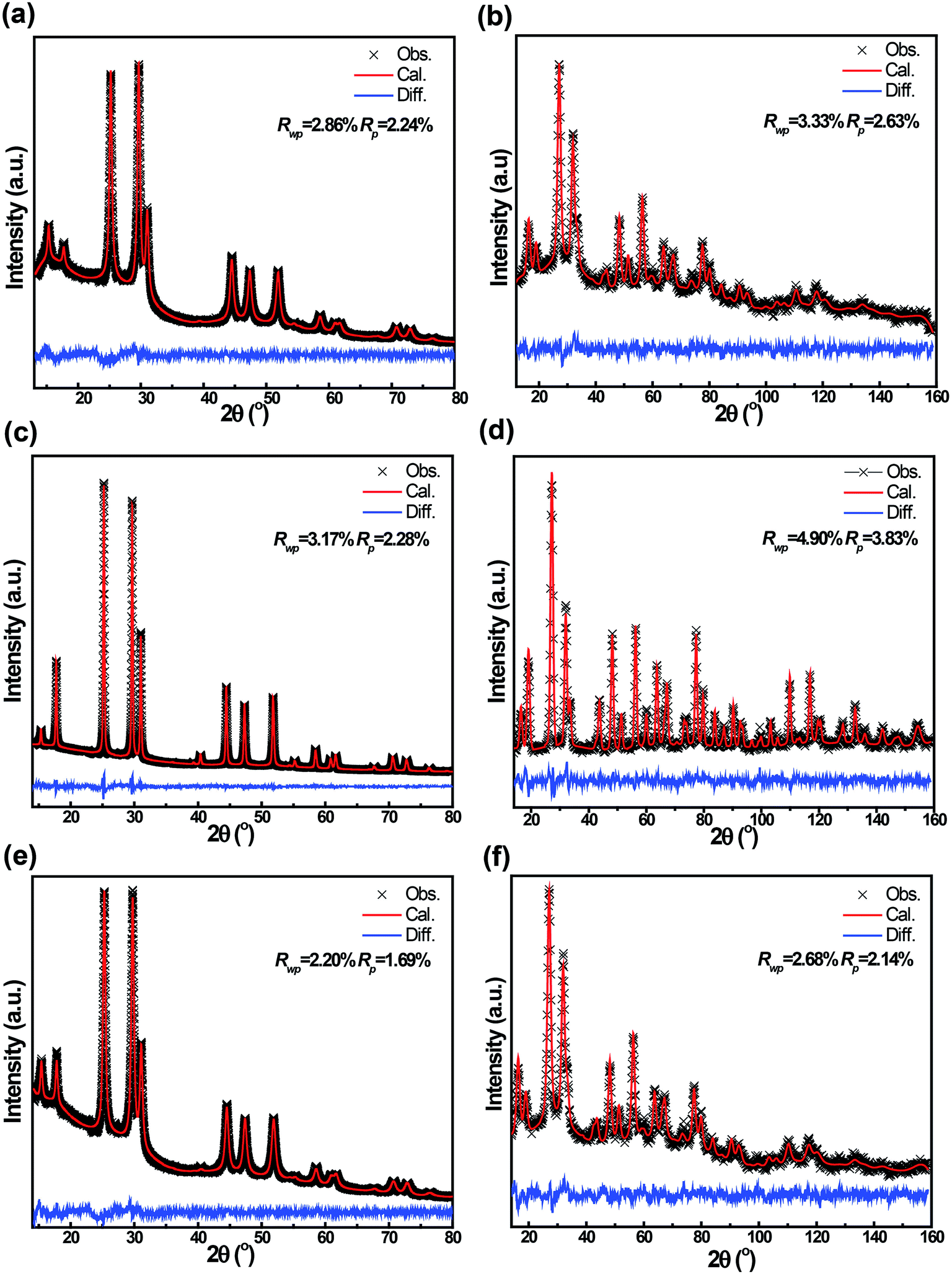 | ||
| Fig. 2 Room temperature X-ray and neutron diffraction patterns including Rietveld refinement of the (a and b) Li6PS5Br, (c and d) AN-Li6PS5Br, and (e and f) AN-BM-Li6PS5Br, respectively. | ||
The simultaneous refinement of the X-ray and neutron diffraction data allows us to determine the average amount of Br and S on the 4a (0,0,0) and 4c (0.25,0.25,0.25) crystallographic sites. Similar to what was found for Li6PS5Cl argyrodite,14 there is a slight preference for Br to occupy the 4c site as shown in Table S1 of the ESI.† Interestingly, upon annealing, the Br occupancy on the 4c site increases significantly, while decreasing on the 4a site, keeping the total Br content close to the intended Li6PS5Br composition. Note that during the fitting the total occupancy of the 4a site and of the 4c site by Br and S is restricted to unity. The increase of the average Br4c occupancy in the AN-Li6PS5Br sample indicates that the Br4c occupancy in the amorphous phase of the Li6PS5Br sample, which turns crystalline upon annealing, is larger compared to the crystalline fraction of the Li6PS5Br sample. Vice versa the Br4a occupancy decreases upon annealing, keeping the total Br occupancy, and hence the Br concentration approximately constant and close to the intended composition as shown in Table S1.†
Subsequent ball milling, going from the AN-Li6PS5Br to the AN-BM-Li6PS5Br sample, decreases the Br4c occupancy (and increases the Br4a occupancy) again to a value still larger (smaller for Br4a) compared to that of the Li6PS5Br sample. Assuming that upon annealing the amorphous material crystallizes around the crystalline core of the Li6PS5Br sample, it is found that the 4c (4a) occupancy in the center of the particles is larger (smaller for Br4a) compared to that at the surface of the particles. Hence these results indicate that, although the Br appears to be homogeneously distributed through the particles, the relative 4a/4c site occupancy is inhomogeneous throughout the particles.
Recent molecular dynamics simulations have that the distribution of the halogen dopant over the 4a and 4c sites strongly influences the Li-ion conductivity.21 Therefore, the suggested inhomogeneous site distribution of the Br dopant in the present argyrodite materials is likely to result in an inhomogeneous Li-ion conductivity, control over which may be an important parameter for the design of optimal solid electrolytes.
Fig. 3a and c show the bright-field TEM (BF-TEM) images of the Li6PS5Br and AN-BM-Li6PS5Br samples respectively and Fig. 3b and d show the electron diffraction patterns of Li6PS5Br and AN-BM-Li6PS5Br samples, acquired from the particles shown in 3a and 3c respectively. The presence of a ring-like diffraction pattern for the Li6PS5Br sample, without distinct diffraction spots, indicates the presence of a significant fraction of the amorphous material. After subsequent annealing and ball milling, the diffraction spots for AN-BM-Li6PS5Br become more distinct, indicating that the material becomes more crystalline, as shown in Fig. 3d. The Scanning Tunnel Microscope-Energy Dispersive X-ray (STEM-EDX) mapping of the Li6PS5Br sample (Fig. 3f) shows a homogeneous distribution of S, P and Br, indicating that this mechanical milling route is an effective synthesis method to prepare Li6PS5Br. Since the AN-Li6PS5Br and AN-BM-Li6PS5Br are prepared starting from the Li6PS5Br sample with subsequent annealing and ball-milling processes, the S, P and Br in those samples are also expected to remain homogeneously distributed. SEM images displayed in Fig. S2 of the ESI,† shows an interconnected platelet like structure for the Li6PS5Br and AN-Li6PS5Br materials, whereas the final ball milling step destroys this morphology resulting in micron sized isotropic particles. EDS in Fig. S3† shows that S, P and Br are homogeneously distributed for the three materials indicating that the first ball milling preparation step already results in a homogeneous mixture.
 | ||
| Fig. 3 High-magnification bright-field TEM images and the corresponding electron diffraction patterns of Li6PS5Br (a and b) and AN-BM-Li6PS5Br (c and d). EDX mapping for the Li6PS5Br sample (e and f). The yellow, green and red dots represent the distribution of S, Br, and P. | ||
Solid-state NMR relaxation experiments
Measurement of the 7Li spin–spin (T2) and spin–lattice (T1) relaxation times as a function of temperature provides insight into the mobility of Li-ions.25–27 The decreasing FWHM of the static 7Li NMR resonances with increasing temperature observed in Fig. 4(a–c) for the three Li6PS5Br samples indicates increased Li-ion mobility, an effect that is referred to as motional narrowing. At lower temperatures i.e. 173 K the 7Li–7Li dipolar interactions are responsible for the broad resonances observed, which are averaged out when the temperature is increased because the Li-ion hopping frequency exceeds the dipolar interaction strength.28 The low temperature static spectrum can be consistently fitted with a Gaussian line shape. The 7Li resonance of the AN-Li6PS5Br shows a much narrower line width than that of Li6PS5Br, at 173 K, suggesting that a larger fraction of mobile Li-ions is present in the AN-Li6PS5Br material. The AN-BM-Li6PS5Br material has a larger line width than AN-Li6PS5Br and narrower line width than Li6PS5Br. A contributing factor towards the broader lines observed for both the ball-milled samples may also be the decreased crystallinity of both the Li6PS5Br and AN-BM-Li6PS5Br samples. This potentially results in a distribution of chemical shifts, induced by the high energy ball-milling. Interestingly, comparing AN-Li6PS5Br to previously studied Li6PS5Cl,17 the Li6PS5Br 7Li NMR resonance is shifted up-field to approximately −3 ppm. This is most likely caused by the increased shielding of the Li-ions by the neighboring Br dopants. The motional narrowing curves of Li6PS5Br, AN-Li6PS5Br, and AN-BM-Li6PS5Br measured versus temperature (see Fig. 4d) are in good agreement with that published by Epp and co-workers for the Li6PS5Br material.29 Epp et al. measured NMR spectra at 13 K and obtained an onset temperature for the Li-ion mobility of 87 K. Because of the temperature limitations of the instrument utilized for the present investigation, the measurements do not reach the so-called rigid-lattice regime where Li-ion hopping is frozen. Above room temperature, the line shape can be deconvoluted into a narrow Lorentzian and a broad Gaussian which are assigned to mobile and static Li-ion populations respectively (see ESI Fig. S1†).30 This allows us to evaluate the fraction of mobile Li-ions (Af) in Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br as a function of temperature, as shown in the inset of Fig. 4d. At 183 K, an Af value of 25% was determined for Li6PS5Br, while that for AN-Li6PS5Br and AN-BM-Li6PS5Br is approximately 50%, indicating that annealing increases the amount of mobile Li-ions significantly. | ||
| Fig. 4 7Li NMR motional narrowing spectra of (a) Li6PS5Br, (b) AN-Li6PS5Br and (c) AN-BM-Li6PS5Br. (d) Evolution of the FWHM of the static 7Li NMR resonance with temperature for Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br. The inset shows the fraction of the mobile Li-ions in the three materials versus temperature. | ||
To quantitatively determine the local Li-ion temperature-dependent mobility, 7Li static spin–lattice relaxation (SLR) rates in the laboratory frame, 1/T1, and in the rotating frame 1/T1ρ are measured, the results of which are shown in Fig. 5. Provided that the variation of the SLR is only induced by Li-ion mobility, the temperature dependence of the SLR rate in the laboratory frame, 1/T1, can be used to quantify the Li-ion jump frequency and the corresponding activation energy.27,31–33, The longer range ionic motion taking place at a larger time scale can be probed by SLR NMR measurements in the rotating frame.27,31–33 The hopping frequency 1/τ, where τ is the residence time, is assumed to be equal to the correlation time between hops. When 1/T1(ρ) reaches a maximum as a function of temperature, the hopping frequency 1/τ is in the order of the Larmor frequency (ωo) or spin lock frequency (ω1).27,31–33 In the laboratory frame, the longitudinal relaxation rate 1/T1 reaches a maximum near ωo2τc2 ≈ 1, a consequence of the effective energy transfer between the system with the diffusing atoms. The maximum relaxation rate 1/T1 is reached at 383 for Li6PS5Br and 358 K for both AN-Li6PS5Br and AN-BM-Li6PS5Br. With a Larmor frequency of ωo/2π = 155.506 MHz, the Li-ion jump frequency 1/τc for the Li6PS5Br (at 383 K) and for AN-Li6PS5Br and AN-BM-Li6PS5Br (at 358 K) can be determined to be approximately 9.8 108 s−1, similar to what was reported previously for Li6PS5Br.29 Because this jump rate is achieved at a lower temperature for both AN-Li6PS5Br and AN-BM-Li6PS5Br, this indicates that these materials have a higher local Li-ion mobility compared to the Li6PS5Br material. Taking into account the diffraction results, the origin of the higher Li-ion mobility may be from the larger crystallinity and/or the Br distribution over the 4a and 4c sites. The SLR rates, 1/T1 and 1/T1ρ, are directly related to the spectral density functions of the Li-ion jumping processes.34 Assuming a 3D diffusion process, in the high temperature limit, τωo ≪ 1, the SLR rates are proportional to the Li-ion residence time τ and in the low temperature limit, τωo ≫ 1, the SLR rates are proportional to τ−1ωo(ρ)−β (with 1 < β ≤ 2). Assuming an Arrhenius behavior for the Li-ion residence time, τ = τoexp(−Ea/(kBT)), the slope of the high and low temperature regimes of the SLR rates in Fig. 5 can be related to the activation energy for the Li-ion motional processes. The original BPP behaviour28 predicts symmetric peaks, β = 2, in the SLR rate as a function of temperature which does not take into account correlation effects and disorder in diffusion.34 In particular the 1/T1 rate appears to be asymmetric, most likely the result of Li-ion diffusional processes taking place at different length scales, illustrated by the different transitions predicted by MD simulations.21 In both the low-temperature and high-temperature flanks, the jump rates exhibit Arrhenius behavior, yielding activation energies of 0.10(9) and 0.15(0) eV for Li6PS5Br, 0.10(5) and 0.14(0) eV for AN-Li6PS5Br, and 0.10(0) and 0.15(3) eV for AN-BM-Li6PS5Br, respectively. The asymmetry, quantified by β = 1.73, 1.78 and 1.67 for Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br, is slightly higher than the previous results reported for annealed Li6PS5Br probed by temperature dependent SLR NMR (1/T1 and 1/T1ρ).29 The low temperature flank represents a short range motional process whereas the high temperature flank most likely represents longer range Li-ion resulting in actual diffusion.34 The activation energies for all three Li6PS5Br in the high temperature flank are close to that deduced by temperature-dependent ionic conductivity measurements reported by Rayavarapu et al.15i.e. 0.17 eV from 300 to 600 K, and are slightly smaller in comparison with the 0.2 eV reported by Epp et al.29 The activation energy for three Li6PS5Br samples in the low temperature flank, representing local motional processes, is approximately 0.1 eV, which is comparable to the values for the 48h–48 h and intra-cage local Li-ion motional processes predicted by MD simulations for Li6PS5Br.21 This suggests that the 1/T1 rates in the low temperature flank reflect local Li-ion movement inside Li6PS5Br similar to what was found for Li6PS5Cl.17
 | ||
| Fig. 5 Temperature dependence of the 7Li spin–lattice relaxation NMR rates T1(ρ)−1 measured in both the laboratory and the rotating frame of reference for the Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br materials. The Larmor frequency is 155.506 MHz (T1−1) and the spin lock frequency is 20 kHz (T1ρ−1). | ||
The 7Li SLR measurements in the rotating frame (1/T1ρ), also shown in Fig. 5, were performed to probe Li-ion mobility on a larger length and time scale in all three Li6PS5Br materials to complement the local hopping probed by 1/T1 relaxation. In the rotating frame at a lock frequency ωlock, the maximum condition for the lithium jump rate is ωlockτ ≈ 0.5.35 The corresponding jump rates in this rotating frame (lock frequency 20 kHz) are the same, 2.512 × 105 s−1 at 228 K for the three Li6PS5Br materials. This indicates that the time scale of Li-ion diffusion on a longer length scale (several hops) is comparable for all three materials. However, the activation energies for the high temperature flank of the 1/T1ρ rates, observed in Fig. 5, are considerably higher at 0.33–0.34 eV, in comparison with the 0.14–0.15 eV of the high temperature flank of the 1/T1 relaxation rates. The larger diffusion time probed by the measurement in the rotating frame suggests that not only the local bulk kinetics are probed, but also Li-ion diffusion through grain boundaries is observed. The slightly smaller activation energies of the AN-Li6PS5Br and AN-BM-Li6PS5Br materials deduced from the 1/T1ρ rates in the high temperature flank suggest that the annealing process promotes Li-motion in Li6PS5Br over longer length scales. This could suggest better mobility over grain boundaries, consistent with the lower grain boundary resistances observed for these materials with room temperature impedance spectroscopy, Fig. 1c. In summary, the 7Li NMR relaxation experiments indicate slightly improved Li-ion mobility for the annealed materials both for local Li-ion hopping, and longer range diffusion, although the activation energies are very similar. The much higher activation energy for the long range diffusion, as observed from the spin-locked SRL experiments, suggests that these experiments probe the Li-ion diffusion over grain boundaries.
Electrochemical performance
When combining a cathode material with a solid electrolyte, it is essential to investigate the chemical and electrochemical stability. Nano-Li2S was selected as the cathode material for this study and the stability and conductivity of the cathode mixture with the AN-Li6PS5Br solid electrolyte were investigated by cyclic voltammetry (CV), powder XRD and AC impedance spectroscopy, the results of which are shown in ESI Fig. S4 and S5.† In the diffraction patterns of the Li2S and AN-Li6PS5Br mixture before and after milling and annealing, no additional diffraction peaks are detected, suggesting that nano-Li2S is stable with the annealed Li6PS5Br solid electrolyte up to 180 °C. Another important factor to consider is the overall conductivity of the cathode–electrolyte mixture,36,37 which for the present mixture resulted in a resistance of 750 Ω yielding a Li+ conductivity of ∼10−4 S cm−1, indicating that this nano-Li2S–Li6PS5Br cathode mixture is a viable solid-state electrode. Initially, it has been suggested that sulfide solid electrolytes, including the presently investigated argyrodite Li6PS5Br, have a very wide electrochemical stability window. However, recent calculations38,39 and experimental observations indicate otherwise.40–42 The performance of the all-solid-state batteries will depend on the formation of oxidation and/or reduction products at the interfaces between the electrode material and the solid electrolyte, and to what extent this hinders Li-ion transport. Previous results16,17 on the chloride doped argyrodite Li6PS5Cl solid electrolyte have shown that the (dis)charge voltage window has a significant influence on the electrochemical performances of these solid-state batteries. At present, the optimal (dis)charge voltage window was determined for the nano-Li2S/AN-Li6PS5Br/In solid-state batteries by systematic variation of the voltage window for cyclic voltammetry (CV) measurements, as shown in Fig. S5 of the ESI.† This indicates that, to obtain the highest apparent coulombic efficiency, the optimum charge/discharge window for this assembled nano-Li2S/AN-Li6PS5Br/In solid-state cell is between 0 and 3.5 V vs. In (from 0.6 to 4.1 V vs. Li/Li+). The finding that the higher cutoff voltage during charge (i.e. 4.0 V vs. In) results in a lower coulombic efficiency, compared to the optimal cutoff (3.5 V vs. In), is most likely the result of more severe oxidation of the solid electrolyte, in line with the limited electrochemical stability obtained by thermodynamic predictions38,39 and experimental analysis after charging of Li6PS5Cl in positive electrode mixtures.43To investigate the impact of annealing and subsequent ball milling treatment on the solid-state battery performance, cathodic mixtures of the nano-Li2S cathode were prepared with the Li6PS5Br, AN-Li6PS5Br and AN-BM-Li6PS5Br solid electrolyte materials. In Fig. 6a, the first 4 charge/discharge curves are compared at the same current density of 0.064 mA cm−2 in the earlier determined optimal voltage window of 0–3.5 V vs. In (0.62–4.12 V vs. Li+/Li). The differences in capacity retention are striking, where the solid-state cell using AN-BM-Li6PS5Br shows a significantly better electrochemical performance when compared to that for both the AN-Li6PS5Br and Li6PS5Br solid electrolyte materials. For the solid-state cell using Li6PS5Br as the electrolyte, the (dis)charge curves during the first four cycles show no obvious (dis)charge plateau and a very low discharge capacity, smaller than 50 mA h g−1. In contrast, the charge curves of the solid-state cells with the AN-Li6PS5Br and AN-BM-Li6PS5Br solid electrolytes deliver two distinct charge plateaus, located at 1.8 and 2.3 V vs. In, respectively and a discharge plateau located at approximately 1.4 V vs. In. Although the AN-Li6PS5Br solid-state cell results in a promising initial discharge capacity, it degrades to below 400 mA h g−1 within a few cycles. The best capacity retention is achieved with the AN-BM-Li6PS5Br solid electrolyte, resulting in a capacity of approximately 500 mA h g−1 retained over 25 cycles. The initial discharge capacity is comparable to that demonstrated by Li2S cathodes in combination with organic liquid electrolytes, which is reported to be 785 mA h g−1 at a higher rate i.e. 0.2C.44 The present solid-state cell provides a higher discharge capacity and better cyclability compared to most reported comparable solid-state cells employing composite Li2S electrodes in combination with argyrodite and 80Li2S–20P2S5 solid electrolytes.19,27 However, the electrochemical performance reported in this work is not as good as that for the solid-state cell in combination with a mixed-conductive Li2S nanocomposite cathode and Li6PS5Cl electrolyte45 most likely due to the better distribution of Li2S, Li6PS5Cl and carbon in the cathode mixture.
 | ||
| Fig. 6 (a) Galvanostatic voltage curves for the first 4 cycles of the assembled nano-Li2S/Li6PS5Br (Li6PS5Br or AN-Li6PS5Br or AN-BM-Li6PS5Br)/In solid-state batteries at a current density of 0.064 mA cm−2 applied between 0 and 3.5 V vs. In (0.62–4.12 V vs. Li+/Li). The solid, dashed, dotted and dashed dotted lines represent cycles 1–4 respectively. (b) Capacity retention on cycling of the solid-state batteries at a current density of 0.064 mA cm−2 applied between 0 and 3.5 V vs. In (0.62–4.12 V vs. Li+/Li). (c) The corresponding coulombic efficiency for the assembled nano-Li2S/Li6PS5Br (Li6PS5Br or AN-Li6PS5Br or AN-BM-Li6PS5Br)/In solid-state batteries. The coulombic efficiency is defined as the ratio of discharge capacity to charge capacity. | ||
During the first 5 cycles, the discharge capacity increases per cycle, which is attributed to the activation process for the Li2S cathode mixture. However, a higher charge capacity than discharge capacity is observed over many cycles. This may indicate that not only the Li2S cathode is oxidized but also the Li6PS5Br electrolyte at the interface with the Li2S may be oxidized. Calculations indicate a very narrow stability window for argyrodite Li6PS5Cl,38,39 resulting in S, P2S5 and LiCl oxidation products above 2.3 V vs. Li/Li+. For Li6PS5Br at the Li2S interface, this suggests that cycling up to 4.12 V vs. Li+/Li during charge leads to the formation of S, P2S5 and LiBr in the present cells, potentially explaining the larger charge capacity observed during cycling. It should be noted that this potentially increases the amount of S in the cell, acting as a positive electrode material that may participate in subsequent (dis)charge cycles. This demonstrates a compromise made in using a large cut-off potential during charge. The large cut-off potential is necessary to overcome the large internal resistance, most likely dominated by the Li6PS5Br–Li2S interface, and utilize the Li2S capacity. At the same time, this initiates the oxidation of the Li6PS5Br solid electrolyte. This suggests that the poor capacity retention of the AN-Li6PS5Br material, shown in Fig. 6b, is related to the larger overpotentials during charge and discharge, observed in Fig. 6a, causing more severe electrolyte decomposition. As the bulk and grain boundary conductivities of the AN-Li6PS5Br and AN-BM-Li6PS5Br do not differ very much, which was concluded from impedance spectroscopy and the solid state NMR relaxation experiments, the larger overpotentials of the AN-Li6PS5Br material must be the consequence of the larger crystallite size providing a smaller interfacial area with the Li2S cathode and hence causing a larger interfacial resistance. Fig. 6c shows the corresponding coulombic efficiency of the assembled nano-Li2S/Li6PS5Br (Li6PS5Br or AN-Li6PS5Br or AN-BM-Li6PS5Br)/In solid-state batteries. Assuming that the larger charge capacity is the result of the oxidation of the Li6PS5Br solid electrolyte,43 the low coulombic efficiency of the Li6PS5Br sample, having the smallest crystallite size (see Table S1†) and largest fraction of the amorphous phase (see Fig. 2), may indicate that these factors promote oxidation. Consistent with this, the coulombic efficiency increases for AN-BN-Li6PS5Br, which has a slightly larger crystallite size and a smaller surface area (see SEM in Fig. S2†) and is less amorphous. Finally, AN-Li6PS5Br results in the largest coulombic efficiency, having the largest crystallite size and smallest amorphous fraction. For AN-Li6PS5Br, the coulombic efficiency even exceeds 100% starting from the 8th cycle, however, at the same time the capacity drops dramatically, making it hard to assess what is the origin of this effect.
In contrast, the larger interfacial area provided by the AN-BM-Li6PS5Br material results in lower overpotentials. This appears to cause the stabilization of the Li6PS5Br, as the differences of (dis)charge capacity in Fig. 6 become smaller upon cycling. Hence, despite the suggested formation of an oxidation layer at the Li6PS5Br–Li2S interface, as well as the large volumetric changes that occur upon Li2S oxidation, these solid-state cells provide a relatively constant capacity retention, at least up to 25 cycles.
Conclusions
The impedance spectra in Fig. 1c indicate a significant increase in the bulk conductivity upon annealing the Li6PS5Br material (AN-Li6PS5Br) prepared by ball milling. The NMR relaxation experiments in Fig. 4 and 5 show that the bulk Li-ion mobility is only marginally improved, and that the increase in conductivity should be mainly attributed to the larger fraction of mobile Li-ions. This increase is most likely due to the presence of the increased crystalline fraction shown by XRD and TEM in Fig. 2 and 3, which is known to display a better mobility.46 Additionally, the simultaneous refinements of the neutron and X-ray diffraction data suggest that the conductivity may be inhomogeneous due to differences in the Br dopant distribution throughout the Li6PS5Br particles. Although the Br dopant appears to be homogeneously distributed over the material, it is not homogeneously distributed over the two crystallographic sites. For the annealed materials, AN-Li6PS5Br and AN-BM-Li6PS5Br, the site distribution appears to be more favorable for a high conductivity in the crystalline fraction that forms due to the annealing process. An interesting future research direction may be to investigate the role of these distributions on solid-state battery performance, and synthesis routes to control the site distribution of the halogen dopant.Annealing lowers the grain boundary resistance dramatically, whereas subsequent ball milling results in a slight increase in grain boundary resistance. Compared to the T1 NMR relaxation experiments, the T1ρ experiments probe a longer diffusion length scale. The much larger activation energy deduced from the T1ρ experiments suggests that this includes diffusion across grain boundaries. However, these larger activation energies changed only marginally with the annealing and the ball milling treatment, suggesting that this does not change the Li-ion dynamics across the grain boundary regions. This also suggests that at the Li6PS5Br–Li6PS5Br interfaces, a larger crystalline fraction is beneficial for the conductivity, most likely increasing the fraction of mobile ions at the interface, rather than the mobility itself. Additionally, we anticipate that the inhomogeneous distribution of Br over the two crystallographic sites may be at least partially responsible for the larger Li-ion mobility at the interfaces.
Both impedance spectroscopy and NMR experiments indicate a very similar bulk and grain boundary Li-ion mobility for the annealed (AN-Li6PS5Br average crystallite size 74 nm) and additionally ball milled (AN-BM-Li6PS5Br average crystallite size 22 nm) materials. However, the solid-state battery with the AN-BM-Li6PS5Br electrolyte provides much better capacity retention upon cycling. The smaller particles of the annealed and ball milled material, AN-BM-Li6PS5Br, appear essential for stable capacity retention, most likely through the establishment of more abundant interfacial contacts with the Li2S cathode resulting from the higher surface area of the small solid electrolyte particles, lowering the interfacial resistance. These results indicate a difference in demands for the solid electrolyte in the solid electrolyte region of the cell compared to the solid electrolyte in the cathode mixture. In the solid electrolyte region, the lowest overall resistance dominated by the grain boundary resistance is achieved for the annealed material (AN-Li6PS5Br), whereas smaller solid electrolyte particle sizes are required to provide sufficient solid electrolyte–cathode interfaces, to minimize the resistance of this rate limiting Li-ion transport step.
In conclusion, the internal resistance of the solid electrolyte is dominated by the solid electrolyte–solid electrolyte Li6PS5Br interfaces, lowering of which can be achieved by annealing treatment. The results indicate that an inhomogeneous distribution of the Br dopant over the crystallite sites may play an important role in the conductivity at the interfaces. The largest contributor to the internal resistance of the Li2S/Li6PS5Br/In solid-state battery appears to exist at the Li6PS5Br/Li2S interface. To utilize a majority of the Li2S capacity, a large overpotential is required, even at the low currents. This compromises the electrochemical stability of the Li6PS5Br solid electrolyte, most likely leading to oxidation products. Despite the electrochemical instability at the Li6PS5Br/Li2S interface and the large volumetric changes upon charging Li2S, the capacity retention of this all-solid-state battery is promising, most likely because of the abundant Li6PS5Br/Li2S interfaces that provide sufficient Li-ion conductivity and the large fraction of Li6PS5Br in the positive electrode mixture. Evidently, the improvement of these solid-state battery systems requires focusing on the stabilization of the solid electrolyte–electrode interfaces, to prevent both electrochemical instabilities and volumetric changes. Additionally, the present results indicate the importance of inhomogeneity in the conductivity and the importance of distinguishing between the electrolyte properties only in the electrolyte region and the electrolyte in the cathode mixture.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. [307161] of M.W. Support from the Dutch organization of scientific research (NWO) for the solid-state NMR facility for advanced materials science in Nijmegen is gratefully acknowledged. The technical assistance of Hans Janssen, Gerrit Janssen and Jan Schoonbrood is gratefully acknowledged. The solid-state NMR facility for advanced material science at the Radboud University is greatly acknowledged for supporting this research. Neutron diffraction assistance of Kees Goubitz is also gratefully acknowledged.References
- J. W. Fergus, J. Power Sources, 2010, 195, 4554–4569 CrossRef CAS.
- P. Knauth, Solid State Ionics, 2009, 180, 911–916 CrossRef CAS.
- Y. S. Jung, D. Y. Oh, Y. J. Nam and K. H. Park, Isr. J. Chem., 2015, 55, 472–485 CrossRef CAS.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart and P. Lamp, Chem. Rev., 2015, 116, 140–162 CrossRef PubMed.
- F. Mizuno, A. Hayashi, K. Tadanaga and M. Tatsumisago, Adv. Mater., 2005, 17, 918–921 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- H.-J. Deiseroth, S.-T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiß and M. Schlosser, Angew. Chem., Int. Ed., 2008, 47, 755–758 CrossRef CAS PubMed.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. y. Adachi, J. Electrochem. Soc., 1990, 137, 1023–1027 CrossRef CAS.
- J. G. Kim, B. Son, S. Mukherjee, N. Schuppert, A. Bates, O. Kwon, M. J. Choi, H. Y. Chung and S. Park, J. Power Sources, 2015, 282, 299–322 CrossRef CAS.
- C. Cao, Z.-B. Li, X.-L. Wang, X.-B. Zhao and W.-Q. Han, Frontiers in Energy Research, 2014, 2, 25 CrossRef.
- K. Takada, Acta Mater., 2013, 61, 759–770 CrossRef CAS.
- Y. Seino, T. Ota, K. Takada, A. Hayashi and M. Tatsumisago, Energy Environ. Sci., 2014, 7, 627–631 CAS.
- R. P. Rao, N. Sharma, V. K. Peterson and S. Adams, Solid State Ionics, 2013, 230, 72–76 CrossRef CAS.
- P. Rayavarapu, N. Sharma, V. Peterson and S. Adams, J. Solid State Electrochem., 2012, 16, 1807–1813 CrossRef CAS.
- C. Yu, S. Ganapathy and M. Wagemaker, Electrochim. Acta, 2016, 215, 93–99 CrossRef CAS.
- C. Yu, S. Ganapathy, N. J. de Klerk, I. Roslon, E. R. Van Eck, A. P. Kentgens and M. Wagemaker, J. Am. Chem. Soc., 2016, 138, 11192–11201 CrossRef CAS PubMed.
- S. Boulineau, M. Courty, J.-M. Tarascon and V. Viallet, Solid State Ionics, 2012, 221, 1–5 CrossRef CAS.
- M. Chen, R. P. Rao and S. Adams, Solid State Ionics, 2014, 262, 183–187 CrossRef CAS.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2015, 28, 266–273 CrossRef.
- N. J. de Klerk, I. Roslon and M. Wagemaker, Chem. Mater., 2016, 28, 7955–7963 CrossRef CAS.
- L. van Eijck, L. D. Cussen, G. J. Sykora, E. M. Schooneveld, N. J. Rhodes, A. A. van Well and C. Pappas, J. Appl. Crystallogr., 2016, 49, 1398–1401 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- S. Ujiie, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2012, 211, 42–45 CrossRef CAS.
- M. Wagemaker, Structure and Dynamics of Lithium in Anatase TiO2, TU Delft, Delft University of Technology, 2003 Search PubMed.
- M. Wagemaker, R. van de Krol, A. P. M. Kentgens, A. A. van Well and F. M. Mulder, J. Am. Chem. Soc., 2001, 123, 11454–11461 CrossRef CAS PubMed.
- M. Wilkening and P. Heitjans, ChemPhysChem, 2012, 13, 53–65 CrossRef CAS PubMed.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- V. Epp, Ö. Gün, H.-J. Deiseroth and M. Wilkening, J. Phys. Chem. Lett., 2013, 4, 2118–2123 CrossRef CAS.
- A. Gupta, R. Murugan, M. P. Paranthaman, Z. Bi, C. A. Bridges, M. Nakanishi, A. P. Sokolov, K. S. Han, E. W. Hagaman, H. Xie, C. B. Mullins and J. B. Goodenough, J. Power Sources, 2012, 209, 184–188 CrossRef CAS.
- V. Epp and M. Wilkening, in Handbook of Solid State Batteries, World Scientific, 2nd edn, 2015, pp. 133–190, DOI:10.1142/9789814651905_0005.
- A. Kuhn, M. Kunze, P. Sreeraj, H. D. Wiemhoefer, V. Thangadurai, M. Wilkening and P. Heitjans, Solid State Nucl. Magn. Reson., 2012, 42, 2–8 CrossRef CAS PubMed.
- A. Kuhn, S. Narayanan, L. Spencer, G. Goward, V. Thangadurai and M. Wilkening, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094302 CrossRef.
- P. Heitjans and J Kärger, Diffusion in Condensed Matter - Methods, Materials, Models, Springer, Berlin, 2005 Search PubMed.
- A. Kuhn, J.-Y. Choi, L. Robben, F. Tietz, M. Wilkening and P. Heitjans, Z. Phys. Chem., 2012, 226, 525–537 CrossRef CAS.
- C. Yu, H. Wang, X. Guan, J. Zheng and L. Li, J. Alloys Compd., 2013, 546, 239–245 CrossRef CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CAS.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Chem. Mater., 2010, 22, 949–956 CrossRef CAS.
- A. Sakuda, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, J. Power Sources, 2011, 196, 6735–6741 CrossRef CAS.
- H. Visbal, Y. Aihara, S. Ito, T. Watanabe, Y. Park and S. Doo, J. Power Sources, 2016, 314, 85–92 CrossRef CAS.
- J. Auvergniot, A. Cassel, J.-B. Ledeuil, V. Viallet, V. Seznec and R. Dedryvère, Chem. Mater., 2017, 29, 3883–3890 CrossRef CAS.
- Z. W. Seh, H. Wang, P.-C. Hsu, Q. Zhang, W. Li, G. Zheng, H. Yao and Y. Cui, Energy Environ. Sci., 2014, 7, 672–676 CAS.
- F. Han, J. Yue, X. Fan, T. Gao, C. Luo, Z. Ma, L. Suo and C. Wang, Nano Lett., 2016, 16, 4521–4527 CrossRef CAS PubMed.
- A. Hayashi, K. Noi, N. Tanibata, M. Nagao and M. Tatsumisago, J. Power Sources, 2014, 258, 420–423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta05031c |
| This journal is © The Royal Society of Chemistry 2017 |