
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring pores in graphene-based materials: from generation to applications
Tieshan
Yang
 a,
Han
Lin
a,
Xiaorui
Zheng
a,
Kian Ping
Loh
bc and
Baohua
Jia
*a
a,
Han
Lin
a,
Xiaorui
Zheng
a,
Kian Ping
Loh
bc and
Baohua
Jia
*a
aCentre for Micro-Photonics, Faculty of Science, Engineering and Technology, Swinburne University of Technology, PO Box 218, Hawthorn, VIC 3122, Australia. E-mail: bjia@swin.edu.au
bDepartment of Chemistry, National University of Singapore, Singapore 117543, Singapore
cCentre for Advanced 2D Materials and Graphene Research Centre, National University of Singapore, Singapore 117546, Singapore
First published on 12th July 2017
Abstract
Combining the merits from both porous material and graphene, porous graphene-based materials have received significant attention due to their unique porous structures, large surface areas and prominent electrical conductivity. The access and tuning of both the in-plane pores of a monolayer graphene and the interlayer spacing (interlayered pores) of multilayered graphene-based materials offer additional selective mechanisms. These extraordinary properties enable porous graphene-based materials to serve as critical components in molecular separation, including water desalination, gas separation and bioseparation, and electrochemical energy storage, such as supercapacitors, lithium–O2/ion/sulfur batteries and fuel cells. The state-of-the-art pore-fabrication methods as well as the interlayered pores tuning approach for graphene-based materials are summarized. The insights between structures, properties and the broad applications of these materials are reviewed and discussed. The remaining challenges and future perspectives of porous graphene-based materials for wide implementations are provided.
 Tieshan Yang | Tieshan Yang received his Bachelor's degree in Applied Physics from Ludong University, China in 2012 and his Master's degree in Optical Engineering from the Beijing University of Technology, China in 2015. He is now a PhD student under the supervision of Professor Baohua Jia at the Swinburne University of Technology, Australia. His research interests focus on laser nanofabrication on 2D materials for functional photonics devices and molecular separation. |
 Han Lin | Han Lin received his BSc (2005) and MSc (2008) degrees from Xiamen University, China. He was awarded a PhD (2013) from the Swinburne University of Technology, Australia. He has dedicated interest and experience on optical system design and dynamic control of light–matter interaction, vectorial diffraction theory and super-resolution. He is currently working as a Postdoctoral Research Fellow at the Swinburne University of Technology. His research interests focus on light–matter interaction on 2D materials and its applications in the energy storage devices and molecular separation. |
 Xiaorui Zheng | Xiaorui Zheng received his BSc degree in Physics from Wuhan University, China in 2009 and MSc degree at the Institute of Physics, Chinese Academy of Sciences, China in 2012. In 2016, he was awarded a PhD from the Swinburne University of Technology, Australia. He is currently working as a Postdoctoral Research Associate at the City University of New York sponsored by the SwissLitho Postdoctoral Fellowship. His current research interests lie in the nanophotonics of low-dimensional materials for both the functional photonic and biomedical applications by using novel thermochemical nanolithography techniques. |
 Kian Ping Loh | Kian Ping Loh is Provost's Chair Professor at the National University of Singapore. He received his BSc degree (1994) from the National University of Singapore. He obtained his D.Phil (1996) from Oxford University and then did his postdoctoral work at the National Institute for Materials, Tsukuba, Japan between 1997 and 1998. Now he is the Head of 2D Materials Research at the Centre for Advanced 2D Materials in the National University of Singapore. His current research interests focus on graphene chemistry and physics of 2D materials; synthesis and applications of 2D organic materials. |
 Baohua Jia | Baohua Jia is a full Professor and Research Leader at Swinburne University of Technology. She received her BSc (2000) and MSc (2003) degrees from Nankai University, China. She was awarded a PhD (2007) from Swinburne University of Technology, Australia. She is the Head of Laser and Nanomaterial Interaction (LNI) Group. She uses light to develop various functional nanostructures to effectively harness and store clean energy and boost the performance of communication and imaging devices. |
1. Introduction
Porous materials have received tremendous attention in both academia and industry due to their broad applications in environmental protection, water cleaning, energy storage and conversion, etc.1–5 Porous materials offer unique properties allowing interactions with various organic or inorganic species both at the surfaces and through their nanostructures.The size of porous materials can be classified into micropores (<2 nm), mesopores (2–50 nm) and macropores (>50 nm).1 The distribution of sizes, shapes and volumes of the void spaces in porous materials directly determines their ability to perform the desired functions for special applications. Hence, tailoring the unique porous structures with different textural and porous properties beyond nature represents an attractive effort for various high performance devices.
Among diverse porous materials, porous carbon is extremely appealing for its low density and high strength-to-weight ratio.6 In addition, it is possible to bond it with various atoms by different hybrid orbitals (sp, sp2 and sp3), enabling porous structures with desired morphology and intriguing properties. The demand to create uniformity within the pore size, shape and volume has increased over the past years, since it can lead to superior application properties.
Graphene, a free-standing atomic-thin layer of sp2 carbon atoms, has emerged as the most significant carbon material over the last decade not only due to its unprecedented physical and chemical properties, but also for its unique two-dimensional (2D) morphology and ultra-large surface area.
Graphene-based porous materials have many apparent merits over other porous carbon materials.6–9 (1) The high mechanical strength of graphene can ensure the stability of porous frameworks, and thus prevents the deformation of the porous structures. (2) The prominent chemical and thermal stabilities of graphene enable this porous material to withstand rigorous environments. (3) The in-plane pores and interlayer spacing (interlayered pores) are suitable for the rapid diffusion of ions and molecules. (4) The narrow pore size distribution range allows accurate control of the pore size for targeted applications. (5) The electrical conductivity of graphene makes it an ideal current collector for the fast transportation of charge carriers within the porous structures. (6) Graphene derivatives, such as graphene oxide (GO) and reduced graphene oxide (rGO), contain abundant oxygen functional groups and can work as appealing substrates for modifying various organic or inorganic species towards graphene-based porous materials with different complicacies. However, a perfect graphene monolayer is impermeable to protons.10 In order to fully utilize the unique properties of pristine graphene and graphene-based materials for molecular separation,10–40 high-performance electrochemical energy storage and conversion devices,41–63 it is imperative to generate pores with accurately controlled narrow size distributions.
In general, graphene-based porous materials can be classified to monolayer porous graphene (also referred as holey graphene with in-plane pores),64 2D laminar porous graphene (interlayered pores)10 and 3D porous graphene networks (including both in-plane and 3D interconnected pore structures),51 as shown in Fig. 1a. In Fig. 1b, we present the most popular applications of porous graphene and their unique pore size ranges according to three categories. For molecular separation, well-defined interlayered pores play a significant role, since they can allow targeted molecules to pass through the membrane precisely with a high permeability and selectivity. For electrochemical energy storage, they enable ions pass through the porous membrane to interact with the electrodes for a high energy density. For the applications mentioned above, pore sizes required are much smaller than the conventional pores of the naturally available materials. Generating such small, well-defined pores represents a major challenge because it approaches the technological limit. In this review, the state-of-the-art pore-generation and tailoring methods for graphene-based materials are summarized and compared. The insight into the structures, properties and applications of these materials in molecular separation, electrochemical energy storage and conversion, and proton conductors is discussed. Through summarizing the outstanding challenges and future perspectives in the field, we aim at stimulating discussions and ideas accelerating the development of porous graphene-based materials for diverse functional device applications.
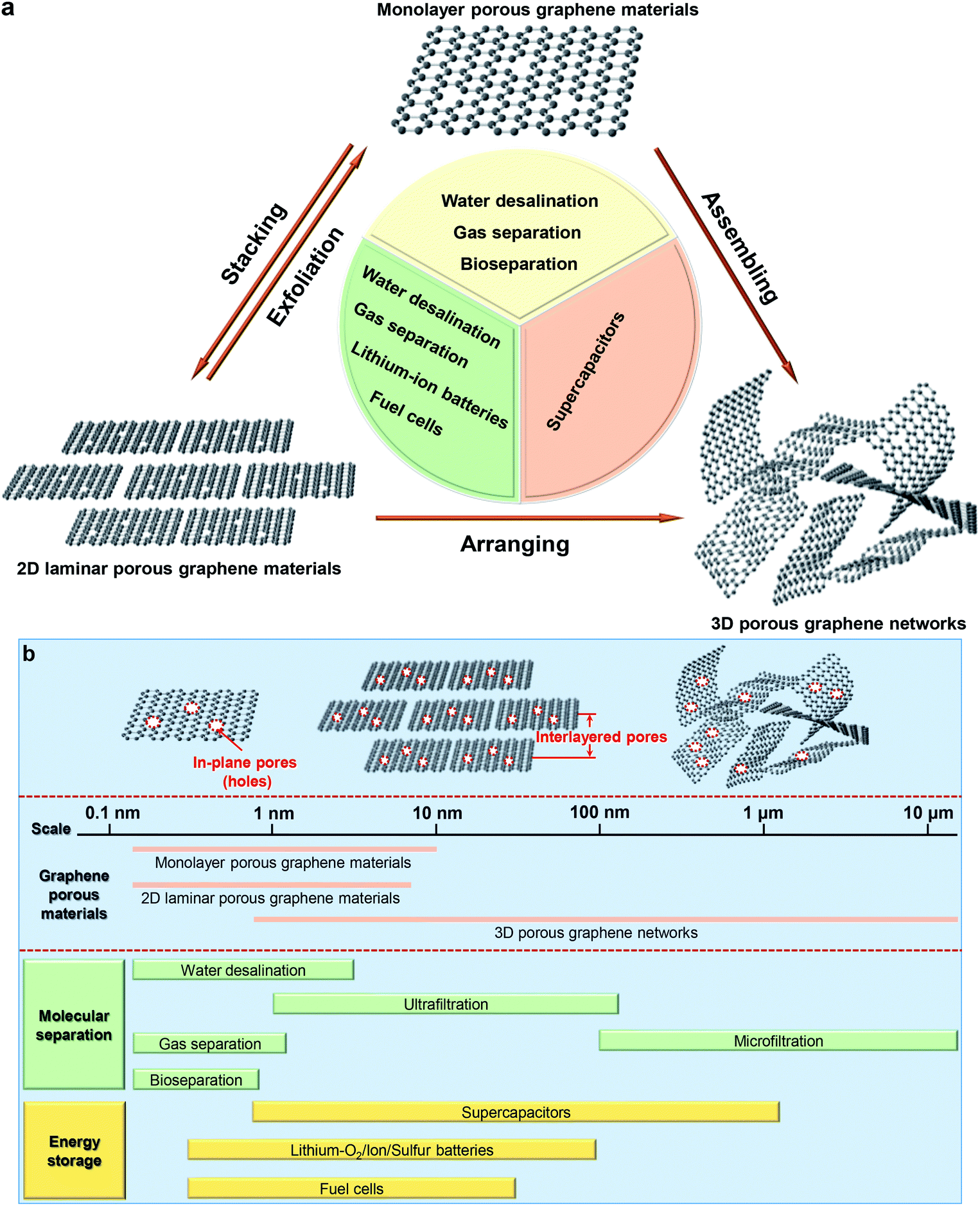 | ||
| Fig. 1 Type of nanoporous graphene-based materials and their broad applications. (a) Nanoporous graphene-based materials and their applications. (b) Classification of nanoporous graphene-based materials, definition of in-plane pores and interlayered pores in nanoporous graphene-based materials and broad applications based on pore sizes. | ||
2. Structure and properties of graphene-based materials
2.1 Structure and properties of graphene
Graphene is a densely packed 2D sheet of sp2-hybridized carbon atoms arranged in a honeycomb lattice, which composes of two equivalent sub-lattices of carbon atoms bonded together with σ bonds, as shown in Fig. 2a.65–68 Each carbon atom in the lattice has a π orbit that contributes to a delocalized network of electrons. The most intriguing aspect of graphene is its extraordinary properties. It has a large theoretical specific surface area (2630 m2 g−1), high intrinsic mobility (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1), high Young's modulus (∼1.0 TPa), high thermal conductivity (∼5000 W m−1 K−1), high optical transmittance (∼97.7%) and good electrical conductivity.9,66,69–74 The large surface area, barrier properties, atomic thickness, excellent mechanical strength, and chemical stability makes graphene an outstanding material for various applications. However, a pristine monolayer of graphene is impermeable to molecules.10 Nanopore generation is essential to enable application in molecule separation and energy storage.
000 cm2 V−1 s−1), high Young's modulus (∼1.0 TPa), high thermal conductivity (∼5000 W m−1 K−1), high optical transmittance (∼97.7%) and good electrical conductivity.9,66,69–74 The large surface area, barrier properties, atomic thickness, excellent mechanical strength, and chemical stability makes graphene an outstanding material for various applications. However, a pristine monolayer of graphene is impermeable to molecules.10 Nanopore generation is essential to enable application in molecule separation and energy storage.
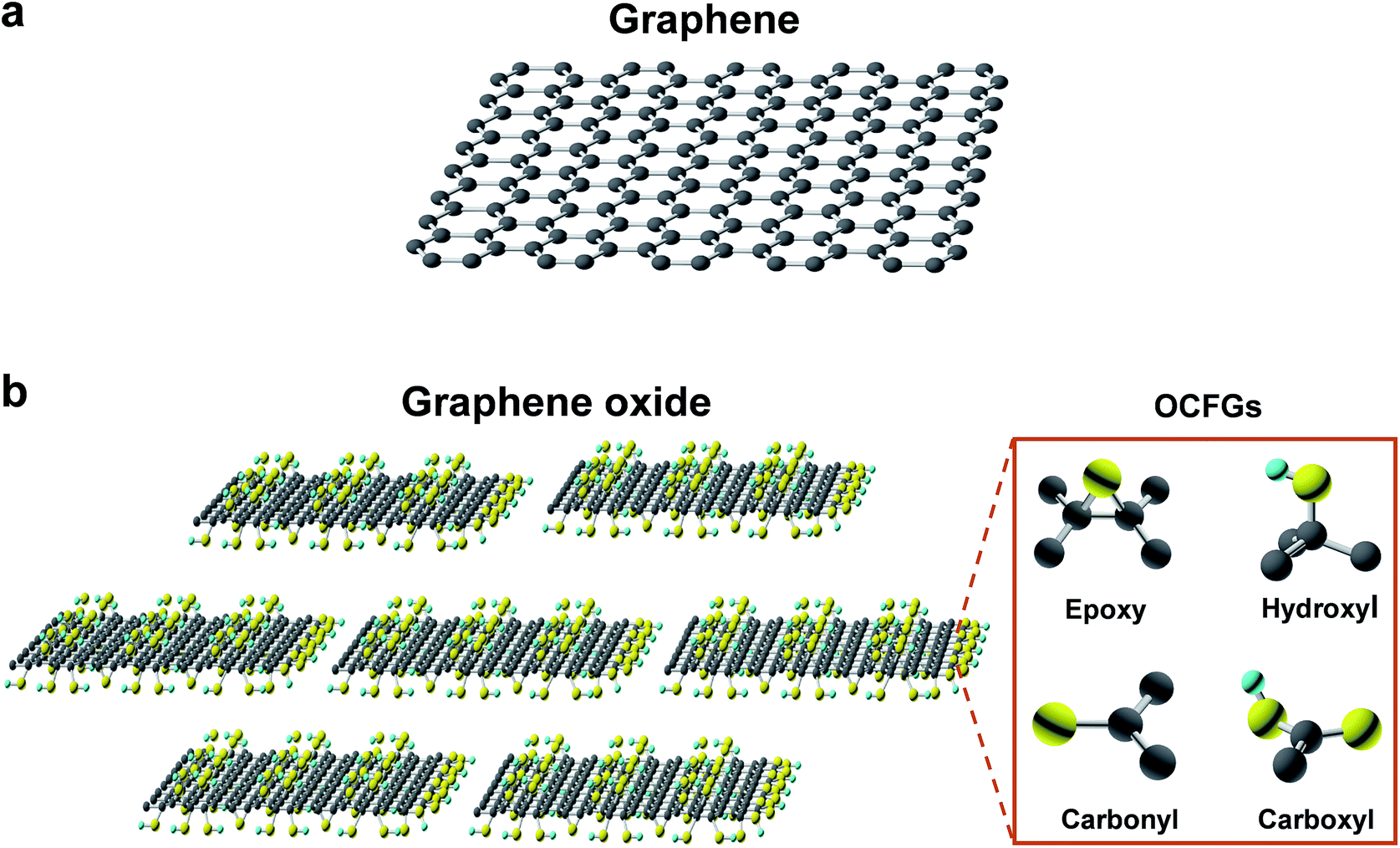 | ||
| Fig. 2 Schematics of the molecular structure of graphene (a) and graphene oxide (b). | ||
2.2 Structure and properties of graphene oxide
As one of the most important derivatives of graphene, GO is a monolayer-thin graphene with several oxygen-containing functional groups (OCFGs), such as epoxy, hydroxyl, carbonyl and carboxyl groups, decorating the basal planes and edges of GO, as shown in Fig. 2b.67,75–77 Due to these OCFGs, GO exhibits a series of distinguished properties.67,75,78–81 On one hand, GO can be readily dispersed in an aqueous medium and can form well-dispersed aqueous colloids without the help of any surfactants agents.40,82 Therefore, solution based film synthesis methods can be applied, such as filtration, spin-coating, drop casting and self-assembly. On the other hand, GO sheets are highly negatively charged when dispersed in water, as a result of the ionization of the carboxyl groups on the edges.67 Additionally, those carbon atoms bonded to oxygen atoms in the form of epoxy, hydroxyl, carbonyl and carboxyl groups tend to form amorphous regions due to distortions from the high proportion of sp3 C–O bonds.75,83 As a result, nanoscale wrinkles and structural defects are formed in the basal plane of the GO sheets, which provide primary passages for molecular transport when GO sheets are assembled into membranes. Moreover, these OCFGs provide many reactive handles for various surface-modification reactions, which can be used to develop a series of functionalized GO based membranes with significantly enhanced performances.84,85 Compared to monolayer porous graphene membranes, where in-plane nanopores provide transport channels, the 2D channels between stacked GO nanosheets allow molecules to pass through, which provide another means to separate molecules.10,36 These OCFGs on GO nanosheets, not only make GO dispersible in water, but also provide convenient sites for enhancing interactions with transport components.3. Methods for pore generation in graphene-based materials
3.1 In-plane pore generation in monolayer graphene materials
Graphene monolayer was first obtained by Geim et al. in 2004 through mechanical exfoliation of graphite.68,69 Since then, research on this novel material has increased rapidly. Many methods in addition to mechanical exfoliation have been developed, for instance, epitaxial growth on substrates such as SiC86 and SiO2,87 including atomic layer deposition (ALD) and chemical vapor deposition (CVD)88 and thermal oxidation of graphite.89 These methods are able to produce large-scale pristine graphene with high quality.Recently, continuous experimental endeavors have been applied to explore various methods for introducing in-plane pores (holes) in graphene monolayer towards high performance devices, which include both physical methods, such as, focused electron beam (FEB) ablation,90,91 focused ion beam (FIB) irradiation,23 ultraviolet (UV)-induced oxidative etching,21 ion bombardment followed by chemical oxidative etching,22 oxygen plasma etching,24 UV light or laser irradiation92,93 and chemical methods, such as surface-assisted aryl–aryl coupling of designed block94 and MnO2 etching.95 Depending on the production techniques used, the pore size (the diameter of the pores) ranges from atomic precision to nanometer scale. As a result of the pores in the graphene plane, porous graphene exhibits properties distinct from those of pristine graphene, enabling many potential applications in numerous fields.
Fischbein et al. showed the formation of pores in suspended graphene sheets by controlled exposure of the sheets to a FEB irradiation of a TEM.90 The pores were created with controlled sizes (∼3.5 nm, Fig. 3a) and a close proximity to each other (Fig. 3b). The holey graphene was very stable. However, this method is inefficient and not suitable for fabricating large-area holey graphene with high pore density. Then, Fox et al. produced pores with a diameter of 5.9 nm ± 0.4 nm in graphene sheets using a low energy FEB in a SEM apparatus with N2 gas, as shown in Fig. 3c and d.91 This method has a relative low-cost due to the use of a low energy electron microscope. However, the serial processing manner makes it not suitable for large-scale manufacturing of porous graphene.
 | ||
| Fig. 3 (a) TEM image of the nanopores generated by FEB in monolayer graphene. (b) Multiple nanopores made in close proximity to each other by FEB. Reproduced with permission.90 Copyright 2008, American Institute of Physics. (c) SEM image of nine holes etched in a graphene flake. (d) Plasmon energy loss image with material thickness contrast, the dot at the hole center shows the nominal diameter of the SEM electron beam used to etch the hole (5.9 nm ± 0.4 nm). Reproduced with permission.91 Copyright 2011, American Institute of Physics. | ||
Koenig et al. created pores in micrometre-sized graphene flakes using UV-induced oxidative etching and showed selective gas transport through porous, atomically thin graphene.21 The approach is scalable but it is challenging to control the pore size distribution (∼0.49–10 nm) and the pore density (a small number of pores in the 5 μm membrane). As a result, it is limited for practical gas separation. O'Hern et al. used ion bombardment to introduce isolated and reactive defects on macroscopic monolayer graphene and further enlarged these defects to pores by controlled chemical oxidative etching, as shown in Fig. 4a.22 The resulting porous graphene possesses high-density pores with diameters of 0.40 ± 0.24 nm. By simply changing the etching time, different pore sizes can be obtained. This work presented the first milestone towards the production of large-area porous graphene membrane with well-defined pore size distribution. However, the performance of porous graphene membranes is limited by the intrinsic tears and defects of pristine monolayer graphene. Celebi et al. developed an optimized CVD growth approach followed by a FIB approach to realize perforation of graphene with narrowly distributed pore sizes ranging from <10 nm to 1 μm and a large number of pores (∼103 to 106 per membrane with a diameter of 4 μm), as shown in Fig. 4b.23 This approach is tunable with well-defined pore sizes, but it is limited to small area (∼12.57 μm2). Surwade et al. created stable, nanometre-sized pores (∼0.5–1 nm) in a suspended, monolayer graphene membrane using an oxygen plasma etching approach, which could tune the size of the pores, as shown in Fig. 4c.24 This method can treat large-area samples with tunable pore sizes, but the density of pore distribution is moderate (∼one pore per 100 nm2).
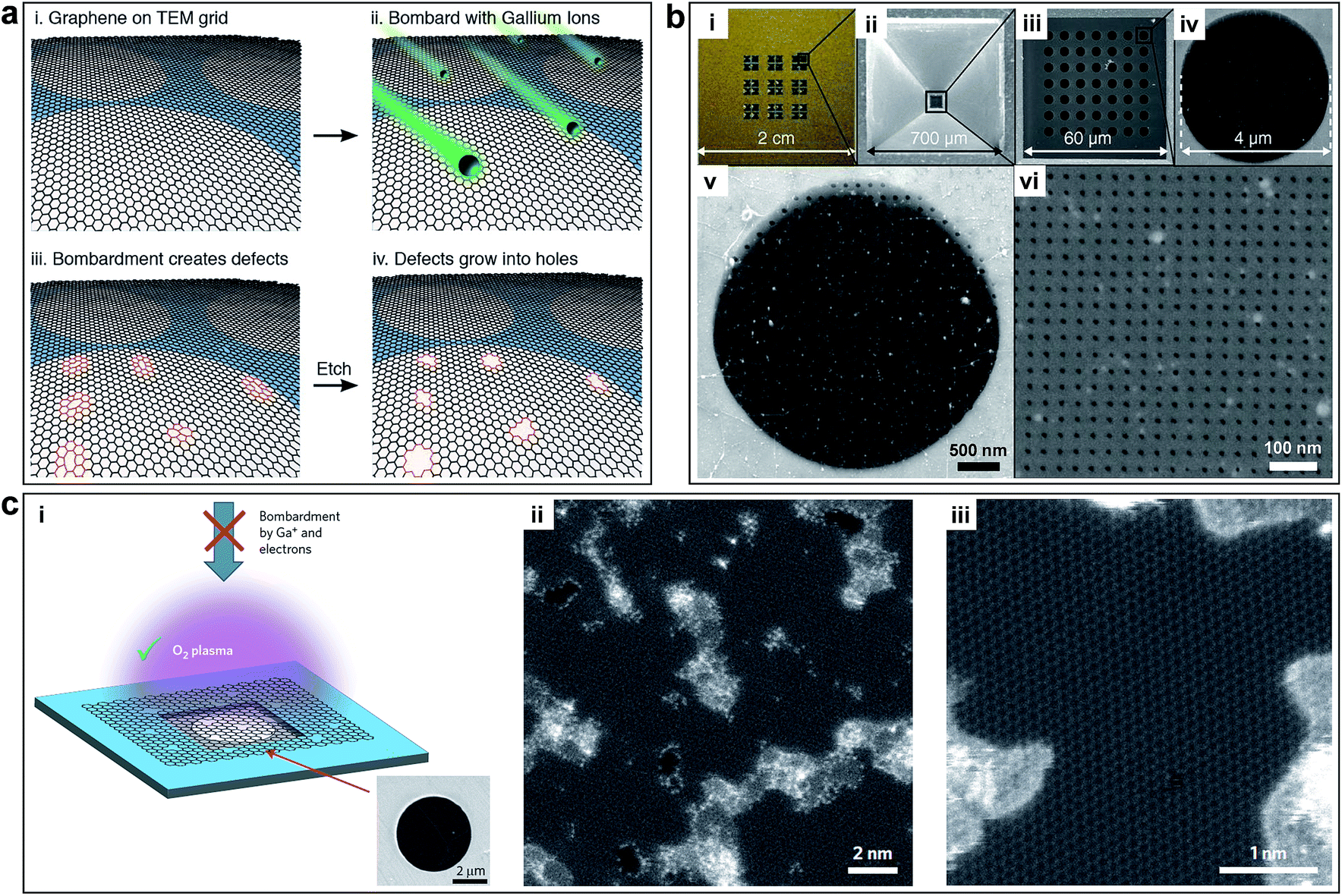 | ||
| Fig. 4 (a) Process to create controlled pores using ion bombardment in graphene membranes. Controlled sub-nanometer pores in graphene are created by ion bombardment followed by chemical oxidation. Reproduced with permission.22 Copyright 2014, American Chemical Society. (b) Membrane fabrication using FIB. (i) Photograph of a full membrane structure. (ii) Bottom view SEM image of the SiNx membrane. (iii to vi) Top view SEM images of (iii) porous freestanding SiNx window before graphene transfer, (iv) freestanding graphene transferred on one of the 4 μm-wide SiNx open pores, (v) 50 nm-wide apertures FIB-drilled on the freestanding graphene (Ga FIB), and (vi) 7.6 nm-wide apertures perforated in the similar way (He FIB). Reproduced with permission.23 Copyright 2014, American Association for the Advancement of Science. (c) Pore generation using oxygen plasma etching. (i) Schematic and SEM image of monolayer graphene suspended on a 5 μm diameter hole. (ii) Aberration corrected STEM images of graphene after 1.5 s exposure to oxygen plasma. (iii) Pores with characteristic dimensions of ∼1 nm are seen clearly. Reproduced with permission.24 Copyright 2015, Nature Publishing Group. | ||
Chemical methods are able to generate graphene in-plane pores on a large scale with low cost.38 Bieri et al. first synthesized porous graphene by using surface-assisted aryl–aryl coupling of specifically designed molecular building blocks (e.g. hexaiodo-substituted macrocycle cyclohexa-m-phenylene (CHP)), yielding regular 2D polyphenyl networks with single-atom wide (∼0.4 nm) pores and sub-nanometre (7.4 Å) periodicity, as shown in Fig. 5a.94 This method may be extended to directly tailor any porous structures with various pore sizes by the proper design of the molecular building units. However, achieving a high uniformity of pores and transferring from a metal substrate are challenging. Another example using the chemical method to obtain porous graphene was shown by Fan et al. They produced porous graphene (holey graphene) by etching carbon atoms on the graphene sheets via MnO2, as shown in Fig. 5b.95 Exfoliated GO was selected as a support to deposit nanoscaled MnO2 as follows: 4MnO4− + 3C + H2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) 4MnO2 + CO32− + 2HCO−. The carbon atoms served as a sacrificial reductant and the MnO4− was converted to insoluble MnO2, which remained on the surface of the sheet. The resulting sheets had numerous mesopores with the sizes of ∼2.4 nm.
4MnO2 + CO32− + 2HCO−. The carbon atoms served as a sacrificial reductant and the MnO4− was converted to insoluble MnO2, which remained on the surface of the sheet. The resulting sheets had numerous mesopores with the sizes of ∼2.4 nm.
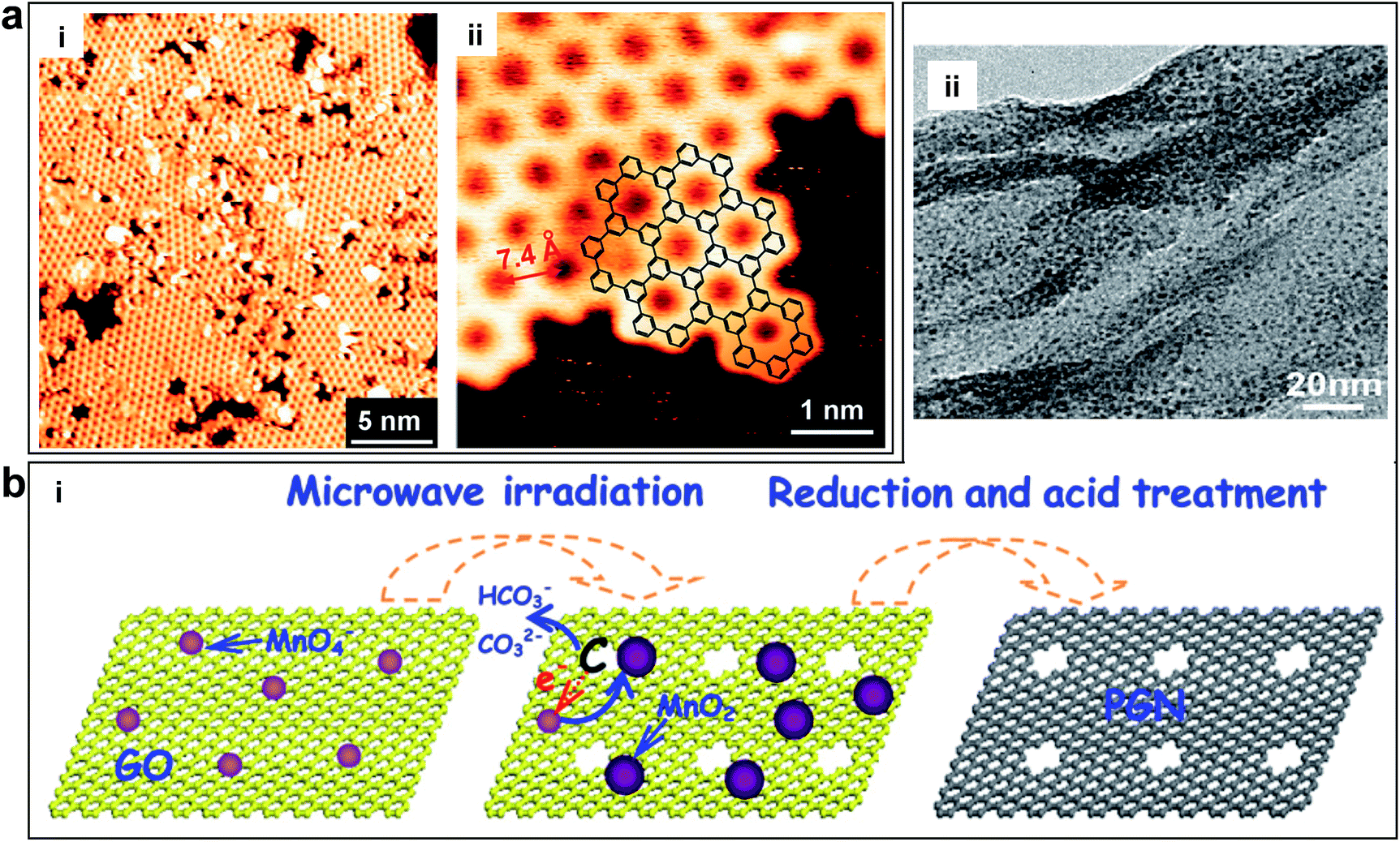 | ||
| Fig. 5 (a) Porous graphene obtained by using surface-assisted aryl–aryl coupling of specifically designed molecular building blocks. (i) STM image of the polyphenylene super honeycomb network on Ag (111) formed after polymerization of CHP precursors at 805 K. (ii) STM image of an edge of the polymer network, where the structure of the CHP backbone (overlaid chemical structure) can be recognized. Reproduced with permission.94 Copyright 2009, Royal Society of Chemistry. (b) Porous graphene nanosheets obtained by etching carbon atoms on the graphene sheets via MnO2. (i) Schematic of the formation of porous graphene material with pores on the surface of the sheets. (ii) TEM image of porous graphene nanosheets. Reproduced with permission.95 Copyright 2012, American Carbon Society. | ||
In summary, in-plane pore in monolayer graphene materials can be achieved via physical or chemical methods. Physical methods are able to tune the pore sizes from sub-nanometer to nanometer by changing the ion and electron doses. FEB irradiation can make a defined pore size, but it is not scalable. Oxygen plasma etching and ion bombardment followed by chemical oxidative etching methods generate porous graphene with tunable pore size in a large area with a moderate pore density. FIB irradiation can also generate a well-defined pore size, but it is limited to a small area. In comparison, direct synthesis of porous graphene-based materials by low-cost and scalable chemical methods exhibit regular pore distribution and precision.38 The start-of-the-art methods of generating in-plane pores in graphene are summarized in Table 1.96,97 The employment of appropriate methods depend on the requirements of specific applications.
| Methods | Pore size (nm) | Area (μm2) | Distribution | Advantages | Limitations | Author (year) | |
|---|---|---|---|---|---|---|---|
| Physical | Focused electron beam (FEB) irradiation | ∼3.5 | — | — | Tunable and well-defined pore size | Small area | Fischbein et al. (2008)90 |
| Nitrogen-assisted electron beam irradiation | 5.9 ± 0.4 | — | — | Controlled pore size | Small area | Fox et al. (2011)91 | |
| UV-induced oxidative etching | 0.4–10 | ∼19.63 | — | Large-area samples | Wide size distribution | Koenig et al. (2012)21 | |
| Ion bombardment and chemical oxidative etching | 0.40 ± 0.24 | — | — | Large-area, controlled pore sizes | Moderate pore density | O'Hern et al. (2012)22 | |
| Focused ion beam (FIB) irradiation | 5–100 | ∼12.57 | 103 to 106 per membrane | Tunable and well-defined pore size | Small area | Celebi et al. (2014)23 | |
| Oxygen plasma etching | ∼1 | ∼12.57 | One/100 nm2 | Large-area samples, tunable pore size | Moderate pore density | Surwade et al. (2015)24 | |
| Chemical | Surface-assisted aryl–aryl coupling of the designed block | ∼0.4 | — | — | Simple, cost-effective | Difficult to transfer | Bieri et al. (2009)94 |
| MnO2 etching | ∼2.4 | — | — | Simple | Difficult to control pore sizes | Fan et al. (2012)95 |
3.2 Interlayered pore generation and tuning in laminar 2D graphene materials
Monolayer graphene has only one selective mechanism for molecular separation (Fig. 1b), which is the in-plane pores. By stacking the monolayer porous graphene into laminar 2D porous graphene materials, another selective mechanism is introduced, namely interlayered pores.10 The advantage of this mechanism is the extremely narrow interlayered pore distribution. The interlayered pores depend on the molecular thickness of the material or the size of the spacer layer, which can be easily and accurately controlled by the molecular structure and the type of the spacer.To synthesize laminar 2D porous graphene materials, it is challenging to use the vacuum based film preparation methods, such as CVD and ALD due to the following two reasons: (1) the stacking of graphene layers will turn the whole structure into graphite, whose layer spacing cannot be controlled. (2) It is extremely difficult to accurately generate in-plane pores in each layer of a multilayer graphene structure. Thus, it is preferred to produce laminar 2D porous graphene materials by using GO material.
GO nanosheets are single-atom thick with lateral dimension as high as tens of micrometers. They contain plenty of OCFGs, which are both on the edges and the basal planes of the GO nanosheets.67,75,83,98 GO nanosheets can be mass-produced in a cost-effective way by chemical oxidization and ultrasonic exfoliation of graphite. The OCFGs modify the surface properties (e.g. the charge properties and hydrophilicity) of GO. By the attaching of OCFGs, the hydrophobic graphene is converted to hydrophilic GO. Meanwhile, the surface of GO becomes negatively charged. As a result, GO flakes can be well dissolved in water, which enable the applications of low-cost solution-based film synthesis technique.75 In addition, it is feasible to modify the GO film structure to control the chemical, electrical and mechanical properties due to the OCFGs. These unique features enable GO to be an attractive platform for building well-defined multifunctional nanostructures.
GO nanosheets can be assembled into laminar structures by vacuum filtration,11,14,15,99 drop-casting,16 spin-coating,10,27 layer-by-layer (LBL)13 deposition and other methods. Vacuum filtration is the most often used method to fabricate GO membranes on substrates. The thickness of the membrane can be controlled by the amount of GO in the solution. Drop-casting is usually used to fabricate free-standing GO membranes, which has a limitation in the composite system because of insufficient interfacial adhesion between the GO layer and the substrate. Spin-coating can arrange GO nanosheets in a fast assembly process. The LBL method is able to precisely control the GO film thickness at the molecular level.36,39
During the assembly of GO nanosheets into laminar membranes, interlayered pores have been proved to play a crucial role in molecular transport.10,36 The interlayered pores in the GO laminates, if well manipulated, provide additional selective pathways for molecules, which are promising for separation. Therefore, precise tuning of the interlayer pores becomes a fundamental method to achieve high-performance GO laminar membranes.
The interlayered pores can be modulated by physical (e.g. stacking GO sheet with nanoscale wrinkles,36 laser irradiation,100,101 thermal treatment26 and epoxy physical confinement12) or chemical methods (e.g. cross-linking by nanoparticle or nanowires,15,102 chemical reduction103,104). Mi summarized the methods to synthesis GO membranes (Fig. 6a-i).28 The GO membranes prepared by vacuum filtration might lack sufficient bonding between GO nanosheets. As shown in Fig. 6b, one can clearly see the laminar structure of the GO membrane.10 However, LBL assembly is ideal for introducing an interlayer stabilizing force via covalent bonding, electrostatic interaction, or both effects.28 Several methods to obtain controllable interlayered pores and various applications are also provided by Mi, as shown in Fig. 6a-ii.28 As for the physical methods, laser irradiation or thermal treatment has been developed to stabilize GO membranes, which can change the coverage of OCGFs of the GO,105–109 thus modulating the interlayered pores for precise molecular separation.26 Immersing GO membranes in water leads to intercalation of 2–3 layers of water molecules between individual GO sheets, which results in GO membrane swelling.11 In order to achieve smaller interlayered pores for specific ion rejection, Abraham et al. obtained GO membranes with tunable interlayered pores varied from ≈6.4 to 9.8 Å using an epoxy physically confined method, as shown in Fig. 6c.12 As for the chemical methods, cross-linking using nanoparticle or nanowires has been used to stabilize the GO membrane and change the interlayered pores.15,102,110 Huang et al. used Cu(OH)2 nanochannels with a diameter of 2.5 nm as sacrificial templates to prepare nanostrand-channelled GO membranes by electrostatic attraction.15 A network of nanochannels with a narrow size distribution of 3–5 nm was formed after the nanochannels were removed by an acid solution, as shown in Fig. 6d.
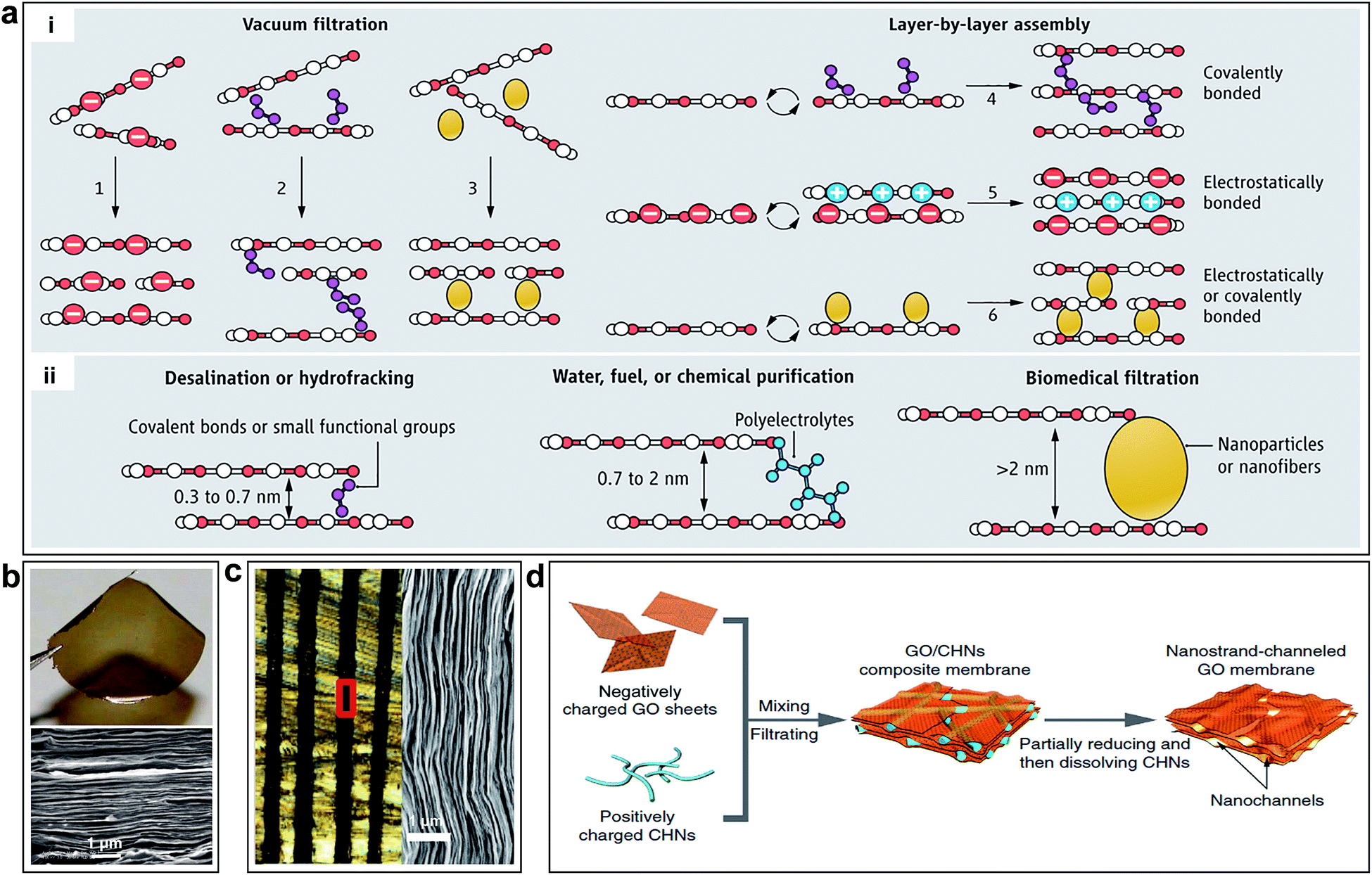 | ||
| Fig. 6 (a) Schematic of the tuning of interlayer galleries of laminar membranes. (i) Several methods to synthesize GO membranes have been reported or envisioned; GO nanosheets can be packed physically by vacuum filtration (options 1 to 3), or they can be stabilized by covalent bonds, electrostatic forces, or both (options 4–6) during LBL assembly. (ii) Separation capability of GO membrane is tunable by adjusting the nanochannel size. Reproduced with permission.28 Copyright 2014, American Association for the Advancement of Science. (b) Photo of a 1 μm-thick GO film peeled off from a Cu foil and SEM of the film's cross section. Reproduced with permission.10 Copyright 2014, American Association for the Advancement of Science. (c) Optical micrograph of the cross-sectional area of epoxy physically confined GO, which shows 100 μm-thick GO laminates (black) embedded in epoxy. Right: SEM image of the marked region in left. Reproduced with permission.12 Copyright 2015, Nature Publishing Group. (d) Illustration of the fabrication process of nanostrand-channelled GO membranes. Reproduced with permission.15 Copyright 2015, Nature Publishing Group. | ||
In summary, GO nanosheets offer an exciting opportunity to assemble a brand new class of ultrathin and high-flux membranes because of their unique 2D and atomic-thin structure, outstanding mechanical strength and good flexibility as well as their facile and large-scale production. Interlayered pores of GO membrane can be precisely tuned due to the OCFGs.
3.3 3D porous networks in graphene-based materials
Although it is possible to accurately control the interlayered pores of laminar 2D porous graphene materials to achieve molecular separation, the tuning range is relatively limited (from sub-nanometre to several nanometre range), which is insufficient for electrochemical applications. To further enlarge the pore size, it is necessary to rearrange the layered structure to generate 3D porous networks with both in-plane and 3D interconnected pore structures. In this way, the pore size is controlled by the overall structure of the 3D networks, which can be in the range from tens of nanometres up to tens of microns. Additionally, the specific surface area (SSA), which plays a key role in electrochemical energy storage, can be controlled by the pore sizes. 3D porous networks in graphene-based materials have a large SSA, which are crucial for electrochemical energy storage devices.6El-Kady et al. used a standard LightScribe DVD optical drive to conduct direct laser reduction of GO to graphene (Fig. 7a).41 During the preparation process, the GO material is reduced by the laser beam converting to laser scribed graphene (LSG), and the gas generated during the laser reduction process generate 3D porous nanostructures. The network structure with open pores and the large accessible SSA (1520 m2 g−1) were optimized for electrolyte ionic diffusion in LSG electrodes. Zhu et al. reported the synthesis of activated graphene with a SSA of up to 3100 m2 g−1via KOH efficient chemical activation of microwave exfoliated GO (MEGO) (Fig. 7b).51 The as-made MEGO powder was placed in KOH solution, followed by filtration and drying, to form a series of MEGO/KOH mixtures. The activation process was subsequently executed by putting the mixture in a tube furnace under flowing argon at a pressure of ∼400 Torr and heating at 800 °C for 1 h. This sp2-bonded carbon had a continuous 3D network of highly curved (Fig. 7b-ii), atomic-thin walls that formed primarily 0.6 to 5 nm-width pores, and substantial curvatures of the single-carbon sheets are visible. Meanwhile, the in-plane crystallinity was preserved. Thus, it appeared the chemical activation not merely digested the MEGO but also largely restructured it.
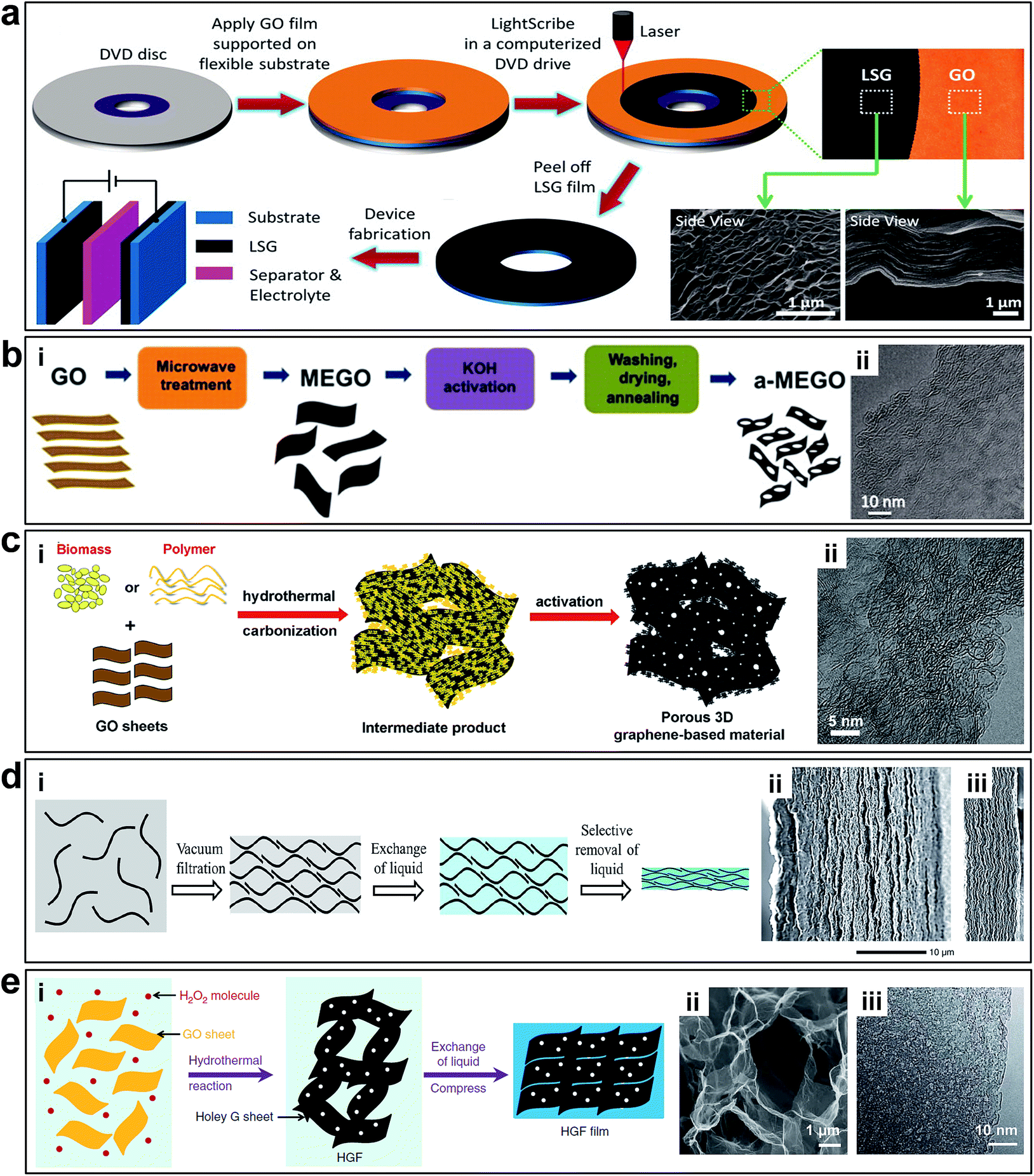 | ||
| Fig. 7 (a) Schematic of the fabrication of LSG-based supercapacitors. Reproduced with permission.41 Copyright 2013, American Association for the Advancement of Science. (b) Schematic illustration of MEGO via KOH efficient chemical activation. (i) Schematic illustration of the fabrication of MEGO with a high surface area. (ii) High-resolution phase contrast electron micrograph of the thin edge of an MEGO sample. Reproduced with permission.51 Copyright 2011, American Association for the Advancement of Science. (c) Schematic illustration of synthesizing porous graphene materials. (i) Schematic of the simple and green process of synthesizing porous graphene-based materials. (ii) HRTEM image of 3D graphene-based bulk materials showing a 3D porous structure with a highly wrinkled surface. Reproduced with permission.52 Copyright 2013, Nature Publishing Group. (d) Schematic illustration of fabrication EM-CCG. (i) Schematic showing a soft chemistry route to fabricate electrolyte-mediated chemically converted graphene (EM-CCG). (ii and iii) SEM images of cross sections of the obtained EM-CCG films. Reproduced with permission.53 Copyright 2013, American Association for the Advancement of Science. (e) Preparation and structural characterization of holey graphene frameworks (HGFs). (i). Schematic illustration of the preparation process of HGFs and HGF films. (ii) SEM image of interior microstructures of HGFs. (iii) TEM image of holey graphene sheets in HGFs. Reproduced with permission.111 Copyright 2014, Nature Publishing Group. | ||
Highly conductive graphene sheets are excellent building blocks for preparing sandwich-like porous carbon layer/graphene hybrids.54 Zhang et al. introduced a simple and green but very efficient method using in situ hydrothermal polymerization and carbonization of the mixture of cheap biomass or industry carbon sources with GO to first get the 3D hybrid precursor materials, followed by a chemical activation process (Fig. 7c).52 Structural and morphology analyses demonstrated that these materials consist of almost entirely defected/wrinkled monolayer graphene sheets in the dimensional size of 4–6 nm (Fig. 7c-ii), with ultrahigh SSA (up to 3523 m2 g−1) and excellent bulk conductivity (up to 303 S m−1).
Taking advantage of the 2D intrinsic micro-corrugated configuration and self-assembly behaviour of chemically converted graphene (CCG), Yang et al. synthesized porous yet highly compact carbon electrodes with a continuous ion transport network by capillary compression of adaptive graphene gel films in the presence of a non-volatile liquid electrolyte (Fig. 7d-i).47,53 A flexible CCG hydrogel film was created by exchanging water with a miscible mixture of volatile and non-volatile liquid electrolytes followed by the volatile liquid removal using vacuum evaporation. The graphene sheets in the films stacked in a nearly face-to-face fashion, and the thickness were nearly proportional to the volumetric fraction of incorporated non-volatile liquids trapped in the gels. The cross-sectional SEM images (Fig. 7d-ii and iii) revealed that the electrolyte-mediated chemically converted graphene (EM-CCG) films had a rather uniform multi-layered microstructure.
Multi-layered graphene sheets with aligned in-plane pores can also facilitate ion transfer through the 2D plane of graphene. Instead of having random in-plane pores, the 3D structure contains well-aligned graphene sheets with holes (Fig. 7e). Xu et al. reported a 3D holey graphene framework (HGF) with a hierarchical porous structure as a high-performance binder-free supercapacitor electrode.111 The HGFs were prepared through a one-step process with simultaneous low temperature etching of nanopores in graphene and self-assembly of graphene into 3D networks, as shown in Fig. 7e-i. A controlled amount of H2O2 aqueous solution was added into a well-dispersed GO solution. The mixture was sealed in a Teflon-lined autoclave and heated at 180 °C for 6 h to yield mechanically strong monolithic HGFs. During the hydrothermal process, GO sheets were reduced and self-assembled into hydrogels with an interconnected 3D macroporous network (Fig. 7e-ii) with the pore sizes ranging from sub-micrometres to several micrometres and the pore walls consisting of single- or few-layer graphene sheets. The creation of nanopores in holey graphene sheets can boost the SSA up to 1560 m2 g−1 with an excellent electrical conductivity of ∼1000 S m−1, which can be used as supercapacitors electrodes directly.
4. Applications
As mentioned in the previous sections, porous graphene-based materials are very attractive for efficient molecular separation, electrochemical energy storage and conversion devices. In the following sections, we will demonstrate several popular applications of porous graphene-based materials in water desalination, gas separation, bioseparation, supercapacitors, lithium–O2/ion/sulfur batteries and fuel cells. Moreover, the relationships between the critical parameters of the porous structures and the performance of these graphene-based devices will be analyzed, and conclusions and recommendations will be provided.4.1 Molecular separation
Various materials, such as polymers and ceramics (e.g. carbon nanotubes, zeolites) have been exploited as membranes for desalination. However, graphene-based materials have recently emerged as promising candidates with distinguished desalination characteristics. Pristine graphene is extremely impermeable to liquid and gases; therefore the possibilities to create pathways through the material are either the generation of nanopores in a monolayer of graphene or the stacking of GO sheets.
Molecular dynamics (MD) simulations conducted by Cohen-Tanugi et al. have predicted that monolayer graphene with sub-nanometre in-plane pores could effectively separate salt ions from water. They concluded that salt ion rejection could be controlled by chemical functionalization of the nanopores.20 Hydrophilic pores (28 Å2) provided higher water flux but lower salt rejection than that of hydrophobic pores (23 Å2). Theoretically, the porous graphene can reject salt ions with a water permeability of 66 L per cm2 per day per MPa, which is 2–3 orders of magnitude higher than that of commercially available reverse osmosis membranes. The work highlighted the potential of atomically thin, functionalized, porous graphene as a highly permeable desalination membrane.
Free-standing graphene membranes were fabricated by Celebi et al. via drilling millions of pores with narrowly distributed diameters ranging from <10 nm to 1 μm using FIB, as shown in Fig. 4b.23 Attributed to the ultimate atomic thickness, the nanoporous graphene membranes show orders of magnitude enhancement permeability/selectivity of gas and water vapour, compared with the other membranes such as acrylic, cellulosics, and polysulfone, as shown in Fig. 8a.
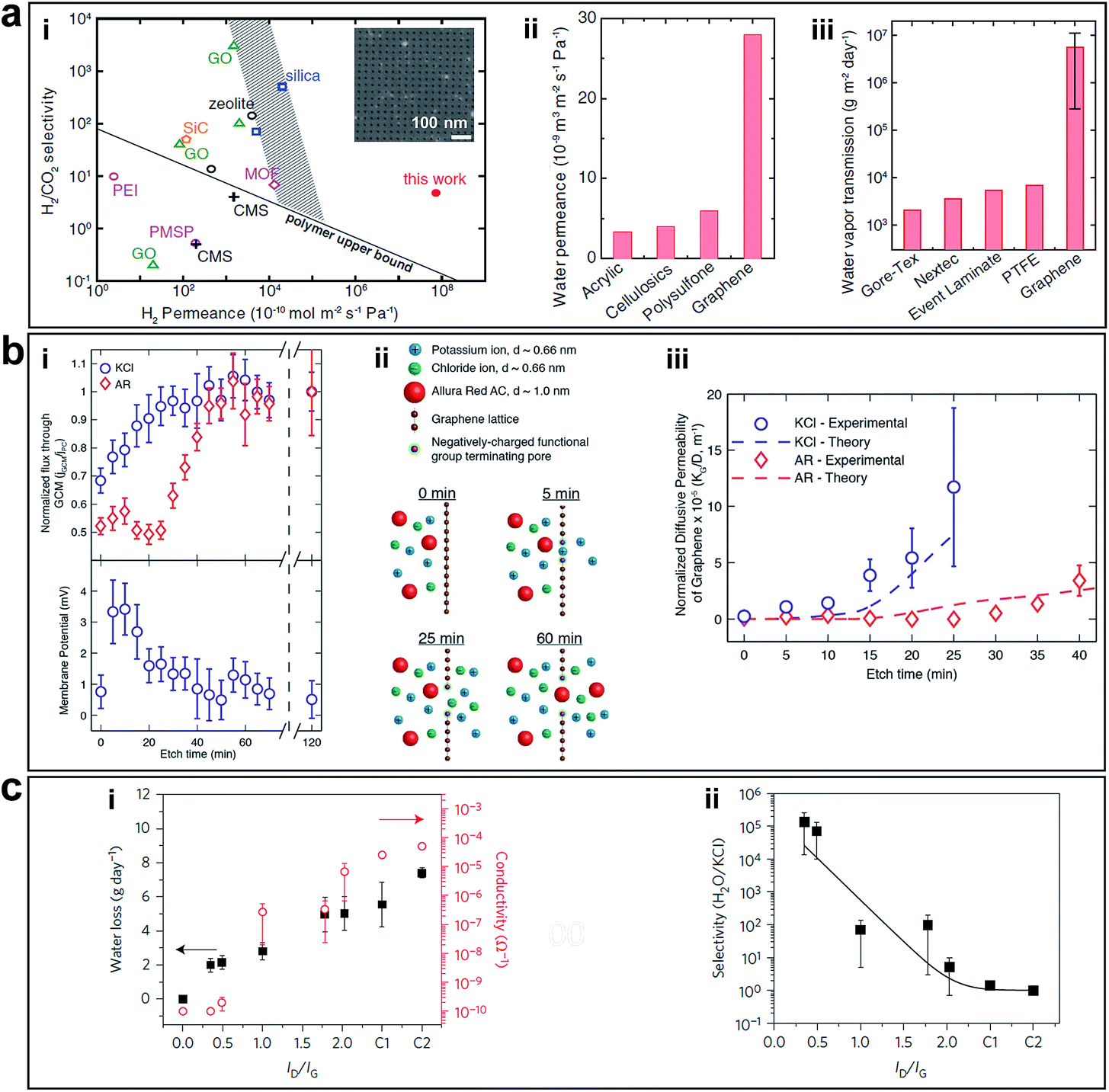 | ||
| Fig. 8 (a) Separation performance of nanoporous graphene obtained using FIB. (i) Comparison of H2/CO2 separation performance of nanoporous graphene (7.6 nm pore diameter, with 4.0% porosity) with other separation membranes. The inset shows an SEM image of nanoporous graphene membrane. (ii) Water permeance values for porous graphene (50 nm pore diameter, with 4.7% porosity) and other ultrafiltration membranes. (iii) Comparison of water vapour transmission rate of porous graphene membranes (five 400 nm-pore membrane samples, with porosity ranging from 3.6 to 11.5%) and waterproof-yet-breathable textile membranes. Reproduced with permission.23 Copyright 2014, American Association for the Advancement of Science. (b) Separation performance of nanoporous graphene obtained by ion bombardment followed by chemical oxidative etching. (i) Diffusion flux versus etching time through nanoporous graphene membrane normalized by the flux at 120 min etch time and the corresponding membrane potential measurements (0.5 M KCl/0.1667 M KCl). (ii) Schematics of selective transport through graphene membranes at various etching times. (iii) Comparison of the normalized diffusive permeability calculated from experimentally measured diffusive transport and the theoretical permeability based on the pore size distribution from STEM imaging suggests that the created pores are responsible for the selective nature of the membrane. Reproduced with permission.22 Copyright 2014, American Chemical Society. (c) Desalination performance of nanoporous graphene obtained using oxygen plasma etching. (i) Water loss after 24 h and ionic conductivity through nanoporous graphene membranes exposed to O2 plasma for various times. C1 and C2 are controls with large tears and completely broken membranes. (ii) Water/salt selectivity as a function of ID/IG ratio in Raman spectra of nanoporous graphene membranes etched at various exposure times, which showed exceptionally high selectivity for a short etching time. Reproduced with permission.24 Copyright 2015, Nature Publishing Group. | ||
O'Hern et al. used ion bombardment to introduce defects on monolayer CVD grown graphene and enlarged these defects to in-plane pores via controlling chemical oxidative etching, which resulted in porous graphene possessing high-density nanopores with <0.2 nm size distribution (Fig. 4a).22 The pores formed in a short etching time are caution-selective, which is attributed to the electrostatic repulsion from the negatively charged functional groups (e.g., carboxyl groups) terminating the pore edges. In a longer etching time, the steric size exclusion effect dominated the permeability of the membrane, allowing transport of salt but prevented transport of larger organic molecules (Fig. 8b). The ability to tune the selectivity of graphene through controlled generation of sub-nanometer pores addresses a significant challenge in the development of advanced nanoporous graphene membranes for desalination, gas separation, and other applications. Following this, experimental work using nanoporous graphene in water desalination was conducted by Surwade et al.24 They created pores with a nanometre-size in a monolayer of graphene via oxygen plasma etching (Fig. 4c) and showed that nanoporous graphene membranes exhibit a salt rejection of almost 100% while allowing for fast water transport (Fig. 8c), which indicates that nanoporous graphene membranes are definitely favourable for water desalination.
In summary, the above examples indicate that by controlling the sizes, and chemical functional groups at the edges of nanopores in graphene, the resulting nanoporous graphene membranes with in-plane pores afford excellent selectivity in water desalination. In order to realize economic and industrial-scale nanoporous graphene membranes for water desalination, the CVD approach is suitable for growing monolayers of graphene with large area and high quality. However, a more advanced non-destructive transfer method needs to be developed to avoid wrinkles and cracks, which inevitably degrades the mechanical strength of graphene.37,97 Therefore, it is critical to develop a novel method to realize the defect-free transfer of large-area CVD graphene onto porous substrates.
GO has a high density of OCFGs and few vacancy defects distributed in the carbon lattice. It can be produced on a large scale using a facile way. GO membrane for separation was first developed by Nair et al., and they found that sub-micrometer thick GO membranes allow a unique water permeation pathway but selectively hinder the motion of gases and non-aqueous solutions, as shown in Fig. 9a-i and ii.10 The ultrafast water permeation due to the formation of sp2 nanocapillaries within GO laminates, through which water molecules experienced an ultralow friction (Fig. 9a-iii). After that, there were several attempts towards the advancement of GO based membranes. Joshi et al. studied the permeation of different ionic charges through micrometer-thick, free-standing GO membranes (Fig. 9b).11 They measured the permeability of various ions and organic molecules and figured out filtration is exclusively related to the hydration radii of particles and not dependent on the charge of ions passing through the system. However, when immersed in ionic solutions, GO nanosheets experienced hydration that subsequently increased the interlayered pores to ∼9 Å. In this case, any ionic species with a hydrated radius >4.5 Å was blocked, while ionic species with hydrated radius <4.5 Å passed through the hydrophobic channels.11 The above examples show that GO membranes have exceptional molecular permeation properties. However, their applications in ion sieving are limited by a permeation cut-off of ∼9 Å, which is larger than the diameters of hydrated ions of common salts. The cut-off is determined by the interlayered pores of ∼13.5 Å, typical for GO laminates that swell in water. Achieving smaller interlayered pores for the laminates immersed in water has proved to be a challenge. Abraham et al. used epoxy physical confinement to modulate interlayered pores, ranging from ∼9.8 to 6.4 Å (Fig. 9c-i), which provided a sieve size smaller than the diameters of hydrated ions.12 They achieved accurate and tunable ion sieving. Permeation rates decreased exponentially with decreasing sieve size but water transport was affected weakly (by a factor of <2), as shown in Fig. 9c-ii and iii.
 | ||
| Fig. 9 (a) Permeation properties of GO membranes. (i) He permeations through a freestanding sub-micrometer-thick GO membrane and a polyethylene terephthalate (PET) film. (ii) Weight loss for a container sealed with a GO film (thickness ≈ 1 μm; aperture's area ≈ 1 cm2). No loss was detected for ethanol, hexane, etc., but water evaporated from the container as freely as through an open aperture (blue curves). (iii) Schematic diagram of the permeation through GO laminates. Reproduced with permission.10 Copyright 2012, American Association for the Advancement of Science. (b) (i) Photograph of a GO membrane and schematic of the experimental setup. (ii) Permeation through a 5 μm-thick GO membrane from the feed compartment with a 0.2 M solution of MgCl2. (Inset) Permeation rates as a function of concentration in the feed solution. (iii) Solute size sieving through the GO membrane (1 M feed solution). Reproduced with permission.11 Copyright 2014, American Association for the Advancement of Science. (c) Permeation properties of epoxy physically confined GO (PCGO) membranes. (i) Humidity-dependent interlayer spacing. (ii) Permeation rates through PCGO membranes with different interlayer distances. (iii) Permeation rates of K+ and Na+ depend exponentially on the interlayer distance (left axis). Water permeation varied only linearly with d (blue squares, right axis). Reproduced with permission.12 Copyright 2017, Nature Publishing Group. | ||
In order to achieve tunable interlayered pores in a relatively large range in GO membranes, nanoparticles or nanowires with various sizes can be intercalated between GO sheets. As shown in Fig. 6d, Huang et al. prepared nanostrand-channelled GO (NSC-GO) membranes with numerous interlayered pores with diameters of 3–5 nm and superior separation performance of small molecules and ultrafast water permeation (695 ± 20 L m−2 h−1 bar−1), as shown in Fig. 10a.15 The water flux increased by 10 times without sacrificing the rejection rate compared with pristine GO membranes because of highly porous structure and remarkably reduced channel length. Additionally, these newly formed and well-defined interlayered pores also exhibited excellent mechanical properties when pressure is applied (Fig. 10b). Note that the elastic deformation of the nanochannels depending on applied pressure leads to an abnormal phenomenon in separation performance, providing enormous possibilities for controlling permeance and the rejection rate, particularly in pressure-driven separation application. Moreover, the deformed interlayered pores can quickly be restored to their original state based on MD simulation results, as shown in Fig. 10c.15
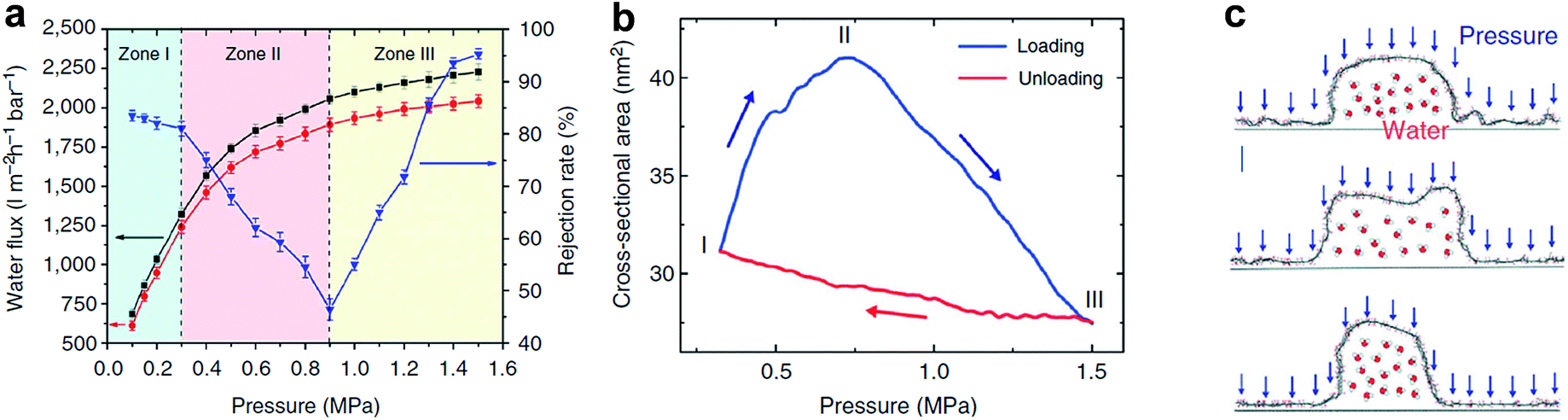 | ||
| Fig. 10 Response of NSC-GO membranes to the applied pressure. (a) Pressure-dependent flux and rejection of EB molecules of the NSC-GO membrane under different pressures. The black solid square and red solid circle curves represent the flux variation during the first and third pressure-loading processes, respectively. The blue solid triangle curve denotes the rejection rate of the EB during the first pressure-loading process. (b) Simulated changes in the cross-sectional area of the nanochannel by varying the applied pressure. (c) The response of a half cylindrical GO nanochannel modelled in MD simulation. Reproduced with permission.15 Copyright 2013, Nature Publishing Group. | ||
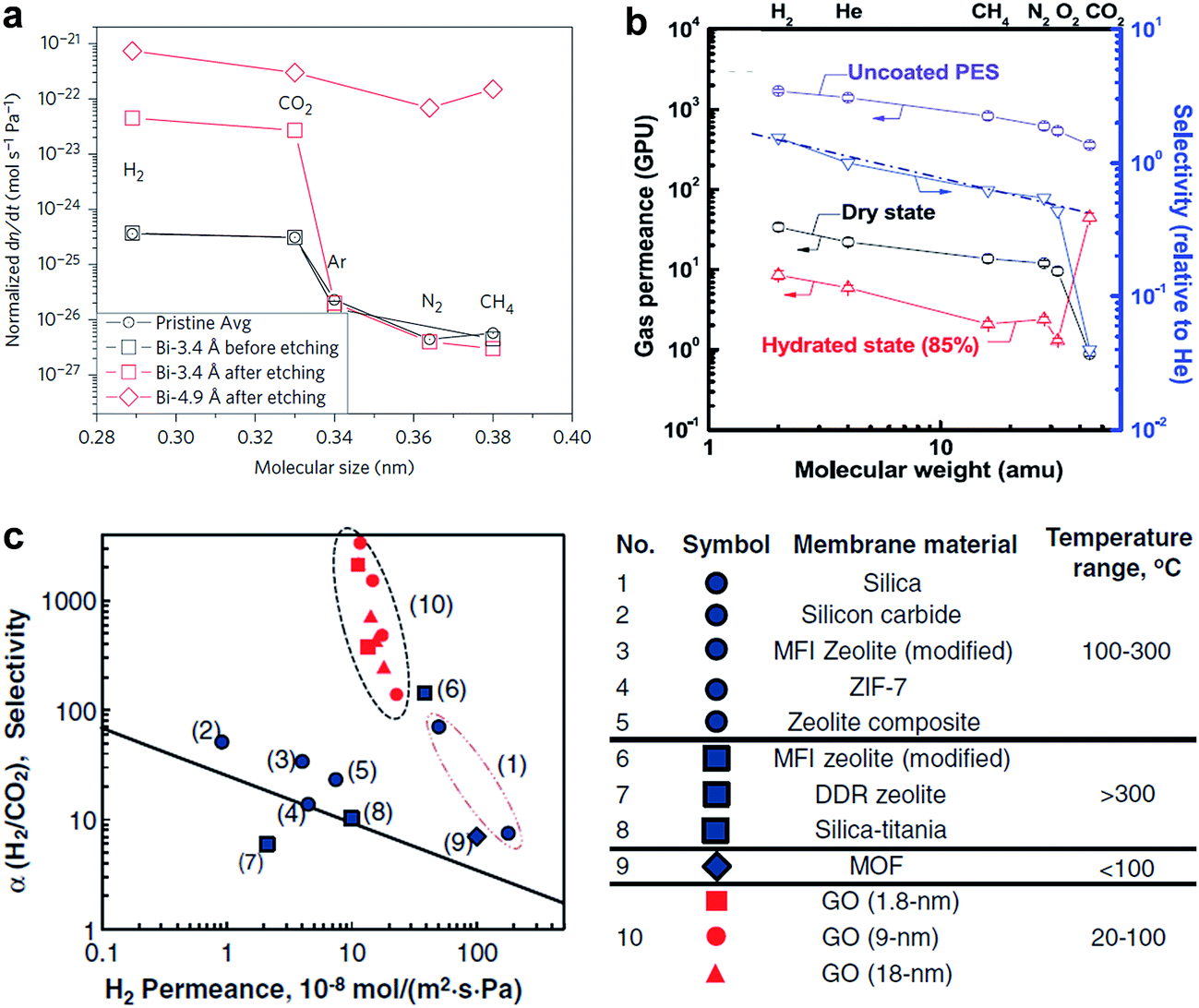 | ||
| Fig. 11 (a) Compilation of measured leak rates. Leak rates out of the microcavity for the Bi-3.4 Å membrane before etching and after etching, the Bi-4.9 Å membrane after etching. Reproduced with permission.21 Copyright 2012, Nature Publishing Group. (b) Gas permeances of GO membranes as a function of molecular weight. (c) Relations between H2 permeability and H2/CO2 selectivity of the GO membranes in comparison with other existing separation membranes. (b) Reproduced with permission.26 (c) Reproduced with permission.27 Copyright 2013, American Association for the Advancement of Science. | ||
GO membranes also show excellent selectivity for various gas mixtures. Kim et al. showed that selective gas diffusion depended on different GO stacking modes.26 They revealed the well-interlocked few layered GO membranes (∼5 nm) can afford high CO2/N2 selectivity in a high hydrated state (Fig. 11b). Li et al. prepared ultrathin (∼1.8 nm) GO membranes by a filtration approach and showed that these membranes can provide separation selectivity as high as 3400 and 900 towards H2/CO2 and H2/N2 mixtures, respectively (Fig. 11c).27 Both figured out that the high gas selectivity was due to the structural defects within GO. But Nair et al. showed that GO membranes are impermeable to vapours, and gases, except water.10 These seemingly inconsistent results was due to the different thickness of GO membranes. In the work by Kim et al. and Li et al., few layers of GO flakes were stacked to form the GO laminates and the continuous 2D nanocapillary networks were not formed. Selective gas permeation occurred mostly through the structural defects (e.g. tears and holes). While, in the work by Nair et al., sub-micrometer-thick (∼1 μm) GO membranes was used, in which GO flakes were stacked together, and continuous nanocapillary networks were formed, which were mainly responsible for water molecule transport.
In summary, by creating in-plane pores in monolayer graphene, it could be used as an effective separation membrane due to its chemical and mechanical stability, flexibility and atomic thickness. The separation performance of GO membranes can be easily affected by the state of OCFGs on the GO sheets, which provide much more straightforward strategies to tune the interlayered pores of GO. Indeed, in-plane pores and interlayered pores have an important role in molecular separation, since they can provide little resistance to flow, yet block the passage of species that are larger than the pores. From the examples, porous graphene-based materials worked as an efficient ultrafast membrane with high permeability and selectivity based on their unique properties than state-of-the-art membranes.23,26,27,37
Graphene provides one of the best choices for DNA sequencing with its remarkable properties satisfying the stringent requirements. Fig. 12a shows a DNA molecule passing through a nanoporous graphene membrane. The changes in ionic current or transverse conductance of the nanoribbon as DNA passing through the nanopore can be used to identify the sequence of bases in the DNA.119,121 Fischbein et al. first fabricated nanopores (single and arrays) in graphene membranes (Fig. 3a and b), and conducted sensing experiments, in which ionic current blockage was detected while passing through molecules.90 Subsequently, three groups led by Colovchenko at Harvard University,123 Dekker at the Delft University of Technology116 and Drndic' at the University of Pennsylvania124 all detected the translocation of individual single-stranded or double-stranded DNA chains through nanopores graphene. Only one nucleotide can reside in the ultrathin graphene pore at a time, so it should be possible to sequence by observing the translocation of consecutive nucleotides through the nanopore. Recently, Garaj et al. fabricated nanopores in monolayer graphene that matched the diameter of a double-stranded DNA molecule.125 Ionic current signals during electrophoretically driven translocation of DNA through these nanopores were experimentally studied and theoretically modelled. These nanopores usually have high sensitivity (0.65 nA Å−1) to extremely small changes in the translocating molecule's outer diameter. Such graphene nanopores with atomic-size can also resolve nanoscale-spaced molecular structures along the length of a polymer. But high sensitivity can only be achieved when the pore and molecule diameters are well matched. Simulation indicated the most closely matched pores have an inherent resolution of ≤0.6 nm along the length of the molecule. However, in these cases, only nonfolded, partially or fully folded DNA could be distinguished. Until now, DNA sequencing with nanoporous graphene-based devices has not been demonstrated in reality. There still exist at least two main theoretical challenges. (1) The structural similarity of purines and pyrimidines limits the identification of nucleobases by monitoring ionic currents. (2) DNA molecule translocates through the nanopores (∼30 base per μs) at a fast speed, which is difficult for instruments to ‘read’ the signals of individual bases. Recently, MD simulations conducted by Wells et al. have indicated DNA sequencing is realizable by measuring the ionic current blockades in nanoporous graphene (Fig. 12b).122 Recent progress in slowing down DNA translocation via nanoporous graphene paved a viable way towards DNA sequencing.126 Nanoporous graphene can also be used for real-time sensing of single molecules.8,127
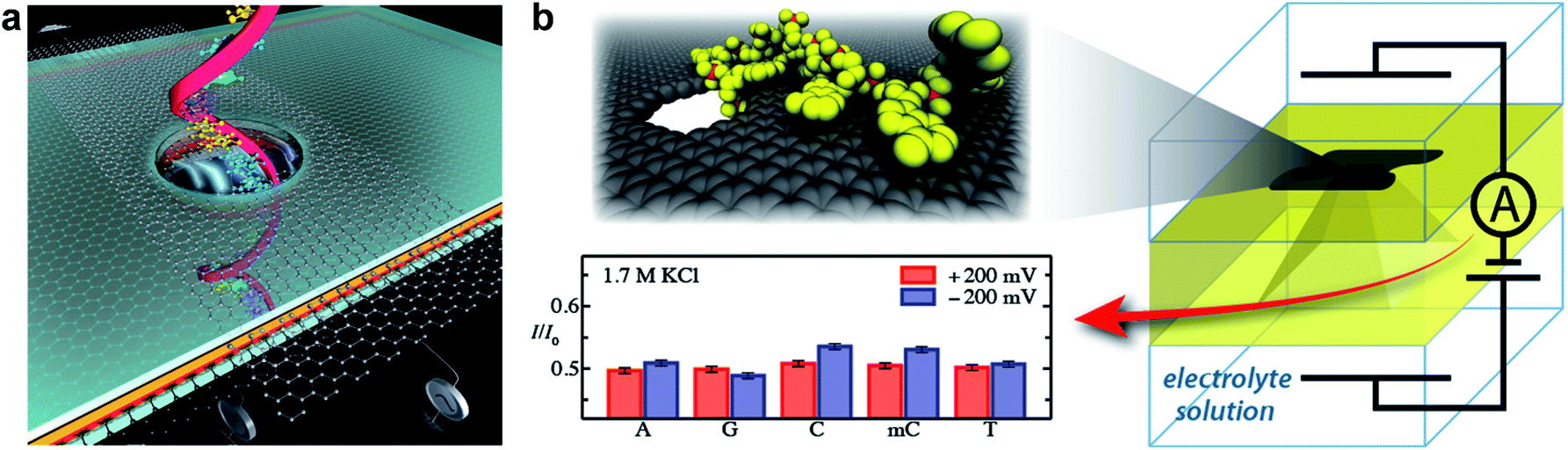 | ||
| Fig. 12 (a) Schematic showing DNA passing through a solid-state nanopore with an embedded graphene gate and a graphene nanoribbon on the membrane containing the nanopore; both the gate and the nanoribbon contain circular openings for the DNA to pass through. The graphene gate could be used to induce either p-type or n-type behaviour in the nanoribbon and to electrostatically slow down DNA. Reproduced with permission.119 Copyright 2011, Nature Publishing Group. (b) Schematic of a graphene nanopore-based device for sequencing DNA. As the DNA transits the nanopore, the ionic current is modulated by the nucleotides in the nanopore, revealing the DNA sequence. Reproduced with permission.122 Copyright 2012, American Chemical Society. | ||
4.2 Energy storage
In principle, graphene has a theoretical SSA of 2675 m2 g−1 and an electric double layer capacitance of 550 F g−1.46,47,133,134 However, the physically measured values are much lower than the theory. Earlier reports using chemically modified graphene as the electrode exhibited the specific capacitance of 130 F g−1 and 99 F g−1 with aqueous and organic electrolytes, respectively.51 Furthermore, specific capacitance values reported by other graphene-derivatives were also much lower than expected. One major reason for this is aggregation and restacking between the individual graphene sheets driven by strong inter-planar π–π interactions and van der Waals forces during the fabrication process.47 This spontaneous graphene layer restacking reduces the SSA of the graphene membranes and the diffusion of electrolyte ions, which strongly decreases the practical surface available for charge storage.45–47,49 Therefore, various strategies have been devoted in order to increase SSA and promote transport of electrolyte ions. The naturally existing 3D aggregate of graphene into porous graphene structures is an effective approach with which to counter the stacking behaviour of graphene sheets and to obtain graphene materials with high SSAs.
Recently, Zhu et al. reported a facile chemical activation method to produce high-quality activate microwave exfoliated GO (a-MEGO) with numerous micropores, extremely large SSA (∼3100 m2 g−1) of highly curved graphene walls that prevent restacking during cycling, high electrical conductivity of 500 S m−1, and low oxygen content (C/O atomic ratio ∼35), as shown in Fig. 7b-i.51 Cyclic voltammetry (CV) showed rectangular curves from 0 to 3.5 V over a wide range of voltage scan rates. The specific capacitances achieved as a result of the porous GO were as high as 165, 166, and 166 F g−1 at current densities of 1.4, 2.8, and 5.7 A g−1, respectively, as shown in Fig. 13a-i and ii. The large gravimetric capacitance was proposed to be resulted from the sp2 bonded carbon with a continuous 3D network of highly curved, atomic-thin graphene walls that form 0.6–5 nm pores and the low oxygen and hydrogen content.
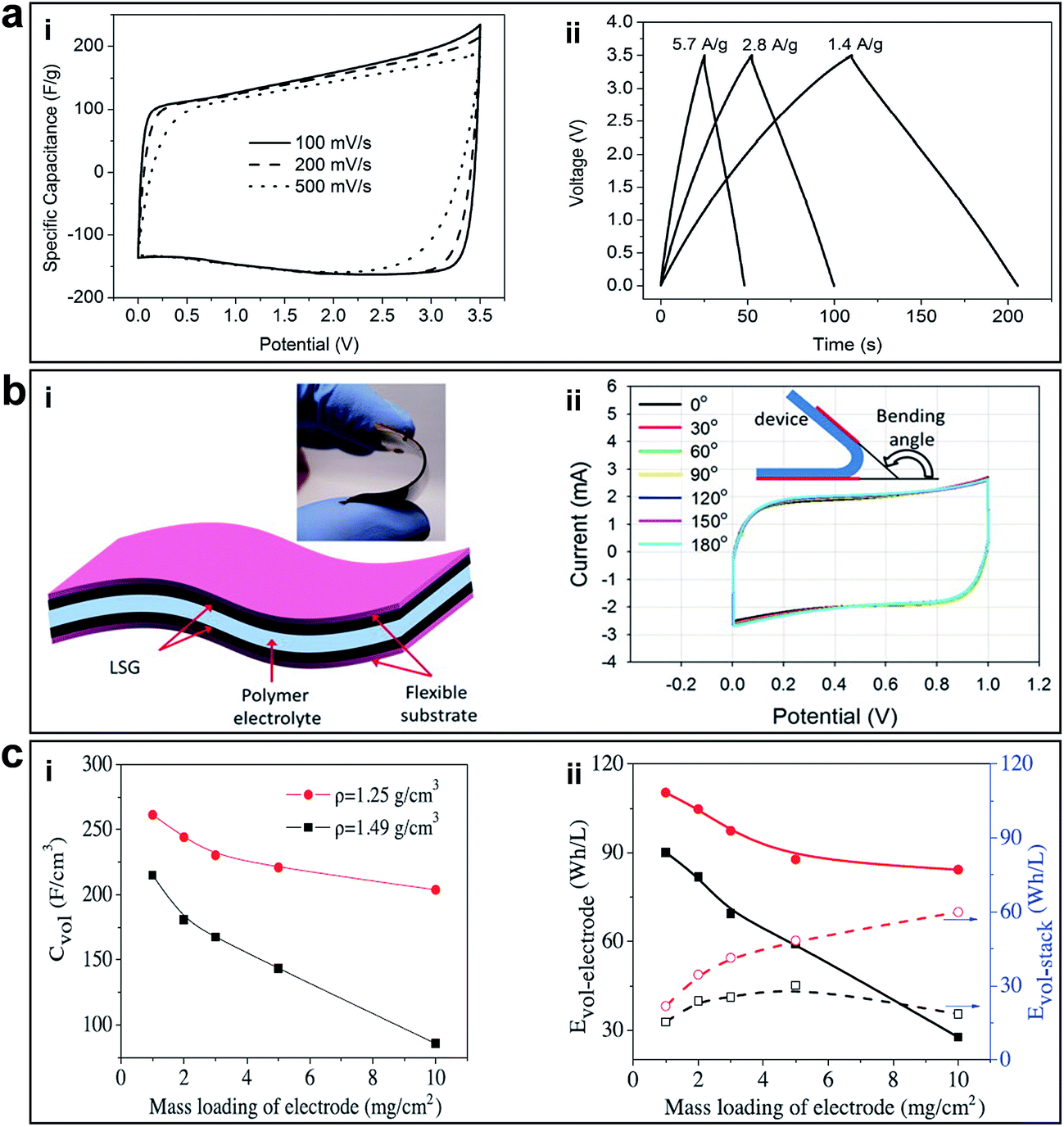 | ||
| Fig. 13 (a) Supercapacitor performance of a-MEGO in the BMIM BF4/AN electrolyte. (i) CV curves for different scan rates. Rectangular shapes indicate the capacitive behavior. (ii) Galvanostatic charge/discharge curves of a-MEGO-based supercapacitor under different constant currents. Reprinted with permission.51 Copyright 2011, American Association for the Advancement of Science. (b) Supercapacitor performance of LSG. (i) A schematic diagram of the LSG supercapacitor illustrates that the gelled electrolyte can serve as both the electrolyte and the separator. (Inset) A digital photograph showing the flexibility of the device. (ii) Bending the device has almost no effect on its performance. Reproduced with permission.41 Copyright 2012, American Association for the Advancement of Science. (c) Volumetric performance of capacitors based on EM-CCG film electrodes. (i) Volumetric capacitance and (ii) energy density as a function of the areal mass loading of an EM-CCG film (1.25 g cm−3) and a dried CCG film (1.49 g cm−3) at a current density of 0.1 A g−1. Reproduced with permission.53 Copyright 2013, American Association for the Advancement of Science. | ||
In order to further improve the energy density and large-scale application of flexible supercapacitors, El-Kady et al. reported a simple solid-state laser scribed strategy to fabricate 3D porous graphene electrodes via a LightScribe DVD optical drive (Fig. 7a-i).41 The network structure with open pores and the large SSA (1520 m2 g−1), high electrical conductivity (1738 S m−1) was optimized for electrolyte ionic diffusion in LSG electrodes, which resulted in sizeable charge storage capacity and generated high power. All-solid-state supercapacitors were assembled using two identical LSG electrodes and combined with a PVA–H3PO4 polymer gel electrolyte. In order to evaluate the potential of this flexible supercapacitor under real conditions, mechanical strain was exerted on the device and the performance was tested. The CV curves tested under different bending conditions are shown in Fig. 13b-i and ii. The bending had negligible effects on the capacitive behaviour; it can be bent arbitrarily without degrading performance. Furthermore, the stability of the device was tested for more than 1000 cycles while in the bent state, with only 5% change in the device capacitance. The flexible LSG supercapacitor maintained 97% of the initial capacitance after 10000 cycles. Moreover, the ionic liquid EMIMBF4 was used as the electrolyte in order to broaden the potential window. As a result, the LSG supercapacitor exhibited an area specific capacitance as high as 5.02 mF cm−2 (gravimetric specific capacitance of 276 F g−1) in a wider potential window of 4 V and a superior volumetric energy density of 1.36 mW h cm−3, approximately two times higher than that of a commercial activated-carbon supercapacitor. Given the simplicity, cost-effectiveness, scalable synthesis, high energy storage capacity and flexibility, these LSG supercapacitors are expected to be promising candidates for flexible energy storage devices.
Highly conductive graphene sheets are good building blocks for constructing sandwich-like porous carbon layer/graphene hybrids. Porous 3D graphene-based bulk materials with exceptional high SSA (3523 m2 g−1) and excellent conductivity (up to 303 S m−1) were fabricated, as shown in Fig. 7c-i.52 Due to the defected and wrinkled graphene sheets in the dimensional size of 4–6 nm, these materials showed superior supercapacitor performance in ionic liquid with a specific capacitance and energy density of 231 F g−1 and 98 W h kg−1, respectively.
In terms of advancement made on the energy density increasing for supercapacitors, another important work worth mentioning is the porous yet densely packed carbon electrodes with high ion-accessible SSA and low ion transport resistance reported by Yang et al., as shown in Fig. 7d.53 The packing density of these flexible EM-CCG films can be controlled from 0.13 to 1.33 g cm−3 by changing the ratio of volatile and non-volatile liquids. The packing density of completely dried CCG films was 1.49 g cm−3. After being fabricated into supercapacitors, the highly compact EM-CCG films (density from 1.25 to 1.33 g cm−3) yielded a high specific capacitance of 255.5 F cm−3 in an aqueous electrolyte and 261.3 F cm−3 in an organic electrolyte at a current density of 0.1 A g−1 with a maximum energy density of 59.9 W h L−1 (Fig. 13c-i and ii). These were among the highest capacitances achieved using pure carbon materials. These results also suggested that the key technical specifications of CCG-based electrochemical capacitors could be customized to satisfy different applications by simply tuning the packing density of the CCG electrodes.
Graphene sheets with in-plane pores can also speed up the ion transport across the 2D plane of graphene. Graphene sheets in a 3D network are highly interconnected and interlocked together to prevent them from restacking and to maintain a highly porous monolithic graphene framework. Xu et al. reported a 3D HGF with a hierarchical porous structure as a high-performance binder-free supercapacitor electrode. A HGF was prepared through one-step process with a low temperature H2O2 etching of nanopores in graphene and self-assembly of graphene into 3D monolithic network (Fig. 7e-i). A 140 μm HGF electrode with a high packing density of 0.71 g cm−3 was synthesized by compressing a piece of 1 cm thick HGF with a packing density of 12 mg cm−3. With large ion-accessible SSA, efficient electron and ion transport pathways as well as high packing density, the HGF electrode can deliver a gravimetric capacitance of 298 F g−1 and a volumetric capacitance of 212 F cm−3 in organic electrolytes. Furthermore, they showed that a fully packaged device stack can deliver gravimetric and volumetric energy densities of 35 W h kg−1 and 49 W h L−1, respectively, approaching those of lead acid batteries. The nanopores in holey graphene sheets are large enough to function as the ion diffusion shortcuts between different layers of graphene to greatly accelerate the ion transportation across the entire film and facilitate ion access to the entire surface area, which is not possible with non-holey graphene frameworks.
To give a brief summary, nanoporous graphene-based materials used as electrodes can exhibit exciting characteristics, such as high electrical conductivity, enhanced SSA, high capacitance, excellent rate capability and stability. Graphene-based materials can serve as mechanically flexible scaffolds for constructing high performance flexible electrodes.6,45,135 The 3D porous graphene networks with both in-plane and 3D interconnected pore structures suppress agglomeration/re-stacking of the graphene sheets and ensure good electrical conductivity of the electrodes. Although many remarkable achievements have been made in graphene-based supercapacitors, there is still significant work outstanding.45,47,54 (1) Reasonably design and generate the 3D porous structures inside the supercapacitor electrodes to form hierarchical interconnected porous networks and avoid the collapse of the porous structures. (2) Control the interfacial interactions between graphene nanosheets and pseudo-capacitive materials to achieve a well-designed flexible structure with high electrochemical performance. (3) Deep understanding of the energy storage mechanism with an aim to increase energy density, high capacitance, excellent rate and cycling stability towards practical applications.
Li–O2 batteries (LOBs) are considered to be one of the most promising candidates as next generation energy storage systems due to their high theoretical specific energy (∼3500 W h kg−1).139 The requirements of an O2 electrode of the LOBs include SSA, high electrical conductivity and strong electrocatalytic activity to the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER). Nanoporous graphene-based materials can be used in the LOBs, demonstrating the versatility of graphene-based materials.139–142 Zhang et al. reported a facile and controllable way to synthesize nanoporous graphene (graphene foams) by the electrochemical leavening process of graphite paper in aqueous solution of Na2SO4 and used these graphene foams as O2 electrodes of LOBs. It was found that the defects were detrimental to the electrochemical performance of the graphene foam electrodes in the LOBs. Higher defect level (ID/IG value) samples exhibited weakened round-trip efficiencies, cycling stabilities and generated more side products. The graphene foams with a high ID/IG value of 0.71 depicted a round-trip efficiency of only 0.51 and a 20th-cycle discharge capacity of only 340 mA h g−1 at a current density of 100 mA g−1. However, the graphene foam electrode annealed at 800 °C with ID/IG = 0.07 delivered a roundtrip efficiency of up 80% with a stable discharge voltage at ∼2.8 V and a stable charge voltage below 3.8 V for 20 cycles. The superior electrochemical properties of graphene foams can be ascribed to the 3D structure and the lower amount of structural defects. The structural defects in the nanoporous graphene are detrimental to the structural stability of the O2 electrode and can slow down the kinetics of charge transfer. This degrades the electrochemical performance of oxygen electrodes. Moreover, these defects lead to the faster growth of side products of the LOBs during cycling, which further deteriorates the electrochemical performance.
Graphene materials with 3D porous structures are favourable for lithium-ion batteries (LIBs).6,54,57,136,143,144 As showed in one typical case, macroporous rGO aerogel loaded with Fe3O4 particles showed a SSA of 95.2 m2 g−1 and pore size distribution ranging from 2 to 100 nm with the pore volume of 0.28 cm3 g−1, standing in contract to commercial Fe3O4 particles (∼2 m2 g−1).143 The rGO/Fe3O4 hybrid showed a high capacity of ∼990 and ∼730 mA h g−1, respectively. The robust 3D graphene framework provided highly conductive networks with large SSA and short diffusion length for the transport of lithium ions.6,54
The separator is a crucial component of electrochemical energy-storage devices, which is a permeable membrane placed between the anode and cathode of a battery.58 An ideal battery separator should be perm-selective, which means it should allow specific species to transport while restricting the passage of other species that can reduce the reversibility of the battery. GO membrane with negative charges is expected to be permeable for lithium ions but reject anions.55 Compared to conductive carbon interlayers that serve as an additional carbon scaffold for physical barrier, GO membranes are able to block polysulfide by electrostatic repulsion and steric exclusion, which are more efficient in long-term inhabitation of the shuttle effect. The unique mass-transport and highly tunable functionalization properties enable graphene-based membranes to act as blocking barriers in lithium–sulfur batteries.37 Recently, Huang et al. applied the ultrathin GO membranes in lithium–sulfur battery systems (Fig. 14a) and found that the lithium ions can easily transport through cationic perm-selective GO membranes, whereas the polysulfides were largely inhibited in the cathode side.55 With the incorporation of a perm-selective GO membrane (loading amount of 0.12 mg cm−2), the lithium–sulfur batteries afforded an improved coulombic efficiency from 67–75% to over 95–98% in 100 cycles at 0.1C, and a slightly higher initial discharge capacity of 920 mA h g−1 was obtained (Fig. 14b and c). The overcharge phenomenon in the cell with the GO membrane was greatly reduced (Fig. 14d). As the GO membrane blocked the diffusion of polysulfide, it was also with merits of anti-self-discharging properties. Compared to the fast open circuit voltage drop of cells with a routine membrane within 5.0 h, the cell with the GO membrane presented a stable open circuit voltage of 2.5 V after 30 h (Fig. 14e). The perm-selectivity of lithium ions over polysulfide anions resulted from both the physical inhibition effect and the chemical effect with oxygenated functional groups on the GO flakes. Additionally, this work also offers a general strategy to combine the GO membrane with a rechargeable battery, which is crucial for illustrating the potential of a functional membrane for energy storage and revealing the dynamic changes on the electrode.55
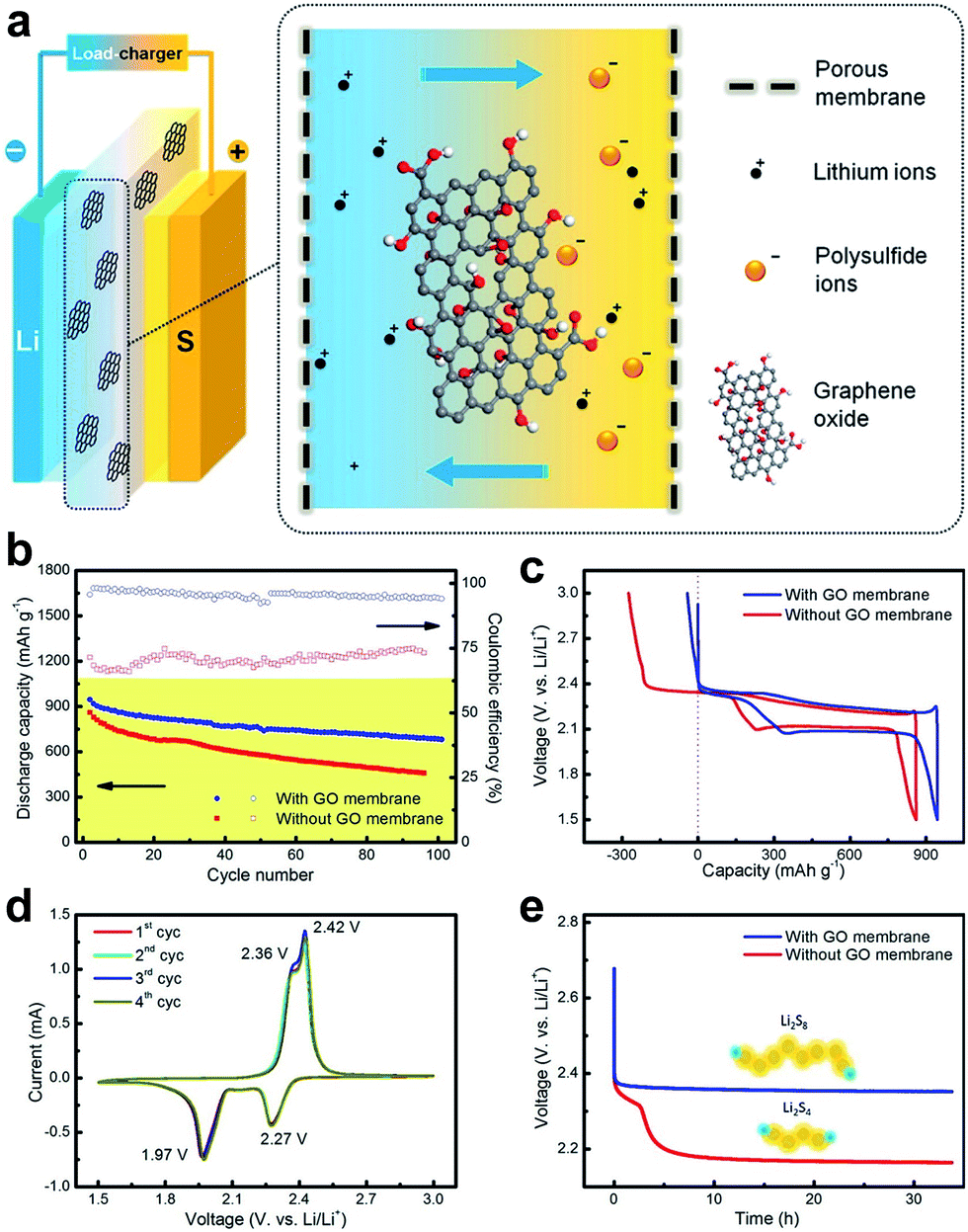 | ||
| Fig. 14 Schematic of a GO membrane incorporates a lithium–sulphur battery and electrochemical performance of the GO-membrane-incorporated lithium–sulphur batteries. (a) GO membrane was sandwiched between cathode and anode electrodes, which may efficiently prohibit the shuttle of polysulfides through the membrane. (b) Cycling performance at a rate of 0.1C with/without a GO membrane. (c) Galvanostatic charge–discharge profiles at a rate of 0.1C. (d) CV profiles with a GO membrane at a scan rate of 0.1 mV s−1. (e) Open circuit voltage profiles showing the self-discharge behavior. Reproduced with permission.55 Copyright 2015, American Chemical Society. | ||
Hatakeyama et al. showed the in-plane proton conductivities of multilayer GO films are thickness-dependent and higher than that of monolayer GO by several orders of magnitude, owing to the enhancement of conduction paths, hydration dynamics and water content.145,146 They concluded that epoxide groups make major contributions to efficient conductivity of proton. These results may lay a foundation for GO-based membranes in fuel cells. They further showed that the proton and electron conductivities of GO can be controlled via photo-irradiation and thermal reduction processes. The proton conductivity decreases when epoxide groups and interlayer distances decrease, whereas the electron conductivity drastically increases with decreasing oxygen content. Both the electron and proton conduction mechanisms for GO were discussed based on the concentrations of various functional groups and defects, changes in the interlayered pores, and the activation energy associated with proton conduction. Finally, a good mixed conductor of protons and electrons can be achieved by reducing GO to the most suitable degree, enabling GO to act as both an electrode and a hydrogen separation membrane that functions at the room temperature.
Recently, Gao et al. treated the GO dispersion with ozone to introduce a higher level of oxidation to GO flakes, and showed the applicability of GO as a proton exchange membrane for fuel cells.62Fig. 15 shows the ozonation, proton hopping in ozonated GO (OGO) flake, conductivity as function of relative humidity and a schematic of the polymer electrolyte fuel cell with the GO/OGO membrane.35 Using vacuum filtration to prepare free-standing OGO membranes, they found that the protonic conductivity of OGO was significantly improved compared to that of GO, which was due to the higher content of oxygenated functional groups and the morphology changes in GO caused by ozonation that provide more hopping sites for proton conduction. A series of hydrogen/air fuel cell tests to assess the feasibility were performed using freestanding OGO and GO films as proton-conducting membranes. It was found that the OGO membranes possess superior fuel cell performance compared to GO, due to the higher proton conductivity of OGO membranes.
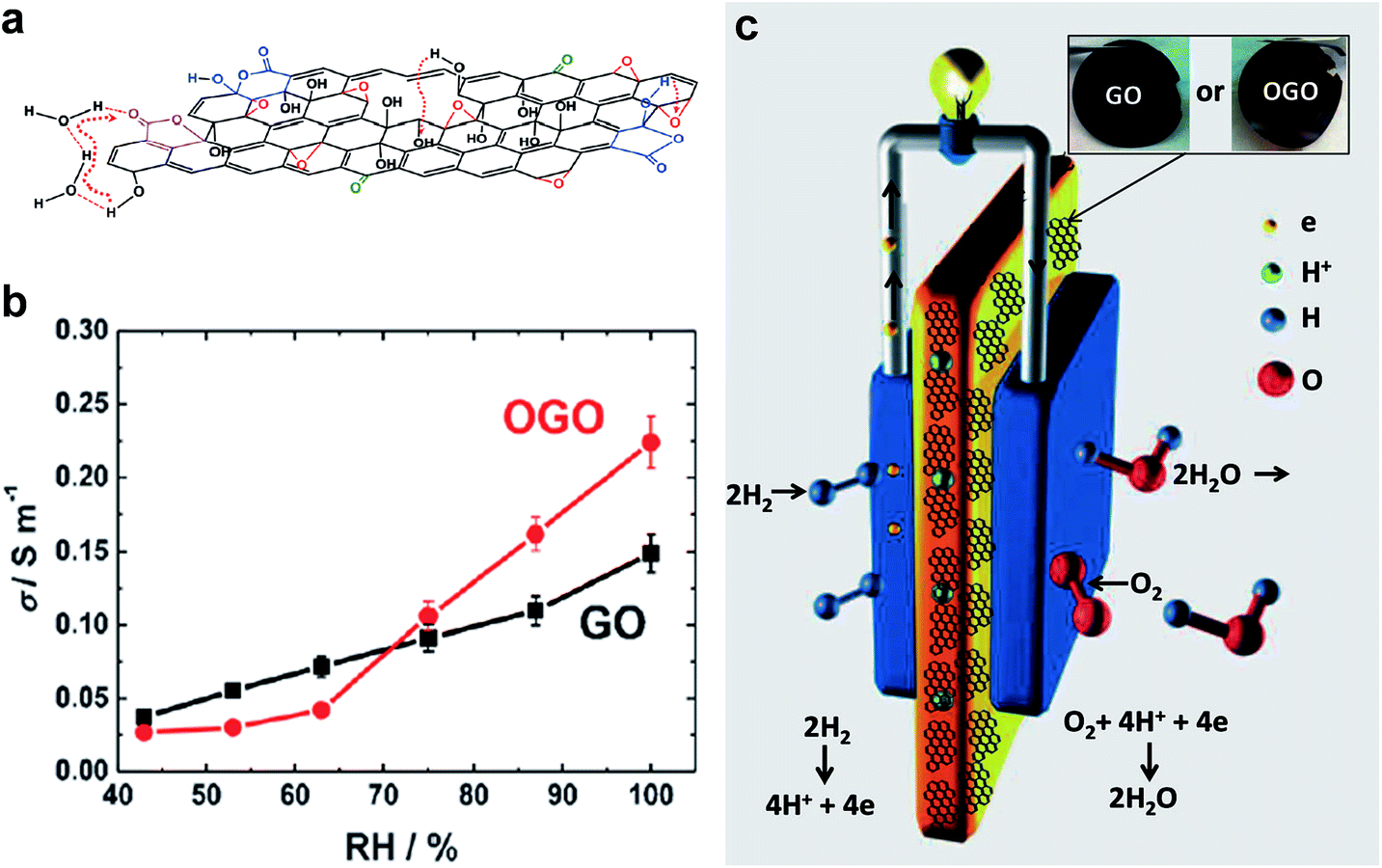 | ||
| Fig. 15 (a) Schematic shows the path of proton hopping in OGO flake. (b) Variation of conductivity with relative humidity for GO and OGO membranes. (c) Schematic of the polymeric electrolyte fuel cell with GO/OGO, inset shows the photographs of GO and OGO membranes. Reproduced with permission.62 Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
5. Conclusions and outlook
As summarized above, due to the unique porous structures, large surface areas and excellent properties, porous graphene-based materials have received significant attention and undergone rapid advancement in recent years. In this review, we have summarized the state-of-the-art pore generation and tuning methods, including both physical and chemical methods that have been used in graphene-based materials. Each method has its advantage and limitation for pore generation serving for specific applications. For molecular separation, electrochemical energy storage, etc., we have figured out that porous graphene structures are essentially important. The relationships between structures, properties and the applications of these materials have been revealed and discussed.Despite the significant advances in the synthesis and applications of porous graphene-based materials, several key challenges remain to be addressed. Firstly, in broad terms, low-cost and facile methods for future production of porous graphene-based materials with controllable structures (e.g. pore size and distribution) and large-areas are still challenging. It is also demanding to control the size and functional groups of graphene and its derivatives in order to tune the membrane structures and chemical properties. For monolayer graphene, femtosecond laser with high photon energy may provide a selective strategy for generating in-plane pores with high density. For GO laminates, whose interlayered pores are important for molecular separation, femtosecond laser can remove OCFGs and reduce GO to rGO to some extent, which enable precisely tuning the interlayered pores, especially realizing sub-nanometer channels. For 3D graphene porous networks, a cost-effective and efficient method should be developed to generate 3D porous graphene networks with large surface areas in a large scale to improve the power and energy density. Secondly, the combination of various pores (i.e. micropores, mesopores and macropores) is of vital importance for porous graphene-based materials in order to take advantages of the synergic effects of different sizes.6 Therefore, further real-life applications of porous graphene-based materials still require the proper realization of their structures.
Acknowledgements
Baohua Jia acknowledges the support from the Australia Research Council through the Discovery Project scheme (DP150102972).Notes and references
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2003, 15, 1049–1059 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- K. M. Thomas, Catal. Today, 2007, 120, 389–398 CrossRef CAS.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Adv. Mater., 2014, 26, 849–864 CrossRef CAS PubMed.
- D. Zhou and B. H. Han, Adv. Funct. Mater., 2010, 20, 2717–2722 CrossRef CAS.
- W. Yuan, J. Chen and G. Shi, Mater. Today, 2014, 17, 77–85 CrossRef CAS.
- P. Xu, J. Yang, K. Wang, Z. Zhou and P. Shen, Chin. Sci. Bull., 2012, 1–8 CAS.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T. Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K. Geim and R. R. Nair, Nat. Nanotechnol., 2017 DOI:10.1038/nnano.2017.21.
- M. Hu and B. Mi, Environ. Sci. Technol., 2013, 47, 3715–3723 CrossRef CAS PubMed.
- Y. Han, Z. Xu and C. Gao, Adv. Funct. Mater., 2013, 23, 3693–3700 CrossRef CAS.
- H. Huang, Z. Song, N. Wei, L. Shi, Y. Mao, Y. Ying, L. Sun, Z. Xu and X. Peng, Nat. Commun., 2013, 4, 2979 Search PubMed.
- P. Sun, M. Zhu, K. Wang, M. Zhong, J. Wei, D. Wu, Z. Xu and H. Zhu, ACS Nano, 2013, 7, 428–437 CrossRef CAS PubMed.
- H. Huang, Y. Mao, Y. Ying, Y. Liu, L. Sun and X. Peng, Chem. Commun., 2013, 49, 5963–5965 RSC.
- S. Blankenburg, M. Bieri, R. Fasel, K. Mullen, C. A. Pignedoli and D. Passerone, Small, 2010, 6, 2266–2271 CrossRef CAS PubMed.
- H. Du, J. Li, J. Zhang, G. Su, X. Li and Y. Zhao, J. Phys. Chem. C, 2011, 115, 23261–23266 CAS.
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS PubMed.
- S. P. Koenig, L. Wang, J. Pellegrino and J. S. Bunch, Nat. Nanotechnol., 2012, 7, 728–732 CrossRef CAS PubMed.
- S. C. O'Hern, M. S. Boutilier, J. C. Idrobo, Y. Song, J. Kong, T. Laoui, M. Atieh and R. Karnik, Nano Lett., 2014, 14, 1234–1241 CrossRef PubMed.
- K. Celebi, J. Buchheim, R. M. Wyss, A. Droudian, P. Gasser, I. Shorubalko, J. I. Kye, C. Lee and H. G. Park, Science, 2014, 344, 289–292 CrossRef CAS PubMed.
- S. P. Surwade, S. N. Smirnov, I. V. Vlassiouk, R. R. Unocic, G. M. Veith, S. Dai and S. M. Mahurin, Nat. Nanotechnol., 2015, 10, 459–464 CrossRef CAS PubMed.
- E. N. Wang and R. Karnik, Nat. Nanotechnol., 2012, 7, 552–554 CrossRef CAS PubMed.
- H. W. Kim, H. W. Yoon, S.-M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J.-Y. Choi and H. B. Park, Science, 2013, 342, 91–95 CrossRef CAS PubMed.
- H. Li, Z. Song, X. Zhang, Y. Huang, S. Li, Y. Mao, H. J. Ploehn, Y. Bao and M. Yu, Science, 2013, 342, 95–98 CrossRef CAS PubMed.
- B. Mi, Science, 2014, 343, 740–742 CrossRef CAS PubMed.
- Z. Zheng, R. Grunker and X. Feng, Adv. Mater., 2016, 28, 6529–6545 CrossRef CAS PubMed.
- J. Shen, M. Zhang, G. Liu and W. Jin, RSC Adv., 2016, 6, 54281–54285 RSC.
- A. R. Koltonow and J. Huang, Science, 2016, 351, 1395–1396 CrossRef CAS PubMed.
- M. Lozada-Hidalgo, S. Hu, O. Marshall, A. Mishchenko, A. N. Grigorenko, R. A. W. Dryfe, B. Radha, I. V. Grigorieva and A. K. Geim, Science, 2016, 351, 68–70 CrossRef CAS PubMed.
- J. Shen, G. Liu, K. Huang, Z. Chu, W. Jin and N. Xu, ACS Nano, 2016, 10, 3398–3409 CrossRef CAS PubMed.
- M. Fathizadeh, W. L. Xu, F. Zhou, Y. Yoon and M. Yu, Adv. Mater. Interfaces, 2017, 4, 1600918 CrossRef.
- Y. You, V. Sahajwalla, M. Yoshimura and R. K. Joshi, Nanoscale, 2016, 8, 117–119 RSC.
- G. Liu, W. Jin and N. Xu, Chem. Soc. Rev., 2015, 44, 5016–5030 RSC.
- P. Sun, K. Wang and H. Zhu, Adv. Mater., 2016, 28, 2287–2310 CrossRef CAS PubMed.
- H. W. Yoon, Y. H. Cho and H. B. Park, Philos. Trans. R. Soc., A, 2016, 374, 20150024 CrossRef PubMed.
- G. Liu, W. Jin and N. Xu, Angew. Chem., Int. Ed., 2016, 55, 13384–13397 CrossRef CAS PubMed.
- H. Huang, Y. Ying and X. Peng, J. Mater. Chem. A, 2014, 2, 13772–13782 CAS.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- J. Hou, Y. Shao, M. W. Ellis, R. B. Moore and B. Yi, Phys. Chem. Chem. Phys., 2011, 13, 15384–15402 RSC.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 CAS.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef CAS PubMed.
- W. Lv, Z. Li, Y. Deng, Q.-H. Yang and F. Kang, Energy Storage Materials, 2016, 2, 107–138 CrossRef.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- Y. Shao, M. F. El-Kady, L. J. Wang, Q. Zhang, Y. Li, H. Wang, M. F. Mousavi and R. B. Kaner, Chem. Soc. Rev., 2015, 44, 3639–3665 RSC.
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 CrossRef.
- A. Ambrosi, C. K. Chua, N. M. Latiff, A. H. Loo, C. H. Wong, A. Y. Eng, A. Bonanni and M. Pumera, Chem. Soc. Rev., 2016, 45, 2458–2493 RSC.
- B. Xu, S. Yue, Z. Sui, X. Zhang, S. Hou, G. Cao and Y. Yang, Energy Environ. Sci., 2011, 4, 2826 CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 CrossRef PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- Y. Dong, Z.-S. Wu, W. Ren, H.-M. Cheng and X. Bao, Sci. Bull., 2017, 62, 724–740 CrossRef.
- J.-Q. Huang, T.-Z. Zhuang, Q. Zhang, H.-J. Peng, C.-M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS PubMed.
- P. Sun, F. Zheng, M. Zhu, K. Wang, M. Zhong, D. Wu and H. Zhu, Sci. Rep., 2014, 4, 5528 CrossRef CAS PubMed.
- R. Xu and K. Zhao, J. Electrochem. Energy Convers. Storage, 2016, 13, 030803 CrossRef.
- M. F. El-Kady, Y. Shao and R. B. Kaner, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- G. Zhou, F. Li and H.-M. Cheng, Energy Environ. Sci., 2014, 7, 1307–1338 CAS.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS PubMed.
- W. Gao, G. Wu, M. T. Janicke, D. A. Cullen, R. Mukundan, J. K. Baldwin, E. L. Brosha, C. Galande, P. M. Ajayan, K. L. More, A. M. Dattelbaum and P. Zelenay, Angew. Chem., Int. Ed., 2014, 53, 3588–3593 CrossRef CAS PubMed.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 CAS.
- Y. Lin, Y. Liao, Z. Chen and J. W. Connell, Mater. Res. Lett., 2017, 5, 209–234 CrossRef CAS.
- A. Fasolino, J. H. Los and M. I. Katsnelson, Nat. Mater., 2007, 6, 858–861 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. C. Neto, F. Guinea, N. M. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef.
- C. Soldano, A. Mahmood and E. Dujardin, Carbon, 2010, 48, 2127–2150 CrossRef CAS.
- P. Avouris, Nano Lett., 2010, 10, 4285–4294 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- Q. Bao and K. P. Loh, ACS Nano, 2012, 6, 3677–3694 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. Krishnan, F. Kim, J. Luo, R. Cruz-Silva, L. J. Cote, H. D. Jang and J. Huang, Nano Today, 2012, 7, 137–152 CrossRef CAS.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef CAS PubMed.
- X. Zheng, B. Jia, X. Chen and M. Gu, Adv. Mater., 2014, 26, 2699–2703 CrossRef CAS PubMed.
- X. Zheng, H. Lin, T. Yang and B. Jia, J. Phys. D: Appl. Phys., 2016, 50, 074003 CrossRef.
- D. Pacilé, J. Meyer, A. F. Rodríguez, M. Papagno, C. Gómez-Navarro, R. Sundaram, M. Burghard, K. Kern, C. Carbone and U. Kaiser, Carbon, 2011, 49, 966–972 CrossRef.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- Z. An, O. C. Compton, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2011, 23, 3842–3846 CAS.
- F. Kim, L. J. Cote and J. Huang, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS PubMed.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912–19916 CrossRef CAS.
- M. Ishigami, J. H. Chen, W. G. Cullen, M. S. Fuhrer and E. D. Williams, Nano Lett., 2007, 7, 1643–1648 CrossRef CAS PubMed.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 3087 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- M. D. Fischbein and M. Drndić, Appl. Phys. Lett., 2008, 93, 113107 CrossRef.
- D. Fox, A. O'Neill, D. Zhou, M. Boese, J. N. Coleman and H. Z. Zhang, Appl. Phys. Lett., 2011, 98, 243117 CrossRef.
- Y. H. Ding, P. Zhang, Q. Zhuo, H. M. Ren, Z. M. Yang and Y. Jiang, Nanotechnology, 2011, 22, 215601 CrossRef CAS PubMed.
- P. Russo, A. Hu, G. Compagnini, W. W. Duley and N. Y. Zhou, Nanoscale, 2014, 6, 2381–2389 RSC.
- M. Bieri, M. Treier, J. Cai, K. Ait-Mansour, P. Ruffieux, O. Groning, P. Groning, M. Kastler, R. Rieger, X. Feng, K. Mullen and R. Fasel, Chem. Commun., 2009, 6919–6921, 10.1039/b915190g.
- Z. Fan, Q. Zhao, T. Li, J. Yan, Y. Ren, J. Feng and T. Wei, Carbon, 2012, 50, 1699–1703 CrossRef CAS.
- F. Perreault, A. Fonseca de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- L. Huang, M. Zhang, C. Li and G. Shi, J. Phys. Chem. Lett., 2015, 6, 2806–2815 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- K. Huang, G. Liu, Y. Lou, Z. Dong, J. Shen and W. Jin, Angew. Chem., Int. Ed., 2014, 53, 6929–6932 CrossRef CAS PubMed.
- Y.-L. Zhang, Q.-D. Chen, H. Xia and H.-B. Sun, Nano Today, 2010, 5, 435–448 CrossRef CAS.
- Y. Zhou, Q. Bao, B. Varghese, L. A. L. Tang, C. K. Tan, C. H. Sow and K. P. Loh, Adv. Mater., 2010, 22, 67–71 CrossRef CAS PubMed.
- W. Wang, E. Eftekhari, G. Zhu, X. Zhang, Z. Yan and Q. Li, Chem. Commun., 2014, 50, 13089–13092 RSC.
- H. Liu, H. Wang and X. Zhang, Adv. Mater., 2015, 27, 249–254 CrossRef CAS PubMed.
- Y. Su, V. G. Kravets, S. L. Wong, J. Waters, A. K. Geim and R. R. Nair, Nat. Commun., 2014, 5, 4843 CrossRef CAS PubMed.
- L. Guo, H.-B. Jiang, R.-Q. Shao, Y.-L. Zhang, S.-Y. Xie, J.-N. Wang, X.-B. Li, F. Jiang, Q.-D. Chen and T. Zhang, Carbon, 2012, 50, 1667–1673 CrossRef CAS.
- Y. L. Zhang, L. Guo, H. Xia, Q. D. Chen, J. Feng and H. B. Sun, Adv. Opt. Mater., 2014, 2, 10–28 CrossRef.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 Search PubMed.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- A. Nicolai, B. G. Sumpter and V. Meunier, Phys. Chem. Chem. Phys., 2014, 16, 8646–8654 RSC.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CAS.
- D.-e. Jiang, V. R. Cooper and S. Dai, Nano Lett., 2009, 9, 4019–4024 CrossRef CAS PubMed.
- A. W. Hauser and P. Schwerdtfeger, Phys. Chem. Chem. Phys., 2012, 14, 13292–13298 RSC.
- H. Liu, S. Dai and D.-e. Jiang, Nanoscale, 2013, 5, 9984–9987 RSC.
- A. W. Hauser, J. Schrier and P. Schwerdtfeger, J. Phys. Chem. C, 2012, 116, 10819–10827 CAS.
- G. F. Schneider, S. W. Kowalczyk, V. E. Calado, G. Pandraud, H. W. Zandbergen, L. M. Vandersypen and C. Dekker, Nano Lett., 2010, 10, 3163–3167 CrossRef CAS PubMed.
- Z. S. Siwy and M. Davenport, Nat. Nanotechnol., 2010, 5, 697–698 CrossRef CAS PubMed.
- G. F. Schneider, Q. Xu, S. Hage, S. Luik, J. N. H. Spoor, S. Malladi, H. Zandbergen and C. Dekker, Nat. Commun., 2013, 4, 2619 Search PubMed.
- B. M. Venkatesan and R. Bashir, Nat. Nanotechnol., 2011, 6, 615–624 CrossRef CAS PubMed.
- M. Wanunu, Phys. Life Rev., 2012, 9, 125–158 CrossRef PubMed.
- T. Nelson, B. Zhang and O. V. Prezhdo, Nano Lett., 2010, 10, 3237–3242 CrossRef CAS PubMed.
- D. B. Wells, M. Belkin, J. Comer and A. Aksimentiev, Nano Lett., 2012, 12, 4117–4123 CrossRef CAS PubMed.
- S. Garaj, W. Hubbard, A. Reina, J. Kong, D. Branton and J. Golovchenko, Nature, 2010, 467, 190–193 CrossRef CAS PubMed.
- C. A. Merchant, K. Healy, M. Wanunu, V. Ray, N. Peterman, J. Bartel, M. D. Fischbein, K. Venta, Z. Luo, A. T. C. Johnson and M. Drndić, Nano Lett., 2010, 10, 2915–2921 CrossRef CAS PubMed.
- S. Garaj, S. Liu, J. A. Golovchenko and D. Branton, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12192–12196 CrossRef CAS PubMed.
- C. Hyun, H. Kaur, R. Rollings, M. Xiao and J. Li, ACS Nano, 2013, 7, 5892–5900 CrossRef CAS PubMed.
- F. Haque, J. Li, H.-C. Wu, X.-J. Liang and P. Guo, Nano Today, 2013, 8, 56–74 CrossRef CAS PubMed.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668–674 CAS.
- S. Vivekchand, C. S. Rout, K. Subrahmanyam, A. Govindaraj and C. Rao, J. Chem. Sci., 2008, 120, 9–13 CrossRef CAS.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- H.-J. Choi, S.-M. Jung, J.-M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS.
- D. A. Brownson and C. E. Banks, Analyst, 2010, 135, 2768–2778 RSC.
- K. Chen, S. Song, F. Liu and D. Xue, Chem. Soc. Rev., 2015, 44, 6230–6257 RSC.
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 3480–3498 CrossRef CAS PubMed.
- P. Guo, H. Song and X. Chen, Electrochem. Commun., 2009, 11, 1320–1324 CrossRef CAS.
- H. Xiang, Z. Li, K. Xie, J. Jiang, J. Chen, P. Lian, J. Wu, Y. Yu and H. Wang, RSC Adv., 2012, 2, 6792–6799 RSC.
- N. Li, Z. Chen, W. Ren, F. Li and H.-M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360–17365 CrossRef CAS PubMed.
- W. Zhang, J. Zhu, H. Ang, Y. Zeng, N. Xiao, Y. Gao, W. Liu, H. H. Hng and Q. Yan, Nanoscale, 2013, 5, 9651–9658 RSC.
- J. Han, G. Huang, Y. Ito, X. Guo, T. Fujita, P. Liu, A. Hirata and M. Chen, Adv. Energy Mater., 2017, 7, 1601933 CrossRef.
- B. Sun, X. Huang, S. Chen, P. Munroe and G. Wang, Nano Lett., 2014, 14, 3145–3152 CrossRef CAS PubMed.
- F. Li, T. Zhang and H. Zhou, Energy Environ. Sci., 2013, 6, 1125–1141 CAS.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- X. Li, X. Huang, D. Liu, X. Wang, S. Song, L. Zhou and H. Zhang, J. Phys. Chem. C, 2011, 115, 21567–21573 CAS.
- K. Hatakeyama, H. Tateishi, T. Taniguchi, M. Koinuma, T. Kida, S. Hayami, H. Yokoi and Y. Matsumoto, Chem. Mater., 2014, 26, 5598–5604 CrossRef CAS.
- K. Hatakeyama, M. S. Islam, K. Michio, C. Ogata, T. Taniguchi, A. Funatsu, T. Kida, S. Hayami and Y. Matsumoto, J. Mater. Chem. A, 2015, 3, 20892–20895 CAS.
| This journal is © The Royal Society of Chemistry 2017 |