
Improved cyclic redox reactivity of lanthanum modified iron-based oxygen carriers in carbon monoxide chemical looping combustion
 †a
Liang-Shih
Fan
*a
†a
Liang-Shih
Fan
*a
Abstract
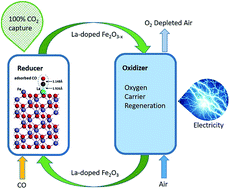
Chemical looping combustion (CLC) technology converts fuel into heat or electricity with complete CO2 capture and does not require air separation units. CLC is realized through redox reactions of gas or solid fuels with oxygen carriers at high temperatures. The oxygen carriers are required to have high reactivity and recyclability with low cost. In this work, we use a low concentration of the lanthanum dopant to dramatically change the reactivity of oxygen carriers in CLC with CO fuel. Lanthanum is a rare earth element and is chosen because of its high catalytic activity. It is found that the conversion rates of lanthanum-doped oxygen carriers are universally higher than those of undoped iron oxide oxygen carriers from 550 °C to 780 °C. The reactivity increases significantly in doped oxygen carriers at these temperatures favourable for CLC and decreases in these carriers at temperatures above approximately 800 °C due to enhanced sintering. Density functional theory (DFT) is used to calculate the surface configuration and energy barriers in CLC reactions and shows dramatically reduced energy barriers in lanthanum-doped iron oxide reactions with CO and oxygen carrier regeneration. This methodology provides substantial reactivity improvements of CLC with oxygen carriers that are relatively simple to fabricate, and it will have an impact on chemical looping particle design and modification.
 Please wait while we load your content...
Please wait while we load your content...

