
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fe3O4 nanoparticles coated with a guanidinium-functionalized polyelectrolyte extend the pH range for phosphate binding†
Laura
Paltrinieri
ab,
Min
Wang
a,
Sumit
Sachdeva
 a,
Nicolaas A. M.
Besseling
a,
Ernst J. R.
Sudhölter
a and
Louis C. P. M.
de Smet
*abc
a,
Nicolaas A. M.
Besseling
a,
Ernst J. R.
Sudhölter
a and
Louis C. P. M.
de Smet
*abc
aDelft University of Technology, Department of Chemical Engineering, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
bWetsus – European Centre of Excellence for Sustainable Water Technology, Oostergoweg 9, 8932 PG Leeuwarden, The Netherlands
cWageningen University & Research, Laboratory of Organic Chemistry, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: louis.desmet@wur.nl
First published on 4th July 2017
Abstract
In this work commercially available Fe3O4 NPs were coated with polyallylamine hydrochloride (PAH) and PAH functionalized with guanidinium groups (PAH–Gu) for investigating the phosphate adsorption properties under alkaline conditions. The coating can be prepared easily and rapidly and results in Fe3O4 NPs with improved properties related to phosphate binding and colloidal stability. At a low initial phosphate concentration (2 mg L−1), the novel Fe3O4@PAH–Gu material was able to remove phosphate rather independently of the pH condition (4.0, 3.6 and 3.7 mg g−1 at pH = 5, 8 and 10, respectively), whereas for the uncoated Fe3O4 NPs the amount of adsorbed phosphate drops by >75% upon changing from acidic to alkaline conditions (0.84 mg g−1 at pH = 10). Under alkaline conditions, the fastest adsorption was observed for Fe3O4@PAH–Gu followed by Fe3O4@PAH and Fe3O4. This can be related to the additional interaction forces due to the presence of primary amine groups (in PAH and PAH–Gu) and Gu groups (in PAH–Gu only) in coatings. Over 80% of the phosphate adsorbed on the novel Fe3O4@PAH–Gu material was successfully desorbed and the coated NPs were re-used over three adsorption/desorption cycles. This work will stimulate the design and preparation of functionalized polyelectrolytes for an extended area of applications, especially for the selective removal of target compounds from wastewater.
Introduction
The uncontrolled discharge of phosphate-containing products as present in aqueous streams from agricultural and cosmetic sectors1,2 has increased eutrophication processes, i.e. the rapid growth of aquatic algae in lakes and rivers.3,4 These processes contribute to an unbalanced aquatic ecology and to a decrease of water quality. Phosphate is therefore considered to be one of the most critical contaminants present in wastewater. Many countries have set a standard for the discharge of phosphate into water.5 For instance, the European Union recently regulated a maximum value of 0.07 μg P per L for rivers and 100 μg P per L for lakes to reduce the risk of eutrophication.6 In order to meet such strict requirements and to manage the high phosphorus demand at the same time,7,8 the recovery of phosphorus from phosphate-contaminated aqueous media has been recognized as a challenging key strategy. For this purpose different technologies have been developed, including biological treatments,9 membrane-based processes,10,11 crystallization,12,13 flotation,14 and adsorption-based processes.15 From this list of well-known techniques, adsorption processes have high potential. This is mainly related to their low operational costs, high efficiency, low energy consumption and their versatility to be applicable in different wastewater sources.15Among candidates for phosphate adsorbents, iron oxides are considered to be highly promising.16 This is because of (1) their high selectivity to bind phosphate in the presence of competing anions and (2) their easy introduction in municipal wastewater treatment plants (WWTPs). Furthermore, a good adsorbent is identified by, amongst others, the available specific adsorption area. For this reason, a lot of attention is now paid to develop new nano-sized adsorbents, because of their high-surface-area-to-volume ratio.17 Nanoparticles of iron oxide (Fe3O4 NPs) fulfill these conditions and even possess magnetic properties, making easy separations possible by using external magnetic fields.18 Phosphate adsorption onto Fe3O4 NPs occurs through an inner-sphere complex, due to the presence of surface hydroxyl groups.16,19 When the pH is lower than the point of zero charge (PZC), the surface of the iron oxide nanoparticles is positively charged, which promotes binding and surface adsorption of phosphate anions. The lower the pH, the more charge on the surface and therefore a higher binding capacity.20,21 However, at lower pH values the amount of phosphate anions decreases, as they are converted to phosphoric acid.22 This becomes significantly below pH < pKa1 = 2.1. The pH of water streams in WWTPs is typically 6–8,23,24i.e., around the PZC of the Fe3O4 NPs. At such pH values, the surface charge is slightly positive, neutral or slightly negative, which has a large negative impact on the phosphate anion binding capacity. In the mentioned pH range, the phosphates are monoanionic and partly dianionic (pKa2 = 7.2).22 Moreover, in this pH range the NPs aggregate to precipitate, due to the decreased inter-particle electrostatic repulsions. Thus, for phosphate separation processes at pH values around the PZC of Fe3O4 NPs, there is room for improvement. For this reason, different types of chemical surface modifications have been applied by the attachment of specific ligands, including amino groups,25 metal organic frameworks (MOFs),19 polymers,26,27 layered double hydroxides (LDHs)28 and graphene.29 These examples illustrate well the effectiveness of surface functionalization in terms of controlling the affinity for a specific target species. Yet, it would be interesting to further employ these surface modification strategies in order to extend the use of iron oxide nanoparticles for phosphate anion binding at higher pH values, where unmodified iron oxide is otherwise less effective.
Receptor-functionalized polyelectrolytes (PEs) can bind to surfaces of opposite charge30–32 and can contribute to nanoparticle stabilization,33 while the receptor groups introduce selectivity for binding certain targets. Recent advances in this direction resulted in the availability of polyelectrolytes that were functionalized with, e.g., biotin, fluorescent probes and guanidinium groups to address chelation and the selective capture of His-tagged proteins,34,35 biosensing,36 fingermark visualization,37 and ion selectivity.38 Interestingly, polyelectrolyte functionalization and the subsequent modification of NPs do not require complicated chemical steps and can be performed rapidly in aqueous media.
In the current study, we present the concept of a simple surface modification of commercially available Fe3O4 NPs using polyelectrolytes functionalized with phosphate-receptors. For the receptor we have chosen the guanidinium moiety, which is able to coordinate phosphate ions in a wide range of pH values.39,40 The Gu-functionalized polyelectrolyte was applied to modify the Fe3O4 NPs. The thus-obtained NPs are characterized in terms of their morphology, thermal stability and surface properties. The effect of the pH on the phosphate adsorption is investigated in detail, as well as the kinetics of the process. The obtained results were compared with those of bare Fe3O4 NPs as well as Fe3O4 NPs coated with a non-functionalized polyelectrolyte.
Experimental
Materials
Poly(allylamine hydrochloride) (PAH, Mw ∼15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) guanidine acetic acid (GAA, 99%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, commercial grade) and N-hydroxysuccinimide (NHS, 98%) were purchased from Sigma-Aldrich and used without any further purification. 1 M HCl and 1 M NaOH (analytical reagent grade, obtained from Fluka, Germany) were used for pH adjustments. Sodium dihydrogen phosphate (NaH2PO4·H2O, Acros Organics) was used in phosphate removal experiments. Commercial iron oxide nanoparticles (Fe3O4 NPs, 8 nm in diameter as determined by TEM) were purchased as a 3 wt% acidic aqueous dispersion from PlasmaChem GmbH (Germany) without any organic stabilizer. All aqueous solutions were prepared using Milli-Q water (Milli-Q Ultrapure Water System, Millipore 22 μm).
000 Da) guanidine acetic acid (GAA, 99%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, commercial grade) and N-hydroxysuccinimide (NHS, 98%) were purchased from Sigma-Aldrich and used without any further purification. 1 M HCl and 1 M NaOH (analytical reagent grade, obtained from Fluka, Germany) were used for pH adjustments. Sodium dihydrogen phosphate (NaH2PO4·H2O, Acros Organics) was used in phosphate removal experiments. Commercial iron oxide nanoparticles (Fe3O4 NPs, 8 nm in diameter as determined by TEM) were purchased as a 3 wt% acidic aqueous dispersion from PlasmaChem GmbH (Germany) without any organic stabilizer. All aqueous solutions were prepared using Milli-Q water (Milli-Q Ultrapure Water System, Millipore 22 μm).
Preparation of PAH–Gu, Fe3O4@PAH and Fe3O4@PAH–Gu
PAH–Gu (Scheme 1, top right) was obtained by the reaction of GAA with a part of the amino groups of PAH following the procedure published earlier by our group.381H-NMR was used to confirm the chemical structure (ESI, Fig. S1†) and to calculate the degree of amino group functionalization by guanidinium (Gu) moieties; it was found to be ∼30% for the batch used in the current work. In order to study the effect of present Gu groups, non-functionalized PAH was used as a reference. | ||
| Scheme 1 A schematic showing the three different NPs (top left) and the interactions between amino-phosphate and guanidinium-phosphate, shown for the monovalent anion (top right). Brief stepwise description of the coating process of Fe3O4 NPs with (functionalized) polyelectrolytes (in the grey-colored box, bottom left), and pictures of different NP suspensions at pH = 9.5 (bottom right). | ||
Next, the Fe3O4 NPs were modified with PAH or PAH–Gu via the following procedure.41 Aqueous solutions of PAH and PAH–Gu (2.5 g L−1) were prepared by sonication using a probe sonicator (Cole-Parmer CPX750, 30% power, 750 watts) for 20 min and simultaneous cooling by placing the tube in ice. Similarly, a Fe3O4 NP suspension (0.5 g L−1) was prepared in MilliQ water and sonicated under the same conditions. After sonication, the pH of all solutions was adjusted to 9.5 by the addition of drops of concentrated HCl or NaOH (1 M). At this pH value the Fe3O4 NPs have a negative surface charge, while the polyelectrolytes are positively charged. The Fe3O4 NP suspension was added drop-wise to the polyelectrolyte solutions and stirred for 24 h at room temperature (RT) to ensure complete adsorption at the Fe3O4 NP surface. The functionalized NPs were separated from the excess of PEs by three cycles of centrifugation, decantation and washing (Heraeus instrument D-37520 Osterode, Germany) at 17000 rpm (20 min at 20 °C). The product was finally re-dispersed in 40 mL of MilliQ water to maintain the initial concentration and then sonicated to obtain uniform solutions of Fe3O4@PAH and Fe3O4@PAH–Gu. A schematic overview of the coating procedure and the different types of Fe3O4 NPs is presented in Scheme 1.
Characterization
The 1H NMR spectrum of the PAH–Gu polymer was obtained using a Bruker AVANCE 400 NMR spectrometer with D2O as solvent.Modified NPs were studied with Fourier Transform InfraRed spectroscopy (Nicolet 8700 FT-IR Spectrometer) by mixing the NPs with KBr and pressing pellets. The spectral range of FT-IR was from 4000 cm−1 to 500 cm−1 with a resolution of 4 cm−1.
X-ray Photoelectron Spectroscopy (XPS, Thermo Fisher Scientific, K-Alpha model) was used to determine the atomic composition of the modified NP surfaces. In more detail, a monochromatic Al Kα X-ray source was used with a spot size of 400 μm at a pressure of 10−7 mbar. A constant pass energy of 200 eV for the survey spectra and 50 eV for the detailed high-resolution spectra was used. The flood gun was turned on during the measurement to compensate for potential charging of the surface. The peak position was adjusted based on the internal standard C1s peak at 284.8 eV, with an accuracy of ±0.05 eV. Avantage processing software was used to analyze all spectra.
ThermoGravimetric Analysis (TGA) measurements were performed with Thermal Analysis (TA) Instruments equipment from RT to 550 °C at a heating rate of 10 °C min−1 under continuous air purging.
The size and morphology of the unmodified and modified NPs were studied using a Transmission Electron Microscope (TEM, JEOL JEM-1400 Plus, USA) operated at 120 kV. A holey carbon support film (200 meshes, Quantifoil®) was dipped into the NP-containing solution and then dried at room temperature overnight. TEM images were analyzed by using Image J software and the mean size value of each NP system was calculated based on 20 separately determined diameters.
The hydrodynamic diameter (Dh) of NPs was determined at 25 °C by Dynamic Light Scattering (DLS) using a Zetasizer Nano ZS900 (Malvern, UK). The instrument was operated at a backscattering angle of 173° with a laser beam with a wavelength of 633 nm. The same instrument was used to measure the ζ-potential at 25 °C for all samples and measurements were performed in triplicate. To this end, an aqueous suspension of Fe3O4 NPs (0.5 mg mL−1) was prepared by adding 167 μL of the original concentrated NP solution into 10 mL MilliQ water. Samples for ζ-potential measurements were prepared by diluting 80 μL of the above-prepared NP suspension (0.5 mg mL−1) to 10 mL using MilliQ water. The solution was sonicated using a probe sonicator (30%, 750 watts, cooling in an ice bath, 6 min) to break the existing aggregates. In the last step, the pH was adjusted to the desired values by using 1 M NaOH and 1 M HCl. The same procedure was used to determine the ζ-potential of all NP systems, as well of the pure PEs (PAH and PAH–Gu), where a solution of 0.5 mg mL−1 in MilliQ water was used. All the measurements were done 5 min after the sonication procedure to minimize possible differences due to colloidal instability.
Batch adsorption experiments
Phosphate adsorption experiments were performed for Fe3O4, Fe3O4@PAH, and Fe3O4@PAH–Gu NPs. All desired phosphate solutions, including the standard known concentration of phosphate for calibration measurements, were prepared by diluting a stock solution (1000 mg L−1 of NaH2PO4 in 250 mL). The phosphate adsorption was studied as a function of time starting with an initial phosphate concentration of 2 mg L−1, taken from the stock solution, and an adsorbent solution of 0.5 g L−1 in 30 mL. The adsorbed amount was deduced from the reduction of the phosphate concentration according to the work optimized by Yoon et al.42 In contrast to other studies,42,43 we have decided to keep a fixed initial adsorbent concentration and to focus on the effect of pH on the adsorption process. In this study the pH conditions have a great impact not only on the stability of the suspension of NPs and their surface charges, but also on the type of speciation of phosphate involved in the adsorption process. Before starting the experiments, the pH of both the adsorbent solution and phosphate solution was adjusted to the desired value. Phosphate was added to NP solutions, followed by stirring at RT for 24 h. Samples were taken at different times and centrifuged (Eppendorf AG, Germany) at 13000 rpm for 1 h. The phosphate adsorption efficiency was measured through UV-vis spectroscopy (UVIKON XL, Beun De Ronde) by using the ascorbic acid method.44,45
Adsorption and desorption cycles
Fe3O4@PAHGu NPs were subjected to three adsorption and desorption cycles to test the reversibility of the binding process as well as the reusability of the coated NPs. In more detail, a solution containing 5 mg L−1 of NaH2PO4 was added to 3 mL of a 0.5 g L−1 Fe3O4@PAHGu suspension at pH = 5. The adsorption experiment was performed by stirring at RT for 24 h followed by a centrifugation step (1 h at 13000 rpm). The desorption experiment was done by adding a 10 mM NaCl solution to the Fe3O4@PAHGu NPs that were obtained after the centrifugation step of the first adsorption. The solutions were stirred for 3 h at RT and the resulting solution was separated from the NPs by centrifugation (1 h at 13000 rpm). Before the 2nd and 3rd adsorption cycle, Fe3O4@PAHGu NPs were washed twice with MilliQ water while stirring for 30 min each time after which the water was removed by centrifugation (1 h at 13000 rpm, each time). All phosphate-containing solutions were analysed through Ion Chromatography (930 Compact IC Flex, 150 mm A Supp 5 column, Metrohm).
Results and discussion
Given the importance of electrostatic interactions in the surface modification using polyelectrolytes,33,46 we first present and discuss the ζ-potential data of the PAH and PAH–Gu separately, and Fe3O4 NPs as such. Next, FTIR, XPS, TGA and TEM data are given to characterize the bare and modified Fe3O4 NPs. The phosphate sorption studies of the NPs are split into three different topics: (i) pH effects, (ii) kinetic study and (iii) reversibility of phosphate binding.ζ-Potential investigation of PEs and Fe3O4 NPs
Fig. 1 shows the ζ-potential of bare Fe3O4 NPs as well as of PAH and PAH–Gu in aqueous solutions as a function of the solution pH. It is observed that for all cases the zeta potential becomes less positive with increasing pH value. | ||
| Fig. 1 ζ-Potential as a function of solution pH for (a) an aqueous suspension of unmodified Fe3O4 NPs (0.5 g L−1, black squares), (b) an aqueous solution of 0.5 g PAH per L (blue circles), and (c) an aqueous solution of 0.5 g PAH–Gu per L (red triangles). | ||
For the unmodified Fe3O4 NPs the zeta potential changes from a positive (Fe–OH2+ groups are in excess) to a negative (Fe–O− groups are in excess) sign around pH = 7, reflecting the PZC as has been reported in the literature.47
In contrast, PAH and PAH–Gu polyelectrolyte solutions remain positive over the whole investigated pH region. PAH–Gu shows a higher positive surface charge compared to the (unfunctionalized) PAH. This can be easily understood in terms of the respective pKa values of PAH and Gu, which are 8–9 for the primary amine of PAH48,49 and 13 for the guanidinium group present in PAH–Gu.50 Furthermore, for PAH–Gu the ζ-potential data at pH < 6.5 show a plateau behavior, which is absent for PAH and the Fe3O4 NPs in the studied pH window. This indicates that the overall surface-charge density of PAH–Gu at pH < 6 is constant. This difference may be associated with the differences in the PZC of the respective materials, including a shift of the apparent dissociation constant of PAH (pKa(app)) due to local changes of the electrostatic environment51 and, for PAH–Gu a saturation of chargeable groups under acidic pH conditions.
To conclude this part, the results show that within the pH window of ∼7 to ∼9.5 the unmodified Fe3O4 NPs are negatively charged, while both PEs are positively charged. In addition, from the literature it is known that Fe3O4 NPs are maximally covered by weak polyelectrolytes (like PAH) if the pH is similar to the polyelectrolyte pKa value.52 We have therefore chosen to perform our experiments at a pH of 9.5, the pKa value of PAH, for both PAH and PAH–Gu modifications.
Characterization of coated Fe3O4 NPs
The FTIR spectra of bare Fe3O4 NPs, Fe3O4@PAH, and Fe3O4@PAH–Gu as well as those of the pure PEs are shown in Fig. 2. The data show that the modified NPs are covered with PAH or PAH–Gu. In all cases a large contribution between 3404 cm−1 and 3017 cm−1 is observed, which can be associated with the O–H bond stretching. Its broadness originates from H-bridge formation with physically adsorbed water, which was used as a solvent and can be entrapped between the polymeric chains (see also TEM and TGA analysis; vide intra).53 The presence of iron oxide is confirmed by the observed stretching of Fe–O at 577 cm−1 in the cases of Fe3O4 NPs (black, dashed), Fe3O4@PAH (blue, dashed) and Fe3O4@PAH–Gu (red, dashed).54 The success of the PAH coating process becomes clear from the typical peaks at 2918 cm−1 and 2850 cm−1 that are associated with C–C stretching and two peaks at 1575 cm−1 and 1541 cm−1 of the C–N and N–H bending, which compare well with bands present in the FTIR spectrum of PAH (blue). Finally, the bands at 1604 cm−1 and 1506 cm−1 can be assigned to the bending vibration related to the amino group.55 Likewise, in agreement with the bare PAH–Gu spectrum (red), the coating of NPs with PAH–Gu is confirmed by the presence of two peaks at 2918 cm−1 and 2850 cm−1 for C–C stretching, a peak at 1631 cm−1 assigned to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond from the Gu group and a peak at 1537 cm−1 of the N–H bending.38,56 It should be mentioned that in both spectra of Fe3O4@PAH and Fe3O4@PAH–Gu, the positions of the characteristic peaks of primary amine and amide bonds shift to some extent with respect to the corresponding bare PEs. This can be explained by the interaction between iron ions and charged groups of PEs and the formation of amino complexes.57
N bond from the Gu group and a peak at 1537 cm−1 of the N–H bending.38,56 It should be mentioned that in both spectra of Fe3O4@PAH and Fe3O4@PAH–Gu, the positions of the characteristic peaks of primary amine and amide bonds shift to some extent with respect to the corresponding bare PEs. This can be explained by the interaction between iron ions and charged groups of PEs and the formation of amino complexes.57
 | ||
| Fig. 2 FTIR spectra of (a) bare Fe3O4 NPs (black, dashed), (b) Fe3O4@PAH (blue, dashed), (c) PAH polymer (blue, solid), (d) Fe3O4@PAH–Gu (red, dashed), and (e) PAH–Gu polymer (red, solid). | ||
XPS was used to further map the surface chemistry of NPs before and after modification (Table 1). The successful NP functionalization is evident from the N/Fe ratio that increases upon the preparation of the coating from 0 (bare Fe3O4 NPs) to 0.30 and 0.60 for Fe3O4@PAH and Fe3O4@PAH–Gu, respectively. Moreover, with respect to the bare NPs, the C/Fe ratio is higher in the presence of the PEs and this can be associated with the alkyl polymer backbone and methylene groups in the side chains. Oxygen is measured in all samples, which can be related to OH groups present at the Fe3O4 NP surface and the CO in the Fe3O4@PAH–Gu system.
| C/Fe | O/Fe | N/Fe | |
|---|---|---|---|
| Fe3O4 | 1.67 | 1.64 | — |
| Fe3O4@PAH | 6.02 | 4.69 | 0.30 |
| Fe3O4@PAH–Gu | 2.44 | 2.52 | 0.60 |
Carbon is detected in Fe3O4 as well, and this can be related to hydrocarbon surface contamination often observed on surfaces.58 While the C/Fe ratio of Fe3O4@PAH is higher than that of Fe3O4@PAH–Gu, the contribution of carbon contamination makes it hard to draw any conclusion on the degree of coverage based on C/Fe. An indication of the amount of PEs bound to the NP surface can be deduced from the N/Fe ratio. Taking into account a degree of Gu group substitution of 30% (see the chemical structure reported in Scheme 1), the calculated amount of N per repeating unit in PAH–Gu is 1.9 times higher than that for PAH (considering 0.3 × 4(N) + 0.7 × 1(N)). From XPS analysis a ratio of (0.60/0.30) = 2 was observed, indicating that a similar amount of both polyelectrolytes is bound to the NPs.
Additional evidence of the changed surface chemistry of the NPs was obtained from TGA analysis (Fig. 3). Bare Fe3O4 (line a) showed hardly any weight loss for the indicated temperature range (residual of 98%). This small weight reduction can be attributed to the loss of water physically adsorbed at the NP surface combined with the loss of condensed groups at temperatures higher than 100 °C.59 In contrast, two degradation steps clearly appear for Fe3O4@PAH (line b) and Fe3O4@PAH–Gu (line c). The first step at 30–120 °C refers to the loss of water. The presence of water is due to both physically adsorbed water at modified NP surfaces and the hydration shell of ions (ammonium and chlorine) of the polyelectrolyte chains, which is found to be almost the same for both systems (in accordance with the FTIR spectra). The second weight loss at 250–400 °C can be related to the breakdown of the PEs. The residuals of Fe3O4@PAH and Fe3O4@PAH–Gu overall drop to 87% and 84%, respectively. The weight drop can be attributed to the bonded polyelectrolyte at the NP surface. The difference between the drop for Fe3O4@PAH and Fe3O4@PAH–Gu is due to the Gu modification, considering that the average mass per monomer unit is larger for PAH–Gu than that for PAH.
 | ||
| Fig. 3 TGA curves showing the fractional weight loss of (a) bare Fe3O4 (black), (b) Fe3O4@PAH (blue), and (c) Fe3O4@PAHGu (red). | ||
The morphology of NPs was examined with TEM; the images of Fe3O4 NPs, Fe3O4@PAH, and Fe3O4@PAH–Gu are shown in Fig. 4. In the absence of a polymeric coating, Fe3O4 NPs show a typical spherical shape.60 The same spherical shape can also be observed in images (b) and (c); in addition, a smooth and transparent layer is seen around the NPs, likely due to the presence of the polymeric coating (c).61Table 2 lists the mean diameters of the NPs as obtained from TEM analysis. Fe3O4 NPs were found to have a diameter of 8 ± 2 nm, confirming the specifications given by the supplier. The diameters of Fe3O4@PAH and Fe3O4@PAH–Gu, including the additional smooth layer, are 11 ± 2 nm, indicating an adsorbed polyelectrolyte layer thickness of ca. 3 nm (TEM-based size distribution plots are presented in ESI Fig. S2†).62
 | ||
| Fig. 4 TEM images of (a) bare Fe3O4, (b) Fe3O4@PAH NPs, and (c) Fe3O4@PAH–Gu NPs. | ||
Table 2 summarizes the size data of our investigated NPs as obtained from TEM and DLS (hydrodynamic diameters) and zeta potential measurements. At pH = 9.5, the unmodified and polyelectrolyte-modified NPs have hydrodynamic diameters much larger than the sizes of single particles observed by TEM. This is due to the agglomeration of these NPs in solution. This agglomeration is reduced for the NPs modified with a polyelectrolyte: 86 nm observed for the unmodified Fe3O4 NPs, compared to 65 nm and 41 nm for the Fe3O4@PAH and Fe3O4@PAH–Gu, respectively. The difference is related to the colloidal stability, which is increased for polyelectrolyte-modified NPs, thus preventing aggregation.29,30,58,63,64 We also observed a stable suspension for both PE-modified NPs, while the unmodified Fe3O4 NPs precipitated after 24 h at pH = 9.5 (Scheme 1, bottom left and ESI Fig. S3 and S4†).
The stability of the NP suspension at pH = 9.5 as observed from DLS was confirmed with ζ-potential, which changes sign upon modification to +26 mV and +32 mV for the PAH and PAH–Gu coatings, respectively. The positive ζ-potential values strongly confirm the presence of polycations at the NP surface.61 Moreover, it should be noted that the magnitude of the surface potential reflects the level of electrostatic repulsion between NPs. A higher zeta potential gives more repulsion and therefore a more stable suspension. From these zeta potential measurements it is now clearly understood why the Fe3O4 NPs start to agglomerate, while the PE-modified NPs are still stable. From the results obtained, it is evident that the addition of a Gu moiety altered the Fe3O4 properties; this is in terms of not only reversing the surface charge to a positive value (as is the case for Gu-free PAH), but also increasing the absolute charge density, leading to an increased colloidal stability. Again, this can be explained by the differences in the PZC between the amino-PAH and Gu moiety.65 Images of the NP suspension at different pH values after 24 h and Fe3O4@PAH–Gu after 1 week are shown in Fig. S2 and S3 in the ESI.†
Phosphate adsorption: effect of pH
In order to map the pH-dependency of phosphate adsorption at our (modified) NPs, three pH values were chosen for the adsorption experiments: pH = 5, pH = 8 and pH = 10. Within the pH window from 5 to 10 the degree of dissociation of phosphoric acid decreases accordingly, thus at pH = 5 H2PO4− is predominant, at pH = 8 H2PO4− and HPO42− are equally present, while at pH = 10 mostly HPO42− can be expected.22 The adsorption experiments were performed at a fixed concentration of 0.5 g (modified) NP per L and 2 mg NaH2PO4 per L; thus there is always an excess of adsorbent. Fig. 5 shows the results of the phosphate adsorption as a function of pH for Fe3O4 NPs, Fe3O4@PAH and Fe3O4@PAH–Gu after equilibration for 24 h at RT as determined by the ascorbic acid/UV method.45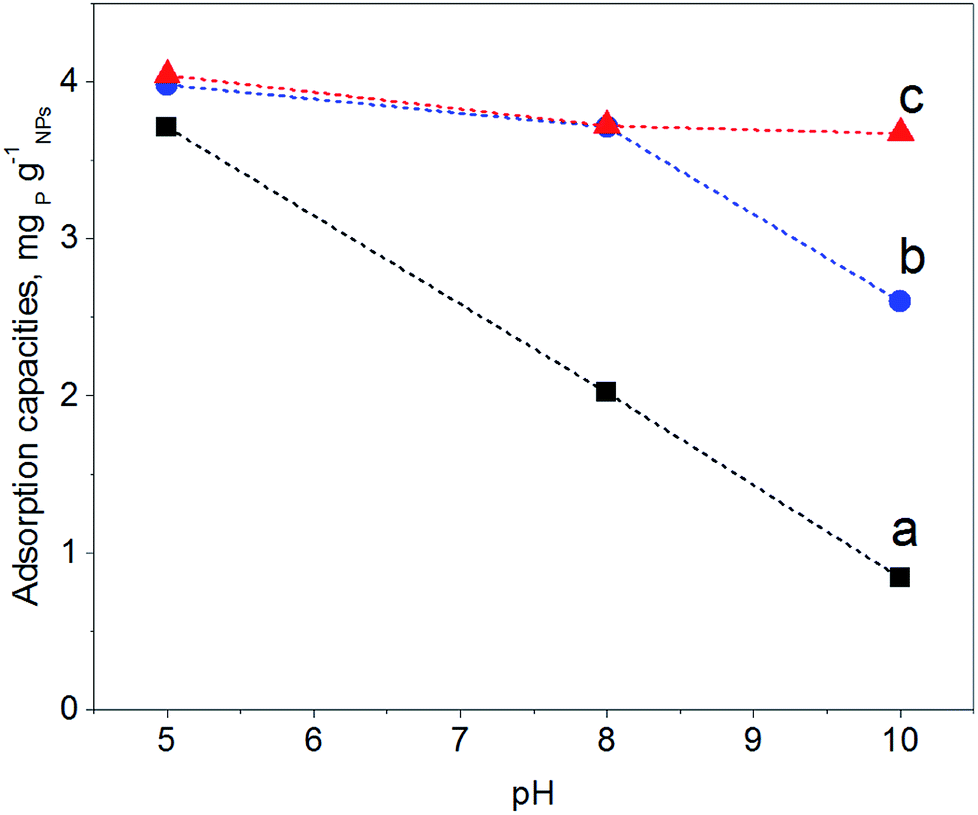 | ||
| Fig. 5 Amount of phosphate adsorbed (mgP gNPs−1) after equilibration for 24 h at RT for (a) Fe3O4 NPs (black), (b) Fe3O4@PAH (blue), and (c) Fe3O4@PAH–Gu NPs (red). The dashed lines serve as a guide to the eye. Measurements were performed in triplicate and all errors were found to be <0.05%. | ||
At pH = 5 all NPs show a similar amount of phosphate adsorbed. Under these conditions, the phosphate is predominantly present as the mono-anion (pKa1 = 2.1 and pKa2 = 7.2) and the Fe3O4 NPs are below their PZC and therefore will have a net positive charge. For the PAH and PAH–Gu modified Fe3O4 NPs also the net surface charge is positive. The phosphate mono-anion will therefore bind to the unmodified Fe3O4 NPs, as reported in the literature.19 Since there is hardly any extra effect of the PAH and PAH–Gu modifications on the adsorbed phosphate amount it is suggested that the Fe3O4 NP surface determines the adsorption under these conditions. Despite the positive charges at the Fe3O4 NP surface, the stability of the PE coating under this pH condition can be related to the presence of neutral amino groups in the PAH and PAH–Gu chains. It is likely that both positive charges and neutral hydroxyl groups present on the Fe3O4 NP surface interact with the unprotonated amino groups of PEs.
Increasing the pH from 5 to 8 and 10 shows a reduction of phosphate adsorption by the unmodified Fe3O4 NPs of 46% and 77%, respectively. At these pH values, the surface charge has turned to a negative value and the adsorption of phosphate monoanions or dianions is suppressed by electrostatic repulsion. Yet, at pH = 10 the phosphate adsorption is not reduced to 0; instead, it is still 0.85 mg g−1. Thus, the adsorption of phosphate onto iron-oxide surfaces occurs both by electrostatic interactions, absent at pH = 10, and by a chemisorption process.66,67 The latter involves the formation of Fe–O–P bonds through a ligand exchange reaction between OH groups at the NP surface and phosphate oxygen. This may explain the P adsorption detected at pH = 10.
A very clear difference is observed for pH = 8 and pH = 10, if PAH or PAH–Gu is present. The amount of adsorbed phosphate is now higher than that observed for unmodified Fe3O4 NPs and more or less similar to the adsorbed amount observed at pH = 5 for the three investigated NPs. Clearly, the reduced affinity of the (unmodified) Fe3O4 NP surface at pH = 8 is compensated nearly completely by the PAH and PAH–Gu modifications. For pH = 10, it is seen that the phosphate adsorption for PAH is decreased compared to that of the PAH–Gu modified surface. For PAH–Gu still a phosphate adsorption of 3.67 mg g−1 is observed. This difference nicely reflects the difference of the pKa values of PAH (8–9) and PAH–Gu (Gu groups pKa = 13) due to which the latter has a higher positive charge density at pH = 10.
In addition, the increased stability of the colloidal suspension may contribute to the uptake of phosphate, because a higher contact area is available compared to the aggregated state. Increased colloidal stability is supported by ζ-potential measurements: at pH = 10 a zeta potential of +21.1 mV and +3.8 mV is found for Fe3O4@PAH–Gu and Fe3O4@PAH, respectively. As mentioned previously, the adsorption of phosphate slightly decreases upon increasing pH. Under alkaline conditions, OH− groups are abundant and they might compete with phosphate in the adsorption process.19,28
Effect of contact time and adsorption kinetics
Phosphate adsorption was monitored as the decrease of the phosphate concentration over time at pH values of 5, 8 and 10 (Fig. 6). At pH = 5 all NPs show a very fast adsorption behavior. Equilibrium was reached within 5 min. Due to our experimental setup we are not able to accurately monitor the adsorption increase within that time frame. However, it is clear that at pH = 8 and pH = 10 the adsorption process is slower, making monitoring of the adsorption increase possible. Equilibrium is now obtained within 1 h. This is similar to observations made by others.29,68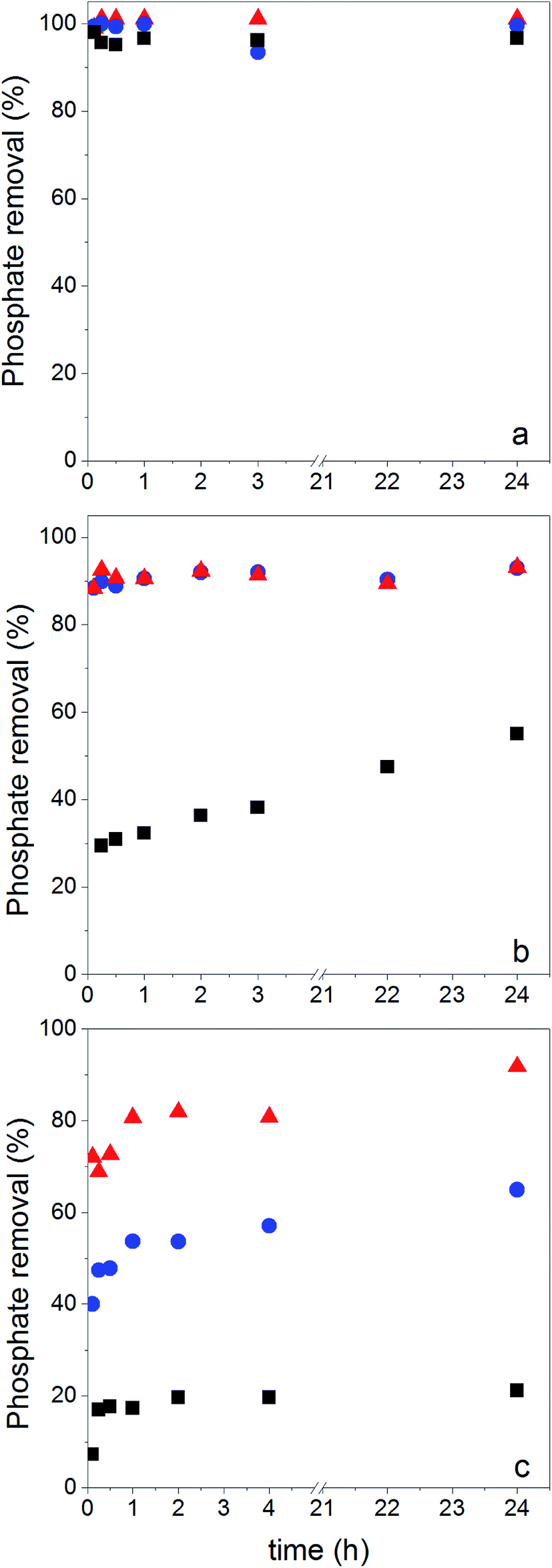 | ||
| Fig. 6 Phosphate adsorption vs. time at (a) pH = 5, (b) pH = 8 and (c) pH = 10. Data were collected for bare Fe3O4 (black squares), Fe3O4@PAH (blue spheres) and Fe3O4@PAH–Gu (red triangles). Measurements were done in triplicate and all errors were found to be lower than 0.05%. | ||
The monitored increase of phosphate adsorption as a function of time can be nicely fitted with a pseudo-second-order kinetic equation.69,70
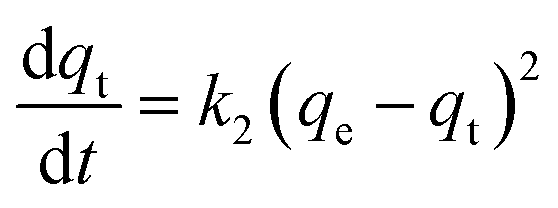 | (1) |
 | (2) |
In Table 3 the results of the fitting qe and k2 are compiled together with the calculated value of the initial rate at t → 0 (h in mg g−1 min−1),
| h = k2qe2 | (3) |
| pH 5 | pH 8 | pH 10 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| q e exp mg g−1 | q e exp mg g−1 | q e mg g−1 | k 2 mg g−1 min−1 | h mg g−1 min−1 | R 2 | q e exp mg g−1 | q e mg g−1 | k 2 mg g−1 min−1 | h mg g−1 min−1 | R 2 | |
| Fe3O4 | 3.7 | 2.0 | 1.5 | 0.28 | 0.63 | 0.99 | 0.84 | 0.82 | 0.15 | 0.10 | 0.99 |
| Fe3O4@PAH | 4.0 | 3.7 | 3.7 | 0.52 | 7.0 | 0.99 | 2.6 | 2.3 | 0.13 | 0.68 | 0.99 |
| Fe3O4@PAH–Gu | 4.0 | 3.7 | 3.6 | 2.2 | 29 | 0.99 | 3.7 | 3.7 | 0.20 | 2.1 | 0.99 |
As said, under conditions of pH = 5 the process is too fast for monitoring adsorption increase data and therefore we only report here experimental values of qe. Under conditions of pH = 8 and 10 the monitored data of increased adsorption fitted very well with the second-order kinetic equation as deduced from the obtained coefficients of determination close to unity. The observed second-order behavior is a net result of the combination of adsorption and desorption processes occurring simultaneously.71 While it is realized that the pseudo-second-order kinetics is often ascribed to a double-site interaction,42,70 we point to the derivation of the pseudo-second-order rate equation from the Langmuir kinetics as described by Liu and Shen.71 Double-site adsorption would be a correct physical interpretation, only if the binding sites involved can move independently over the surface and need to be close in order to bind one phosphate. However, the work of Liu and Shen71 demonstrates that the combination of the simultaneous adsorption and desorption processes also leads to apparent second-order kinetics when the total amount of binding sites per unit of volume is larger than both the initial concentration of the adsorbate and the inverse of the equilibrium binding constant. The fact that we observe second-order kinetics implies that these conditions are met.
No physical meaning can be attributed to k2,72 but the values for the initial adsorption rate (h) and the amount of adsorbed phosphate at equilibrium (qe) can be interpreted. At both pH = 8 and pH = 10, h increases from Fe3O4 NPs to Fe3O4@PAH to Fe3O4@PAH–Gu. At pH = 10, qe also increases in this order. In contrast, at pH = 8 for Fe3O4@PAH and Fe3O4@PAH–Gu a similar order of qe is observed, which is higher than that of Fe3O4 NPs. It is suggested therefore that at pH = 8 the adsorption capacity of the two investigated polyelectrolytes is similar. The difference between the two polyelectrolytes becomes visible at pH = 10, in favor of Fe3O4@PAH–Gu, showing a pH-independent value of qe. This is likely due to the differences in the pKa of the PAH and PAH–Gu PEs; the Gu moieties are still protonated at pH = 10, while for PAH the degree of protonation is reduced compared to the situation at pH = 8. From the results shown in Table 3, it is also clearly seen that at pH = 8 and pH = 10 the phosphate adsorption is dictated by the present PEs and that the dominant role observed for Fe3O4 at pH = 5 is now tempered. An additional difference between Fe3O4@PAH and Fe3O4@PAH–Gu (not shown here) is the selectivity for phosphate binding for the Gu containing polyelectrolytes, which we have shown in our previous study.38
Reversibility of phosphate binding
The reversibility of the adsorption process is highly relevant when it comes to practical applications. Initially we run desorption experiments under high alkaline conditions43 (pH = 12.9) to weaken the electrostatic interaction between PAH–Gu and phosphate. While a high phosphate desorption was obtained this way (>80%), the NPs were found to agglomerate and it was difficult to get them re-dispersed. Hence, the second adsorption cycle was unsuccessful and the NPs were not reusable, likely caused by a partial removal of the PAH–Gu coating, which acts as a coagulant for Fe3O4 NPs.54 However, phosphate could be removed successfully from Fe3O4@PAH–Gu NPs by a regeneration process using 10 mM of NaCl solution through an anion-exchange mechanism. Phosphate adsorption and desorption were monitored over three cycles (Fig. 7) and high levels of phosphate recovery were reached in good agreement with previous work reported in the literature.26,42 After the first cycle, the adsorption of phosphate was decreased in the next cycle by almost 20%, which might be explained by phosphate being irreversibly bound to (and/or physically entrapped in) the NPs and PAH–Gu network or to a decrease of the available active surface caused by a partial NP aggregation after the centrifugation steps.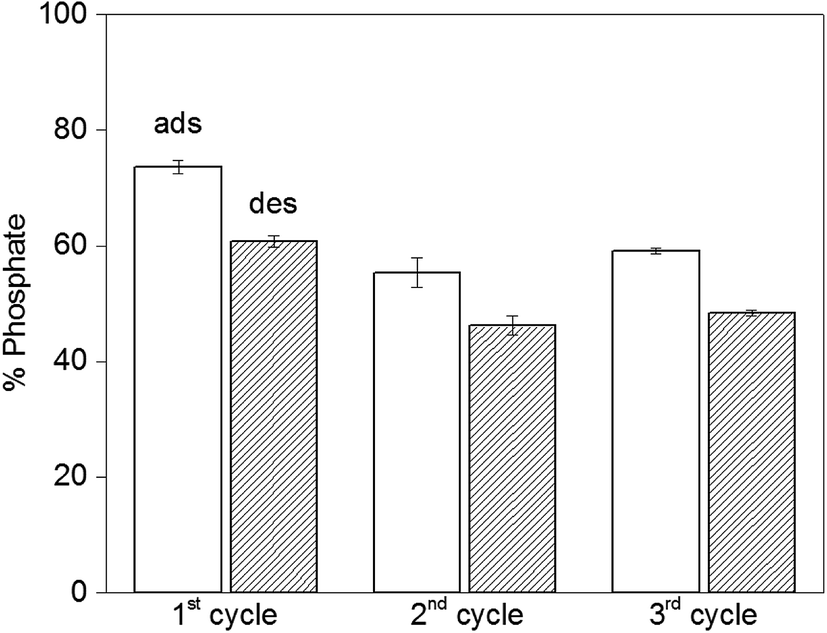 | ||
| Fig. 7 Recovery of phosphate for Fe3O4@PAH-Gu over three cycles; empty columns give the adsorption data after 24 h, while the filled columns show the desorption results. NPs were regenerated with a 10 mM NaCl solution at RT and pH = 5, the adsorbent dosage was 0.5 g L−1 and the initial phosphate concentration was 5 mg L−1. | ||
Conclusions
While several nanomaterials have been investigated for the removal of phosphate from aqueous (wastewater) streams, it remains a challenge to develop new systems operable under alkaline conditions. This study shows the results of a simple surface modification method applied to commercially available Fe3O4 NPs by using a polyelectrolyte functionalized with guanidinium groups for phosphate anion binding. The surface modification was confirmed by thermal, morphological and surface analysis measurements (FTIR, XPS and ζ-potential analysis). The PAH–Gu modified Fe3O4 NPs showed good phosphate adsorption (3.7 mg g−1) up to pH = 10, where the phosphate adsorption ability of the PAH-modified Fe3O4 (2.3 mg g−1) and unmodified Fe3O4 (0.82 mg g−1) is reduced. The initial rate of phosphate adsorption increased from 2.1 to 29 mg g−1 min−1 for PAH–Gu coated Fe3O4 NPs upon switching the pH from 10 to 8. The observed second-order adsorption kinetics can be explained as the net result of simultaneous adsorption and desorption processes at the NP surface. At the same time, the colloidal stability was enhanced upon coating the NPs with polyelectrolytes. Finally, the reversibility of phosphate binding to the novel Fe3O4@PAH–Gu NPs was studied over three cycles of adsorption and desorption, showing the reusability of the NPs. While already most (>80%) of the bound phosphate could be released again, we believe that the efficiency can be further improved by additional advanced surface modification strategies, e.g. by covalently binding the coating to the NPs and capping the remaining surface hydroxyl groups.Acknowledgements
The authors thank Wetsus – European centre of excellence for sustainable water technology (Leeuwarden, the Netherlands) for financial support and Dr Henk Miedema (Wetsus) and Mr Willem van Baak (FUJIFILM Manufacturing Europe BV, The Netherlands) for their support and fruitful discussions. This study was partly supported by the European Research Council (ERC Consolidator Grant 682444, E-motion, PI De Smet). The authors thank Dr Wiel Evers and Mr Yi Song (both TU Delft) for obtaining the TEM images and experimental support, respectively.References
- J. Schröder, D. Cordell, A. L. Smit and A. Rosemarin, Sustainable use of phosphorus, 2010 Search PubMed.
- FAO, Current world fertilizer trends and outlook to 2016, 2012 Search PubMed.
- J. J. Hudson, W. D. Taylor and D. W. Schindler, Nature, 2000, 400, 55–56 Search PubMed.
- G. E. Conley, D. J. H. Paerl, W. Howarth, R. W. Boesch, D. F. Seitzinger, S. P. Havens, K. E. Lancelot and C. Likens, Science, 2009, 323, 1014–1015 CrossRef PubMed.
- E. Levlin, M. Löwén, B. Stark and K. Hultman, Water Sci. Technol., 2002, 46, 435–440 CAS.
- European Commission, Common implementation strategy for the water framework directive (200/60/EC), 2009 Search PubMed.
- E. Desmidt, K. Ghyselbrecht, Y. Zhang, L. Pinoy, B. Van der Bruggen, W. Verstraete, K. Rabaey and B. Meesschaert, Crit. Rev. Environ. Sci. Technol., 2015, 45, 336–384 CrossRef CAS.
- D. P. Van Vuuren, A. F. Bouwman and A. H. W. Beusen, Global Environ. Change., 2010, 20, 428–439 CrossRef.
- T. Mino, M. C. M. Van Loosdrecht and J. J. Heijnen, Water Res., 1998, 32, 3193–3207 CrossRef CAS.
- Y. Zhang, B. Van der Bruggen, L. Pinoy and B. Meesschaert, J. Membr. Sci., 2009, 332, 104–112 CrossRef CAS.
- S. U. Hong, L. Ouyang and M. L. Bruening, J. Membr. Sci., 2009, 327, 2–5 CrossRef CAS.
- B. Tran, A. T. K. Tran, A. T. K. Zhang, Y. De Corte, D. Hannes, J. B. Ye, W. Mondal and P. Van Der Bruggen, J. Cleaner Prod., 2014, 77, 140–151 CrossRef.
- X. Hao, C. Wang, M. C. M. Van Loosdrecht and Y. Hu, Environ. Sci. Technol., 2013, 47, 4965–4966 CrossRef CAS PubMed.
- K. Karageorgiou, M. Paschalis and G. N. Anastassakis, J. Hazard. Mater., 2007, 139, 447–452 CrossRef CAS PubMed.
- S. Sen Gupta and K. G. Bhattacharyya, Adv. Colloid Interface Sci., 2011, 162, 39–58 CrossRef CAS PubMed.
- P. Wilfert, P. Suresh Kumar, L. Korving, G.-J. Witkamp and M. C. M. Van Loosdrecht, Environ. Sci. Technol., 2015, 49, 9400–9414 CrossRef CAS PubMed.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- K. Mandel, A. Drenkova-Tuhtan, F. Hutter, C. Gellermann, H. Steinmetz and G. Sextl, J. Mater. Chem. A, 2013, 1, 1840–1848 CAS.
- Y. Li, Q. Xie, Q. Hu, C. Li, Z. Huang, X. Yang and H. Guo, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- H. Qu, D. Caruntu, H. Liu and C. J. O'Connor, Langmuir, 2011, 27, 2271–2278 CrossRef CAS PubMed.
- Y. P. Sun, X. Q. Li, W. X. Zhang and H. P. Wang, Colloids Surf., A, 2007, 308, 60–66 CrossRef CAS.
- V. Sarapulova, E. Nevakshenova, N. Pismenskaya, L. Dammak and V. Nikonenko, J. Membr. Sci., 2015, 479, 28–38 CrossRef CAS.
- H. Huang, J. Yang and D. Li, Bioresour. Technol., 2014, 172, 253–259 CrossRef CAS PubMed.
- D. Guaya, M. Hermassi, C. Valderrama, A. Farran and J. L. Cortina, J. Environ. Chem. Eng., 2016, 4, 3519–3526 CrossRef CAS.
- H. Wang, X. Xu, R. Fei and B. Gao, RSC Adv., 2016, 6, 47237–47248 RSC.
- A. Abo Markeb, A. Alonso, A. D. Dorado, A. Sánchez and X. Font, Environ. Technol., 2016, 3330, 1–14 Search PubMed.
- J. Zhang, J. Han, M. Wang and R. Guo, J. Mater. Chem. A, 2017, 5, 4058–4066 CAS.
- L. Guo Yan, K. Yang, R. Shan, T. Yan, J. Wei, S. Jun Yu, H. Qin Yu and B. Du, J. Colloid Interface Sci., 2015, 448, 508–516 CrossRef PubMed.
- D. N. H. Tran, S. Kabiri, L. Wang and D. Losic, J. Mater. Chem. A, 2015, 3, 6844–6852 CAS.
- J. J. Richardson, M. Bjornmalm and F. Caruso, Science, 2015, 348, 1–11 CrossRef PubMed.
- R. Femmer, A. Mani and M. Wessling, Sci. Rep., 2015, 5, 1–12 Search PubMed.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS.
- Y.-W. Choi, H. Lee, Y. Song and D. Sohn, J. Colloid Interface Sci., 2015, 443, 8–12 CrossRef CAS PubMed.
- S. Wijeratne, M. L. Bruening and G. L. Baker, Langmuir, 2013, 29, 12720–12729 CrossRef CAS PubMed.
- S. Wijeratne, W. Liu, J. Dong, W. Ning, N. D. Ratnayake, K. D. Walker and M. L. Bruening, ACS Appl. Mater. Interfaces, 2016, 8, 10164–10173 CAS.
- D. Ullien, P. J. Harmsma, S. M. C. Abdulla, B. M. de Boer, D. Bosma, E. J. R. Sudhölter, L. C. P. M. de Smet and W. F. Jager, Opt. Express, 2014, 22, 16585–16594 CrossRef PubMed.
- L. van der Mee, E. S. Y. Chow, L. C. P. M. de Smet, M. de Puit, E. J. R. Sudhölter and W. F. Jager, Anal. Methods, 2015, 7, 10121–10124 RSC.
- Z. Cao, P. I. Gordiichuk, K. Loos, E. J. R. Sudhölter and L. C. P. M. de Smet, Soft Matter, 2016, 12, 1496–1505 RSC.
- K. Schug and W. Lindner, Chem. Rev., 2005, 105, 67–114 CrossRef CAS PubMed.
- Z. Xiong, Y. Chen, L. Zhang, J. Ren, Q. Zhang, M. Ye, W. Zhang and H. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 22743–22750 CAS.
- F. Caruso, Chem.–Eur. J., 2000, 6, 413–419 CrossRef CAS.
- S. Y. Yoon, C. G. Lee, J. A. Park, J. H. Kim, S. B. Kim, S. H. Lee and J. W. Choi, Chem. Eng. J., 2014, 236, 341–347 CrossRef CAS.
- O. Eljamal, A. M. E. Khalil, Y. Sugihara and N. Matsunaga, Chem. Eng. J., 2016, 293, 225–231 CrossRef CAS.
- Laboratory Research in Environmental Engineering, Phosphorus Determination using the Colorimetric Ascorbic Acid Technique, 2001 Search PubMed.
- Water Environment Federation, Standard Methods for the Examination of Water and Wastewater, 1999 Search PubMed.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Soft Matter, 2014, 10, 2479–2502 RSC.
- G. Lamanna, M. Kueny-Stotz, H. Mamlouk-Chaouachi, C. Ghobril, B. Basly, A. Bertin, I. Miladi, C. Billotey, G. Pourroy, S. Begin-Colin and D. Felder-Flesch, Biomaterials, 2011, 32, 8562–8573 CrossRef CAS PubMed.
- J. Choi and M. F. Rubner, Macromolecules, 2005, 38, 116–124 CrossRef CAS.
- B. Jachimska, T. Jasiski, P. Warszyski and Z. Adamczyk, Colloids Surf., A, 2010, 355, 7–15 CrossRef CAS.
- A. Pantos, I. Tsogas and C. M. Paleos, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 811–823 CrossRef CAS PubMed.
- S. E. Burke and C. J. Barrett, Langmuir, 2003, 19, 3297–3303 CrossRef CAS.
- P. Maroni, F. J. Montes Ruiz-Cabello, C. Cardoso and A. Tiraferri, Langmuir, 2015, 31, 6045–6054 CrossRef CAS PubMed.
- J. E. Wong, A. K. Gaharwar, D. Müller-Schulte, D. Bahadur and W. Richtering, J. Nanosci. Nanotechnol., 2008, 8, 4033–4040 CrossRef CAS PubMed.
- T. Wang, L. Zhang, H. Wang, W. Yang, Y. Fu, W. Zhou, W. Yu, K. Xiang, Z. Su, S. Dai and L. Chai, ACS Appl. Mater. Interfaces, 2013, 5, 12449–12459 CAS.
- F. Tristán, G. Palestino, J.-L. Menchaca, E. Pérez, H. Atmani, F. Cuisinier and G. Ladam, Biomacromolecules, 2009, 10, 2275–2283 CrossRef PubMed.
- X. Lin, L. Wu, Y. Liu, A. L. Ong, S. D. Poynton, J. R. Varcoe and T. Xu, J. Power Sources, 2012, 217, 373–380 CrossRef CAS.
- A. Szpak, G. Kania, T. Skorka, W. Tokarz, S. Zapotoczny and M. Nowakowska, J. Nanopart. Res., 2013, 15, 1372 CrossRef PubMed.
- J. S. Stevens, A. C. De Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238–1246 CrossRef CAS.
- L. Li and S. Zheng, Ind. Eng. Chem. Res., 2015, 54, 171–180 CrossRef CAS.
- Y. Lai, W. Yin, J. Liu, R. Xi and J. Zhan, Nanoscale Res. Lett., 2009, 5, 302–307 CrossRef PubMed.
- L. Zhang, N. Zhou, B. Wang, C. Liu and G. Zhu, J. Colloid Interface Sci., 2014, 421, 1–5 CrossRef CAS PubMed.
- P. Liu, X. Li, B. Mu, P. Du, X. Zhao and Z. Zhong, Ind. Eng. Chem. Res., 2012, 51, 13875–13881 CrossRef CAS.
- D. L. Wilcox and M. Berg, MRS Proceedings, 1994, 372, 3 CrossRef.
- R. Davies, G. A. Schurr, P. Meenan, R. D. Nelson, H. E. Bergna, C. A. S. Brevett and R. H. Goldbaum, Adv. Mater., 1998, 10, 1264–1270 CrossRef CAS.
- S. D. Sajjad, Y. Hong and F. Liu, Polym. Adv. Technol., 2014, 25, 108–116 CrossRef CAS.
- R. Chitrakar, S. Tezuka, A. Sonoda, K. Sakane, K. Ooi and T. Hirotsu, J. Colloid Interface Sci., 2006, 298, 602–608 CrossRef CAS PubMed.
- J. Kim, W. Li, B. L. Philips and C. P. Grey, Energy Environ. Sci., 2011, 4, 4298–4305 CAS.
- Z. Zhu, H. Zeng, Y. Zhu, F. Yang, H. Zhu, H. Qin and W. Wei, Sep. Purif. Technol., 2013, 117, 124–130 CrossRef CAS.
- D. Robati, J. Nanostruct. Chem., 2013, 3, 1–6 Search PubMed.
- Y. Ho, J. Hazard. Mater., 2006, 136, 681–689 CrossRef CAS PubMed.
- Y. Liu and L. Shen, Langmuir, 2008, 24, 11625–11630 CrossRef CAS PubMed.
- Y. Miyake, H. Ishida, S. Tanaka and S. D. Kolev, Chem. Eng. J., 2013, 218, 350–357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR of the synthesized PAH–Gu (Fig. S1), pictures showing the stability of several suspensions (Fig. S2 and S3). Pseudo second-order linear curves (Fig. S4) and pseudo second-order non-linear curve fitting parameters (Table S1). See DOI: 10.1039/c7ta04054g |
| This journal is © The Royal Society of Chemistry 2017 |