
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancing the n → π* electron transition of carbon nitride nanotubes for H2 photosynthesis†
Guigang
Zhang
 *,
Aleksandr
Savateev
,
Yubao
Zhao
,
Lina
Li
and
Markus
Antonietti
*,
Aleksandr
Savateev
,
Yubao
Zhao
,
Lina
Li
and
Markus
Antonietti
Department of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Potsdam, 14476, Germany. E-mail: guigang.zhang@mpikg.mpg.de
First published on 24th May 2017
Abstract
Melon-based carbon nitride (g-C3N4) is a promising metal-free and sustainable material for photocatalytic water splitting. In principle, pristine carbon nitride only exhibits moderate activity due to insufficient visible light absorption and fast charge recombination. Enhancement of the solar-to-energy conversion efficiency of g-C3N4 depends on the rational design of its morphology and electronic structure. Herein, we report the self-assembly of g-C3N4 nanotubes by co-polycondensation of urea and oxamide with their similar structure and reactivity to optimize the textural and electronic properties. Unlike pristine g-C3N4, the obtained copolymers exhibit clear optical absorption above 465 nm, which is ascribed to the n → π* electron transition involving lone pairs of the edge nitrogen atoms of the heptazine units. Besides, the charge carrier mobility was also optimized in the spatially separated nanotube structure, which contributes to the generation of more hot electrons. The optimized copolymers show dramatically enhanced H2 evolution activities especially with green light. The achieved apparent quantum yield (AQY) of optimal CN-OA-0.05 for H2 evolution with a green LED (λ = 525 nm) reaches 1.3%, which is about 10 times higher than that of pure CN with state-of-the-art activity in this wavelength region.
Introduction
The development of efficient visible light photocatalysts, namely being cost-acceptable, environmentally benign, and stable, has long been pursued as the key issue to convert abundant solar energy into storable and sustainable chemical fuels.1–4 Conjugated polymers, which are commonly composed of C, N, O, and H, i.e. light non-metal elements, can be optimized by varying the composition, structure and properties of building blocks and are considered by many researchers as the most promising candidates for visible light photocatalytic H2 evolution to meet the future energy demand.5–16As a prototypical conjugated polymer, melon-based carbon nitride has drawn particular research interest due to its advantages such as suitable HOMO and LUMO positions, robust stability, and accessible fabrication. However, similar to most polymers, pristine carbon nitride without modification only shows moderate activity due to the insufficient optical absorption and rapid charge carrier recombination. To obtain sufficient solar-to-energy conversion efficiency, it is reasonable to optimize the structural, textural and electronic properties of the polymer. Such modifications have indeed been certified effective in improving the photocatalytic activities.17–22 However, it is still a great challenge to synthesize carbon nitride polymers that could present sufficient visible light activities with light wavelengths longer than 500 nm.
In principle, the band gap of photocatalysts should be at least narrower than 2.48 eV in order to achieve sufficient visible light absorption above 500 nm. The normal way to enhance the visible light absorption is copolymerization with another monomer to mediate the polymerization process and thus to tailor the structural and electronic properties of the as-prepared polymers. For instance, by copolymerization with conjugated monomers, the optical absorption of the as-prepared carbon nitride copolymers could be extended to 650 nm. Consequently, the optimized copolymers showed evidently enhanced visible light H2 evolution activities.23,24 Apart from the enhanced optical absorption, the polymers should bear an optimized architecture to accelerate the bulk and surface charge carrier separation and transfer. For instance, carbon nitride with a well-designed morphology, e.g., nano-rods,25 hollow spheres,26,27 and mesopores,28 indeed could provide a spatially modified architecture to liberate the charge transfer and thus promote the photocatalytic activities. We therefore imagine that the simultaneous modification in the architecture and the localized electronic hybridization of the polymers would potentially advance the charge carrier behaviour and the visible light capture ability, thus achieving sufficient visible light activity even above 500 nm.
To this end, oxamide (OA), which by structure and reactivity is rather similar to urea, was selected as a suitable monomer to incorporate with urea for tailoring the structural and electronic properties of g-C3N4 polymers. The two molecular compounds could be interacted by a hydrogen bond to form a cross-linked complex and subsequently generate unique carbon nitride with a well-designed architecture after thermal condensation (see Scheme S1†). It is thus desirable to create new promising carbon nitride polymers with optimized morphology and optical properties to improve the visible light H2 evolution activities beyond the white and blue light region.
Results and discussion
Typically, carbon nitride prepared from urea only (as a reference) is denoted as CN for simplicity and OA-modified CN polymers are labelled CN-OA-x, where x represents the initial mass fraction of OA (x = 0.01, 0.03, 0.05 and 0.1).As shown in Fig. 1a, the local structures of all samples are indeed very similar, characterized by two distinct diffraction peaks located at about 2θ = 13.0° and 27.4°, respectively. The first peak corresponds to the in-plane periodic repeat units of heptazine, while the latter one is attributed to the interlayer (002) aromatic stacking of the graphitic layer.28–31 The vibration modes shown in Fig. 1b reflect that all the samples also possess a very similar skeleton structure. The strong peaks located at 1200 to 1600 cm−1 are mainly attributed to the feature-distinctive stretching mode of the heptazine cycles (C6N7), while the sharp peaks appearing at about 810 cm−1 belong to the breathing mode of the triazine units.32,33 Both of these peaks are classical variations of g-C3N4, further revealing the successful evolution of heptazine-based CN heterocycles. The broad peaks located at 2900–3300 cm−1 are the typical stretching modes of N–H and O–H, which are ascribed to the un-condensed surface residual N–H group and absorbed H2O molecules, respectively.34,35 Besides, the proposed tri-s-triazine based chemical structure was further confirmed by solid-state 13C CP-MAS NMR analysis. As shown in Fig. S1,† two distinct peaks located at 163.1 and 156.5 ppm were observed for both CN and CN-OA-0.05. The first one is attributed to C(e) atoms in the form of N2–CN or terminal CN2–NHx, whereas the latter one is related to C(i) atoms in the form of C–N3. It is to be noted that no other new peaks were viewed, once again confirming that copolymerization with oxamide does not change the intrinsic building units of carbon nitride.
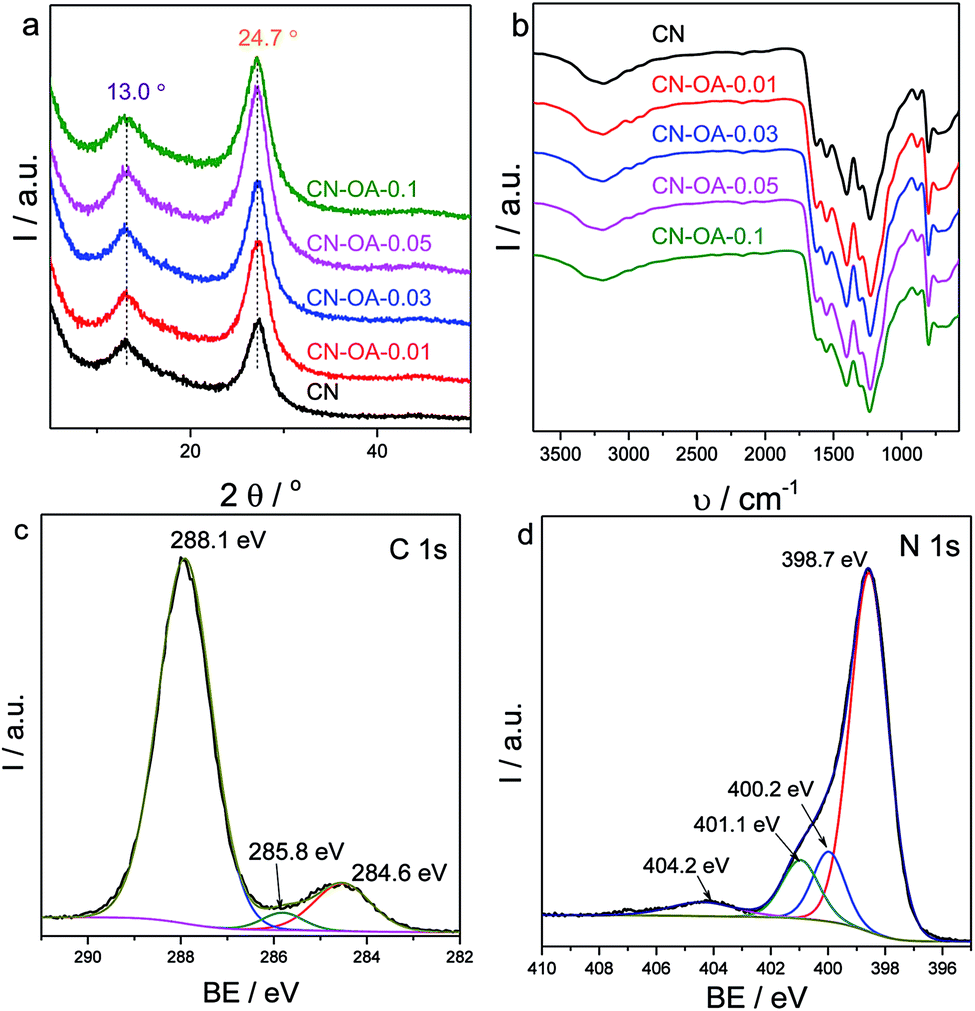 | ||
| Fig. 1 (a) Powder XRD patterns, (b) FT-IR spectra of CN and CN-OA-x samples; high-resolution XPS spectra of (c) C 1s and (d) N 1s for CN-OA-0.05. | ||
To gather information about more local binding environments of the polymers, XPS analysis was further conducted. Carbon, nitrogen, and small amounts of oxygen co-existed (Fig. S2†). The high resolution XPS spectra of C 1s and N 1s of CN-OA-0.05 are shown in Fig. 1c and d. Two evident peaks at 284.6 and 288.1 eV were observed for C 1s, which are attributed to the standard carbon being present as a disturbance in the sample and sp2-hybridized carbon (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) in the nitrogen rich heterocyclic environment of carbon nitride, respectively.36,37 Besides, an additional peak at 285.8 eV assigned to the C–O bond could be viewed, which we attribute to the residual C–O and surface adsorbed CO2. The N 1s peak can be de-convoluted into four distinct peaks at 398.7, 400.2, 401.1 and 404.2 eV. The major peak at 398.7 eV is related to the sp2-hybridized nitrogen in the form of C–NC, whereas the second peak at 400.2 eV belongs to the N–(C)3 group.30 These two types of nitrogen together with the sp2-hybridized carbon make up the key skeleton of the heptazine heterocyclic (C6N7) units of CN polymers. The weak peak at 401.1 eV is assigned to the surface residual C–N–H bond, while the broad peak at 404.2 eV results from charging effects or positive charge localization in the heterocycles.28 It is to be noted that the XPS analysis results are very similar to those of the reference CN in the previous literature.32–35 The C/N molecular ratio of the samples is listed in Table S1.† Expectedly, only minor changes in the C/N ratio are obtained (from 0.71 for CN to 0.77 for CN-OA-0.1) after OA modification. We therefore summarize those analytical data that the structural changes in the semiconductor system by partial doping with OA are indeed minor.
N) in the nitrogen rich heterocyclic environment of carbon nitride, respectively.36,37 Besides, an additional peak at 285.8 eV assigned to the C–O bond could be viewed, which we attribute to the residual C–O and surface adsorbed CO2. The N 1s peak can be de-convoluted into four distinct peaks at 398.7, 400.2, 401.1 and 404.2 eV. The major peak at 398.7 eV is related to the sp2-hybridized nitrogen in the form of C–NC, whereas the second peak at 400.2 eV belongs to the N–(C)3 group.30 These two types of nitrogen together with the sp2-hybridized carbon make up the key skeleton of the heptazine heterocyclic (C6N7) units of CN polymers. The weak peak at 401.1 eV is assigned to the surface residual C–N–H bond, while the broad peak at 404.2 eV results from charging effects or positive charge localization in the heterocycles.28 It is to be noted that the XPS analysis results are very similar to those of the reference CN in the previous literature.32–35 The C/N molecular ratio of the samples is listed in Table S1.† Expectedly, only minor changes in the C/N ratio are obtained (from 0.71 for CN to 0.77 for CN-OA-0.1) after OA modification. We therefore summarize those analytical data that the structural changes in the semiconductor system by partial doping with OA are indeed minor.
As OA has a linear molecular structure, it could connect with urea by a hydrogen bond to form a linear complex (see Scheme S1†). Besides, OA possesses more carbon and oxygen components, and it tends to decompose with evolution of CO2 and NH3 when heated in an oven. Consequently, the fast evolution of CO2 gas may greatly accelerate the sluggish deamination process and will optimize the texture and morphology of the as-prepared copolymers. The morphologies of the polymers were then characterized by SEM and TEM. As shown in Fig. 2a and S3,† pure CN displays the typical graphitic packed-layer structure. When the initial mass fraction of OA was increased from 0.01 to 0.1, the morphology of the samples indeed changes from nanosheets to weakly ordered, hollow nanotubes with some pores, which we attribute to the formation of a cross-linked linear complex by a hydrogen bond and fast evolution of CO2 and NH3 gases during the thermal treatment of urea and oxamide at high temperatures.38,39 This nanotube architecture in fact could provide better access and transport for the consecutive heterogeneous catalytic reactions. The SEM elemental mapping analysis further confirms that the as-prepared CN-OA-0.05 sample was composed of C, N and small amounts of O (Fig. S4†). Energy-dispersive X-ray spectroscopy (EDS) in Fig. S5† of CN and CN-OA-0.05 samples shows that the C/N molecular ratio is slightly increased from 0.71 to 0.75, which is in good accordance with the previous elemental analysis. The BET specific surface area of CN-OA-x samples was also examined (Table S1†). It could be found that the specific surface area only slightly changes from 56 (CN) to 64 m2 g−1 (CN-OA-0.1), revealing the fact that the enhanced activity was not ascribed to the surface area. We then characterized the structural details of the polymers in order to gather more useful information to better understand the structure–activity relationship. The TEM pictures of CN and CN-OA-0.05 are shown in Fig. 2c and d. It could be observed that pure CN exhibits the typical 2D nanosheet like structure, whereas the CN-OA-0.05 polymer presents a hollow-tube like morphology with tube thicknesses of less than 10 nm. This hollow structure may benefit the visible light absorption due to the multiple diffuse reflectances inside the optimized nano-architecture and it could also provide more active sites for the reactive species. Both of the enhanced light absorption and the open construction system probably contribute to improve the interfacial photocatalytic activities.
 | ||
| Fig. 2 SEM images of (a) CN and (b) CN-OA-0.05 samples. TEM images of (c) CN and (d) CN-OA-0.05 samples. | ||
To evaluate the effect of OA on the optimization of the optical properties, the light absorption behaviour of CN and CN-OA-x samples was then examined by UV-Vis diffusive reflection spectra (DRS). In Fig. 3a, pure CN only shows light absorption below 460 nm, i.e. it can only be excited by white and blue light. On the contrary, the OA-modified polymers showed greatly enhanced visible light absorption especially in the light region above 450 nm, which can be verified by the colour change of the samples from pale yellow for CN to brownish yellow for CN-OA-0.1 (the inset of Fig. 3a). The intrinsic absorption bands below 450 nm are a result of π → π* electron transitions in the conjugated aromatic system.23 However, the newly emerged absorption bands located at approximately 520 nm are probably assigned to n → π* electron transitions involving lone pairs of the edge nitrogen atoms of the heptazine units.40 It is to be noted that n → π* transitions are spatially forbidden for perfectly symmetric and planar units, implying the generation of an asymmetric planar structure for the OA-copolymerized photocatalysts. Evidently, the nanotube architecture is investigated to spatially accelerate the charge carrier transfer and subsequently improve the photocatalytic activities.28 The band gap energies estimated from the Tauc plot of pure CN is calculated to be 2.75 eV (Table S1†), whereas that of CN-OA-x is narrowed from 2.64 to 2.21 eV, supplying sufficient light absorption needed for photocatalytic H2 evolution with blue and green light. As the actual conduction and valence band positions are normally closely related to the reduction and oxidation capacity of the semiconductors, it is necessary to make clear the band position after OA-modification. Therefore, the flat potential of the samples was estimated by the traditional Mott–Schottky methods (Fig. 3c and d). It could be observed that the flat potential of CN-OA-0.05 was −1.1 V vs. Ag/AgCl (pH 6.6), which is slightly positive compared with that of pristine CN (−1.41 V, vs. Ag/AgCl). The relative band positions of the polymers are described in Fig. 3b. It is observed that the conduction band was slightly decreased to −1.1 V after modification while quite a few changes could be viewed in the valence band. Although the reduction capacity of the conduction electrons in CN-OA-0.05 is reduced, it is still negative enough for proton reduction (EH+/H2: −0.59 V vs. Ag/AgCl, pH 6.6). We then speculate that the overall band structure was really modified, while not only the additional interband state with lower or no activity was added.
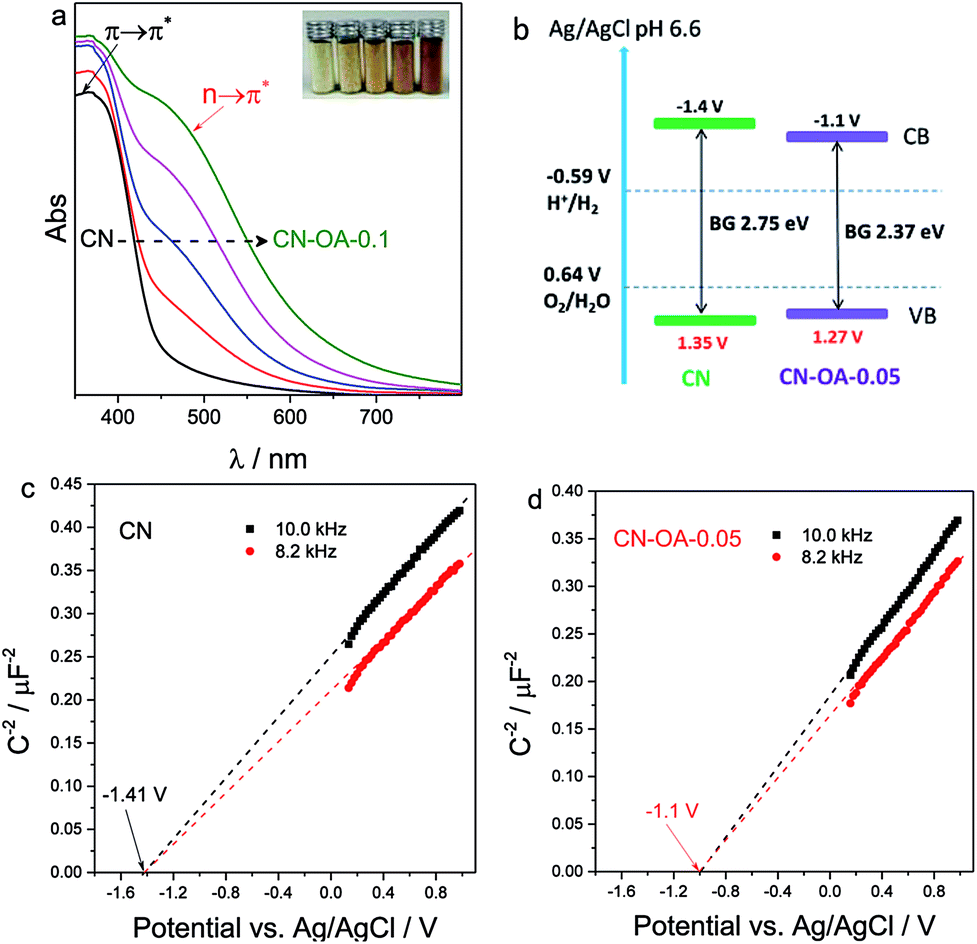 | ||
| Fig. 3 (a) UV-vis diffraction reflectance spectra (DRS), inset: apparent picture of the samples; (b) conduction and valence band positions of CN and CN-OA-0.05; (c) and (d) Mott–Schottky plots of CN and CN-OA-0.05 samples, experimental conditions: 0.2 M Na2SO4 and pH 6.6. | ||
Optimizing the morphology and optical properties leads to substantial suppression of radiative electron–hole recombination in carbon nitride nanotubes, as indicated by the greatly weakened PL peak in Fig. 4a. For the pristine CN, it shows a strong emission peak at 462 nm, which originates from the band-to-band recombination of the electrons and holes.41,42 In principle, in comparison with CN, the PL emission peak intensity of CN-OA-x is however dramatically decreased, indicating that the radiative charge carrier recombination is suppressed in the slightly irregular construction, which may favor the potential interfacial redox reactions.25 Moreover, it could be observed that a clear red-shift of the emission peak from approximately 462 nm to 522 nm was generated after OA-modification. In principle, the restrained charge carrier recombination could be further verified by the time-resolved transient PL decay of the samples. As shown in Fig. 4b, the average PL lifetime decreases from 15.37 ns for CN to 8.12 ns for CN-OA-0.05. The remarkable shortening of the PL lifetime is probably attributed to efficient exciton dissociation in the nanotube structure, which favours the hot charge carrier transfer and yield. The huge advantage of OA-doping in optimizing the electronic performance could also be evaluated by Electrochemical Impedance Spectra (EIS) and transient generation of photocurrent. With EIS analysis (Fig. 4c), it is clearly observed that the optimized copolymers present much smaller resistances than pristine CN due to the modified surface zeta potentials (Table S1†). This results also in a larger transient photocurrent (Fig. 4d), when comparing CN-OA-0.05 with CN, further illustrating the advanced charge carrier behaviour. Both of these electronic properties indicate the fast transfer of charge carriers owing to the well optimized texture, optical and electronic properties from the OA-modification, which is certainly favourable to promote the photocatalytic activities.
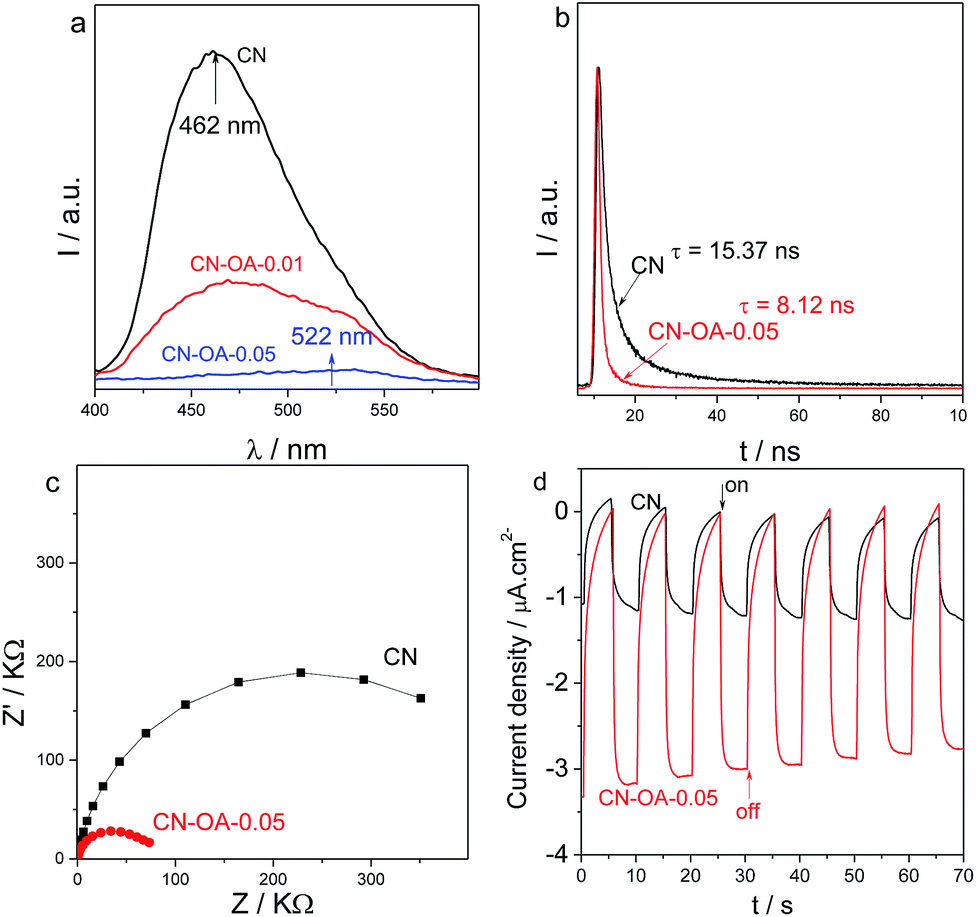 | ||
| Fig. 4 (a) Room temperature steady state photoluminescence (PL) emission spectra of CN and CN-OA-x samples with an excitation wavelength of λ = 400 nm; (b) time-resolved transient PL decay of CN and CN-OA-0.05; (c) electrochemical impedance spectroscopy (EIS) Nyquist plots and (d) transient photocurrent (λ > 420 nm, applied potential: −0.2 V) for CN and CN-OA-0.05 in 0.2 M Na2SO4 aqueous solution (pH 6.6). | ||
The photocatalytic activities of CN and the CN-OA-x series were then evaluated by an assay of photocatalytic H2 evolution from water. All the lamps used here are LED lamps. Compared with the traditional xenon lamp (with a work power of 300 W), the LED lamp (50 W) consumes less energy and generates less heat while maintaining robust stability for a long time course reaction, which is more resource efficient for evaluating the photocatalytic activities. As shown in Fig. 5a, all the polymers are indeed active for photocatalytic H2 evolution with white light irradiation. It is noteworthy to observe that all CN-OA-x polymers exhibit enhanced activities in comparison with the pure CN, among which CN-OA-0.03 presents the highest rate, two times higher than that of CN. The H2 evolution activities can indeed be further enhanced when the polymerization temperature was increased to 600 °C (Fig. S6†). This enhancement is attributed to an enhanced degree of polycondensation, which can also be certified by DRS, XRD and FTIR (Fig. S7–S9†).
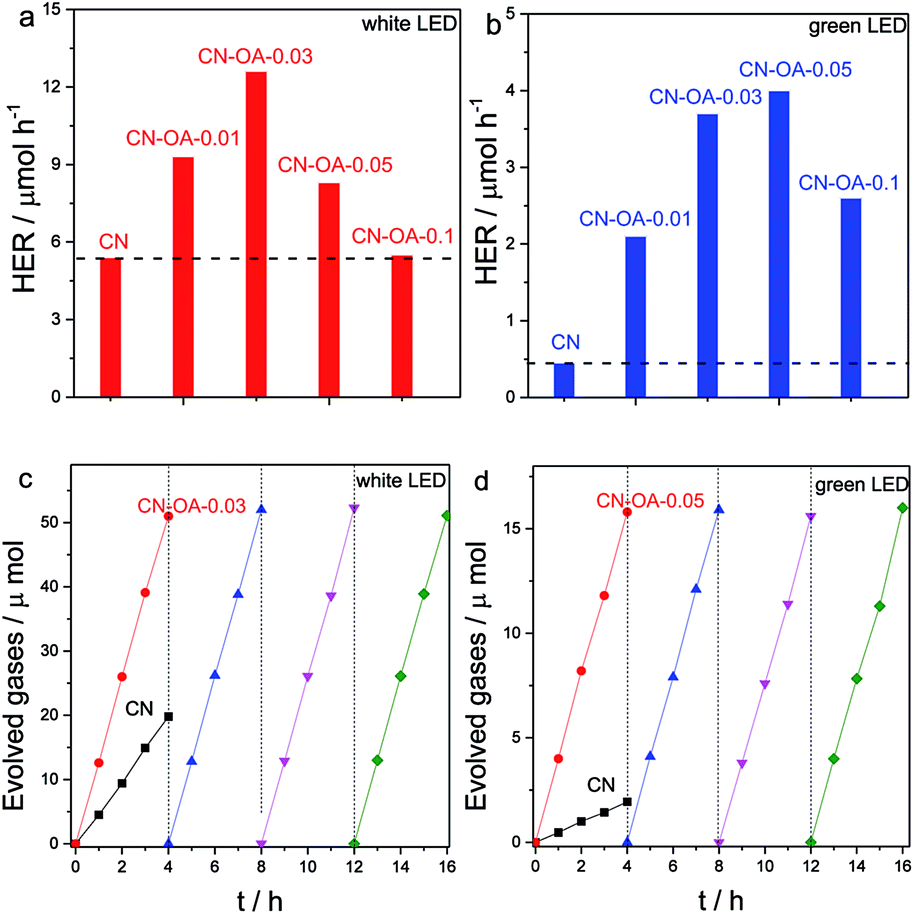 | ||
| Fig. 5 Photocatalytic H2 evolution activities of CN and CN-OA-x samples with (a) white LED and (b) green LED light irradiation. Long-time course photocatalytic H2 evolution of CN and CN-OA-x samples: (c) white LED and (d) green LED light irradiation. | ||
As indicated by the optical absorption spectra, CN shows only negligible optical absorption at λ > 500 nm, while the absorption band of CN-OA-x could be extended up to 650 nm. Typically, the light absorption at 420 nm was observed from 0.49 to 1.02, while the value was clearly increased from 0.05 to 0.66 at 525 nm (Table S2†). This is why we further evaluated the H2 evolution activities of the samples under green (λ = 525 nm) LED light irradiation. Fig. 5b shows that all CN-OA-x polymers exhibit enhanced H2 evolution activities in comparison with CN. The best sample for this purpose, CN-OA-0.05, presents about ten times faster H2 evolution rate than CN, which is in good accordance with the optical absorption properties. The fact that photoactivity goes with light absorption supports our concept that in this case indeed the overall band structure was modified, while not only the additional interband state with lower or no activity was added.
In addition, both CN and CN-OA-0.05 samples express robust stability at long reaction times. In Fig. 5c and d, no obvious decrease of H2 evolution was observed under both visible light and green light irradiation even after 16 hours of persistent reaction, implying the robust stability of the copolymers towards long time course solution- and light-corrosion. No obvious change in the structure could be observed when the polymers were collected and characterized by XRD and FT-IR (Fig. S10 and S11†) after full photocatalytic cycles. Besides, the nanotube structure remains well constructed even after the heterogeneous reaction (see the SEM picture in Fig. S12†), once again certifying the good stability towards solution and light corrosion. In a last step, in order to better understand and compare the activities with the previously reported studies, the apparent quantum yield (AQY) of CN and the optimal CN-OA-0.05 under green light (λ = 525 nm) was examined and found to be 0.12% and 1.3%, respectively. This significant enhancement of H2 evolution activity with green light can be ascribed to the higher optical absorption, improved electronic properties and a better micro- and nanostructure.
Conclusions
Promising carbon nitride nanotubes were synthesized by self-assembly polymerization of oxamide and urea. It was found that the as-prepared CN-OA-x polymers show only very minor differences in composition, while the morphology, electronic and optical properties were clearly optimized. The optimal CN-OA-0.05 polymer shows largely increased H2 evolution activities especially under green light irradiation. The AQY for H2 evolution of CN-OA-0.05 with green light (λ = 525 nm) was quantified to be 1.3%, which is about 10 times higher than that of the pure CN. This study highlights the self-assembly of carbon nitride nanotubes to spatially promote the charge transfer in the well-designed architecture and also enhance the visible light absorption above 500 nm, which is certainly beneficial to improve the visible light photocatalytic activities. It opens up a pathway to simultaneously optimize the morphology and optical properties of semiconductors without using any sacrificial template, making it very attractive for developing new polymeric materials with advanced structures and properties for promising solar energy conversion applications.Acknowledgements
Dr G. G. Zhang thanks the AvH (Alexander von Humboldt) foundation for a postdoctoral fellowship. We also thank very much the technicians in Max Planck Institute who have greatly supported us for sample characterization. Open Access funding provided by the Max Planck Society.References
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Ioune and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 65031–66570 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- (a) J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed; (b) G. G. Zhang, Z. A. Lan, L. H. Lin, S. Lin and X. C. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- (a) X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed; (b) G. G. Zhang, Z. A. Lan and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed; (c) Z. M. Pan, Y. Zheng, F. S. Guo, P. P. Niu and X. C. Wang, ChemSusChem, 2017, 10, 87–90 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 988 CrossRef CAS PubMed.
- M. G. Schwab, M. Hamburger, X. L. Feng, J. Shu, H. W. Spiess, X. C. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- A. Indra, A. Acharjya, P. W. Menezes, C. Merschjann, D. Hollmann, M. Schwarze, M. Aktas, A. Friedrich, S. Lochbrunner, A. Thomas and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 1653–1657 CrossRef CAS PubMed.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- (a) G. Liu, P. Niu, C. H. Sun, S. C. Smith, Z. G. Chen, G. Q. Lu and H. M. Chen, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed; (b) C. Yang, B. Wang, L. Z. Zhang, L. Yin and X. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 6627–6631 CrossRef CAS PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- T. Y. Ma, J. Y. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- H. Wang, X. S. Sun, D. D. Li, X. D. Zhang, S. C. Chen, W. Shao, Y. P. Tian and Y. Xie, J. Am. Chem. Soc., 2017, 139, 2468–2473 CrossRef CAS PubMed.
- X. D. Zhuang, Y. Y. Mai, D. Q. Wu, F. Zhang and X. L. Feng, Adv. Mater., 2015, 27, 403–427 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- (a) J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394–16401 CrossRef CAS; (b) P. J. Yang, H. H. Ou, Y. X. Fang and X. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992–3996 CrossRef CAS PubMed.
- Z. X. Zhou, J. H. Wang, J. C. Yu, Y. F. Li, Y. Shen, A. R. Liu, S. Q. Liu and Y. J. Zhang, J. Am. Chem. Soc., 2015, 137, 2179–2182 CrossRef CAS PubMed.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2018, 138, 10019–10025 CrossRef PubMed.
- Q. H. Liang, Z. Li, Xi. L. Yu, Z. H. Huang, F. Y. Kang and Q. H. Yang, Adv. Mater., 2015, 27, 4634–4639 CrossRef CAS PubMed.
- Q. Han, B. Wang, Y. Zhao, C. G. Hu and L. T. Qu, Angew. Chem., Int. Ed., 2015, 54, 11433–11437 CrossRef CAS PubMed.
- G. G. Liu, T. Wang, H. B. Zhang, X. G. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. H. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- J. S. Zhang, G. G. Zhang, X. F. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. C. Wang, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS PubMed.
- (a) X. H. Li, J. S. Zhang, X. F. Chen, A. Fischer, A. Thomas, M. Antonietti and X. C. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS; (b) B. H. Long, Y. Zheng, L. H. Lin, K. A. Alamary, A. M. Asiri and X. C. Wang, J. Mater. Chem. A, 2017 10.10391/C6TA09802A.
- D. D. Zheng, X. N. Cao and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef CAS PubMed.
- Y. S. Jun, J. Park, S. U. Lee, A. Thomas, W. H. Hong and G. D. Stucky, Angew. Chem., Int. Ed., 2013, 52, 11083–11087 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe, K. Domen, Y. D. Hou, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- G. G. Zhang, M. W. Zhang, X. X. Ye, X. Q. Qiu, S. Lin and X. C. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlöglb and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- Y. Y. Kang, Y. Q. Yang, L. C. Yin, X. D. Kang, L. Z. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- S. W. Cao, J. X. Low, J. G. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. H. Zhu, L. H. Li, Y. Han, Y. Chen, A. J. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- (a) Q. Han, B. Wang, J. Gao and L. T. Qu, Angew. Chem., Int. Ed., 2016, 55, 10849–10853 CrossRef CAS PubMed; (b) L. H. Lin, H. H. Ou, Y. F. Zhang and X. C. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. J. Du, W. M. Zhang, Z. H. Zhu, S. C. Smith, M. Jaroniec, G. Q. Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- Y. W. Zhang, J. H. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- D. J. Martin, P. J. Reardon, S. J. A. Moniz and J. W. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- P. Niu, L. C. Yin, Y. Q. Yang, G. Liu and H. M. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed.
- Y. Chen, B. Wang, S. Lin, Y. F. Zhang and X. C. Wang, J. Phys. Chem. C, 2014, 118, 29981–29989 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta03777e |
| This journal is © The Royal Society of Chemistry 2017 |