
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the correlation and balance between the miscibility and optoelectronic properties of polymer–fullerene solar cells†
Chaohong
Zhang
 *a,
Stefan
Langner
a,
Alexander V.
Mumyatov
b,
Denis V.
Anokhin
bcd,
Jie
Min
ea,
José Darío
Perea
a,
Kirill L.
Gerasimov
d,
Andres
Osvet
a,
Dimitri A.
Ivanov
cdf,
Pavel
Troshin
*gb,
Ning
Li
*a and
Christoph J.
Brabec
*ah
*a,
Stefan
Langner
a,
Alexander V.
Mumyatov
b,
Denis V.
Anokhin
bcd,
Jie
Min
ea,
José Darío
Perea
a,
Kirill L.
Gerasimov
d,
Andres
Osvet
a,
Dimitri A.
Ivanov
cdf,
Pavel
Troshin
*gb,
Ning
Li
*a and
Christoph J.
Brabec
*ah
aInstitute of Materials for Electronics and Energy Technology (i-MEET), Friedrich-Alexander University Erlangen-Nürnberg, Martensstrasse 7, 91058 Erlangen, Germany. E-mail: chaohong.zhang@fau.de; ning.li@fau.de; christoph.brabec@fau.de
bInstitute for Problems of Chemical Physics of Russian Academy of Sciences, Semenov Prospect 1, Chernogolovka, 142432, Russia
cMoscow Institute of Physics and Technology (State University), Institutskiy per. 9, Dolgoprudny, 141700, Russia
dMoscow State University, Faculty of Fundamental Physical and Chemical Engineering, GSP-1, 1-51 Leninskie Gory, Moscow, 119991, Russia
eThe Institute for Advanced Studies, Wuhan University, Wuhan 430072, China
fInstitut de Sciences des Matériaux de Mullhouse, CNRS UMR 7361, 15 Jean Starcky, F-68051 Mulhouse, France
gSkolkovo Institute of Science and Technology, Skolkovo Innovation Center, Nobel St. 3, Moscow, 143026, Russian Federation. E-mail: p.troshin@skoltech.ru
hBavarian Center for Applied Energy Research (ZAE Bayern), Immerwahrstr. 2, 91058 Erlangen, Germany
First published on 27th July 2017
Abstract
Organic photovoltaics is one of the most promising technologies for sustainable green energy supply. Because of their high electron affinity and superior electron-transporting ability, fullerene-based materials are deemed as very strong electron-accepting components in organic solar cells. However, the most widely used fullerene-based acceptors, such as phenyl-C61-butyric acid methyl ester, exhibit limited microstructural stability and unsatisfactory thermal stability owing to their insufficient compatibility with organic donors. Here, we in-depth investigate the carrier dynamics along with structural evolution and analyze the acceptor loadings in optimized bulk-heterojunction (BHJ) solar cells as a function of the polymer–fullerene miscibility. The polymer–fullerene miscibility has more influential effects than the crystallinity of single components on the optimized acceptor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :donor ratio in polymer–fullerene solar cells. The findings demonstrated in this work suggest that the balance between the miscibility of BHJ composites and their optoelectronic properties has to be carefully considered for future development and optimization of OPV solar cells based on BHJ composites. Miscibility is proposed in addition to crystallinity as a further design criterion for long lived and efficient solar cells.
:donor ratio in polymer–fullerene solar cells. The findings demonstrated in this work suggest that the balance between the miscibility of BHJ composites and their optoelectronic properties has to be carefully considered for future development and optimization of OPV solar cells based on BHJ composites. Miscibility is proposed in addition to crystallinity as a further design criterion for long lived and efficient solar cells.
Introduction
Owing to the advantages of light weight, easy fabrication, low cost and flexible devices, worldwide research efforts have been devoted to thin-film organic photovoltaics (OPV) in the past two decades.1–12 The active layers of organic solar cells are commonly composed of blends of an electron donor and an electron acceptor. After introducing the bulk-heterojunction (BHJ)1,2 concept for the active layers, enormous progress has been made in every aspect of OPV technology.13–27 [6,6]-Phenyl-C61-butyric acid methyl ester (PC61BM) and its corresponding C70 analogue PC71BM have been the most prominent acceptor materials over the last two decades, and achieved the efficiency milestones in combination with various benchmark polymer donors, such as poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylenevinylene] (MDMO-PPV),3,13 poly(3-hexylthiophene-2,5-diyl) (P3HT),4 poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (PCPDTBT),8 poly((3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl)) (PTB7),28 poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-2-carboxylate-2-6-diyl)] (PTB7-Th)11,29 and poly[(5,6-difluoro-2,1,3-benzothiadiazol-4,7-diyl)-alt-(3,3′′′-di(2-octyldodecyl)-2,2′;5′,2′′;5′′,2′′′-quaterthiophen-5,5′′′-diyl)] (PffBT4T-2OD).30 The unique properties of functional fullerene acceptors are mainly related to pristine fullerenes, such as great electron-transporting ability and high electron affinity. The adducted organic group of fullerene-based acceptors can be chemically modified to tune their solubility in organic solvents and compatibility with organic donor materials, which makes the functional fullerene acceptors so successful in the field of OPV technology.20,24,31–33Although great photovoltaic performance has been achieved by utilizing PCBM, engineers further try to understand and improve the device stability of organic solar cells, and chemists also try to create more optimized fullerene acceptors.24,33 To minimize the bandgap-to-open-circuit voltage (Voc) losses and to approach the theoretical VOC limits, a series of bis-adduct fullerene derivatives were designed, such as bis(1-[3-(methoxycarbonyl)propyl]-1-phenyl)-[6,6]C62 (bis-PCBM),34 indene-PC60BM (IPCBM),35 bisadducts of thieno-o-quinodimethane with C60 (bis-TOQC),36 di(4-methylphenyl)methano-C60 bis-adduct (DMPCBA),37 and indene-C60 bisadduct (ICBA).38 To attain fullerene materials with the high-lying lowest unoccupied molecular orbital (LUMO) as well as high isomer purity, mono-adduct fullerenes like methanofullerenes (besides PCBM),39–41 di(organo)fullerenes,42–44 and fulleropyrrolidine45–48 were developed and systematically investigated. To improve morphological stability and long-term stability, fullerene-based oligomers or polymers were developed and applied in solar cells.49–52 Most recently, we introduced two novel fullerene acceptors, pyrrolidinofullerene (PyF5) and methanofullerene (FAP1), which display excellent thermal stability and promising VOC as well as photovoltaic efficiency in combination with two representative polymer donors.53 However, during the device optimization, we found that the attained photovoltaic performance of PyF5- and FAP1-based solar cells is slightly lower than that of the corresponding PC61BM-based devices with respect to reduced short-circuit current densities (JSC) as well as fill factors (FF). Since PyF5 and FAP1 are highly promising alternatives to replace PC61BM for efficient OPV solar cells with excellent thermal stability, it is therefore of major relevance to analyze the photovoltaic losses and to gain valuable insight into future design and development of functional acceptors for highly efficient yet stable OPV solar cells.
In this study, we in-depth investigate the loss mechanism of polymer solar cells based on the amorphous dominated fullerene acceptor PyF5 as well as on the semi-crystalline fullerene acceptor FAP1. Increased fill factors are observed for PyF5- and FAP1-based solar cells with higher fullerene loadings, while the fill factor of PC61BM based devices is quite saturated after certain fullerene loadings. Space-charge-limited current (SCLC) measurements are performed to study the charge carrier transport properties of pristine fullerenes and blends with various fullerene contents. The electron mobilities of these three pristine fullerenes are quite comparable but distinctly differing electron mobilities are found in BHJ composites. We further investigate the morphological evolution of the blend films by means of in situ temperature-dependent grazing-incidence wide-angle X-ray scattering (GIWAXS). Combining our findings and understanding, insights into the balance between miscibility and photovoltaic performance are given in a conclusive picture.
Results and discussion
Photovoltaic properties of amorphous and crystalline fullerene based solar cells
Fig. 1a summarizes the chemical structures of the polymer donor, PTB7-Th and three fullerene acceptors studied in this work. Fig. 1b depicts the normalized absorption of pristine PTB7-Th, PC61BM, Py5 and FAP1. The absorption region of the three fullerenes is mainly located at around 320 nm while PTB7-Th has a strong absorption area between 500 and 800 nm. The photoluminescence (PL) peak of PTB7-Th appears at ∼830 nm. When blended with PC61BM (donor (D):acceptor (A) = 1.5:1), PyF5 (D:A = 1:1 or 1:3), or FAP1 (D:A = 1:1 or 1:3), the PL emission of PTB7-Th is significantly quenched by over 98% for all blends. These phenomena suggest that the exciton dissociation/charge separation process is highly efficient for PTB7-Th:PyF5 and PTB7-Th:FAP1 with either high or low fullerene loadings. We further fabricate solar cells consisting of PTB7-Th as the donor, PC61BM, PyF5 or FAP1 as the acceptor with various D:A ratios. The solar cell architecture is shown in Fig. 1c.
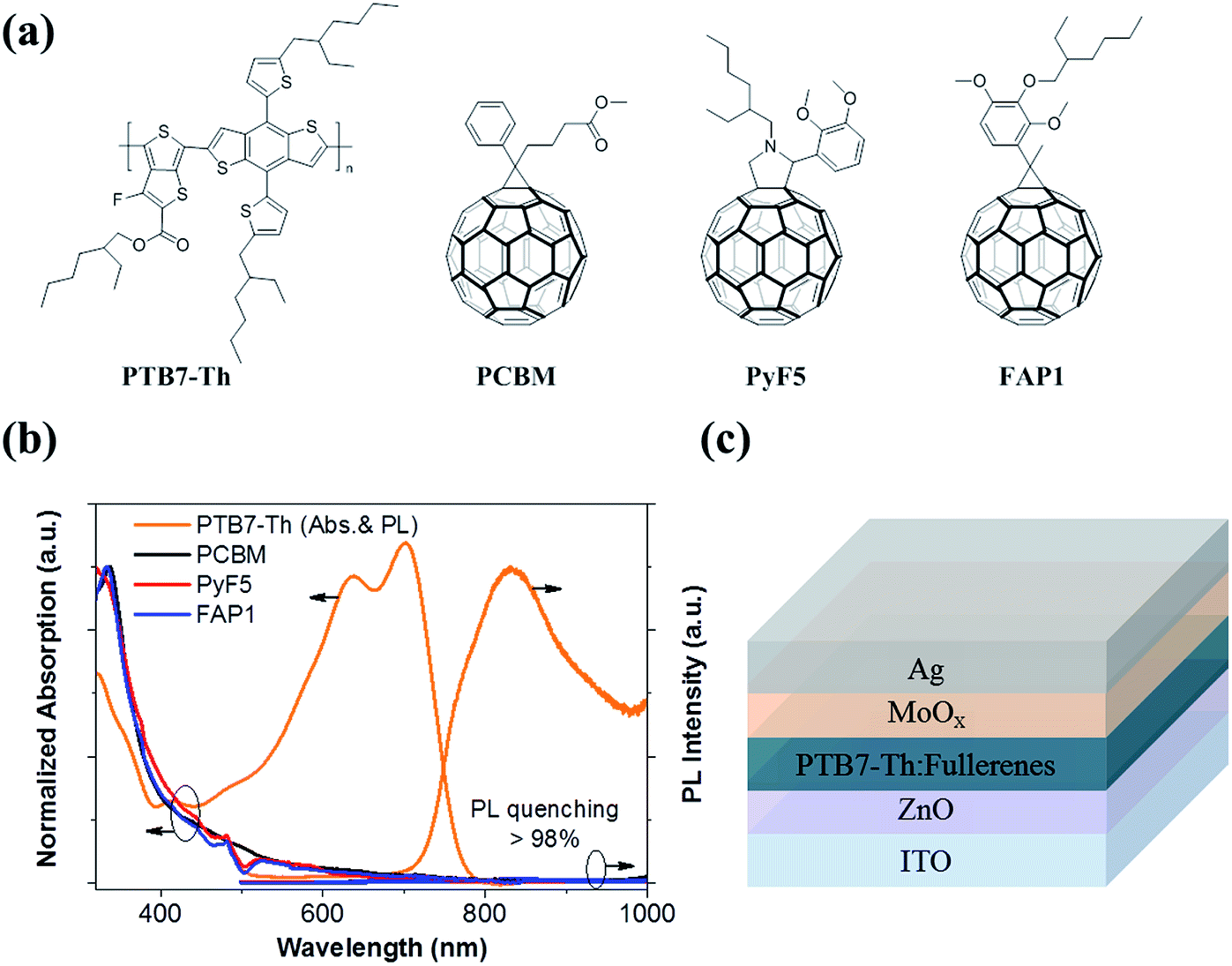 | ||
| Fig. 1 (a) Chemical structures of polymer and fullerenes; (b) absorption of pristine materials and photoluminescence of the polymer and blends; (c) device structure of solar cells. | ||
The optimized photovoltaic performance of PyF5 and FAP1 based devices is slightly lower than that of PC61BM based devices, owing to the slightly lower fill factor and JSC. The photovoltaic characteristics of PTB7-Th:fullerene with different donor:acceptor ratios are summarized in Fig. 2 and Table 1. It is important to point out that PC61BM instead of PC71BM was employed for reference devices to ensure rational comparison with the two C60-based fullerene derivatives. As observed in Fig. 2, when the acceptor loading is low (D:A = 1:0.8), JSC, fill factor and power conversion efficiency (PCE) are much lower than the optimized device performance for each corresponding system. With increasing the fullerene loadings, the photovoltaic parameters of the three systems have a trend to increase. In PC61BM-based BHJ systems, PCE reaches a summit at a D:A ratio of 1:1.5. Further increasing the ratio beyond 1:1.5 only moderately increases the fill factor. In the case of PTB7-Th:PyF5 and PTB7-Th:FAP1, PCE peaks at a D:A ratio of 1:2. Further increasing the fullerene ratio slowly enhances the fill factor beyond 60% at a D:A ratio of 1:3. It is noticed that, at the D:A ratio of 1:3, when the fill factor of PyF5 and FAP1 based devices is above 60%, JSC and PCE are almost the same for all three systems; similar phenomena are observed for pDDP5T-2:PyF5 and pDDP5T-2:FAP1 solar cells (Fig. S1 and Table S1†).
 | ||
| Fig. 2 (a) Current density–voltage (J–V) characteristics of PTB7-Th:fullerenes organic solar cells; (b) PCE and (c) fill factor of the corresponding organic solar cells as a function of D:A ratios. | ||
| Active layer | D:A ratios |
V OC [V] | J SC [mA cm−2] | FF [%] | PCE [%] |
|---|---|---|---|---|---|
| PTB7-Th:PC61BM | 1:0.8 |
0.80 ± 0.04 | 11.8 ± 2.7 | 44 ± 0.7 | 4.1 ± 0.7 |
| 1:1 |
0.81 ± 0.02 | 14.3 ± 1.3 | 57 ± 1.5 | 6.6 ± 0.6 | |
| 1:1.5 |
0.80 ± 0.01 | 14.0 ± 0.6 | 65 ± 0.7 | 7.3 ± 0.3 | |
| 1:2 |
0.80 ± 0.01 | 12.6 ± 0.3 | 68 ± 0.5 | 6.9 ± 0.2 | |
| 1:3 |
0.77 ± 0.01 | 11.1 ± 0.4 | 70 ± 1.2 | 6.0 ± 0.1 | |
| PTB7-Th:PyF5 | 1:0.8 |
0.84 ± 0.01 | 8.9 ± 0.6 | 36 ± 0.4 | 2.7 ± 0.1 |
| 1:1 |
0.84 ± 0.01 | 14.6 ± 0.6 | 45 ± 1.1 | 5.6 ± 0.3 | |
| 1:1.5 |
0.84 ± 0.01 | 13.8 ± 1.1 | 53 ± 1.0 | 6.2 ± 0.5 | |
| 1:2 |
0.84 ± 0.01 | 13.7 ± 0.6 | 56 ± 0.6 | 6.5 ± 0.5 | |
| 1:3 |
0.83 ± 0.01 | 11.2 ± 0.3 | 62 ± 0.8 | 5.8 ± 0.1 | |
| PTB7-Th:FAP1 | 1:0.8 |
0.81 ± 0.01 | 5.7 ± 0.3 | 37 ± 0.3 | 1.7 ± 0.1 |
| 1:1 |
0.83 ± 0.01 | 11.5 ± 0.4 | 38 ± 1.2 | 3.6 ± 0.3 | |
| 1:1.5 |
0.86 ± 0.01 | 12.9 ± 0.2 | 44 ± 1.1 | 4.9 ± 0.1 | |
| 1:2 |
0.87 ± 0.01 | 12.7 ± 0.5 | 55 ± 0.3 | 6.1 ± 0.3 | |
| 1:3 |
0.86 ± 0.01 | 11.2 ± 0.2 | 61 ± 0.1 | 5.9 ± 0.1 |
Carrier dynamics along with structural evolution
To find out the correlation between the photovoltaic parameters, especially the FF, and the fullerene loadings, space-charge-limited current (SCLC) measurements were performed to study the charge carrier transport properties of fullerene:polymer blends for various fullerene loadings. The electron-only devices were constructed with the architecture of ITO/Al/active layer/Ca/Ag for pristine films and ITO/ZnO/active layer/Ca/Ag for blend films. The electron mobility was estimated by fitting the current–voltage curves according to the SCLC modified Mott–Gurney model. As shown in Fig. 3, Table S2 and Fig. S2,† pristine PyF5 and FAP1 demonstrate a high electron mobility of 4.0 × 10−3 cm2 V−1 s−1 and 4.4 × 10−3 cm2 V−1 s−1, respectively, which are very similar to that determined for PC61BM (4.5 × 10−3 cm2 V−1 s−1), suggesting that the electron transporting properties of the functional fullerenes are very comparable to the ones of PC61BM. However, the electron mobilities of PC61BM-based blends remain high (∼10−4 cm2 V−1 s−1) in composites with up to 50 wt% polymer loading. Such high electron mobility in blends is essential to guarantee a high fill factor and a correspondingly high efficiency. In stark contrast, the electron mobility of PTB7-Th:PyF5 (1:1) and PTB7-Th:FAP1 (1:1) BHJ systems was significantly reduced to the level of 10−7–10−6 cm2 V−1 s−1. This is associated with the rather low FF observed for such low fullerene loadings (Table 1). When more PyF5 or FAP1 was added to the blends, the electron mobility was improved by two orders of magnitude to 8.8 × 10−5 cm2 V−1 s−1 for PTB7-Th:PyF5 (1:3) and 9.3 × 10−5 cm2 V−1 s−1 for PTB7-Th:FAP1 (1:3). This is in good agreement with the comparable JSC and PCE for all three systems.
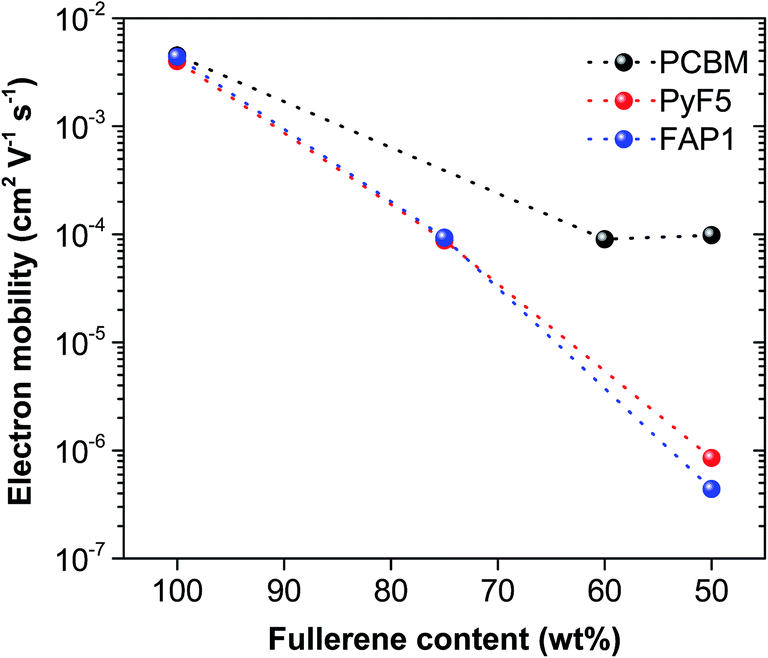 | ||
| Fig. 3 SCLC electron motilities of PTB7-Th:fullerene as a function of fullerene content. The dashed lines are a guide to the eye. | ||
We note that for PTB7-Th:PyF5 and PTB7-Th:FAP1 blends significantly higher fullerene loadings are necessary to reach comparably high electron mobilities as for PTB7-Th:PC61BM blends. Surprisingly, we interpret these findings to indicate that the crystallinity of a fullerene acceptor is not the sole material property dominating the electron mobility of a BHJ composite. Our studies demonstrate that the amorphous-dominated fullerene acceptor PyF5 has the same mobility as the semi-crystalline fullerene acceptor FAP1. When blended with PTB7-Th, at low fullerene loadings, the electron mobility drops dramatically for both fullerene composites. Upon increasing the fullerene loading, the electron mobility increases for both the amorphous and the crystalline fullerene. This observation leads us to the conclusion that crystallinity per se is an important material parameter but probably not the decisive parameter guaranteeing good solar cell performance. This trend is best documented by Fig. 3, where the electron motilities of PTB7-Th:fullerene as a function of fullerene content are presented. The much distinct electron mobility delivered by the polymer:fullerene blends can be mainly attributed to the different microscale BHJ morphology.
In situ temperature-resolved GIWAXS was employed to acquire a deeper insight into the polymer:fullerene morphology. According to the literature, pristine PC61BM and PyF5 films mainly reveal amorphous fingerprints while the FAP1 thin film exhibits highly crystalline reflections with narrow peaks. In Fig. S3,† the presence of an in-plane (100) peak and an out-of-plane (010) peak (polymer π–π stacking) indicates that the polymer chains preferentially form face-on orientation in pristine PTB7-Th. With S as axes, the position of the (100) and (010) peaks of polymer is at 0.048 and 0.255 Å−1 corresponding to 0.30 and 1.60 Å−1 with Q as axes (Q = 2πS), respectively, which is in good agreement with the literature.54 Temperature-resolved GIWAXS measurements were performed in the temperature range from 25 to 150 °C. At each temperature (70 °C and 150 °C, Fig. S4†) the annealing process was performed for 5 minutes before the measurement.
Fig. 4 depicts the 2D GIWAXS patterns, the corresponding in-plane and out-of-plane cuts, and the azimuthal distribution of the PTB7-Th (100) peak of three BHJ blends. PTB7-Th:PC61BM blends present the reflections of the crystal phase of the polymer and amorphous halo of PC61BM. Annealing leads to an increase of the crystalline polymer (100) and (010) peak intensity in both, in-plane and out-of-plane, directions as well as enhanced PC61BM aggregation (the distinction between the polymer π–π stacking and the halo of PC61BM aggregates can be found in Fig. S5†). For PTB7-Th:PyF5 and PTB7-Th:FAP1 blends, after annealing, the polymer out-of-plane (100) peak increases while the polymer in-plane (100) peak as well as the maximum of the azimuthal distribution of the (100) peak slightly decreases. From previous studies, it was found that the photoluminescence of PTB7-Th:PCBM blends significantly increases after thermal annealing, with a strong rise of the singlet emission of PTB7-Th, indicating a strong separation of the donor and acceptor phases. For both PTB7-Th:PyF5 and PTB7-Th:FAP1 systems, the PL spectra remain almost unchanged during thermal annealing, indicating that PCBM and PTB7-Th have a strong tendency to phase separate on a large scale while both PyF5 and FAP1 maintain their good intermixing.53 In summary, all three PTB7-Th:fullerene blends re-organize in the nano-scale during this short-time annealing; polymer packing and fullerene aggregates were observed to increase in PC61BM blends, which indicates a tendency to phase separate, while no such tendency was observed for PyF5- and FAP1-blends.
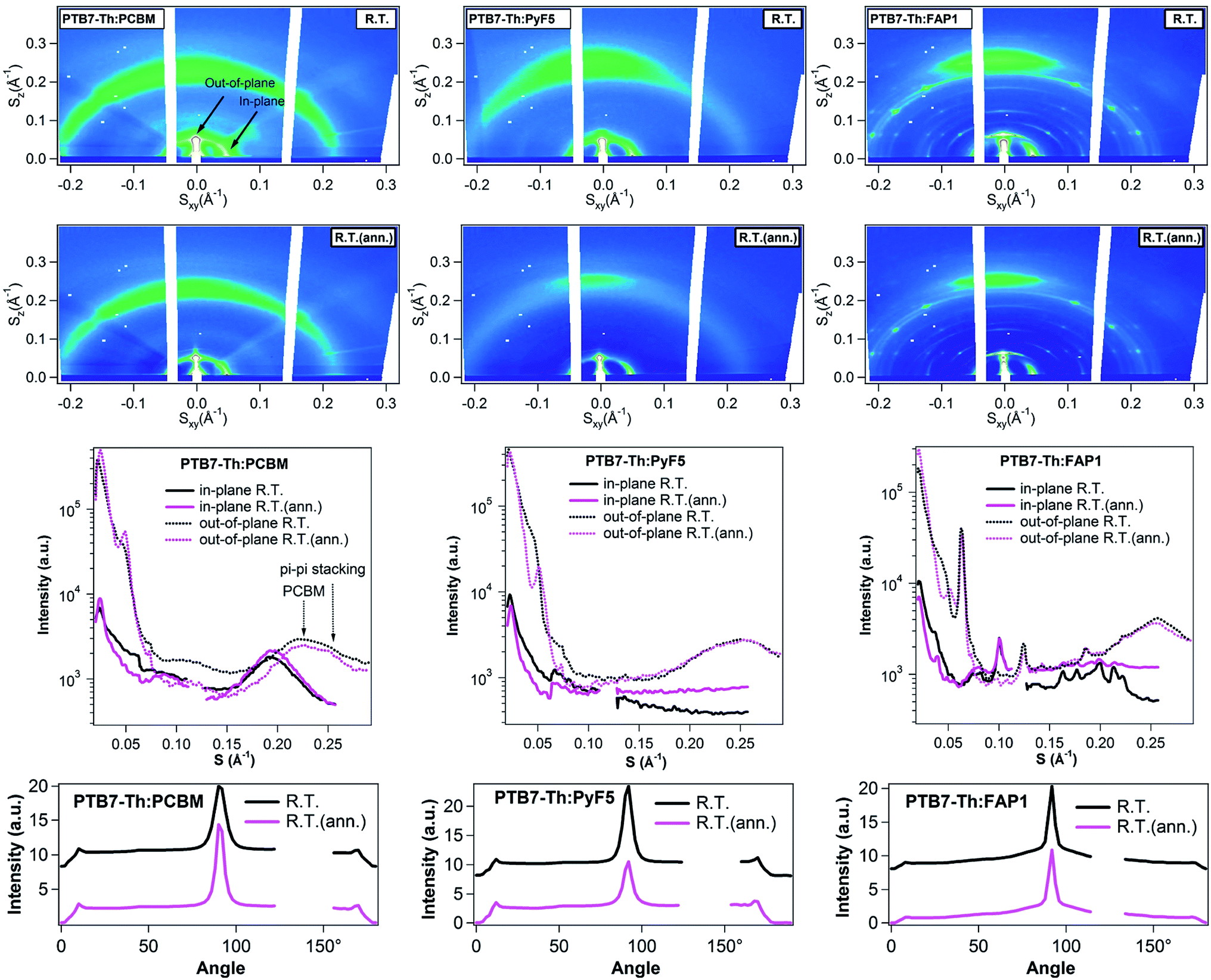 | ||
| Fig. 4 2D GIWAXS patterns, corresponding linecuts, and azimuthal distribution of the polymer (100) peak of PTB7-Th:PC61BM, PTB7-Th:PyF5 and PTB7-Th:FAP1 blends before and after annealing (Q = 2πS). | ||
Morphological evolution of polymer–fullerene BHJ blends
It is essential to explore why the same polymer matrix (PTB7-Th or pDDP5T-2) can show such a distinctly different microstructure evolution for the one fullerene (PC61BM) over the other fullerenes (PyF5 and FAP1). Following this analysis we will identify the miscibility of the single components as one essential parameter influencing photovoltaic performance.In principle, there are three phases in OPV BHJ blends: pristine donor and acceptor domains, and a mixed domain;55–58 the mixed domain should be as inter-mixed as possible to facilitate charge generation and dissociation; the pristine domains should be as pure and crystalline as possible in order to reduce recombination and lead to better charge transportation. Previous studies discussed even more complete pictures of the BHJ microstructure involving 5 or even more phases;59–63 however, we will limit the discussion here to one amorphous mixed phase which allows us to simplify the introduction of the miscibility concept without limiting its generality. Previous research indicates that a certain degree of miscibility between the donor and acceptor is necessary to provide a sufficiently mixed polymer–fullerene domain which is important for charge generation and dissociation. Nevertheless, if the polymer donor and the fullerene acceptor have too fine inter-mixing, full percolation of the fullerene phase gets hampered and the electron mobility starts to break down, resulting in reduced μτ products and worsened fill factors. Higher fullerene loadings favor a more complete percolation of extended fullerene phases and domains and, within specific tolerances, will typically lead to enhanced μτ products and improved photovoltaic performance.55,64–67
The rather low miscibility of PTB7-Th and PCBM typically does result in an unfavorable thin film morphology. Advanced processing recipes relying on additives provide a path to microstructure optimization at rather low fullerene loading;29 nevertheless, this optimized microstructure turns out to be metastable.53 The microstructural instability makes these composites vulnerable against external stress (e.g. heating, light excitation, etc. summarized in Fig. 5a), resulting in the thermal instability of PTB7-Th:PCBM solar cells.68–70 We previously demonstrated that PyF5 and FAP1 are better miscible with PTB7-Th than PCBM, leading to superior thermal stability of the corresponding solar cells.53 In the current study, we find that the PL signal of PTB7-Th is efficiently quenched by adding PCBM, PyF5 or FAP1 (Fig. 1b), indicating equally efficient charge dissociation; however the electron mobilities of PyF5- and FAP1-based blends at a D:A ratio of 1:1 are two orders of magnitude lower than that of the PCBM-based blend, indicating deficient charge transport. At a D:A ratio of 1:3, the electron mobilities as well as the photovoltaic performance of PyF5- and FAP1-based composites advance to a performance level comparable to that of the PCBM-based composite at a mixing ratio of 1:3. From the absorption spectra of thin films (Fig. S6†), the low polymer content in PTB7-Th:PyF5 (1:3) and PTB7-Th:FAP1 (1:3) films results in reduced absorption and JSC, compared to the optimized PTB7-Th:PCBM (1:1.5) blend with the same layer thickness. This fullerene ratio dependence was reported multiple times in the literature and is schematically summarized in Fig. 5b, visualizing that higher fullerene loadings typically lead to an improved percolation.55,71–74
 | ||
| Fig. 5 Schematic of the morphological evolution of polymer–fullerene with (a) poor miscibility and (b) good miscibility. | ||
Balance between miscibility and optoelectronic properties
The current state of the art does discuss the optimum fullerene ratios in relation to the crystallinity of the donor.74–76 It is suggested that polymers with higher crystallinity require lower PCBM loadings to form effective percolation pathways. However, this hypothesis cannot fully explain the optimized PCBM loadings reported for multiple organic solar cells, such as MDMO-PPV:PCBM, PTB7-Th:PCBM or PffBT4T-2OD:PCBM. Both MDMO-PPV and PTB7-Th are dominantly amorphous polymers, but PTB7-Th requires far less PCBM than MDMO-PPV to attain optimized performance. The required fullerene loading for PTB7-Th is very similar to that of the highly crystalline/aggregated PffBT4T-2OD. Thus, we suggest considering that the D:A ratio in BHJ blends does not solely depend on the crystalline nature of the polymer donor but may be governed by other factors. This thought provoking impulse is further motivated by the data from this work where both, PyF5- and FAP1-based, systems require more fullerene loadings than the PCBM-based system independent of the crystalline nature of fullerene acceptors.
It has been reported that the dimensionless Flory–Huggins interaction parameter (x12) can be used for evaluating the miscibility between polymers and small molecules.77,78 As already described in previous publications, the interaction parameters can be determined as follows:77,78
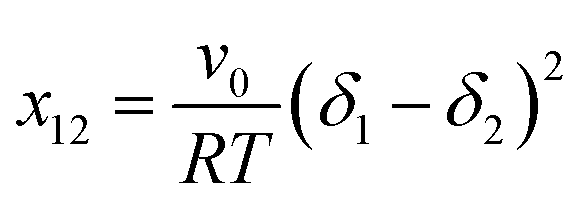 | (1) |
K−1 mol−1) and T is the temperature (298 K).
The Hildebrand parameters as determined by Hansen solubility parameters (HSP) are summarized in Table S3† for a selection of relevant polymer donors and fullerene acceptors. In addition to taking HSP of polymers from the literature, we further determined the HSP for a number of polymers. According to eqn (1), the interaction parameter for various polymer–fullerene combinations was calculated and is summarized in Table S4.† The site volume v0 in eqn (1) must be specified whenever discussing the interaction parameter as it is defined in terms of energy per site.30 When v0 is fixed in eqn (1), the value of (δ1 − δ2)2 is proportional to the interaction parameter x12 which is directly correlated with the polymer–fullerene miscibility. The optimized acceptor:donor ratios of the corresponding polymer–fullerene solar cells were taken from the literature3,7,11,13,30,79–83 (Table S4†).
Fig. 6 summarizes the acceptor:donor ratios for optimized devices as a function of the relative polymer–fullerene miscibility. Most surprisingly we find a well expressed relation between the acceptor:donor ratio and the miscibility. Two separate regions are observed in Fig. 6. Systems with a relatively low miscibility, below the threshold, and a significant tendency to phase separate perform best at low fullerene ratios of 1:1 or 2:1 (acceptor:donor), more or less independent of their crystallinity. On the other hand, composites with high miscibility, above the threshold, show a more expressed dependency on the interaction parameter and exhibit the best performance at high fullerene loadings of 3:1 to 5:1 (acceptor:donor). Although the miscibility of PC61BM and PC71BM was found to be different when blended with the same polymer, the difference is still below the threshold of miscibility as shown in Fig. 6. In this case, the optimized acceptor:donor ratio is expected to be very similar for a certain polymer. Fig. 6 is in good agreement with the observation that polymers having better miscibility with fullerenes require higher fullerene loadings to attain optimized photovoltaic performance, quite independent of their crystallinity. The correlation between the miscibility and optimized acceptor:donor ratio is more clear from Fig. S8,† where the acceptor:donor ratio is plotted logarithmically.
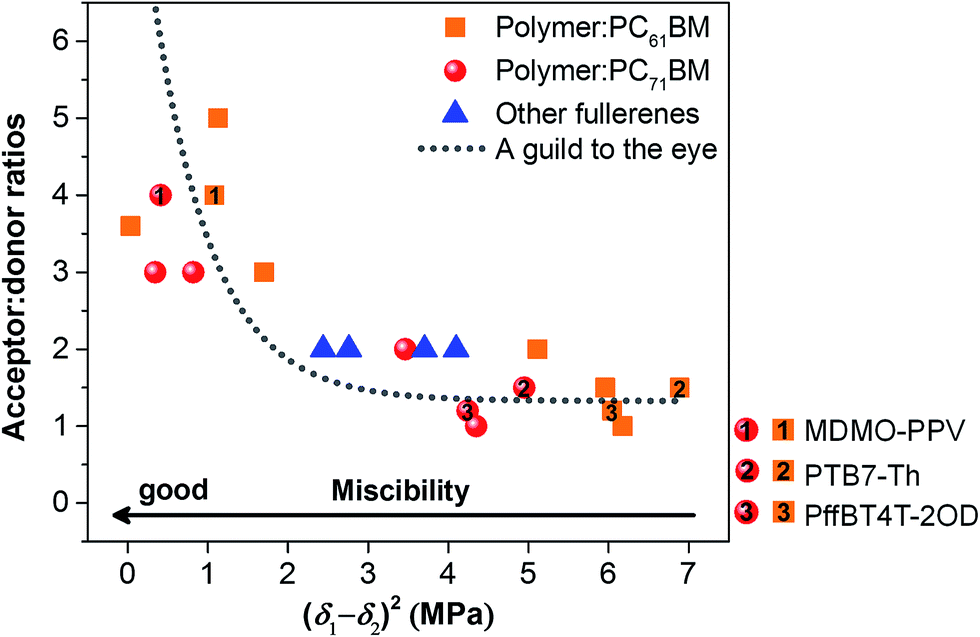 | ||
| Fig. 6 Optimal fullerene acceptor:polymer donor ratios as a function of polymer–fullerene miscibility. | ||
We want to accentuate a final important aspect of miscibility. Polymer–fullerene BHJ composites with high miscibility are promising opportunities for enhanced thermal stability.
On the other hand, high fullerene loadings do result in weak absorption and reduced photo-current generation at equal layer thickness. This is a strong argument to further foster the development of highly absorbing, non-fullerene acceptors,84–86 which have the potential to overcome this limitation.
Conclusion
In summary, we in-depth investigated the carrier transport loss mechanism for PTB7-Th:PCBM, PTB7-Th:PyF5 and PTB7-Th:FAP1 BHJ composites as a function of their microstructure and especially their miscibility. An obvious correlation between the microstructural compatibility of the polymer–fullerene components with the electron transport properties and the ultimate photovoltaic performance of BHJ solar cells was observed. Surprisingly, this interaction is independent of the crystalline nature of the fullerenes but rather depends on the miscibility of the polymer donor with the fullerene acceptor. We further found that the miscibility between the donor and acceptor dominates the optimized acceptor loadings in polymer–fullerene BHJ systems. BHJ composites with good polymer–fullerene miscibility require higher fullerene loadings than composites with a tendency to phase separate. It has to be carefully considered for future design of the donor and acceptor BHJ composites that the miscibility and the optoelectronic properties need to be well balanced in order to maximize the photovoltaic performance of organic BHJ solar cells.Experimental section
Materials
The ZnO-nanoparticle dispersion in isopropyl alcohol was received from Nanograde. pDPP5T-2 (batch no. GKS1-001) was provided by BASF. PTB7-Th and PC61BM (99%) were purchased from One Material and Solenne BV, respectively. PyF5 and FAP1 were synthesized according to the literature.48,87Device fabrication
All solar cells were fabricated in an inverted structure of ITO/ZnO/active layer/MoOx/Ag. The pre-structured ITO coated glass substrates were subsequently cleaned in toluene, acetone and isopropyl alcohol for 10 min each. Then 30 nm ZnO (Nanograde, N10) was doctor bladed on the ITO substrate and annealed at 85 °C in ambient air. The PTB7-Th based active layer from a chlorobenzene:1,8-diiodooctane (100:3) solution was spin coated on top of ZnO in a nitrogen atmosphere. The pDPP5T-2 based active layer from a chloroform:1,2-dichlorobenzene (90:10) solution was bladed under ambient conditions. 10 nm MoOx and 100 nm silver were deposited subsequently under 6 × 10−6 Torr by thermal evaporation through a shadow mask to form an active area of 10.4 mm2. For SCLC devices, a pristine fullerene layer was spin coated under an ambient atmosphere. The BHJ active layer was processed the same as solar cells. 15 nm calcium and 100 nm silver were deposited subsequently under 6 × 10−6 Torr by thermal evaporation through a shadow mask to form an active area of 10.4 mm2.
General device characterization
The current–voltage characteristics of the solar cells were measured under AM 1.5G irradiation on an OrielSollA Solar simulator (100 mW cm−2). The light source was calibrated by using a silicon reference cell. All cells were tested in ambient air.Film absorption was characterized with a UV-vis-NIR spectrometer Lambda 950 from PerkinElmer.
PL
Thin film photoluminescence measurements were conducted under excitation from a 375 nm diode laser. The spectra were recorded with a Si-CCD attached to an iHR320 monochromator (Horiba).GIWAXS
GIWAXS patterns of the pristine fullerenes were recorded on the ID-10 beamline at European Synchrotron Radiation Facilities (Grenoble, France). Diffraction patterns were collected with a Pilatus 300k detector (172 × 172 μm pixel size). The wavelength used was 1.24 Å. The measurements were performed on thin films on a Si substrate at an incidence angle of 0.16°. The modulus of the scattering vector was calibrated using several diffraction orders of silver behenate. In situ heating ramps were performed with a Linkam heating stage. The integration of 2D-WAXS patterns was performed in a home-made routine written in Igor Pro software. The 2D GIWAXS patterns and the corresponding linecuts are presented in coordinates Sxy and Sz . To present the 2D GIWAXS patterns in coordinates Sxy and Sz, the original rectangular images were deformed after recalculation in reciprocal space. White stripes on the images are the gaps between photoelectronic chips. The 1D profiles represent the original distribution of intensity as a function of the wave vector modulus.
. To present the 2D GIWAXS patterns in coordinates Sxy and Sz, the original rectangular images were deformed after recalculation in reciprocal space. White stripes on the images are the gaps between photoelectronic chips. The 1D profiles represent the original distribution of intensity as a function of the wave vector modulus.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
C. Z. would like to acknowledge the financial support from the China Scholarship Council (CSC). S. L. gratefully acknowledges Umweltfreundliche Hocheffiziente Organische Solarzellen (UOS) for financial support. A. O. acknowledges the support of the Cluster of Excellence “Engineering of Advanced Materials”. P. T. and A. M. acknowledge the financial support from the Russian Science Foundation (grant No. 14-13-01031). D. A. K. G. and D. I. acknowledge the Ministry of Education and Science of the Russian Federation for financial support (contract No. 14.587.21.0034 (RFMEFI58716X0034)). J. D. P. is funded by a doctoral fellowship grant of the Colombian Agency COLCIENCIAS. N. L. acknowledges the financial support from the ETI funding at FAU Erlangen-Nürnberg, the DFG grant: BR 4031/13-1, and the Bavarian Ministry of Economic Affairs and Media, Energy and Technology by funding the HI-ERN (IEK11) of FZ Jülich. C. J. B. gratefully acknowledges the financial support through the “Aufbruch Bayern” initiative of the state of Bavaria (EnCN and solar factory of the future), the Bavarian Initiative “Solar Technologies go Hybrid” (SolTech), the SFB 953 (DFG) and the Cluster of Excellence “Engineering of Advanced Materials” (EAM) at FAU Erlangen-Nürnberg. T. Ameri and F. Machui are acknowledged for fruitful discussions on determining solubility parameters.References
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- M. M. Wienk, J. M. Kroon, W. J. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. Janssen, Angew. Chem., 2003, 115, 3493–3497 CrossRef.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- S. Goffri, C. Muller, N. Stingelin-Stutzmann, D. W. Breiby, C. P. Radano, J. W. Andreasen, R. Thompson, R. A. J. Janssen, M. M. Nielsen, P. Smith and H. Sirringhaus, Nat. Mater., 2006, 5, 950–956 CrossRef CAS PubMed.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef CAS PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganas and J. V. Manca, Nat. Mater., 2009, 8, 904–909 CrossRef CAS PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- Z. C. He, B. Xiao, F. Liu, H. B. Wu, Y. L. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- S. Xiao, Q. Zhang and W. You, Adv. Mater., 2017, 29, 1601391 CrossRef PubMed.
- S. E. Shaheen, C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz and J. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841–843 CrossRef CAS.
- T. Ameri, N. Li and C. J. Brabec, Energy Environ. Sci., 2013, 6, 2390–2413 CAS.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- C. Cui, Y. Li and Y. Li, Adv. Energy Mater., 2017, 7, 1601251 CrossRef.
- N. K. Elumalai and A. Uddin, Energy Environ. Sci., 2016, 9, 391–410 CAS.
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821–7861 CrossRef CAS PubMed.
- H. Kang, W. Lee, J. Oh, T. Kim, C. Lee and B. J. Kim, Acc. Chem. Res., 2016, 49, 2424–2434 CrossRef CAS PubMed.
- R. Ganesamoorthy, G. Sathiyan and P. Sakthivel, Sol. Energy Mater. Sol. Cells, 2017, 161, 102–148 CrossRef CAS.
- K. Zhang, Z. Hu, C. Sun, Z. Wu, F. Huang and Y. Cao, Chem. Mater., 2017, 29, 141–148 CrossRef CAS.
- H.-C. Liao, C.-C. Ho, C.-Y. Chang, M.-H. Jao, S. B. Darling and W.-F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- Y. Liu, V. V. Duzhko, Z. A. Page, T. Emrick and T. P. Russell, Acc. Chem. Res., 2016, 49, 2478–2488 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- S. H. Liao, H. J. Jhuo, Y. S. Cheng and S. A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS.
- C.-Z. Li, H.-L. Yip and A. K.-Y. Jen, J. Mater. Chem., 2012, 22, 4161–4177 RSC.
- Y.-Y. Lai, Y.-J. Cheng and C.-S. Hsu, Energy Environ. Sci., 2014, 7, 1866–1883 CAS.
- M. Lenes, G. J. A. H. Wetzelaer, F. B. Kooistra, S. C. Veenstra, J. C. Hummelen and P. W. M. Blom, Adv. Mater., 2008, 20, 2116–2119 CrossRef CAS.
- Y. He, B. Peng, G. Zhao, Y. Zou and Y. Li, J. Phys. Chem. C, 2011, 115, 4340–4344 CAS.
- C. Zhang, S. Chen, Z. Xiao, Q. Zuo and L. Ding, Org. Lett., 2012, 14, 1508–1511 CrossRef CAS PubMed.
- Y.-J. Cheng, M.-H. Liao, C.-Y. Chang, W.-S. Kao, C.-E. Wu and C.-S. Hsu, Chem. Mater., 2011, 23, 4056–4062 CrossRef CAS.
- Y. He, H.-Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- Y. Zhang, H.-L. Yip, O. Acton, S. K. Hau, F. Huang and A. K.-Y. Jen, Chem. Mater., 2009, 21, 2598–2600 CrossRef CAS.
- G. Garcia-Belmonte, P. P. Boix, J. Bisquert, M. Lenes, H. J. Bolink, A. La Rosa, S. Filippone and N. Martín, J. Phys. Chem. Lett., 2010, 1, 2566–2571 CrossRef CAS.
- G. Zhao, Y. He, Z. Xu, J. Hou, M. Zhang, J. Min, H. Y. Chen, M. Ye, Z. Hong and Y. Yang, Adv. Funct. Mater., 2010, 20, 1480–1487 CrossRef CAS.
- Y. Matsuo, Y. Sato, T. Niinomi, I. Soga, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 16048–16050 CrossRef CAS PubMed.
- A. Varotto, N. D. Treat, J. Jo, C. G. Shuttle, N. A. Batara, F. G. Brunetti, J. H. Seo, M. L. Chabinyc, C. J. Hawker and A. J. Heeger, Angew. Chem., Int. Ed., 2011, 50, 5166–5169 CrossRef CAS PubMed.
- Z. Li, F. Wu, H. Lv, D. Yang, Z. Chen, X. Zhao and X. Yang, Adv. Mater., 2015, 27, 6999–7003 CrossRef CAS PubMed.
- S. H. Park, C. Yang, S. Cowan, J. K. Lee, F. Wudl, K. Lee and A. J. Heeger, J. Mater. Chem., 2009, 19, 5624–5628 RSC.
- K. Matsumoto, K. Hashimoto, M. Kamo, Y. Uetani, S. Hayase, M. Kawatsura and T. Itoh, J. Mater. Chem., 2010, 20, 9226–9230 RSC.
- C.-Z. Li, S.-C. Chien, H.-L. Yip, C.-C. Chueh, F.-C. Chen, Y. Matsuo, E. Nakamura and A. K. Y. Jen, Chem. Commun., 2011, 47, 10082–10084 RSC.
- A. V. Mumyatov, F. A. Prudnov, L. N. Inasaridze, O. A. Mukhacheva and P. A. Troshin, J. Mater. Chem. C, 2015, 3, 11612–11617 RSC.
- K. Sivula, Z. T. Ball, N. Watanabe and J. M. J. Fréchet, Adv. Mater., 2006, 18, 206–210 CrossRef CAS.
- Y. J. Cheng, C. H. Hsieh, P. J. Li and C. S. Hsu, Adv. Funct. Mater., 2011, 21, 1723–1732 CrossRef CAS.
- Q. Tai, J. Li, Z. Liu, Z. Sun, X. Zhao and F. Yan, J. Mater. Chem., 2011, 21, 6848–6853 RSC.
- M.-H. Liao, C.-E. Tsai, Y.-Y. Lai, F.-Y. Cao, J.-S. Wu, C.-L. Wang, C.-S. Hsu, I. Liau and Y.-J. Cheng, Adv. Funct. Mater., 2014, 24, 1418–1429 CrossRef CAS.
- C. H. Zhang, A. Mumyatov, S. Langner, J. D. Perea, T. Kassar, J. Min, L. L. Ke, H. W. Chen, K. L. Gerasimov, D. V. Anokhin, D. A. Ivanov, T. Ameri, A. Osvet, D. K. Susarova, T. Unruh, N. Li, P. Troshin and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1601204 CrossRef.
- F. Zhao, Y. Li, Z. Wang, Y. Yang, Z. Wang, G. He, J. Zhang, L. Jiang, T. Wang, Z. Wei, W. Ma, B. Li, A. Xia, Y. Li and C. Wang, Adv. Energy Mater., 2017, 7, 1602552 CrossRef.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364–374 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- P. Westacott, J. R. Tumbleston, S. Shoaee, S. Fearn, J. H. Bannock, J. B. Gilchrist, S. Heutz, J. deMello, M. Heeney, H. Ade, J. Durrant, D. S. McPhail and N. Stingelin, Energy Environ. Sci., 2013, 6, 2756–2764 CAS.
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Güldal, H. Chen, S. Chen, S. Langner, M. Berlinghof, T. Unruh and C. J. Brabec, Nat. Commun., 2017, 8, 14541 CrossRef CAS PubMed.
- A. C. Mayer, M. F. Toney, S. R. Scully, J. Rivnay, C. J. Brabec, M. Scharber, M. Koppe, M. Heeney, I. McCulloch and M. D. McGehee, Adv. Funct. Mater., 2009, 19, 1173–1179 CrossRef CAS.
- E. Buchaca-Domingo, A. J. Ferguson, F. C. Jamieson, T. McCarthy-Ward, S. Shoaee, J. R. Tumbleston, O. G. Reid, L. Yu, M. B. Madec, M. Pfannmoller, F. Hermerschmidt, R. R. Schroder, S. E. Watkins, N. Kopidakis, G. Portale, A. Amassian, M. Heeney, H. Ade, G. Rumbles, J. R. Durrant and N. Stingelin, Mater. Horiz., 2014, 1, 270–279 RSC.
- E. Buchaca-Domingo, K. Vandewal, Z. Fei, S. E. Watkins, F. H. Scholes, J. H. Bannock, J. C. de Mello, L. J. Richter, D. M. DeLongchamp, A. Amassian, M. Heeney, A. Salleo and N. Stingelin, J. Am. Chem. Soc., 2015, 137, 5256–5259 CrossRef CAS PubMed.
- M. Scarongella, J. De Jonghe-Risse, E. Buchaca-Domingo, M. Causa’, Z. Fei, M. Heeney, J.-E. Moser, N. Stingelin and N. Banerji, J. Am. Chem. Soc., 2015, 137, 2908–2918 CrossRef CAS PubMed.
- M. Causa, J. De Jonghe-Risse, M. Scarongella, J. C. Brauer, E. Buchaca-Domingo, J.-E. Moser, N. Stingelin and N. Banerji, Nat. Commun., 2016, 7, 12556 CrossRef CAS PubMed.
- A. M. Ballantyne, T. A. Ferenczi, M. Campoy-Quiles, T. M. Clarke, A. Maurano, K. H. Wong, W. Zhang, N. Stingelin-Stutzmann, J.-S. Kim and D. D. Bradley, Macromolecules, 2010, 43, 1169 CrossRef CAS.
- N. D. Treat, A. Varotto, C. J. Takacs, N. Batara, M. Al-Hashimi, M. J. Heeney, A. J. Heeger, F. Wudl, C. J. Hawker and M. L. Chabinyc, J. Am. Chem. Soc., 2012, 134, 15869–15879 CrossRef CAS PubMed.
- K. Vakhshouri, D. R. Kozub, C. Wang, A. Salleo and E. D. Gomez, Phys. Rev. Lett., 2012, 108, 026601 CrossRef PubMed.
- S. Ulum, N. Holmes, M. Barr, A. D. Kilcoyne, B. B. Gong, X. Zhou, W. Belcher and P. Dastoor, Nano Energy, 2013, 2, 897–905 CrossRef CAS.
- M. Jorgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- L. Derue, O. Dautel, A. Tournebize, M. Drees, H. Pan, S. Berthumeyrie, B. Pavageau, E. Cloutet, S. Chambon and L. Hirsch, Adv. Mater., 2014, 26, 5831–5838 CrossRef CAS PubMed.
- J. W. Rumer and I. McCulloch, Mater. Today, 2015, 18, 425–435 CrossRef CAS.
- D. Chirvase, J. Parisi, J. C. Hummelen and V. Dyakonov, Nanotechnology, 2004, 15, 1317 CrossRef CAS.
- G. Dennler, A. J. Mozer, G. Juška, A. Pivrikas, R. Österbacka, A. Fuchsbauer and N. S. Sariciftci, Org. Electron., 2006, 7, 229–234 CrossRef CAS.
- D. Veldman, Ö. İpek, S. C. J. Meskers, J. Sweelssen, M. M. Koetse, S. C. Veenstra, J. M. Kroon, S. S. van Bavel, J. Loos and R. A. J. Janssen, J. Am. Chem. Soc., 2008, 130, 7721–7735 CrossRef CAS PubMed.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- D. H. Wang, J. K. Kim, J. H. Seo, O. O. Park and J. H. Park, Sol. Energy Mater. Sol. Cells, 2012, 101, 249–255 CrossRef CAS.
- T. Heumueller, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, J. A. Bartelt, T. M. Burke, T. Ameri, C. J. Brabec and M. D. McGehee, Energy Environ. Sci., 2014, 7, 2974–2980 CAS.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- R. A. Orwoll and P. A. Arnold, in Physical Properties of Polymers Handbook, ed. J. E. Mark, Springer New York, New York, NY, 2007, pp. 233–257, DOI:10.1007/978-0-387-69002-5_14.
- S. Alem, R. de Bettignies, J.-M. Nunzi and M. Cariou, Appl. Phys. Lett., 2004, 84, 2178–2180 CrossRef CAS.
- E. Wang, L. Hou, Z. Wang, S. Hellström, F. Zhang, O. Inganäs and M. R. Andersson, Adv. Mater., 2010, 22, 5240–5244 CrossRef CAS PubMed.
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS PubMed.
- V. S. Gevaerts, A. Furlan, M. M. Wienk, M. Turbiez and R. A. Janssen, Adv. Mater., 2012, 24, 2130–2134 CrossRef CAS PubMed.
- N. Li, D. Baran, K. Forberich, F. Machui, T. Ameri, M. Turbiez, M. Carrasco-Orozco, M. Drees, A. Facchetti, F. C. Krebs and C. J. Brabec, Energy Environ. Sci., 2013, 6, 3407–3413 CAS.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Rohr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- N. Liang, W. Jiang, J. Hou and Z. Wang, Mater. Chem. Front., 2017, 1, 1291–1303 RSC.
- A. V. Mumyatov, Institute of Problems of Chemical Physics of the Russian Academy of Sciences Chernogolovka, Moscow Region 142432 (RU), LANXESS Deutschland GmbH 50569 Köln (DE), Germany, 2014.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta03505e |
| This journal is © The Royal Society of Chemistry 2017 |