
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Three-dimensional carbon nanotube networks enhanced sodium trimesic: a new anode material for sodium ion batteries and Na-storage mechanism revealed by ex situ studies†
Xin
Yan
a,
Huan
Ye
b,
Xing-Long
Wu
 *a,
Yan-Ping
Zheng
a,
Fang
Wan
a,
Mingkai
Liu
c,
Xiao-Hua
Zhang
a,
Jing-Ping
Zhang
a and
Yu-Guo
Guo
*b
*a,
Yan-Ping
Zheng
a,
Fang
Wan
a,
Mingkai
Liu
c,
Xiao-Hua
Zhang
a,
Jing-Ping
Zhang
a and
Yu-Guo
Guo
*b
aNational & Local United Engineering Laboratory for Power Batteries, Faculty of Chemistry, Northeast Normal University, Changchun, Jilin 130024, PR China. E-mail: xinglong@nenu.edu.cn
bKey Laboratory of Molecular Nanostructure and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences (CAS), Beijing 100190, PR China. E-mail: ygguo@iccas.ac.cn
cSchool of Chemistry and Chemical Engineering, Jiangsu Normal University, Shanghai Road, Xu Zhou 221116, PR China
First published on 5th June 2017
Abstract
Recently, room temperature sodium ion batteries (SIBs) have attracted considerable attention as one of the promising candidates to replace lithium ion batteries. Nevertheless, achieving high capacity and cycling stability remains a great challenge for the electrode materials of SIBs. Compared to the traditional inorganic electrode materials, organic ones should be more attractive because of their easier sodium (Na)-transport accessibility as well as their diversities of organic skeleton and functional groups. In this work, a new carboxyl-based organic, sodium trimesic (Na3TM), is proposed for the first time as an anode material for SIBs, and its Na-storage properties are significantly enhanced by constructing three-dimensional conductive networks of carbon nanotubes (CNT-NWs) in the Na3TM microparticles. In comparison to the pure Na3TM exhibiting almost inactive Na storage, the prepared CNT-NWs@Na3TM composite delivers a reversible capacity of 214.6 mA h g−1 at 0.1 A g−1, and exhibits excellent rate performance with the specific capacities of 149 and 87.5 mA h g−1 at 1 and 10 A g−1, respectively. The CNT-NWs@Na3TM also exhibit good cycling performance. More importantly, the Na-storage mechanism of CNT-NWs@Na3TM was ascertained using several ex situ technologies of Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy, and 23Na solid-state nuclear magnetic resonance spectroscopy. It is discovered that the two Na uptake/release processes were reversible during cycling and contributed to the Na-storage capacity except for the 1st sodiation process with a three Na uptake.
1. Introduction
In the past two decades, lithium ion batteries (LIBs) have played a significant role in powering diverse electrical equipment, such as portable consumer electronics (including mobile phones, laptops, electronic watches, smart bands, and so on), (hybrid) electric vehicles, and grid-scale energy storage stations.1–5 With the rapid development of these areas, the increasing use of LIBs will inevitably lead to the shortage of Li sources, and thus the continuous price rise of LIBs in the near future.6–9 It is therefore becoming more and more urgent to seek appropriate alternatives for LIBs. Amongst all the potential alternatives, room temperature sodium ion batteries (SIBs)10 have been the subject of much research interest in recent years11–13 because sodium is much more abundant, more environmentally friendly and cheaper than Li,14 and are considered to be the most likely and direct alternative for LIBs because of their almost identical energy storage mechanism.6,15,16For the anode of SIBs, most of the traditional inorganics with superior Li-storage properties17 do not exhibit the expected Na-storage capacity mainly because of the larger radius of the Na ion compared to that of its Li counterpart.18 For example, graphitized carbon materials, is one class of the most widely used anode materials for LIBs with a market share of above 90%, but it is very difficult to achieve the reversible Na-insertion/extraction in the common Na+ electrolyte19–21 and although the silica (Si) anode can alloy with Li to achieve the highest theoretical Li-storage capacity of 4200 mA h g−1,22 it is almost impossible for the Si to uptake Na when used for SIBs.23 Thus, it is still a huge challenge to prepare superior anode materials for SIBs.10 In comparison to the inorganic electrode materials with high crystallinity and rigidity,24 organic ones usually exhibit much easier Na+-transport because of the more flexible skeleton of organics, which means that Na ions are more accessible to the electrochemically active functional groups for Na storage in the organic electrodes.25 Furthermore, the diversities of organic functional groups and countless combinations between them means that there is a vast number of organics that could be selected as electrode materials for SIBs. The organics also have many other benefits for practical applications including recyclability, low cost and nontoxicity. Therefore, organic materials have become one of the most significant research hotspots for SIBs electrodes. In recent years, a lot of Na-storable organics, such as disodium terephthalate (Na2C8H4O4),26,27 sodium 2,5-dihydroxy-1,4-benzoquinone (Na2C6H2O4),28 perylene 3,4,9,10-tetracarboxylic dianhydride,25 monosodium terephthalate (NaC8H5O4), disodium tetrafluoratherphthalate (Na2C8F4O4), 1,2,4,5-tetrasodium terephthalate (Na4C10H2O8),29 and tetrasodium 3,4,9,10-tetracarboxylic dianhydride (Na4C24H8O8),30,31 have been developed as electrode materials for SIBs.
Nevertheless, using organics as the electrodes of SIBs still leaves one major difficulty, namely, low electronic and ionic conductivities, which severely impede the expression of superior Na-storage properties. In order to make organic electrode materials to be practicable, some appropriate strategies must be developed to increase the conductivities, and thus enhance the kinetics of the electrodes. Among these strategies, one of the main methods is to reduce the particles to the nanometer scale,32 because nanomaterials cannot only provide a much shorter ion/electron-transfer distance and time but also disperse in the conductive networks (NWs) more uniformly.17 For example, it has recently been demonstrated that the Na-storage properties of a disodium terephthalate anode can be enhanced by preparing plate-like nanostructures because of their improved electrode kinetics and a new one-step desodiation process.31 However, the nano strategy has its own deficiency, namely, it lacks practical operability.33 In addition, the incorporation of highly conductive carbonaceous materials, such as carbon nanotubes (CNTs)34–36 and graphene,37–41 is another significant method for improving the electrochemical properties of electrode materials.42 Although this method had been widely verified to be effective for many inorganic electrodes,34,43 it is rarely employed to enhance the Na-storage properties of organic electrode materials.27
In this research, not only was a new carboxyl-based organic salt (sodium trimesic, Na3TM) developed for the first time as an anode for SIBs, but its Na-storage properties were also significantly enhanced by constructing three-dimensional (3D) CNT NWs in the Na3TM microparticles. When used as an anode material for SIBs, the Na3TM could deliver a reversible Na-storage capacity of 214.6 mA h g−1 at a low current density of 0.1 A g−1, and exhibited excellent rate performance with the specific capacities of 149 and 87.5 mA h g−1 at high current densities of 1 and 10 A g−1, respectively. It also exhibited good cycling performance. More importantly, the sodiation/desodiation mechanism was preliminarily ascertained using ex situ Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and 23Na solid-state nuclear magnetic resonance (23Na SS-NMR) spectroscopy. It was interesting to discover that two of the Na uptake/release processes were reversible during cycling to contribute to the Na-storage capacity but the 1st sodiation process with three Na uptake was not.
2. Experimental section
2.1 Synthesis
A general acid-base neutralization reaction was employed to synthesize the pure sodium trimesic (Na3TM) material. Specifically, 1 g of trimesic acid (H3TM) was added to 50 mL ethanol (EA) and stirred until dissolved completely to form the transparent Na3TM/EA solution. At the same time, 0.2 g of sodium hydroxide (NaOH) was dissolved in 50 mL of EA to obtain the 0.1 mol L−1 NaOH/EA solution. Subsequently, the prepared NaOH/EA solution was added to the Na3TM/EA solution under stirring, and then stirred for a few hours until the formation of white product, which was Na3TM. In order to collect the Na3TM sample, the as-obtained suspension was centrifuged and washed several times using EA until the pH was ∼7, and then dried at 60 °C for 10 h in a vacuum oven to obtain Na3TM powder.To prepare the proposed CNT-NWs@Na3TM composite, Na3TM aqueous solution was added to the CNTs/EA suspension, which was obtained by dispersing CNTs in EA under ultrasonication. This process resulted in the formation of a precursor suspension for the preparation of CNT-NWs@Na3TM. In the suspension, the added volumetric ratio of EA and water (H2O) was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After the EA/H2O liquid was extracted from the suspension in the rotary evaporator at 60 °C under vacuum, the CNT-NWs@Na3TM bulk could be easily collected. The powder CNT-NWs@Na3TM composite was finally formed by mechanically crushing the as-obtained bulk material. In the prepared CNT-NWs@Na3TM composite, the proportion of CNTs was about 20.2% [calculated from the thermogravimetric analysis (TGA) curves], which was close to the added value of about 25%. The difference between the two values might originate from the experimental loss of CNTs in the preparation processes.
:1. After the EA/H2O liquid was extracted from the suspension in the rotary evaporator at 60 °C under vacuum, the CNT-NWs@Na3TM bulk could be easily collected. The powder CNT-NWs@Na3TM composite was finally formed by mechanically crushing the as-obtained bulk material. In the prepared CNT-NWs@Na3TM composite, the proportion of CNTs was about 20.2% [calculated from the thermogravimetric analysis (TGA) curves], which was close to the added value of about 25%. The difference between the two values might originate from the experimental loss of CNTs in the preparation processes.
2.2 Structural characterizations
Powder X-ray diffraction (PXRD) measurements were carried out using a D/Max 200 PC (Rigaku) with Cu Kα radiation of 1.5406 Å. The proton nuclear magnetic resonance (1H-NMR) spectrums were obtained using an Inova 500 MHz Superconducting NMR spectrometer (Varian). Scanning electron microscopy (SEM) was performed using a 6700F instrument (Jeol) operating at 10 kV, to investigate the morphology and size of the CNT-NWs@Na3TM composite. The nitrogen (N2) adsorption–desorption test was carried out at 77.1 K the results were analyzed using AsiQwin software version 3.01 (Quantachrome Instruments). FTIR was performed using a Nicolet 6700 FTIR spectrometer (Thermo Scientific). In order to implement the ex situ FTIR tests, the electrode materials containing active CNT-NWs@Na3TM materials needed to be first washed with dimethyl carbonate and then scraped off from the current collector of copper (Cu) foils at different charge states (pristine, ST 0.3 V, ST 0.01 V, DT 0.35 V, DT 1 V and DT 2 V versus Na+/Na) in an argon filled glove box and finally analyzed with the Nicolet 6700 spectrometer using normal procedures. XPS spectra were obtained using an ESCALAB MKII X-ray photoelectron spectrophotometer (VG Instruments). 23Na SS-NMR data were collected on an AVIII 400 spectrometer (Bruker) working at a 23Na Larmor frequency of 105.58 MHz. The tests of ex situ XPS and 23Na SS-NMR were carried out using an analogous operation with the ex situ FTIR.2.3 Electrochemical measurements
The working electrode consisting of 80% active materials, 10% acetylene black, 10% poly(vinylidene fluoride), then a few drops of N-methyl-2-pyrrolidinone solvent were added into a mortar before being cast onto a Cu foil current collector. The electrolyte was 1 M sodium perchlorate mixed with ethylene carbonate (EC) and propylene carbonate (PC) (EC:PC 1:1 by volume). The whole the cell was assembled in the argon filled glove box with the concentrations of moisture and oxygen all below 0.1 ppm. The assembled cell was tested using a CT2001A testing system (LAND Instruments) between 0.01 V and 2.0 V with sodium metal as the counter electrodes at room temperature. Electrochemical impedance spectroscopy (EIS) was carried out using a VersaSTAT 3F instrument (Princeton Applied Research) with the frequency ranging from 1 MHz to 100 mHz with an amplitude voltage of 5 mV. The galvanostatic charge–discharge tests were performed on a CT2001A (LAND Instruments) between 0.01 V to 2 V. Cyclic voltammetry (CV) was carried out on a VersaSTAT 3 (Princeton Applied Research) with potential ranges from 0.01 V to 2 V at a scan rate of 0.1 mV s−1.
3. Results and discussion
Fig. 1a and b are representative SEM images of the prepared composite at different magnifications, and they reveal that it is composed of micrometer sized particles of about 6–10 μm. In the microparticles, as the schematic in Fig. 1c and S1†shows, CNTs are well dispersed in the Na3TM substrate and crosslinked with each other to form the 3D conductive networks. Because of the microstructure observed, the as-prepared composite was abbreviated as CNT-NWs@Na3TM, suggesting that the active Na3TM material has been well implanted into the CNT NWs. Such a specific structure will be very helpful in improving the Na-storage properties, and in making the as-prepared CNT-NWs@Na3TM composite exhibit a much enhanced electrochemical performance compared to the control of pure Na3TM microparticles (see Fig. S2†). This is demonstrated in the following electrochemical sections in this paper. | ||
| Fig. 1 (a and b) Representative SEM images and (c) schematic illustration of the prepared CNT-NWs@Na3TM composite. The SEM images are shown at different magnifications. (d) 1H-NMR spectrum of pristine Na3TM used to prepare the CNT-NWs@Na3TM composite. (e) A comparison of the FTIR spectra of the raw material of TMA and the prepared CNT-NWs@Na3TM. (f) The pore size distribution pattern of the CNT-NWs@Na3TM composite calculated from the corresponding N2 adsorption/desorption isotherm (inset of (f)). | ||
In addition to the morphology and microstructures, the chemical composition and crystalline phase of CNT-NWs@Na3TM were further investigated using 1H-NMR, FTIR, and PXRD. In the 1H-NMR spectrum (Fig. 1d), there are no obvious peaks of impurities, except for the ones located at 8.274 and 4.708 ppm for H in Na3TM and deuterium oxide (D2O) solvent, respectively, demonstrating the high purity of Na3TM in the CNT-NWs@Na3TM composite with ∼20.2% CNT by weight (calculated from the curves of TGA as shown in Fig. S3†). Fig. 1e compares the FTIR spectra of CNT-NWs@Na3TM and the raw material of trimestic acid (TMA). For TMA, the peaks located at 1720, 1606, 1454 and 1406 cm−1 originate from the infrared absorption of carboxylic acid (–COOH) groups. After the neutralization reaction between TMA and NaOH, the four peaks redshift to 1620, 1567, 1433 and 1371 cm−1, respectively, with the disappearance of original ones, indicating the full conversion of the –COOH groups in TMA to sodium carboxylate (–COONa) and thus the formation of Na3TM. Furthermore, the Barrett–Joyner–Halenda (BJH, Fig. 1f) pattern calculated from the desorption branch of the N2 adsorption/desorption isotherm (inset of Fig. 1f) demonstrates the presence of hierarchical meso/micropores of 2–100 nm in the CNT-NWs@Na3TM. The pores should be derived from the interspaces between Na3TM and CNTs as denoted by the arrows in the high magnification SEM image of Fig. 1b. For the pure Na3TM material, the pore volume is only 0.014 cm3 g−1 with a few meso/micropores obtained from the BJH (Fig. S4†), which is obviously lower than that of 0.082 cm3 g−1 for the prepared CNT-NWs@Na3TM composite. In the electrochemical processes, the pores can serve as the electrolyte transport channels and buffering reservoirs to facilitate the ion-transport kinetics and thus enhance the Na-storage properties.
In order to evaluate the potential application of CNT-NWs@Na3TM as an anode material for SIBs, its Na-storage properties were first tested in the half cells with metallic Na foils as counter electrodes. Fig. 2a shows the pattern of capacity retention together with the variation of current densities (j) between 0.1 and 10 A g−1. It was shown that the CNT-NWs@Na3TM can deliver a reversible Na-storage capacity of about 214.6 mA h g−1 at a low j of 0.1 A g−1. Note that all the specific capacities for the Na3TM-containing materials were calculated based on the mass of Na3TM. When j increases to 1 and 10 A g−1, the specific capacities delivered are still about 149 and 87.5 mA h g−1, respectively, which shows the excellent rate performance of the as-prepared CNT-NWs@Na3TM composite. More importantly, after the high j (10 A g−1) cycles, the specific capacities can recover to the previous values when j gradually changes back to 0.1 A g−1. Such a characteristic is very significant for practical applications because it can adapt well to the random changes of the working current as cycling. Furthermore, the CNT-NWs@Na3TM also exhibits excellent cycling performance when used as the anode material for SIBs. As shown in Fig. 2b, at a j of 0.5 A g−1, the capacity retention is up to 88.9% after 500 cycles with a decrease of specific capacities from 168.2 to 149.6 mA h g−1. The corresponding capacity attenuation is only about 0.0221% per cycle. The excellent cycling performance is obtained from the high stability of the CNT-NWs@Na3TM electrode during cycling (SEM images after 100 cycles, Fig. S5†). The coulombic efficiencies can rapidly reach 99–100% after the initial low value of ∼48% at the 1st cycle. Fig. 2c shows the Nyquist plots of the EIS at the fresh state and after 20/40 cycles at 0.1 A g−1, from which it is very obvious that the charge-transfer resistances (Rct) of the electrodes remain almost constant during cycling except for the fresh electrode with the obviously much larger Rct value. This demonstrates the outstanding stability of the CNT-NWs@Na3TM electrodes during cycling after the initial activation (the electrolyte infiltration).
 | ||
| Fig. 2 (a) Rate performance of the CNT-NWs@Na3TM composite at different current densities from 0.1 to 10 A g−1. (b) The comparison of cycling performance between CNT-NWs@Na3TM and pure Na3TM cycled between 0.01 and 2 V versus Na+/Na. (c) Nyquist EIS plots of the CNT-NWs@Na3TM electrode in the fresh cell as well as after 20 and 40 cycles. (d) Schematic illustration of the CNT-NWs@Na3TM to show the electronic and ionic transport. | ||
It is worth mentioning that the as-prepared CNT-NWs@Na3TM composite exhibits much better Na-storage properties than either pure Na3TM or CNTs materials. If there is no CNT-NW incorporation in the Na3TM microparticles (Fig. S2†), the reversible Na-storage capacity delivered is less than 8 mA h g−1 even at a low j of 0.1 A g−1 (Fig. S6a† and 2b). Meanwhile, as shown in Fig. S6b,† pure CNTs also deliver a very low Na-storage capacity of 37.5 mA h g−1. The much enhanced Na-storage properties of CNT-NWs@Na3TM composite should benefit from its unique CNT-NWs-incorporating structure in the Na3TM microparticles, in which CNTs act as highly conductive networks to achieve the Na3TM material for Na-storage. As shown in Fig. 2d, during the Na-uptake/release processes, the crosslinked CNTs act as the 3D expressway for electronic conduction, while sodium ions are transferring into/out of the active and porous Na3TM materials. The synergistic processes of electron conduction and ion transport facilitate the electrode kinetics and thus achieve the fast, stable and complete Na storage (superior rate and cycling performance with high Na-storage capacity).
For a new electrode material for SIBs, studying the Na-uptake/release processes to reveal the Na-storage mechanism should be a more significant task than the performance evaluation. Thus, the Na-storage mechanism of CNT-NWs@Na3TM was ascertained by combining the analyses of CV and galvanostatic curves with the ex situ FTIR, XPS and 23Na SS-NMR tests. Fig. 3a shows the representative CV curves of the initial five cycles, disclosing the obvious difference between the 1st sodiation and the following sodiation/desodiation processes. In the 1st sodiation process, there are three clear cathodic peaks at about 0.49, 0.36 and 0.03 V versus Na+/Na, which should be attributed to the stepwise three-electron (3e) sodiation with the one-electron (1e) reaction per step. Interestingly, the anodic/cathodic peaks become two couples and stabilize at about 0.38/0.57 V and 0.03/0.23 V in the subsequent cycles, implying that the overall redox reaction has changed from 3e to two-electron (2e) processes. Furthermore, the cathodic current in the 1st sodiation process is clearly higher compared to the following ones below 1.0 V versus Na+/Na, and this is derived from the formation of stable solid-state interphase (SEI) layers. These inferences were further verified from the galvanostatic curves (Fig. 3b), whose differential capacity plots (inset in Fig. 3b) exhibit almost the same patterns as the CV curves. As shown in Fig. 3b, only two couples of plateaux are reversible in the non-first cycles, and the specific capacity of each charge/discharge plateau is around 100 mA h g−1, which is very close to the theoretical capacity (97.1 mA h g−1) of the 1e reaction for the Na3TM material. Obviously, the specific capacity in the 1st sodiation process is much higher than those in the subsequent cycles. The excess value should be because of one more Na is stored and the formation of SEI layers, which are consistent with the CV tests. In addition, the delivered reversible capacity of CNT-NWs@Na3TM is 214.6 mA h g−1, which is slightly higher than the theoretical value of 194.2 mA h g−1 corresponding to the 2e reaction. The more specific capacity of 20.4 mA h g−1 (the difference between the previous two values) should originate from the Na storage in the interlayers of the benzene rings.28,44,45
 | ||
| Fig. 3 (a) CV profiles and (b) galvanostatic curves of the CNT-NWs@Na3TM electrodes of the initial five cycles. The CV was scanned at the scan rate of 0.1 mV s−1, and galvanostatic test was cycled at the current density of 0.1 A g−1. Inset of (b) the corresponding differential capacity plots. | ||
To confirm the previously mentioned 2e reversible mechanism of the carbonyl groups, the electrodes at different cut-off potentials (sodiation/desodiation states; ST/DT) were further detected using several ex situ technologies. Fig. 4a is the FTIR spectra at the pristine state, the ST of 0.3 V and 0.01 V, and the DT of 0.35 V, 1.0 V and 2.0 V. The selection of the potential values for an improved study of the change of functional groups during sodiation/desodiation processes was mainly based on the plateaux in the galvanostatic curves (Fig. 4b). In the FTIR spectra obtained, the peaks located at 1640 cm−1 and 1080 cm−1, can be assigned to the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bonds, respectively, in the –COO groups. In the pristine state, the ratio of both is theoretically 1:1, namely, the percentage of CO and C–O is the same at 50%. Despite this, the intensity of CO vibration is obviously stronger than that of C–O vibration because of the nature of functional groups for the infrared response. Together with the reduction of cut-off potential in the discharge process, the CO peaks weaken gradually, whereas the C–O one becomes strong, suggesting the gradual decrease/increase of CO/C–O bonds. This mainly originated from the stepwise sodiation of Na3TM material with the gradual transformation of functional groups from
O and C–O bonds, respectively, in the –COO groups. In the pristine state, the ratio of both is theoretically 1:1, namely, the percentage of CO and C–O is the same at 50%. Despite this, the intensity of CO vibration is obviously stronger than that of C–O vibration because of the nature of functional groups for the infrared response. Together with the reduction of cut-off potential in the discharge process, the CO peaks weaken gradually, whereas the C–O one becomes strong, suggesting the gradual decrease/increase of CO/C–O bonds. This mainly originated from the stepwise sodiation of Na3TM material with the gradual transformation of functional groups from 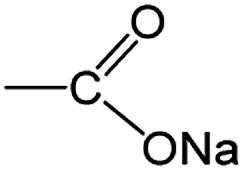 to
to 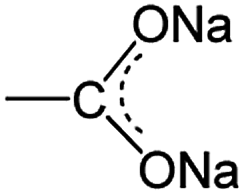 , resulting in the stepwise formation of Na4TM, Na5TM and finally Na6TM. For the opposite charge process, the ratio change of peak intensities is contrary to the previous one for the discharge, demonstrating the reversibility of the sodiation/desodiation reactions. It should be noted that, in comparison to the pristine electrode, the peak intensity of the C–O group could not be reinstated completely when desodiated to 2.0 V, namely, the FTIR peak for C–O groups at DT 2.0 V is stronger than that for the pristine electrode. This suggests the existence of more C–O groups at the DT of 2.0 V compared to the pristine state, because the theoretical percentage of C–O groups has increased to 66.7% because of the formation of partly (1e more) sodiated Na4TM. Additionally, the stronger C–O peaks should be also derived from the formation of the SEI layers.
, resulting in the stepwise formation of Na4TM, Na5TM and finally Na6TM. For the opposite charge process, the ratio change of peak intensities is contrary to the previous one for the discharge, demonstrating the reversibility of the sodiation/desodiation reactions. It should be noted that, in comparison to the pristine electrode, the peak intensity of the C–O group could not be reinstated completely when desodiated to 2.0 V, namely, the FTIR peak for C–O groups at DT 2.0 V is stronger than that for the pristine electrode. This suggests the existence of more C–O groups at the DT of 2.0 V compared to the pristine state, because the theoretical percentage of C–O groups has increased to 66.7% because of the formation of partly (1e more) sodiated Na4TM. Additionally, the stronger C–O peaks should be also derived from the formation of the SEI layers.
 | ||
| Fig. 4 (a) The ex situ FTIR spectra for the CNT-NWs@Na3TM electrodes cut off at the different sodiated/desodiated potentials. (b) The corresponding galvanostatic curves to show the cut-off potential. | ||
Fig. 5a–e further shows the high resolution C 1s XPS spectra of the electrode materials at the same ST/DT given in the FTIR discussion. From the deconvolution results, the peaks for C–O and CO bonds clearly exist in all the spectra, and the C–O/CO ratio changes of both peak intensities and peak areas are consistent with the previously discussed FTIR results obtained during cycling. As shown in Fig. 5a, the intensity difference (ΔI) between the C–O and CO peaks is very small for the pristine Na3TM electrode. In the sodiation processes, such ΔI values increase together with the potential drop and reach the highest one at the lowest voltage, ST = 0.01 V, and then decreases as in desodiation. The ΔI cannot be restored to the pristine value, which can also be attributed to the previously determined inferences of only 2e reversible redox reactions and SEI formation. In addition, as shown in Table S1,† the C–O/CO ratio of the calculated peak area also exhibits the same variation rules, further demonstrating the correctness of the inferred Na-storage mechanism and processes, which is also corroborated by the results of the 23Na SS-NMR tests. As shown in Fig. 5f, the 23Na SS-NMR patterns of two intermediate states at ST 0.3 V and DT 0.35 V are almost the same as the two peaks centered at the chemical shifts of −7.3 ± 0.1 and −5.15 ± 0.05 ppm, respectively, suggesting the reversibility of sodiation/desodiation processes.46 In comparison to such intermediate states, the completely sodiated/desodiated electrodes at ST 0.01 V and DT 2.0 V exhibit the stronger resonance peaks at the high chemical shift of −15 ± 0.5 ppm and larger peak areas, demonstrating the more concentrated Na configurations of –C(ONa)(ONa) and –COONa at ST 0.01 V and DT 2.0 V, respectively.47 Note that the NMR pattern of pure Na3TM is obviously different from the other samples after electrochemical tests, which ought to originate from the electrochemically sodiated carbon black (Super P) and the formation of SEI films on the electrode surface.48
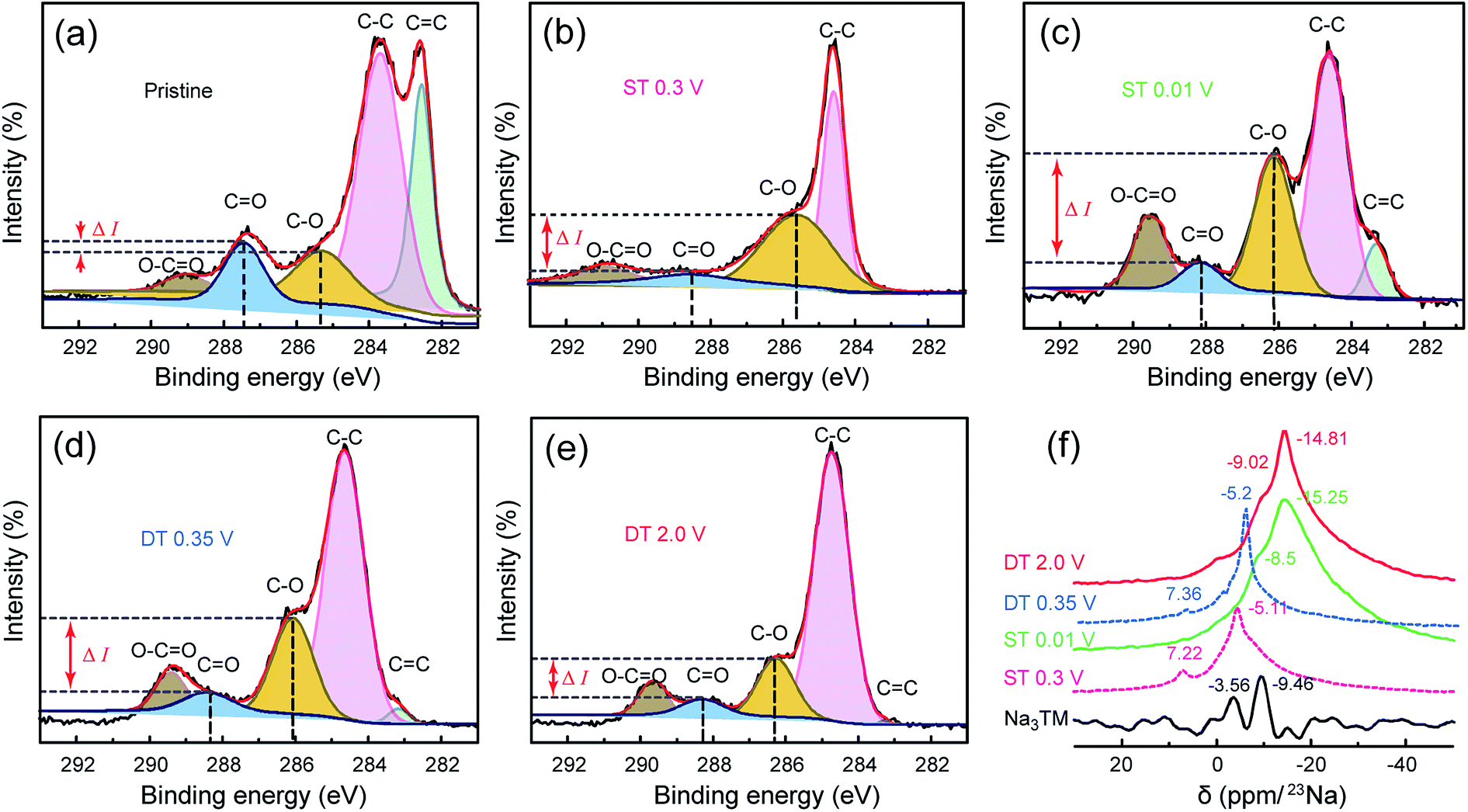 | ||
| Fig. 5 (a–e) The deconvoluted C 1s XPS and (f) 23Na SS-NMR spectra for the CNT-NWs@Na3TM electrodes cut off at the different sodiated/desodiated potentials, which are the same as those shown in Fig. 4. | ||
In order to more clearly show the Na-storage mechanism of the as-prepared CNT-NWs@Na3TM composite, the Na-uptake/release processes are illustrated in Fig. 6 and are based on the previously studied results of CV, galvanostatic curves, ex situ FTIR, XPS, and 23Na SS-NMR spectra. It is shown that the electrochemical reaction of the 1st sodiation is an obvious 3e (3Na+) transferring process with stepwise Na-uptake from the Na3TM raw material to Na4TM, Na5TM, and finally Na6TM. In the following desodiation processes, the Na6TM formed cannot recover to the initial Na3TM instead of the 1e-sodiated Na4TM. After the 1st cycle, the active materials for Na-storage convert reversibly between Na4TM (oxidation product) and Na6TM (reduction product) with the formation of an Na5TM intermediate during the subsequent cycles. In other words, the presently proposed CNT-NWs@Na3TM anode is 2e reversible during cycling except for the 1st sodiation process with 3Na uptake, and the Na-uptake/release processes are stepwise with one Na transfer per step.
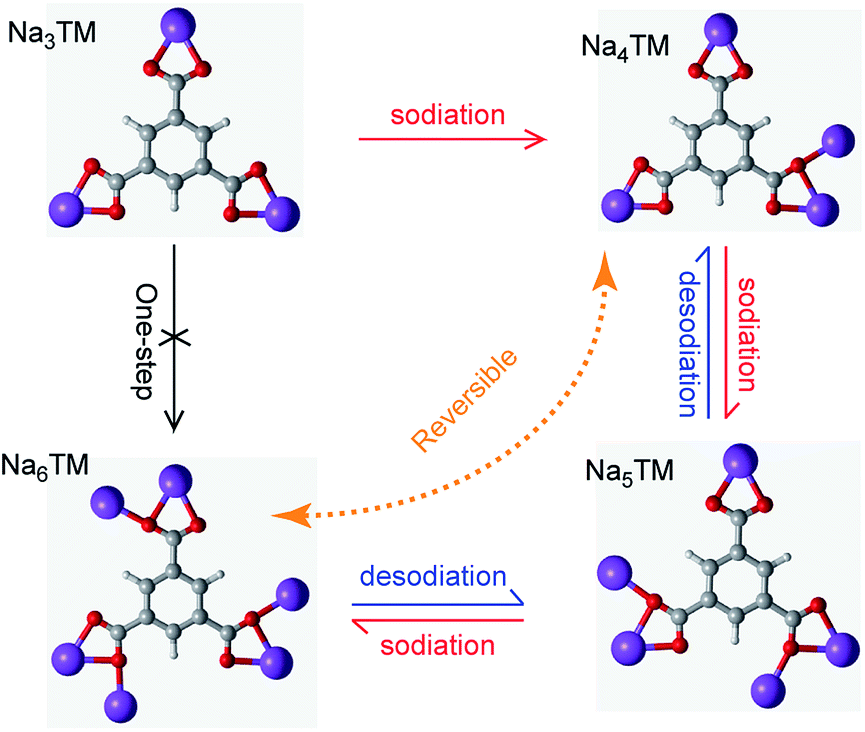 | ||
| Fig. 6 Illustration of the possible sodiation/desodiation mechanism of the as-prepared CNT-NWs@Na3TM composite. | ||
4. Conclusions
In summary, one new anode material, Na3TM, was proposed for SIBs, and the Na-storage properties were significantly enhanced by embedding 3D CNT networks in its microparticles. Although the pure Na3TM was almost inactive for Na storage, the as-prepared CNT-NWs@Na3TM composite could deliver a reversible capacity of 214.6 mA h g−1 at 0.1 A g−1, and exhibit outstanding rate capability (e.g., the specific capacities delivered were still 149 and 87.5 mA h g−1 at the current density of 1 and 10 A g−1, respectively), and good cycling performance. Furthermore, the ex situ tests of FTIR, XPS and 23Na SS-NMR in combination with the analyses of CV and galvanostatic curves were implemented to ascertain the Na-storage mechanism of the new developed CNT-NWs@Na3TM anode. It was disclosed that only 2Na storage was reversible after the initial 3Na uptake, and the Na-uptake/release processes were stepwise with one Na transfer per step. Because of the high conductivity of CNTs and the feasibility of constructing 3D CNT-NWs, the presently proposed strategy of embedding 3D CNT-NWs is expected to be universally used to improve the Na-storage properties of organic electrodes, the cutting-edge but low conductive electrode materials for SIBs as well as LIBs.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51602048, 51225204), the Ministry of Science and Technology of the People's Republic of China (2016YFA0202500), and the Science Technology Program of Jilin Province (20150520027JH).References
- N. J. Dudney and J. C. Li, Science, 2015, 347, 131–132 CrossRef CAS PubMed.
- N.-W. Li, Y.-X. Yin, C.-P. Yang and Y.-G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- Y.-X. Wang, J. Yang, S.-L. Chou, H. K. Liu, W.-X. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6, 8689–8698 CrossRef CAS PubMed.
- K. Kirshenbaum, D. C. Bock, C. Y. Lee, Z. Zhong, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Science, 2015, 347, 149–154 CrossRef CAS PubMed.
- W.-H. Liu and X.-L. Wu, J. Mol. Sci., 2016, 32, 379–395 Search PubMed.
- D. Su and G. Wang, ACS Nano, 2013, 7, 11218–11226 CrossRef CAS PubMed.
- D.-H. Liu, H.-Y. Lu, X.-L. Wu, J. Wang, X. Yan, J.-P. Zhang, H. Geng, Y. Zhang and Q. Yan, Nanoscale Horiz., 2016, 1, 496–501 RSC.
- W. Li, C. Yao, X.-L. Wu, H.-M. Xie, L.-Q. Sun and R.-S. Wang, J. Mol. Sci., 2013, 29, 448–460 CAS.
- Z. Wang, K.-S. Zhao, Y. Sun, D. Zhou and H.-M. Xie, J. Mol. Sci., 2016, 32, 367–378 Search PubMed.
- Y. You, X.-L. Wu, Y.-X. Yin and Y.-G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 CAS.
- H. Gao, T. Zhou, Y. Zheng, Y. Liu, J. Chen, H. Liu and Z. Guo, Adv. Energy Mater., 2016, 6, 1601037 CrossRef.
- J.-Z. Guo, F. Wan, X.-L. Wu and J.-P. Zhang, J. Mol. Sci., 2016, 32, 265–279 Search PubMed.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS PubMed.
- Y. S. Hu, L. Kienle, Y. G. Guo and J. Maier, Adv. Mater., 2006, 18, 1421–1426 CrossRef CAS.
- Y. Li, Y.-S. Hu, M.-M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef.
- G. Gao, Q. Zhang, X.-B. Cheng, J. G. Shapter, T. Yin, R. Sun and D. Cui, Sci. Rep., 2015, 5, 17553 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- H. Kim, J. Hong, Y.-U. Park, J. Kim, I. Hwang and K. Kang, Adv. Funct. Mater., 2015, 25, 534–541 CrossRef CAS.
- Y. Li, L. Mu, Y.-S. Hu, H. Li, L. Chen and X. Huang, Energy Storage Materials, 2016, 2, 139–145 CrossRef.
- X. Zhou, Z. Dai, J. Bao and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 13727–13731 CAS.
- Y.-C. Zhang, Y. You, S. Xin, Y.-X. Yin, J. Zhang, P. Wang, X.-s. Zheng, F.-F. Cao and Y.-G. Guo, Nano Energy, 2016, 25, 120–127 CrossRef CAS.
- Q. Xiao, M. Gu, H. Yang, B. Li, C. Zhang, Y. Liu, F. Liu, F. Dai, L. Yang, Z. Liu, X. Xiao, G. Liu, P. Zhao, S. Zhang, C. Wang, Y. Lu and M. Cai, Nat. Commun., 2015, 6, 8844–8892 CrossRef CAS PubMed.
- D. Yang, X.-Z. Liao, B. Huang, J. Shen, Y.-S. He and Z.-F. Ma, J. Mater. Chem. A, 2013, 1, 13417–13421 CAS.
- H.-G. Wang, S. Yuan, D.-L. Ma, X.-L. Huang, F.-L. Meng and X.-B. Zhang, Adv. Energy Mater., 2014, 4, 1301651 CrossRef.
- L. Zhao, J. Zhao, Y.-S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2, 962–965 CrossRef CAS.
- Y. Park, D.-S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef CAS PubMed.
- X. Wu, S. Jin, Z. Zhang, L. Jiang, L. Mu, Y.-S. Hu, H. Li, X. Chen, M. Armand, L. Chen and X. Huang, Sci. Adv., 2015, 1, e1500330 Search PubMed.
- A. Abouimrane, W. Weng, H. Eltayeb, Y. Cui, J. Niklas, O. Poluektov and K. Amine, Energy Environ. Sci., 2012, 5, 9632–9638 CAS.
- H.-G. Wang, S. Yuan, Z. Si and X.-B. Zhang, Energy Environ. Sci., 2015, 8, 3160–3165 CAS.
- F. Wan, X. L. Wu, J. Z. Guo, J. Y. Li, J. P. Zhang, L. Niu and R. S. Wang, Nano Energy, 2015, 13, 450–457 CrossRef CAS.
- A. de Kergommeaux, M. Lopez-Haro, S. Pouget, J. M. Zuo, C. Lebrun, F. Chandezon, D. Aldakov and P. Reiss, J. Am. Chem. Soc., 2015, 137, 9943–9952 CrossRef CAS PubMed.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- X. L. Wu, Y. G. Guo, J. Su, J. W. Xiong, Y. L. Zhang and L. J. Wan, Adv. Energy Mater., 2013, 3, 1155–1160 CrossRef CAS.
- X. Zhou, L. Yu, X.-Y. Yu and X. W. Lou, Adv. Energy Mater., 2016, 6, 1601177 CrossRef.
- X. Zhou, T. Wu, K. Ding, B. Hu, M. Hou and B. Han, Chem. Commun., 2009, 1897–1899 RSC.
- X. Zhou, Y.-X. Yin, A.-M. Cao, L.-J. Wan and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 2824–2828 CAS.
- F. Wan, Y.-H. Li, D.-H. Liu, J.-Z. Guo, H.-Z. Sun, J.-P. Zhang and X.-L. Wu, Chem.–Eur. J., 2016, 22, 8152–8157 CrossRef CAS PubMed.
- F. Wan, H.-Y. Lü, X.-H. Zhang, D.-H. Liu, J.-P. Zhang, X. He and X.-L. Wu, J. Alloys Compd., 2016, 672, 72–78 CrossRef CAS.
- B.-H. Hou, X.-L. Wu, Y.-Y. Wang, H.-Y. Lü, D.-H. Liu, H.-Z. Sun, J.-P. Zhang and H.-Y. Guan, Part. Part. Syst. Charact., 2015, 32, 1020–1027 CrossRef CAS.
- F. Wan, J.-Z. Guo, X.-H. Zhang, J.-P. Zhang, H.-Z. Sun, Q. Yan, D.-X. Han, L. Niu and X.-L. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 7790–7799 CAS.
- Y. Xu, Y. Zhao, J. Ren, Y. Zhang and H. Peng, Angew. Chem., Int. Ed., 2016, 7979–7982 CrossRef CAS PubMed.
- F. Wan, H.-Y. Lü, X.-L. Wu, X. Yan, J.-Z. Guo, J.-P. Zhang, G. Wang, D.-X. Han and L. Niu, Energy Storage Materials, 2016, 5, 214–222 CrossRef.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552–557 CAS.
- X. Han, F. Yi, T. Sun and J. Sun, Electrochem. Commun., 2012, 25, 136–139 CrossRef CAS.
- R. Turoscy, H. Leidheiser and J. E. Roberts, J. Electrochem. Soc., 1993, 140, 149–154 CrossRef CAS.
- R. Turoscy, H. Leidheiser and J. E. Roberts, J. Electrochem. Soc., 1990, 137, 1785–1788 CrossRef CAS.
- P. K. Allan, J. M. Griffin, A. Darwiche, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, A. J. Morris, P. J. Chupas, L. Monconduit and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 2352–2365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: 10.1039/c7ta03484a |
| This journal is © The Royal Society of Chemistry 2017 |