
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green one-pot synthesis and processing of polyimide–silica hybrid materials†
Lukas
Leimhofer
a,
Bettina
Baumgartner
a,
Michael
Puchberger
a,
Thomas
Prochaska
b,
Thomas
Konegger
 b and
Miriam M.
Unterlass
*a
b and
Miriam M.
Unterlass
*a
aTechnische Universität Wien, Institute of Materials Chemistry, Getreidemarkt 9/BC/2, A-1060 Vienna, Austria. E-mail: miriam.unterlass@tuwien.ac.at; Fax: +43-1-58801-165981; Tel: +43-1-58801-165206
bTechnische Universität Wien, Institute of Chemical Technologies and Analytics, Getreidemarkt 9/164-CTA, 1060 Vienna, Austria
First published on 19th May 2017
Abstract
Inorganic–organic hybrid materials allow for combining features typical of the inorganic component with those of the organic component in one material. Generally, the preparation of organic and inorganic compounds requires considerably different synthesis conditions. Hence, the development of one-pot routes to inorganic–organic hybrid materials is challenging. We herein report a fully green one-pot synthesis of polyimide/silica (PI/SiO2) hybrids. Specifically, we co-condense both components hydrothermally, using nothing but the respective precursors and water. Furthermore, we show that the PI and the SiO2 component can be covalently connected under hydrothermal conditions, using the compatibilizer (3-aminopropyl)-triethoxysilane. We thoroughly investigate the effect of different reaction conditions, including temperature, pH, precursor concentration and reaction time on the morphology and crystallinity of the final materials. The polyimide component, poly(hexamethylene pyromellitimide) was chosen for its thermoplasticity, which allows for processing both the PI and the PI/SiO2via sintering. For being a solvent-free method, sintering qualifies as a green processing technique. This work is the first report of the simultaneous hydrothermal condensation of an inorganic and an organic material.
1 Introduction
The continuously increasing level of technologization of our modern world generates the need for even more sophisticated materials. Unfortunately, the majority of currently employed syntheses of – especially organic – materials, are harsh and toxic. Mankind's growing awareness of human health and the environment creates the demand for less harmful procedures for both producing and processing materials. The development of chemical reactions which minimize environmental impact is summarized by the principles of green chemistry,1 and is a highly timely topic. Green materials chemistry aims at responding to both the need for highly advanced materials, and the need for benign synthesis approaches.2There are a handful of major approaches to introduce a high level of sophistication into materials, amongst which is the preparation of hybrid materials. Hybrids or composites combine at least two chemically different compounds (and thus also their properties) into an intimately mixed or connected novel material.3 Employing this concept, one can for instance couple the low densities and ease of processing found in organic polymers with the mechanical performance of inorganic materials, such as ceramics.4 The resulting compound is a so-called inorganic–organic hybrid material. There are four major synthesis approaches towards inorganic–organic hybrid materials, which are (i) mixing the ready-made inorganic and the ready-made organic component; (ii) synthesizing the organic component in the presence of the pre-fabricated inorganic component; (iii) preparing the inorganic component in the presence of the pre-fabricated organic component; and (iv) synthesizing both components simultaneously.2 In terms of green chemistry, such concurrent one-pot approaches are clearly preferable over the other routes, as the reduced number of synthesis steps automatically minimizes e.g. solvent consumption and purification steps. However, the development of one-pot syntheses of inorganic–organic hybrid materials is intrinsically challenging, since conventional preparation conditions of the two components often vary considerably from each other. For instance, inorganic compounds often require polar reaction media or high temperatures, while syntheses of organic materials are mainly carried out in comparatively apolar environments at rather moderate temperatures.
We have recently started to work on transferring a widely used inorganic synthesis route, hydrothermal synthesis, to the preparation of organic polymers. With hydrothermal polymerization (HTP), we have developed a geomimetic technique to synthesize fully aromatic polyimides (PIs) of high crystallinity.5–8 The fact that a plethora of inorganic compounds, including for instance metal oxides, carbonates and sulfates, can be generated hydrothermally has been known for over 150 years.9 Moreover, such processes are a major naturally occurring formation pathway of crystalline minerals. Since HTP also generates high crystallinity in polyimides, it is referred to as a geomimetic technique. HTP is inherently green, as it does not require anything besides H2O as the reaction medium, and the starting compounds, i.e. the monomers. This is indeed in stark contrast to conventional routes towards polyimides, which require the use of toxic and high-boiling (and hence costly to recycle) solvents such as dimethylformamide or N-methyl-2-pyrrolidone.10 The outstanding crystallinity generated via HTP is a feature that is highly sought after for PIs, since it enhances certain material properties that these high-performance polymers are especially desired for, such as thermal or chemical stability. Consequently, hydrothermally synthesized PIs are typically insoluble in virtually any solvent, including conc. H2SO4 at reflux.5 Moreover, PIs generated from HTP are typically devoid of thermally induced softening via a glass or melting transition.5 Therefore, the advantages of HTP, i.e. the facts that the technique is green and at the same time leads to an enhancement of the properties of materials, go to date hand in hand with the major drawback of non-processability of the obtained PIs.
In this study, we present the first example of a one-pot, fully green synthesis of an inorganic–organic hybrid material. Specifically, we selected a semialiphatic PI as the organic component, and a metal oxide silica as the inorganic component. The target PI was chosen, as semialiphatic PIs are known thermoplastics,11 which we anticipated to allow for processing via sintering. In fact, sintering can be considered a green technique as it does not employ any organic solvents. Moreover, we expected semialiphatic PIs to be colorless, since the conventional orange-brownish color of PIs is believed to be related to those PIs typically comprising alternating electron-rich and electron-poor aromatic rings and their intermolecular packing into charge-transfer complexes.12 The latter is unwanted in e.g. aerospace applications – where PIs are the external coating materials of choice, a consequence of their high resistance to cosmic radiation – since the yellowish coloring generates high solar absorptivities.13 SiO2 was selected as the inorganic component for (i) being the archetype of a metal oxide that can be prepared hydrothermally, and (ii) its white color, from which the target PI/SiO2 materials were expected to additionally profit.
2 Results and discussion
2.1 Hydrothermal synthesis of PI6
Since HTP of semialiphatic PIs has to date not been reported, we decided to concentrate our efforts on one system only: PI6 – the polyimide resulting from the comonomers hexamethylene diamine (HMDA) and pyromellitic acid (PMA), see Fig. 1. The choice of PI6 was based on (i) the fact that it is a well-known, high-melting thermoplastic with a reported melting point Tm of 454 °C,11 and (ii) the low-cost of both comonomers (both are used as starting compounds in industrially relevant syntheses: PMA or its dianhydride is one of the two comonomers of Kapton™; HMDA is one of the two comonomers of nylon 6,6). To first generally check the feasibility of PI6 synthesis via HTP, a dispersion containing HMDA and PMA in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at c = 0.1 mol L−1 in H2O was prepared. This dispersion was filled into a non-stirred autoclave (see the ESI† for the set-up), heated to the reaction temperature TR ≈ 200 °C, and kept there for a reaction time of tR = 24 h. After tR, the autoclave was rapidly cooled back to RT by quenching with tap water. The obtained white dispersion was filtered, rinsed with deionized water, and dried at RT and reduced pressure (see the ESI† for experimental details). The ATR-FTIR spectrum of hydrothermally prepared PI6 (Fig. 2) agrees well with the literature.14 The most prominent modes are
:1 ratio at c = 0.1 mol L−1 in H2O was prepared. This dispersion was filled into a non-stirred autoclave (see the ESI† for the set-up), heated to the reaction temperature TR ≈ 200 °C, and kept there for a reaction time of tR = 24 h. After tR, the autoclave was rapidly cooled back to RT by quenching with tap water. The obtained white dispersion was filtered, rinsed with deionized water, and dried at RT and reduced pressure (see the ESI† for experimental details). The ATR-FTIR spectrum of hydrothermally prepared PI6 (Fig. 2) agrees well with the literature.14 The most prominent modes are ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) s,C
s,C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ≈ 1765 cm−1 and as,CO ≈ 1700 cm−1. Also, the imide ring's (O)C–N stretching mode at ≈1370 cm−1 and the H2C–N stretching mode at ≈1400 cm−1 are clearly present. Moreover, in the higher wavenumber region of 4000–2500 cm−1, solely aromatic and aliphatic C–H modes are present (CAr–H: 3101 and 3035 cm−1; Calkyl–H: 2941, 2858 cm−1). Any heteroatom–H modes (O–H, N–H), including H-bonding, would occur in this region. Hence, we conclude from ATR-FTIR analysis that neither of the possible end-groups NH2, NH3+ or CO2H is present. Two further end-groups are conceivable, namely CO2− and cyclic anhydrides. Neither the characteristic carboxylate CO mode (≈1570 cm−1) nor the characteristic CO vibration of pyromellitic anhydride (≈1820 cm−1) is present, which also excludes these end-groups. Hence, it can be stated that PI6 is fully condensed as judged from ATR-FTIR spectra. Note that as,CO ≈ 1700 cm−1 occurs at almost the same wavenumber as the CO vibration in free PMA would (1698 cm−1, cf.Fig. 2). Consequently the presence of a strong vibration at 1700 cm−1 cannot be viewed as indicative of PMA consumption and imide formation by itself, but must be considered together with s,CO (≈1765 cm−1).
O ≈ 1765 cm−1 and as,CO ≈ 1700 cm−1. Also, the imide ring's (O)C–N stretching mode at ≈1370 cm−1 and the H2C–N stretching mode at ≈1400 cm−1 are clearly present. Moreover, in the higher wavenumber region of 4000–2500 cm−1, solely aromatic and aliphatic C–H modes are present (CAr–H: 3101 and 3035 cm−1; Calkyl–H: 2941, 2858 cm−1). Any heteroatom–H modes (O–H, N–H), including H-bonding, would occur in this region. Hence, we conclude from ATR-FTIR analysis that neither of the possible end-groups NH2, NH3+ or CO2H is present. Two further end-groups are conceivable, namely CO2− and cyclic anhydrides. Neither the characteristic carboxylate CO mode (≈1570 cm−1) nor the characteristic CO vibration of pyromellitic anhydride (≈1820 cm−1) is present, which also excludes these end-groups. Hence, it can be stated that PI6 is fully condensed as judged from ATR-FTIR spectra. Note that as,CO ≈ 1700 cm−1 occurs at almost the same wavenumber as the CO vibration in free PMA would (1698 cm−1, cf.Fig. 2). Consequently the presence of a strong vibration at 1700 cm−1 cannot be viewed as indicative of PMA consumption and imide formation by itself, but must be considered together with s,CO (≈1765 cm−1).
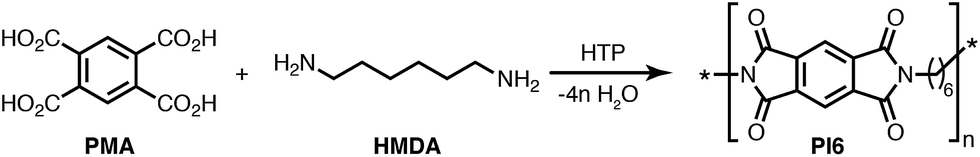 | ||
| Fig. 1 Reaction equation of the hydrothermal polymerization of PI6 from comonomers HMDA and PMA. | ||
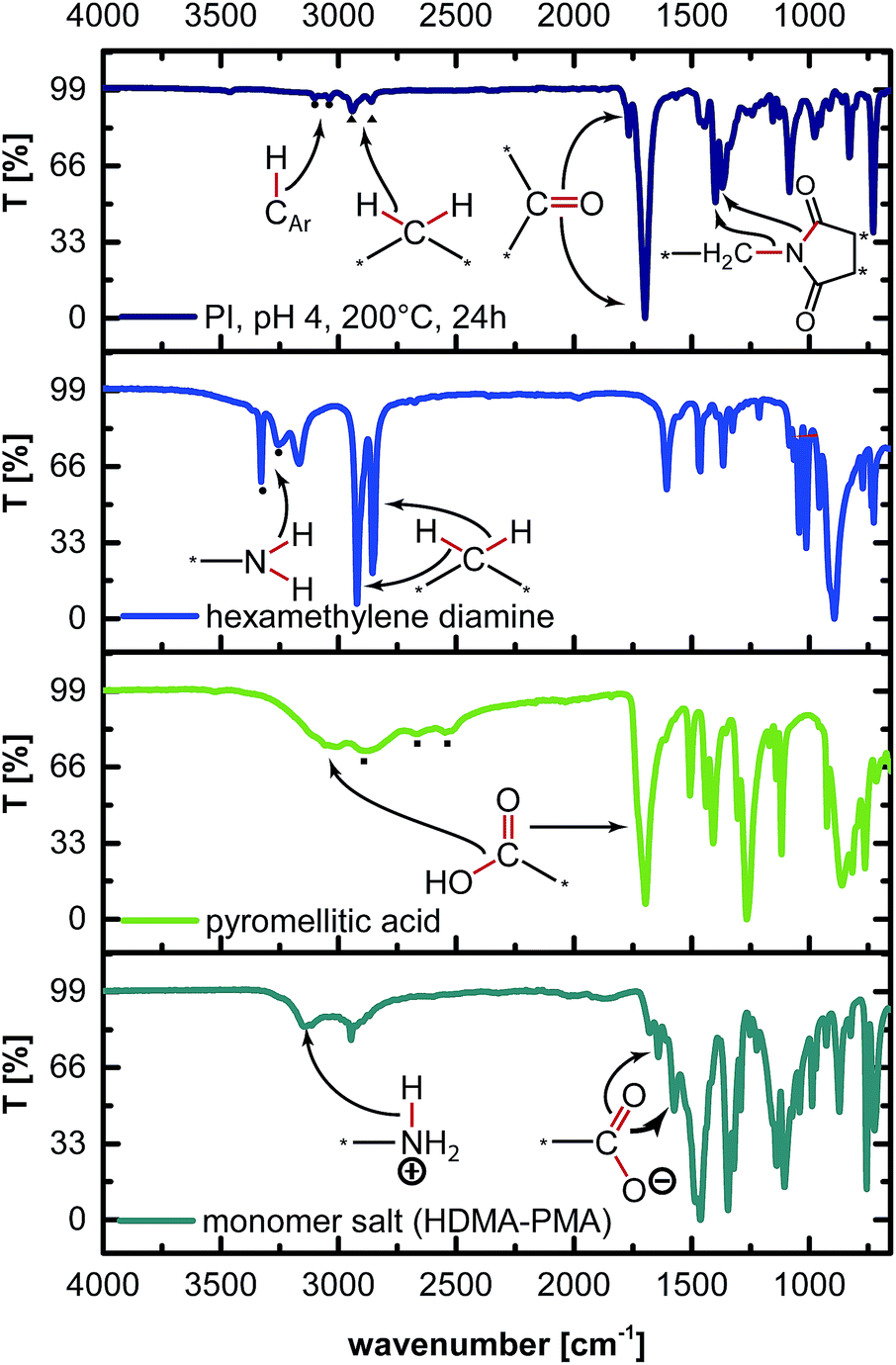 | ||
| Fig. 2 ATR-FTIR spectra of PI6 and monomers. From top to bottom: PI6, HMDA, PMA, and monomer salt. Mode assignments are illustrated by molecule drawings. | ||
Powder X-ray diffraction (PXRD) of PI6 confirms very high crystallinity (Fig. 3A). This is indeed expected, since HTP is a polymerization technique that generates outstandingly crystalline polyimides.5–7 The diffractogram shows barely any amorphous background and the major reflections are rather sharp, cf.Fig. 3A. Specifically, we find four intense reflections at 7.3°, 18.5°, 25.1° and 28.4° (2θ, Cu-Kα), labelled A–D (Fig. 3A). Moreover, we find six smaller and rather broad reflections between 10° and 35° (2θ, Cu-Kα), labelled E–J (Fig. 3A). To our knowledge, to date, there is only one article which reported on the PXRD analysis of PI6.15 The authors prepared PI6 by polycondensation in ethylene glycol (at 130 °C) and by solid-state polycondensation of the monomer salt (at 180 °C). Both polymerizations lead to PI6 of rather poor crystallinity, with diffractograms showing only two reflections, at ≈7° and ≈19° (2θ, Cu-Kα), respectively. We speculate that these reflections correspond to the most intense reflections in PI6 prepared by HTP (A and B in Fig. 3A).
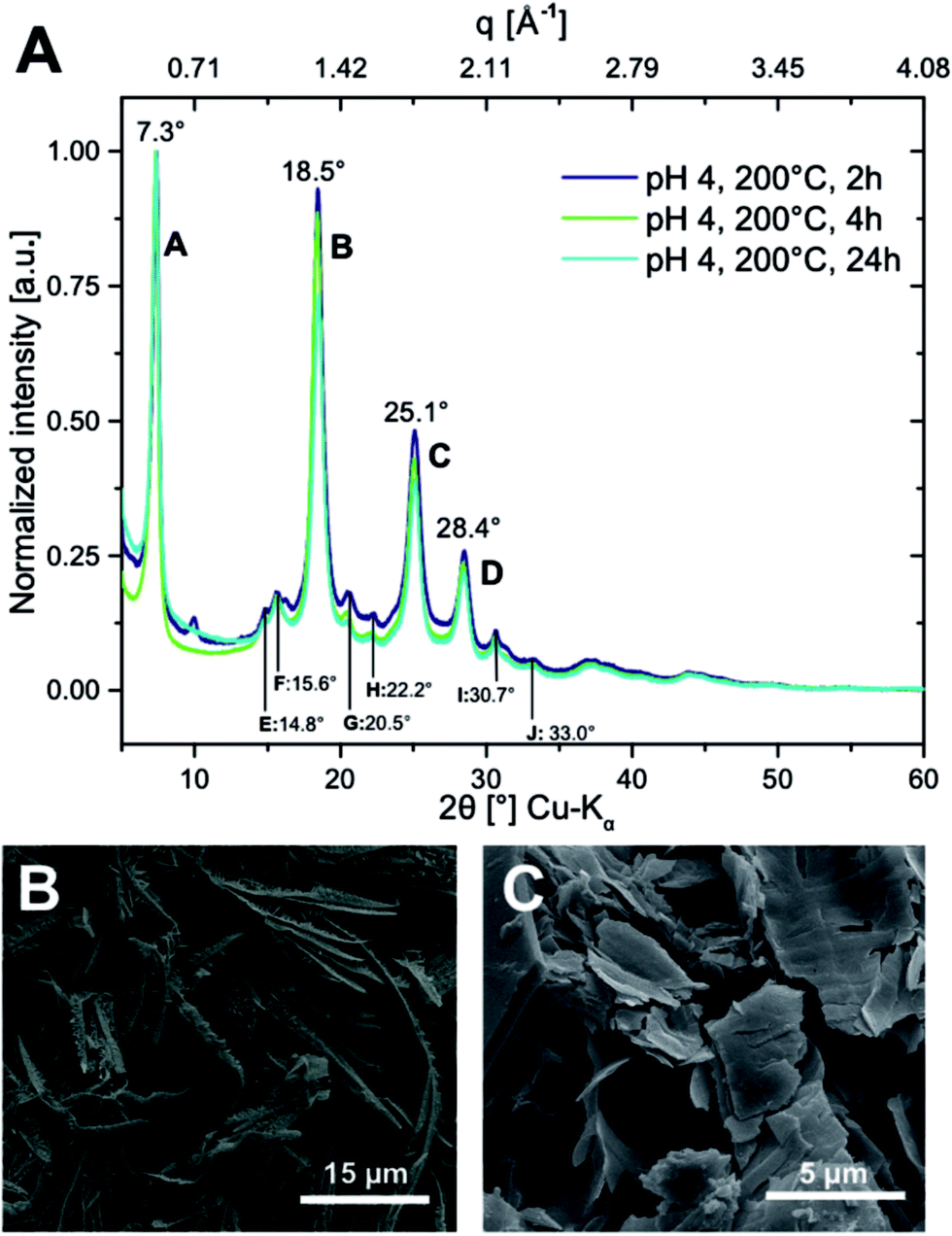 | ||
| Fig. 3 Crystallinity of hydrothermally prepared PI6. (A) PXRD pattern of PI6 prepared at 200 °C and pH = 4 at tR = 2 h (dark blue); 4 h (green) and 24 h (turquoise). Reflections are labelled (A)–(J) with annotated scattering angles 2θ (Cu-Kα). (B) SEM image of PI6 synthesized at 200 °C and pH = 4 for 24 h. (C) SEM image of PI6 synthesized at 200 °C and pH = 1 for 24 h. | ||
Scanning electron microscopy (SEM) evinces a highly homogeneous sample (cf. ESI†), exclusively composed of ribbons of ≈50 μm in length, ≈1–3 μm in width, and ≈100 nm in thickness. These ribbons are additionally covered with highly oriented, smaller ribbons (or scales) growing away from the parent ribbons almost perpendicularly (Fig. 3B). These structures are indicative of dendritic growth.
After this first proof-of-concept, we studied the effect of three parameters on the hydrothermal synthesis of PI6: TR, th and pH. Aiming at lower TR and tR is highly interesting in terms of green chemistry (lowering the energy consumption). The pH was additionally studied, since polycondensations – the formation reaction of both polyimides and SiO2 – can typically be sped up by either acidic or basic catalysis. However, PIs do not subsist basic conditions: both mineral and organic bases tend to attack the cyclic imide moiety and generate oligo-/poly(amic acid)s and oligo-/poly(amic carboxylates).16,17 Therefore, we only investigated acidic conditions. Specifically, we studied two TR, 140 and 200 °C; four different tR, 2, 4, 24, and 48 h; and additional acidification with HCl to pH = 1. Note that a dispersion of the comonomers PMA and HMDA is already acidic (pH = 4), due to the acidity of the tetracarboxylic acid PMA. When studying PI6 formation at the significantly lower reaction temperature TR ≈ 140 °C, from ATR-FTIR spectra (ESI†) it becomes clear that PI6 does not fully form even after 48 h for pH = 1, and appears nearly complete after 48 h at pH = 4 (non-acidified samples). Interestingly, at pH = 1 and TR = 140 °C, a large number of broad modes at 3750–2250 cm−1 is found for any reaction time. These modes are attributed to protonation of e.g. CO2− and NH2 groups, in connection with H-bonding between these groups (cf. ESI†). We hypothesize that these samples contain considerable amounts of protonated low-molecular weight compounds, i.e. monomers, monomer salts and eventually dimers, that did not yet react to the polyimide. This hypothesis is further strengthened by PXRD data: samples prepared at TR = 140 °C at pH = 1 either show many sharp reflections (indicative of the presence of crystalline small molecules) that do not fit the major reflections of PI6 (at tR = 2 h, 4 h), or show both reflections associated with PI6 and many sharp reflections. SEM analysis of all the products of all attempts to synthesize PI6 at 140 °C – no matter if pH = 1 or pH = 4 – corroborates the hypothesis that crystalline small molecule species form rather than the desired PI. Specifically, we find numerous angular or otherwise regularly intergrown objects of forms indicative of crystallinity, of considerable size (up to 0.5 mm). These morphologies are clearly too big to be polyimide crystallites. As PMA is a well-known tecton for supramolecular solid-state assemblies, such as co-crystals and salts,18,19 we speculate that several different compounds of this type form hydrothermally at 140 °C. Consequently, we did not further pursue low TR.
For TR = 200 °C, we only investigated shorter tR, since full condensation and high crystallinity had already been obtained at tR = 24 h (Fig. 2 and 3). ATR-FTIR analysis indicates full condensation even after tR = 2 h and 4 h (ESI†), for both pH = 1 and pH = 4. From PXRD we find high crystallinity in samples obtained at pH = 1: all major (A–D) and minor reflections (E–J) found for tR = 24 h at pH = 4 (Fig. 3A) are present for both 2 h and 4 h. However, PI6 obtained after 2 h shows one additional reflection at approx. 10° (2θ, Cu-Kα), which is most likely due to the presence of shorter oligomers.8 PI6 obtained at pH = 1 at TR = 200 °C appears fully condensed from ATR-FTIR at all tested tR (2 h, 4 h, 24 h) – the spectra (ESI†) fit the literature and the spectrum of PI6 obtained after 24 h at pH = 4 (Fig. 2) well. The PXRD pattern shows the presence of the major reflections (A–D, Fig. 3A). In addition, there are however many strong reflections (tR = 2 h) that we attribute to monomer salts/oligomers and weak (tR = 4 h, 24 h) reflections that are not present in PI6 synthesized at pH = 4 (see the ESI†). To our surprise, we must conclude that the PI6 formation is slower and changed at pH = 1 as compared to pH = 4. This effect could be due to the increased acidity, but could also be related to the presence of Cl− ions in the system. Morphology-wise, we find ribbon morphologies at pH = 4 (cf.Fig. 3B and the ESI†), and platelet-type morphologies at pH = 1 (cf.Fig. 3C and the ESI†), for all tR. To investigate the differences in morphology and crystal structure of PI6 as a function of the pH, we performed energy dispersive X-ray spectroscopy (EDAX) analysis on samples synthesized at TR = 200 °C, th = 24 h, at pH = 4 and pH = 1, respectively. From EDAX spectra (cf. ESI†), it becomes clear that PI6 synthesized at pH = 4 contains exclusively C, N and O (note that H cannot be detected via EDAX), while PI6 synthesized at pH = 1 contains Cl. Therefrom, we infer that despite thorough washing of PI6 samples with aq. dest. prior to drying, HCl is retained by the polyimide. While further investigations would be necessary to clearly identify the nature of the retention, we speculate that HCl forms adducts with either imide carbonyl oxygen or imide nitrogen atoms: for the presence of lone pairs these parts of the PI chains are the only conceivable loci in the polymer where an adduct with HCl can be formed. For the preparation of PI6/SiO2 hybrid materials, we refrained from pursuing HCl addition any further.
2.2 Hydrothermal synthesis of PI6/SiO2 hybrid materials
The silica precursors used for the synthesis of PI6/SiO2 hybrid materials are tetraethoxysilane (TEOS) and (3-aminopropyl)triethoxysilane (APTES). The condensation reaction equations of both TEOS and PI6 in the presence of APTES and TEOS are depicted in Fig. 4. TEOS is a standard sol–gel precursor of SiO2, and APTES is a standard network-modifier that has been used to (i) introduce amino-functionalities in SiO2,20 and (ii) as a compatibilizer between SiO2 and PIs in the preparation of PI/SiO2 hybrids.21,22 Note that other approaches to covalently link PIs to SiO2 include (i) employing epoxy-functionalized silicon alkoxides,23 (ii) the use of PI monomers with pendant OH groups able to co-condense with TEOS,24 (iii) employing pre-formed PMMA-functionalized SiO2 particles, where the PMMA-functionalization allows for crosslinking with the PI via transesterification,25 (iv) to decorate PIs with pendant silane side-groups that are able to co-condense with TEOS,26 or to employ APTES end-capped poly(amic acid) species.27 However, all the PI/SiO2 hybrids and composites reported to date have been prepared employing classical, non-green routes, i.e. by polycondensation in toxic solvents such as DMF or NMP via poly(amic acid) intermediates.21–27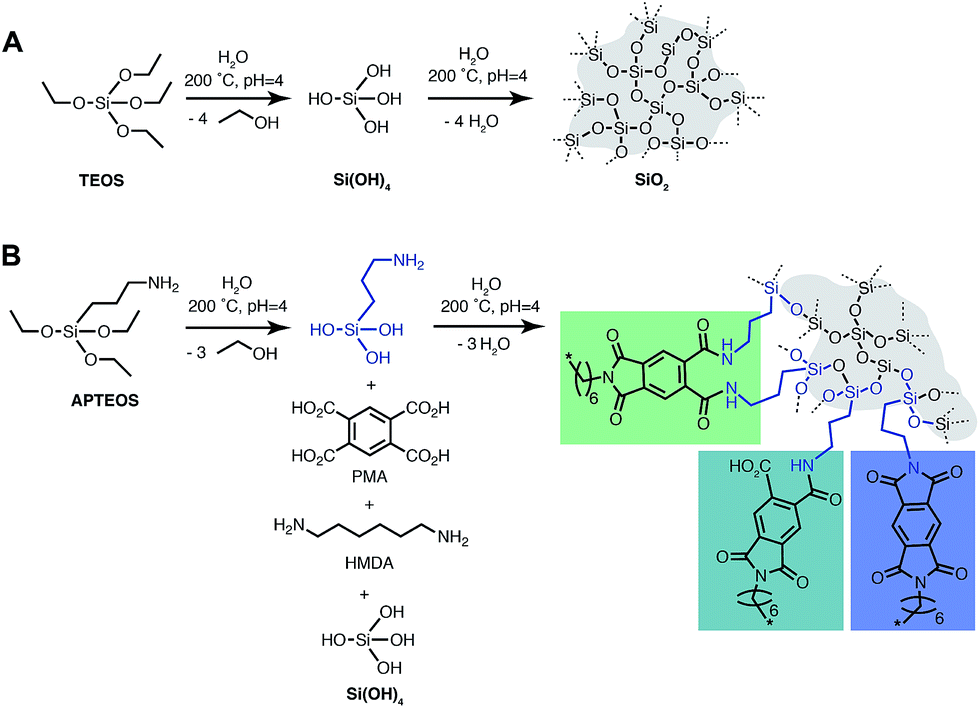 | ||
| Fig. 4 Reaction equation of SiO2 and PI6/SiO2 co-condensation. (A) Hydrothermal hydrolysis and condensation sequence from TEOS to SiO2. (B) Co-condensation of PI6 and SiO2 in the presence of the compatibilizer APTES; possible linkages (1,2-diamide, amic acid; and imide) are shown. | ||
From our studies of HTP of PI6, we restricted the employed reaction parameters to TR = 200 °C, tR = 24 h and the autogenous pH = 4, generated by the acidity of PMA. We investigated three types of precursor mixtures: (i) HMDA/PMA/TEOS (experiments #1–3) (ii) HMDA/PMA/TEOS/APTES (experiments #4–6), and (iii) HMDA/PMA/APTES (experiments #7–9), all summarized in Table 1.
| # | n PMA | n HMDA | n APTES | n TEOS | ∑n(NH2) | ∑n(CO2H) | ∑n(Si) |
|---|---|---|---|---|---|---|---|
| 1 | 1.5 | 1.5 | — | 1.5 | 3.0 | 6.0 | 1.5 |
| 2 | 1.5 | 1.5 | — | 3.0 | 3.0 | 6.0 | 3.0 |
| 3 | 1.5 | 1.5 | — | 7.5 | 3.0 | 6.0 | 7.5 |
| 4 | 1.5 | 1.2 | 0.6 | 1.5 | 3.0 | 6.0 | 2.1 |
| 5 | 1.5 | 0.9 | 0.9 | 1.5 | 3.0 | 6.0 | 2.4 |
| 6 | 1.5 | 0.6 | 1.2 | 1.5 | 3.0 | 6.0 | 1.7 |
| 7 | 1.5 | 1.2 | 0.6 | — | 3.0 | 6.0 | 0.6 |
| 8 | 1.5 | 0.9 | 0.9 | — | 3.0 | 6.0 | 0.9 |
| 9 | 1.5 | 0.6 | 1.2 | — | 3.0 | 6.0 | 1.2 |
The first set of experiments (#1–3) was aimed at a first proof-of-concept that PI6 and SiO2 can be synthesized simultaneously. ATR-FTIR spectra of all three obtained hybrids show all major modes of PI6 and lack modes of unreacted monomers or end-groups (see the ESI†), as discussed previously and shown in Fig. 2. In addition, we find the presence of the characteristic siloxane (Si–O–Si) asymmetric vibration at 1080 cm−1 (ref. 28) that clearly grows in intensity with increasing molar amount of the supplied TEOS (cf. ESI†). However, we do not find silanol modes, which would be expected at approx. 3500–3000 cm−1 (SiO–H) and approx. 960 cm−1 (Si–OH).28 It should be mentioned that the Si–OH mode at ca. 960 cm−1 would be hard to assign, since it overlaps with several peaks in the fingerprint region of the PI6 backbone. Note that the 960 cm−1 peak as well as the characteristic 800 cm−1 (Si–O bond bending vibration)28 are present in SiO2 that we prepared hydrothermally from TEOS at pH = 4 for comparison (see the ESI†). EDAX further confirms the presence of C, N, O, and Si, in all three materials (see the ESI†). From SEM images, it becomes clear that the morphology of PI6 is retained at all three concentrations of TEOS: as for pure PI6, we find ribbon morphologies (see Fig. 5C and the ESI†). In addition, the presence of spherical nanoparticles (approx. 100–200 nm) that are agglomerated (Fig. 5B) and often cover the PI6 ribbons, especially at the high initial amount of TEOS (nTEOS = 7.5 mmol; exp. #3, SEM see the ESI†) is also evidenced. From SEM it becomes apparent that the number of particles increases with increasing initial TEOS concentration. EDAX evinces that these nanoparticles contain mostly Si and O, which allows for the conclusion that they are composed of SiO2.‡ Furthermore, the diffractograms of all three PI6/SiO2 materials show the presence of all (major and minor) reflections of PI6 (Fig. 5C, and compare Fig. 3A). Moreover, with increasing nTEOS, especially evident for nTEOS = 3.0 mmol and 7.5 mmol, a growing amorphous halo becomes apparent, which we attribute to SiO2. Note that at nTEOS = 1.5 mmol, the diffractogram shows two small but sharp reflections at approx. 30 and 32° 2θ, Cu-Kα (highlighted by orange arrows in Fig. 5A). We speculate that these reflections are related to a small amount (as not apparent from ATR-FTIR spectra) of either unreacted monomers or low-molecular weight oligomers, which might have been enclosed in freshly formed PI6. From the analysis of experiments #1–3, we can draw the conclusions that TEOS, HMDA and PMA can simultaneously undergo polycondensation to a mixture of amorphous, spherical colloidal SiO2 particles and crystalline PI6 ribbons. As expected, the products are mixtures of SiO2 and PI6, which are not covalently bound to each other.
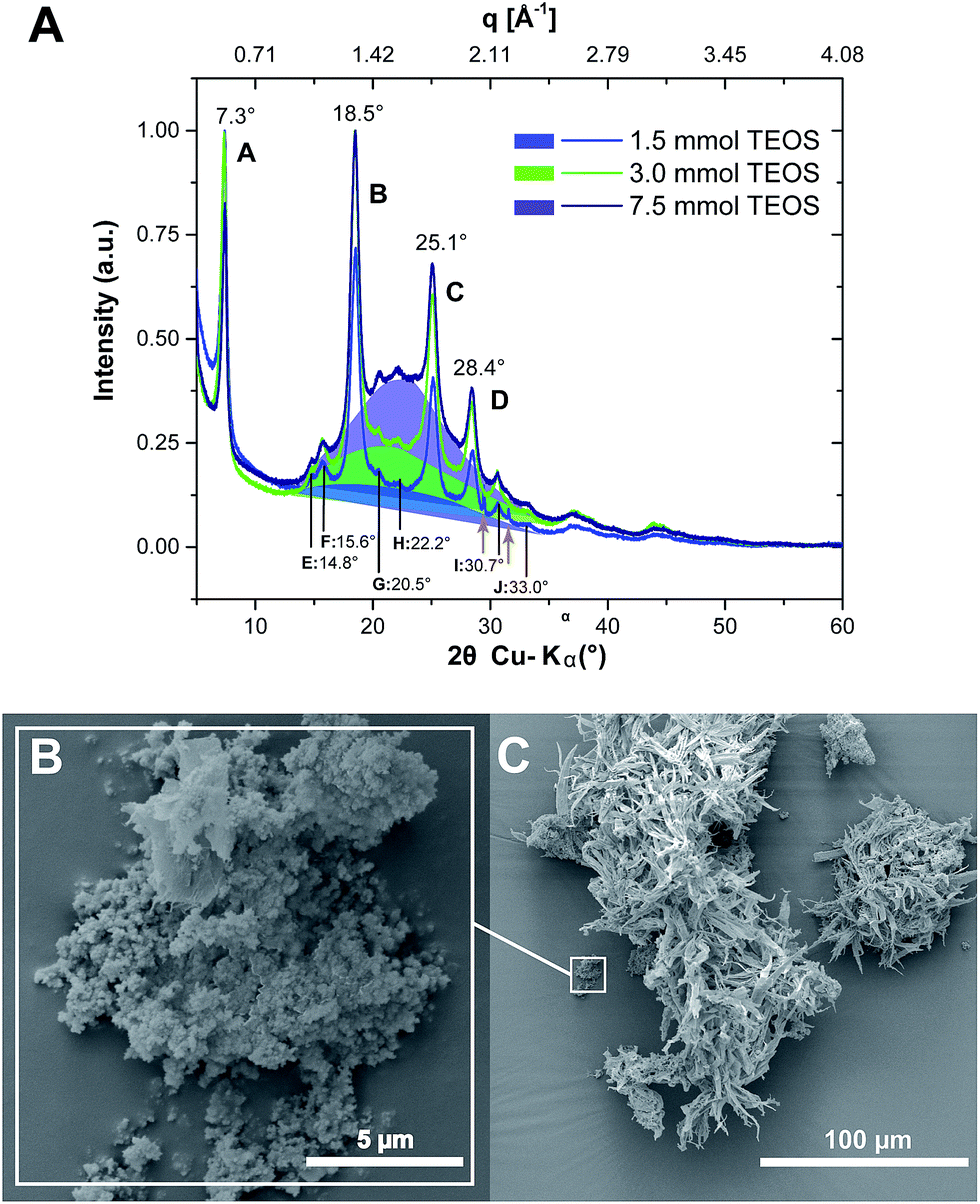 | ||
| Fig. 5 PI6/SiO2 hybrids prepared from HMDA, PMA and TEOS. (A) Diffractograms of PI6/SiO2 prepared using different nTEOS; amorphous backgrounds are highlighted. (B) and (C) SEM images of PI6/SiO2 at nTEOS = 1.5 mmol; SiO2 nanoparticles (B) coexist with PI6 ribbons (C). | ||
In a second set of experiments (#4–6), we employed APTES (Fig. 4) as an additional compatibilizer. APTES has been used as a compatibilizer in several studies on PI/SiO2 hybrids.21,22 On one hand, the presence of Si–OEt moieties allows for hydrolysis to Si–OH, and subsequent co-condensation with Si–OH from hydrolyzed TEOS to Si–O–Si functions. On the other hand, the NH2 functionality of the aminopropyl moiety is able to co-condense with 1,2-dicarboxylic acid moieties of PMA to form imide functions (Fig. 4). Therefore, the use of APTES is known to enable covalent bonding between PI and SiO2 which should allow for evolution from a mere PI/SiO2 mixture to a class II hybrid material (defined as a hybrid in which the different components are connected via covalent or iono-covalent bonds).3 When additionally employing APTES, we adjusted the stoichiometric ratio of reactants such as n(NH2):n(CO2H) = 1:2, which corresponds to equimolarity of amino and 1,2-dicarboxylic acid moieties. This is extremely important, since polyimides are polycondensates obeying Carothers' law, which states that high conversion and degrees of polymerization can only be obtained at ‘ideal’ equimolarity of reacting functions. In experiments employing PMA/HMDA/TEOS/APTES, we tested different nAPTES. Specifically the reactant ratio was adjusted to 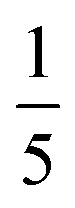 (#4),
(#4), 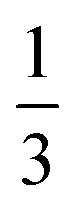 (#5), and
(#5), and 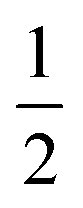 (#6), respectively; imide N atoms would – at full conversion – be covalently bound to SiO2. ATR-FTIR spectra of all three hybrid materials exhibit the presence of imide modes and the Si–O–Si mode, and absence of end-groups or unreacted monomers (see the ESI†). EDAX further underlines the presence of C, N, O and Si in all three samples (see the ESI†). Moreover, we do not find any indication for linking functions besides the imide moiety. Other linking functions that would be conceivable are diamide or amic acid moieties (cf.Fig. 4): no amide modes are observed in ATR-FTIR spectra. PXRD results in completely different patterns than those obtained for PI6/SiO2 mixtures. In all three diffractograms, the minor reflections of PI6 (E–J in Fig. 3) are fully absent (Fig. 6). At the lowest employed APTES amount (nAPTES = 0.6 mmol), the major reflections of pure PI6 (A–D, compare Fig. 6) are clearly visible (Fig. 6A). Note that A–D are situated on top of an amorphous underground, which can be described by two overlapping amorphous halos, centered around approx. 9 and 19° (2θ, Cu-Kα), respectively (orange arrows in Fig. 6). Note that the position of these halos is significantly different from amorphous SiO2 prepared by hydrothermal condensation of TEOS for comparison (grey diffractogram in Fig. 6). Moreover, three new relatively sharp reflections are present (labelled K, L and M in Fig. 6). At nAPTES = 0.9 mmol, K, L and M are also present and found on top of two overlapping amorphous halos, however at changed relative intensities and the major reflections of PI6 (A–D) are fully absent. At nAPTES = 1.2 mmol, no sharp reflections are present anymore – nothing but the two amorphous halos are visible (Fig. 6A, orange arrows). From PXRD results of all three hybrid materials, it becomes clear that the presence of APTES hampers the formation of domains of crystalline PI6, i.e. of the crystal structure observed for pure, hydrothermally prepared PI6. This clearly points at the successful covalent bonding between SiO2 and PI6, especially in comparison to hybrids prepared in the absence of APTES, where the diffractograms reflected a mere mixture of PI6 and SiO2. The presence of some order, as evinced by the new reflections K, L and M, points to segregation in SiO2 and PI6-rich domains, in which the PI6 domains have some degree of ordering that is however different from pure PI6. We believe that this segregation is a consequence of the different polarities (and hence solubilities in high-temperature water) and speeds of condensation of HMDA and PMA, and TEOS and APTES, respectively. Moreover, our experimental set-up, i.e. the use of non-stirred autoclaves (see the ESI†), further supports – or at least does not disturb – the segregation processes. SEM reveals very interesting morphological changes. At nAPTES = 0.6 mmol, spheres of approx. 1–5 μm in diameter coexist with intergrown platelets of approx. 100–200 nm thickness and several microns in lateral extension (Fig. 6B). The amount of spherical objects is rather small with respect to the amount of intergrown platelets (Fig. 6B and ESI†for overview images). At higher amounts of APTES, i.e. nAPTES = 0.6 and 0.9 mmol, approx. 1–5 μm-sized spheres coexist with partly bundled and interwoven bent fibers of ca. 100 nm in diameter and several microns in length. The relative number of spheres with respect to fibers increases with growing nAPTES (Fig. 6B and ESI†). EDAX results show that the relative amount of Si is higher in the spherical objects than in the fibers (see the ESI†). The fact that spherical, Si-rich morphologies coexist with platelets (nAPTES = 0.6 mmol) and fibers (nAPTES = 0.9 mmol and 1.2 mmol) further substantiates the hypothesis of segregated domains in the hydrothermal syntheses of these hybrid materials. Note that there are striking differences in the SiO2-rich morphologies between the absence of APTES and its presence (e.g. #1 vs. #4). While SiO2 nanoparticles form in the absence of APTES, its presence leads to the formation of spherical microparticles. We have currently no explanation for these differences – their understanding would require a more detailed micromorphological study. The factors that might play a role are (i) the decreased apolarity of APTES compared to TEOS (and especially Si(OH)4), (ii) different hydrolysis and condensation rates of APTES vs. TEOS, and (iii) different co-condensation rates of APTES with PI6 vs. APTES with TEOS.
(#6), respectively; imide N atoms would – at full conversion – be covalently bound to SiO2. ATR-FTIR spectra of all three hybrid materials exhibit the presence of imide modes and the Si–O–Si mode, and absence of end-groups or unreacted monomers (see the ESI†). EDAX further underlines the presence of C, N, O and Si in all three samples (see the ESI†). Moreover, we do not find any indication for linking functions besides the imide moiety. Other linking functions that would be conceivable are diamide or amic acid moieties (cf.Fig. 4): no amide modes are observed in ATR-FTIR spectra. PXRD results in completely different patterns than those obtained for PI6/SiO2 mixtures. In all three diffractograms, the minor reflections of PI6 (E–J in Fig. 3) are fully absent (Fig. 6). At the lowest employed APTES amount (nAPTES = 0.6 mmol), the major reflections of pure PI6 (A–D, compare Fig. 6) are clearly visible (Fig. 6A). Note that A–D are situated on top of an amorphous underground, which can be described by two overlapping amorphous halos, centered around approx. 9 and 19° (2θ, Cu-Kα), respectively (orange arrows in Fig. 6). Note that the position of these halos is significantly different from amorphous SiO2 prepared by hydrothermal condensation of TEOS for comparison (grey diffractogram in Fig. 6). Moreover, three new relatively sharp reflections are present (labelled K, L and M in Fig. 6). At nAPTES = 0.9 mmol, K, L and M are also present and found on top of two overlapping amorphous halos, however at changed relative intensities and the major reflections of PI6 (A–D) are fully absent. At nAPTES = 1.2 mmol, no sharp reflections are present anymore – nothing but the two amorphous halos are visible (Fig. 6A, orange arrows). From PXRD results of all three hybrid materials, it becomes clear that the presence of APTES hampers the formation of domains of crystalline PI6, i.e. of the crystal structure observed for pure, hydrothermally prepared PI6. This clearly points at the successful covalent bonding between SiO2 and PI6, especially in comparison to hybrids prepared in the absence of APTES, where the diffractograms reflected a mere mixture of PI6 and SiO2. The presence of some order, as evinced by the new reflections K, L and M, points to segregation in SiO2 and PI6-rich domains, in which the PI6 domains have some degree of ordering that is however different from pure PI6. We believe that this segregation is a consequence of the different polarities (and hence solubilities in high-temperature water) and speeds of condensation of HMDA and PMA, and TEOS and APTES, respectively. Moreover, our experimental set-up, i.e. the use of non-stirred autoclaves (see the ESI†), further supports – or at least does not disturb – the segregation processes. SEM reveals very interesting morphological changes. At nAPTES = 0.6 mmol, spheres of approx. 1–5 μm in diameter coexist with intergrown platelets of approx. 100–200 nm thickness and several microns in lateral extension (Fig. 6B). The amount of spherical objects is rather small with respect to the amount of intergrown platelets (Fig. 6B and ESI†for overview images). At higher amounts of APTES, i.e. nAPTES = 0.6 and 0.9 mmol, approx. 1–5 μm-sized spheres coexist with partly bundled and interwoven bent fibers of ca. 100 nm in diameter and several microns in length. The relative number of spheres with respect to fibers increases with growing nAPTES (Fig. 6B and ESI†). EDAX results show that the relative amount of Si is higher in the spherical objects than in the fibers (see the ESI†). The fact that spherical, Si-rich morphologies coexist with platelets (nAPTES = 0.6 mmol) and fibers (nAPTES = 0.9 mmol and 1.2 mmol) further substantiates the hypothesis of segregated domains in the hydrothermal syntheses of these hybrid materials. Note that there are striking differences in the SiO2-rich morphologies between the absence of APTES and its presence (e.g. #1 vs. #4). While SiO2 nanoparticles form in the absence of APTES, its presence leads to the formation of spherical microparticles. We have currently no explanation for these differences – their understanding would require a more detailed micromorphological study. The factors that might play a role are (i) the decreased apolarity of APTES compared to TEOS (and especially Si(OH)4), (ii) different hydrolysis and condensation rates of APTES vs. TEOS, and (iii) different co-condensation rates of APTES with PI6 vs. APTES with TEOS.
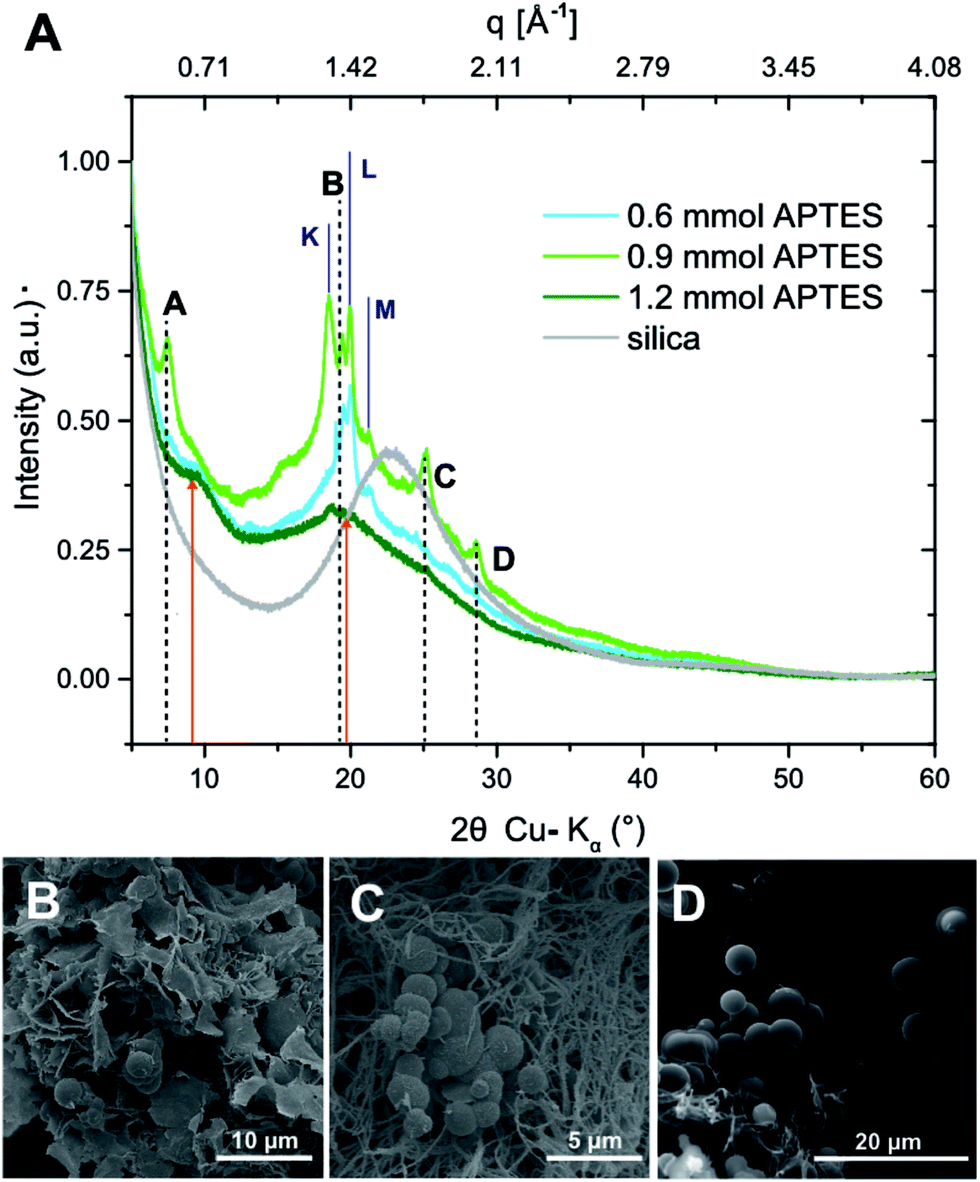 | ||
| Fig. 6 PI6/SiO2 prepared from PMA, HMDA, APTES and TEOS. (A) Diffractograms of the hybrids prepared at different nAPTES, and diffractogram of hydrothermally synthesized SiO2 for comparison (gray). (B)–(D) SEM images of the corresponding hybrids at nAPTES = 0.6 mmol (B), nAPTES = 0.9 mmol, and nAPTES = 1.2 mmol. | ||
In a last set of experiments (#7–9, Table 1), we investigated the co-condensation of PMA, HMDA and APTES in the absence of TEOS. ATR-FTIR spectra show both characteristic imide and siloxane modes, and the absence of modes related to potential end-groups or unreacted monomeric species (see the ESI†). Note that as seen for the previous set of experiments (#4–6), the intensity of Si–O–Si modes increases with growing amount of APTES, which is expected. As for experiments in the presence of APTES and TEOS, we do not find any indication for amide (1,2-diamide or 1-amide-2-carboxylic acid) linkages. PXRD patterns show the same trends as observed in samples #4–6: at the lowest nAPTES (0.6 mmol), we still find the major reflections A–D of PI6, in coexistence with the new reflections K and L, and the previously observed two amorphous halos (see the ESI†). Furthermore, the morphologies observed from SEM images show features as noted for the previous series of hybrids (#4–6): (i) in all three samples #7–9, we find the coexistence of interwoven, bundled fibers with spherical particles of approx. 5 μm in diameter; and (ii) the number of spheres relative to fibers increases with increasing nAPTES (see the ESI†). EDAX shows that the spherical morphologies are by far richer in Si than in the fibers. From the characterization of hybrids in the synthesis of which APTES was used (#4–9), we can conclude that APTES allows for covalently linking SiO2 and PI6, as underlined by the presence of imide and Si–O–Si modes, and the absence of monomer/end-group-related modes in ATR-FTIR spectra. Moreover, employing APTES has important consequences for crystallinity and morphology, while an impact of TEOS on both features is not detected.
2.3 Upscaling of the hydrothermal PI6/SiO2 synthesis and solvent-free processing by sintering
To this point, we have shown that PI6 and SiO2 can be synthesized simultaneously in nothing but hot water from the comonomers HMDA, PMA and the silica source TEOS. Moreover we showed that the well-known compatibilizer APTES also efficiently links PIs and SiO2 under hydrothermal synthesis conditions. However, both crystallinity and morphology of PI6 and SiO2 are strongly influenced when APTES is employed, which is supposed to be additionally supported by using non-stirred autoclaves where the absence of mechanically mixing the reaction mixtures reinforces segregation phenomena.In a last set of experiments, we investigated the upscaling of the hydrothermal PI6/SiO2 synthesis, as well as the processing of the obtained powders into macroscopic objects, specifically into pellets, by solvent-free sintering. Scaled-up syntheses were performed in a 1 L stirred batch autoclave (see the ESI† for the experimental set-up). The reactor was filled with a dispersion containing HMDA, PMA, TEOS and APTES in 400 mL H2O. The used concentrations and reactant ratios were the same as in experiment #5 (cf.Table 1), with resulting employed molar amounts of nPMA = 40 mmol, nHMDA = 24 mmol, nAPTES = 32 mmol, and nTEOS = 32 mmol. Therefore, the PI6/SiO2 synthesis was effectively scaled up by a factor of 26.![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) . For comparison, we also scaled up the HTP of pure PI6 (see the ESI†for experimental details). As for the small scale experiments, hydrothermal syntheses were run for 24 h at 200 °C and the autogenous pH = 4. As for the smaller scale experiments, pure PI6 and PI6/SiO2 were obtained as white powders. ATR-FTIR spectra show the presence of imide modes and lack modes related to unreacted monomers/end-groups for upscaled PI6 – an overlay of the spectra of PI6 from small scale synthesis and upscaled PI6 shows virtually no differences in spectra (see the ESI†). For upscaled PI6/SiO2, we also find both imide and Si–O–Si modes, and the absence of unreacted end-groups/monomers in the ATR-FTIR spectrum. In fact, the spectrum perfectly coincides with the one of the corresponding small scale synthesis (ESI†). PXRD of upscaled PI6 generates exactly the same diffractogram as in the small scale: all major and minor reflections of PI6 are present (see Fig. 7A). For the upscaled hybrid material there are slight differences with respect to the small scale PI6/SiO2. While only three of the four major PI6 reflections persist (B, C, D), the new reflections K, L, M are all present (see Fig. 7B). Moreover, the amorphous underground is now composed of only one halo centered around 19° (2θ, Cu-Kα). In addition, the reflections are less intense and sharper than for the small scale synthesis. This underlines the effect of stirring, leading to less segregation into PI6 and SiO2-rich domains, respectively, and therefore less crystallinity. SEM analysis reveals that the upscaled pure PI6 is still composed of ribbon structures (see Fig. 7E). However, they are not covered with smaller ribbons (which was the case for small scale PI6, cf.Fig. 3), and seem less globally oriented (see Fig. 7F). The upscaled PI6/SiO2 shows two coexisting morphologies: fibers and spherical particles of approx. 1 μm in diameter. Compared to the small scale PI6/SiO2, the upscaled sample is more homogeneous and the observed spherical particles are smaller and less disperse in size. This again underlines the effect of stirring. EDAX of the upscaled PI6/SiO2 evinces that the spherical objects are richer in Si than the fibers (see the ESI†).
. For comparison, we also scaled up the HTP of pure PI6 (see the ESI†for experimental details). As for the small scale experiments, hydrothermal syntheses were run for 24 h at 200 °C and the autogenous pH = 4. As for the smaller scale experiments, pure PI6 and PI6/SiO2 were obtained as white powders. ATR-FTIR spectra show the presence of imide modes and lack modes related to unreacted monomers/end-groups for upscaled PI6 – an overlay of the spectra of PI6 from small scale synthesis and upscaled PI6 shows virtually no differences in spectra (see the ESI†). For upscaled PI6/SiO2, we also find both imide and Si–O–Si modes, and the absence of unreacted end-groups/monomers in the ATR-FTIR spectrum. In fact, the spectrum perfectly coincides with the one of the corresponding small scale synthesis (ESI†). PXRD of upscaled PI6 generates exactly the same diffractogram as in the small scale: all major and minor reflections of PI6 are present (see Fig. 7A). For the upscaled hybrid material there are slight differences with respect to the small scale PI6/SiO2. While only three of the four major PI6 reflections persist (B, C, D), the new reflections K, L, M are all present (see Fig. 7B). Moreover, the amorphous underground is now composed of only one halo centered around 19° (2θ, Cu-Kα). In addition, the reflections are less intense and sharper than for the small scale synthesis. This underlines the effect of stirring, leading to less segregation into PI6 and SiO2-rich domains, respectively, and therefore less crystallinity. SEM analysis reveals that the upscaled pure PI6 is still composed of ribbon structures (see Fig. 7E). However, they are not covered with smaller ribbons (which was the case for small scale PI6, cf.Fig. 3), and seem less globally oriented (see Fig. 7F). The upscaled PI6/SiO2 shows two coexisting morphologies: fibers and spherical particles of approx. 1 μm in diameter. Compared to the small scale PI6/SiO2, the upscaled sample is more homogeneous and the observed spherical particles are smaller and less disperse in size. This again underlines the effect of stirring. EDAX of the upscaled PI6/SiO2 evinces that the spherical objects are richer in Si than the fibers (see the ESI†).
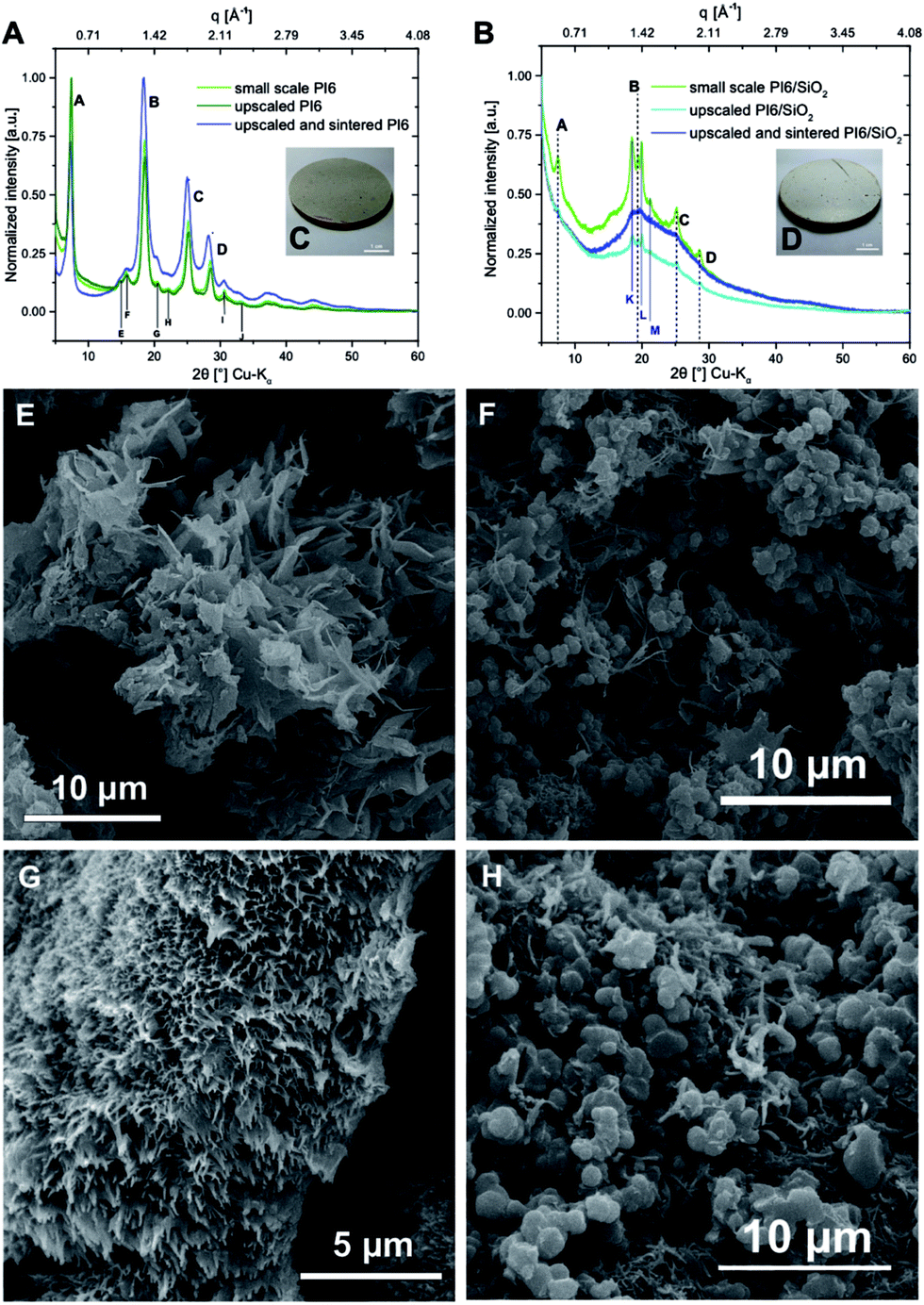 | ||
| Fig. 7 PI6 and PI6/SiO2 upscaling and sintering. (A) and (B) Comparison of the diffractograms of small scale synthesis, upscaled synthesis and material after sintering (PI6: (A) PI6/SiO2: (B)). (C) and (D) Photographs of the PI6 (C), and the PI6/SiO2 pellet from sintering. (E)–(H) SEM images of upscaled PI6 before (E) and after (G) sintering, and of upscaled PI6/SiO2 before (F) and after (H) sintering. | ||
From the upscaled syntheses, we obtained ca. 10 g of PI6 and PI6/SiO2 respectively. 5 g of each was used to prepare pellets by sintering in a hot press, aka compression molding, a technique well suited for the compaction of polymer-based particulate composites.29 Sintering of polyimides is an established technique, which is conventionally used to process semicrystalline polyimides such as Kapton™.30,31 Moreover, sintering techniques have been employed for preparing PI composites with e.g. graphite flakes or microsilica as dispersed reinforcement.32,33 Before sintering, the samples were first finely ground using a cryo-ball mill where powder freezing was performed using liquid N2. Compression molding was subsequently performed for 2 h at 300 °C applying a pressure of 80 MPa, yielding cylindrical specimens with a diameter of 40 mm and a height of around 2.5 mm (see the ESI† for information on the experimental set-up). Photographs of the pure PI6 pellet and the PI6/SiO2 pellet are shown in Fig. 7C and D. Both pellets were shiny and reflective, which is a first indication that (i) the samples had crept during pressing, and (ii) that successful compaction had taken place. Note that the color of both (however more pronounced for the PI6 pellet) changed from white to off-white/grayish. While one might suspect thermo-oxidative phenomena to be responsible for the darkening, ATR-FTIR analysis of the pellets shows spectra that are unchanged as compared to pristine upscaled PI6 and PI6/SiO2, respectively (see the ESI†). Hence, we conclude that the sintering procedure did not lead to major chemical changes. Instead, we speculate that the compaction could have generated weak charge-transfer complexes, e.g. between the electron poor aromatic ring in PI6 and a CO function of a neighboring chain, by bringing the polymer chains closer together. PXRD analysis of the pellets shows that the average order in both materials is retained. The diffractogram of the PI6 pellet shows all major and minor PI6 reflections. However, the reflections are less sharp than before sintering. The diffractogram of the PI6/SiO2 pellet has changed more significantly. The overall shape of the diffraction curve is retained as compared to PI6/SiO2 before sintering. However, the reflections K, L, M and B, C, D are barely visible anymore. SEM analysis of the PI6 pellets (Fig. 7G and the ESI†) shows major morphological changes. The pellet is composed of highly parallel fibers having near-circular cross-sections of 100–200 nm in diameter. For the compactness of the sample, it is difficult to comment on the length of the fibers. Given these important morphological changes, which further corroborate that the sample crept efficiently, it is even more impressive that the crystallinity of PI6 is retained. The PI6/SiO2 pellet shows less pronounced morphological changes than PI6. While the sample is much denser than before sintering and shows similar fibrous structures as the sintered PI6 pellet, the spherical SiO2-rich particles have not merged with the fibrous structures and with each other (Fig. 7H). We believe that this result is a consequence of the sintering temperatures being too low for effective consolidation of the SiO2-containing compound. Indeed, SiO2 sintering is typically carried out at temperatures around 1000 °C.34 Finally, we additionally investigated the chemical nature of upscaled PI6 and PI6/SiO2 hybrids before and after sintering via solid-state magic angle spinning nuclear magnetic resonance spectroscopy (MAS NMR). Specifically, we performed 13C, 15N and 29Si MAS NMR of all four samples (see the ESI† for measurement details). It becomes clear from comparing the spectra that there is virtually no chemical difference between PI6 and PI6/SiO2 before and after sintering (see the ESI† for all MAS NMR spectra). Representative 13C, 15N and 29Si MAS NMR spectra of PI6 and PI6/SiO2 pellets (after sintering) are depicted in Fig. 8. The 13C MAS NMR spectrum of PI6 (Fig. 8A) allows for clear assignment of all carbon atoms. The carbonyl C (labelled A) of the imide ring is the most intense signal found at 160.19 ppm, the aromatic C-atom signals are found at 130.16 ppm (B) and 112.76 ppm (C), the aliphatic C-atoms in α to the imide nitrogen are found at 32.36 ppm (D), and the C-atoms in β and γ give rise to a signal at 20.80 ppm. These shifts are in agreement with the literature.35 Note that the signals attributed to A, B and C give rise to symmetrical spinning sidebands (labelled A*, B* and C*), which is a consequence of the crystallinity of PI6. The 13C MAS NMR spectrum of PI6/SiO2 (Fig. 8B) from sintering shows all the same peaks as pure PI6. Moreover, we find one additional signal in the aliphatic region assigned to the C-atom in α to Si (F), hence arising from covalent binding of APTES to a polyimide segment. Note that the 13C MAS NMR spectrum of PI6/SiO2 also shows spinning side bands. This underlines our previous conclusions drawn from PXRD on PI6/SiO2 still showing higher ordering/crystallinity than a fully amorphous sample. 15N MAS NMR of both PI6 and PI6/SiO2 shows one single signal at 127.18 ppm (PI6) and 127.35 ppm (PI6/SiO2) assigned to the imide nitrogen atom (see the ESI†). From both 13C and 15N MAS NMR, we can conclude that neither unreacted NH2 (or NH3+), nor amide moieties are present. The 29Si MAS NMR spectrum of PI6/SiO2 (Fig. 8C) shows the presence of both Q4 (Si in bulk silica, i.e. Si connected to four OSi groups) and Q3/Q2 (Si connected to three/two OSi groups and one/two OH groups), which arise from the condensation of TEOS. Moreover, we find a strong T band (Si connected to three SiOH/SiOR groups and to one alkyl moiety), which arises from the condensation of APTES. Most interestingly, the observed T band is accompanied by two symmetrical spinning side bands, which can only arise in a (at least to some extent) crystalline sample. Therefrom, we can conclude that Si is covalently linked to crystalline/ordered PI6 chains. Note that a first conjecture that the peak at ca. 0 ppm might be due to a D band (two alkyl moieties), which could arise from APTES containing some extent of di-(3-aminopropyl)diethoxysilane, was disproved by 29Si NMR of the used APTES (see the ESI†). The chemical shifts of T and Q signals are in agreement with the literature.3629Si MAS NMR clearly shows that OH groups are still present in PI6/SiO2. These might arise from incomplete condensation of hydrolyzed APTES or TEOS, but could also fully arise from surfacial Si–OH.36 The observed T signal cannot be so clearly assigned as to conclude if nonhydrolyzed OEt groups are still present or not. However, we would expect to find corresponding signals in 13C MAS NMR in this case. Lastly, neither of the MAS NMR experiments of the sintered materials gives an indication of the side-products of potential thermo-oxidation.
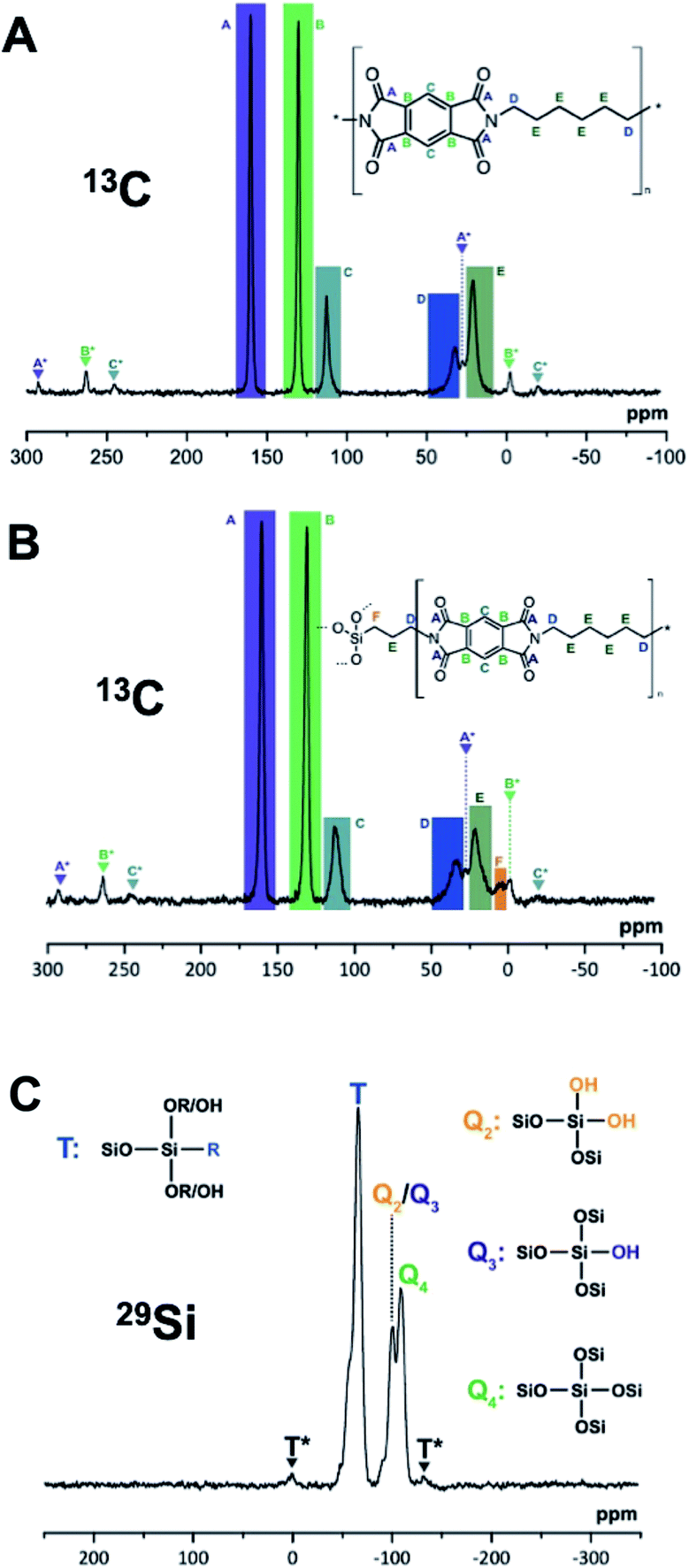 | ||
| Fig. 8 Solid-state MAS NMR spectra. Spinning side-bands are labelled with *. (A) 13C spectrum of PI6 after sintering. (B) 13C spectrum of PI6/SiO2 after sintering. (C) 29Si spectrum of PI6/SiO2 after sintering. | ||
3 Conclusion
In this contribution, we reported the (i) first example of the hydrothermal synthesis of a semialiphatic polyimide, PI6; (ii) the first simultaneous hydrothermal synthesis of an inorganic–organic hybrid material, PI6/SiO2, including its upscaling and (iii) the solvent-free processing of both PI6 and PI6/SiO2via hot pressing. Our investigations show that PI6 and SiO2 are obtained as a mixture when synthesized from the comonomers HMDA and PMA, and TEOS. As the resulting product is a mere mixture of the polyimide and silica, neither the crystallinity nor morphology of either of the components is influenced. Moreover, the organically modified silicon alkoxide APTES can be employed hydrothermally as a compatibilizer. The addition of APTES efficiently allows for generating class II hybrid materials, but hampers the crystallinity of the PI6 phase and has major morphological implications. The more APTES is added, the more PI6 phases change to fibrous morphologies, and SiO2 phases change from nanoparticles to colloidal particles of 1–5 μm. Both the hydrothermal PI6 and PI6/SiO2 synthesis were upscaled from an initial reaction volume of 15 mL to 400 mL using a stirred 1 L batch reactor. Stirring has a negative effect on the morphology of pure PI6, which shows more features indicating crystallinity from non-stirred syntheses, but has a positive effect on the homogeneity of PI6/SiO2. PI6 and PI6/SiO2 from upscaling were used for processing experiments. These first results of PI6 and PI6/SiO2 sintering show that (i) PI6 can be nicely compacted with retention of the initial crystallinity, and (ii) that PI6/SiO2 hot pressing results in densification but lacks sufficient creep of the SiO2 particles. This is attributed to the fact that the employed sintering temperature of 300 °C is most likely too low for silica. Beyond this first proof of concept, further optimization is necessary and will be performed in the near future. All hybrid materials were fully characterized using ATR-FTIR, SEM, EDAX, PXRD and solid-state MAS NMR. The results of these analyses strongly support the drawn conclusions.Both the synthesis of PI6 and PI6/SiO2 can be qualified as green, as solely water is used aside the starting compounds. Moreover, conventional hydrothermal SiO2 syntheses demand for the use of adding mineral acids or bases as condensation promotors. In the reported one-pot route, additional promotors are not necessary, since the comonomer PMA provides sufficient acidity. Processing by sintering also qualifies as green, since no solvents are used at all. As we show in this contribution with the example of PI6/SiO2, hydrothermal synthesis is a potent means of synthesizing inorganic–organic hybrid materials in one pot.
Acknowledgements
The authors acknowledge TU Wien and the Austria Wirtschaftsservice (aws), project no. P1504501 for partially funding this project, and thank Heribert Gerstner and Ana Subotic for preliminary experiments. Powder X-ray diffraction measurements were carried out at the X-ray Center of TU Wien (XRC), and SEM was carried out at the interfaculty electron microscopy facility of TU Wien (USTEM). Werner Artner (XRC) is especially acknowledged for experimental support of the PXRD measurements. The authors acknowledge the TU Wien University Library for financial support through its Open Access Funding Program.References
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed.
- M. M. Unterlass, Eur. J. Inorg. Chem., 2016, 8, 1135–1156 CrossRef.
- P. Judeinstein and C. Sanchez, J. Mater. Chem., 1996, 6, 511–525 RSC.
- U. Schubert and N. Hüsing, Synthesis of Inorganic Materials, John Wiley & Sons, 2012 Search PubMed.
- B. Baumgartner, M. J. Bojdys and M. M. Unterlass, Polym. Chem., 2014, 5, 3771–3776 RSC.
- M. M. Unterlass, Mater. Today, 2015, 5, 242–243 CrossRef.
- B. Baumgartner, M. Puchberger and M. M. Unterlass, Polym. Chem., 2015, 6, 5773–5781 RSC.
- B. Baumgartner, M. J. Bojdys, P. Skrinjar and M. M. Unterlass, Macromol. Chem. Phys., 2016, 217, 485–500 CrossRef CAS.
- A. Rabenau, Angew. Chem., Int. Ed. Engl., 1985, 24, 1026–1040 CrossRef.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- J. Preston and Y. Tropsha, Polym. Eng. Sci., 1994, 34, 305–307 CAS.
- M. Hasegawa and K. Horie, Prog. Polym. Sci., 2001, 26, 259–335 CrossRef CAS.
- K. A. Watson, F. L. Palmieri and J. W. Connell, Macromolecules, 2002, 35, 4968–4974 CrossRef CAS.
- M. Putkonen, J. Harjuoja, T. Sajavaara and L. Niinistö, J. Mater. Chem., 2007, 17, 664–669 RSC.
- S. Watanabe, A. Wakino, T. Namikoshi and M. Murata, High Perform. Polym., 2012, 24, 710–716 CrossRef.
- X. D. Huang, S. M. Bhangale, P. M. Moran, N. L. Yakovlev and J. Pan, Polym. Int., 2003, 52, 1064–1069 CrossRef CAS.
- T. Lee, S. S. Park, Y. Jung, S. Han, D. Han, I. Kim and C.-S. Ha, Eur. Polym. J., 2009, 45, 19–29 CrossRef CAS.
- K. Biradha and M. Zaworotko, Cryst. Eng., 1998, 1, 67–78 CrossRef CAS.
- D. Mrvos-Sermek, Z. Popovic and D. Matkovic-Calogovic, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 2538–2541 CrossRef.
- U. Schubert, N. Hüsing and R. Laine, Materials Syntheses: A Practical Guide, Springer Science, 2008 Search PubMed.
- Y. Chen and J. O. Iroh, Chem. Mater., 1999, 11, 1218–1222 CrossRef CAS.
- S. Kim, S. Ando and X. Wang, RSC Adv., 2015, 5, 40046–40054 RSC.
- B.-K. Chen, T.-M. Chiu and S.-Y. Tsay, J. Appl. Polym. Sci., 2004, 94, 382–393 CrossRef CAS.
- H. U. Rehman, H. Schmidt and Z. Ahmad, J. Macromol. Sci., Part A: Pure Appl. Chem., 2006, 43, 703–717 CrossRef CAS.
- J.-S. Im, J.-H. Lee, S.-K. An, K.-W. Song, N.-J. Jo, J.-O. Lee and K. Yoshinaga, J. Appl. Polym. Sci., 2006, 100, 2053–2061 CrossRef CAS.
- A. Morikawa, Y. Iyoku, M.-A. Kakimoto and Y. Imai, J. Mater. Chem., 1992, 2, 679–689 RSC.
- J. Guo, B. N. Nguyen, L. Li, M. A. B. Meador, D. A. Scheiman and M. Cakmak, J. Mater. Chem. A, 2013, 1, 7211–7221 CAS.
- J. Musgo, J. C. Echeverría, J. Estella, M. Laguna and J. J. Garrido, Microporous Mesoporous Mater., 2009, 118, 280–287 CrossRef CAS.
- T. Konegger, A. Liersch, C. Gierl and M. Scheffler, Adv. Eng. Mater., 2013, 15, 394–406 CrossRef CAS.
- J. L. Throne, Adv. Polym. Technol., 1989, 9, 281–291 CrossRef CAS.
- P. Samyn and G. Schoukens, Eur. Polym. J., 2008, 44, 716–732 CrossRef CAS.
- P. Samyn and G. Schoukens, Carbon, 2008, 46, 1072–1084 CrossRef CAS.
- X.-R. Zhang, X.-Q. Pei and Q.-H. Wang, Mater. Des., 2009, 30, 4414–4420 CrossRef CAS.
- T. Vasilos, J. Am. Ceram. Soc., 1960, 43, 517–519 CrossRef CAS.
- A. Martínez-Richa and R. Vera-Graziano, J. Appl. Polym. Sci., 1998, 70, 1053–1064 CrossRef.
- K. Albert and E. Bayer, J. Chromatogr. A, 1991, 544, 345–370 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials & methods, additional ATR-FTIR, SEM, EDAX, and PXRD results. See DOI: 10.1039/c7ta02498c |
| ‡ For the resolution limit of EDAX being situated at ≈1 μm, we ensured that we always analyzed large agglomerates of the nanoparticles, such as those depicted in Fig. 5B. |
| This journal is © The Royal Society of Chemistry 2017 |