
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired electrocatalysts for oxygen reduction using recombinant silk films†
Trevor D.
Rapson
 *a,
Ryo
Kusuoka
b,
Joseph
Butcher
cd,
Mustafa
Musameh
e,
Christopher J.
Dunn
e,
Jeffrey S.
Church
f,
Andrew C.
Warden
a,
Christopher F.
Blanford
dg,
Nobuhumi
Nakamura
b and
Tara D.
Sutherland
a
*a,
Ryo
Kusuoka
b,
Joseph
Butcher
cd,
Mustafa
Musameh
e,
Christopher J.
Dunn
e,
Jeffrey S.
Church
f,
Andrew C.
Warden
a,
Christopher F.
Blanford
dg,
Nobuhumi
Nakamura
b and
Tara D.
Sutherland
a
aCSIRO, Black Mountain, Canberra, ACT 2601, Australia. E-mail: trevor.rapson@csiro.au
bTokyo University of Agriculture and Technology, Department of Biotechnology and Life Science, Koganei, Tokyo 184-8588, Japan
cSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
eCSIRO, Clayton, Melbourne, VIC 3168, Australia
fCSIRO, Waurn Ponds, Geelong, VIC 3216, Australia
gSchool of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 8th May 2017
Abstract
Fuel cells are a promising avenue for renewable energy production. While oxygen remains the preferred oxidant, its slow reduction kinetics has limited fuel cell performance and it currently requires the use of platinum as the cathode catalyst. In the search for non-platinum cathodes, inspiration has been sought from biological oxygen reduction processes which use heme proteins for respiration. Here, we describe the use of recombinant honeybee silk protein, which can be produced at high scale in E. coli, to generate a heme–protein material. In these solid-state silk materials, a tyrosine residue coordinates directly to the heme iron center. This axial coordination promotes heterolytic O–O bond cleavage, rather than homolytic cleavage, avoiding the generation of destructive hydroxyl radicals. The heme–silk materials can fully reduce oxygen to water with 3.7 electrons transferred to oxygen and only 14% hydrogen peroxide produced. Importantly, the films demonstrate remarkable stability. The films retained activity when used under continuous operation for over 16 hours and retained 85% of their catalytic activity when used at pH 3 for two hours.
Introduction
The electrochemical oxygen reduction reaction for fuel cells continues to stimulate a large amount of research.1–4 The two main reasons as to why oxygen remains the preferred cathodic fuel are its abundance in the earth's atmosphere and the fact that its fully reduced form, water, is non-toxic.5 The performance of fuel cells, however, is limited by the slow reaction kinetics of oxygen reduction. To overcome this limitation, platinum is commonly used as the cathode catalyst.5,6 The expense and scarcity of platinum are two of the major factors limiting the widespread use of fuel cells. Therefore, there is an urgent need for cost-effective and readily available electrodes for oxygen reduction.6 The ideal oxygen reduction catalyst reduces oxygen, in a four electron reaction, directly to water. Such a four electron reduction allows maximum output power, while avoiding the two electron reduction reaction that produces hydrogen peroxide, which would damage the electrodes.1,5More than a billion years before scientists became interested in the oxygen reduction reaction, biological systems developed their own strategies to reduce oxygen.7 Initially, oxygen reduction was used by anaerobes as a detoxification mechanism but more recently, aerobes evolved the ability to use oxygen reduction as a form of energy metabolism in aerobic respiration.6
Biological oxygen reduction is predominantly carried out by proteins containing a heme cofactor,4,6 such as cytochrome c oxidase8–11 and cytochrome bd oxidase.12 For this reason, metalloporphyrins13–16 and related metal complexes such as phthalocyanines17 and corroles18 continue to be of significant interest in fuel cell research. While these studies have provided a greater understanding into the mechanism of biological oxygen reduction, the poor stabilities of these metal macrocycles have prevented their use in fuel cells.6,9,17 One method of improving the stability of metal-macrocycle catalysts is to immobilize them in a matrix.6 The choice of immobilization medium can play an important role in the selectivity of oxygen reduction.15,17
Recently, we reported a new class of biologically inspired materials known as solid-state metalloproteins.19,20 In these systems, heme b (iron protoporphyrin IX) can be immobilized within honeybee silk materials. The heme iron center is coordinated by a tyrosine residue in the silk.19,20 This axial ligation to heme b is of particular interest to this work as amino acid ligands are known to facilitate O–O bond heterolysis, required for reduction of oxygen to water.6 The heme–silk materials have remarkable stability,21 retaining their function when stored dry and at room temperature for over a year. The stability of these materials suggests that heme–silk films could be promising for producing bioelectrodes.
Protein film voltammetry, in which redox proteins are immobilized on the surface of an electrode, has emerged as a powerful technique to gain both fundamental insights into the functioning of redox proteins22–25 and the development of third generation biosensors and biofuel cells.26–28 By immobilizing the protein on an electrode, sluggish diffusion of the protein to the electrode is avoided and sub-picomole amounts of protein can be used.29
One of the challenges in protein film voltammetry is to develop a stable film in which the functional properties of the protein are maintained.30,31 Here, we use recombinant honeybee silk (AmelF3 produced in E. coli)32,33 to make a stable protein film for protein film voltammetry. Following the incorporation of heme groups, the electrochemical properties of the films are investigated to determine if they are suitable for use as alternative non-platinum electrocatalysts for oxygen reduction.
Experimental
Cyclic voltammetry was carried out using a BAS Epsilon potentiostat with a C3 cell stand at room temperature. A three-electrode system was employed comprising a modified glassy carbon electrode as working electrode, Pt wire counter and Ag|AgCl|saturated NaCl reference electrode. Rotating ring disk electrode measurements (RRDE) were conducted using an ALS Model 702B electrochemical analyzer (ALS Inc., Tokyo, Japan) equipped with a rotation apparatus (RRDE-3A, ALS Inc., Tokyo, Japan). All potentials are quoted versus the normal hydrogen electrode (NHE), calculated as Ag/AgCl + 196 mV.Heme–silk electrodes were prepared, firstly by polishing a glassy carbon electrode (BASi, West Lafayette, USA) as per the manufacturer's instructions, using 0.05 μm alumina polish, followed by sonication for 5–10 minutes in milliQ water. Carbon nanotubes (CNT – short 0.5–2 μm MWCNTs, surface area > 100 m2 g−1, Nanostructures and Amorphous Materials Inc, Houston, USA) dispersed in dimethylformamide (1 mg mL−1) were placed on to the electrode. Following air drying at room temperature for 2 hours, the CNT-modified electrode was heated in an oven at 50 °C for 1 hour to remove any remaining DMF.
Recombinant honeybee silk (AmelF3; NCBI accession no: NP_001129680) was produced as described by Sutherland and co-workers.32 A freeze-dried sample of AmelF3 was dissolved in milliQ water (10 μL of 10 mg mL−1 solution) and placed on the MWCNT-modified electrode and the drop was air-dried overnight. Heme was incorporated into the silk films by soaking the electrodes in a solution of 0.5 mg mL−1 hemin (Frontier Scientific) in 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 v/v methanol:water solution for 18–24 hours. Excess heme was rinsed off the electrode by placing the electrode in 70% methanol:water for 3 minutes with gentle agitation. The electrode was then air-dried and stored at room temperature ready for use.
:30 v/v methanol:water solution for 18–24 hours. Excess heme was rinsed off the electrode by placing the electrode in 70% methanol:water for 3 minutes with gentle agitation. The electrode was then air-dried and stored at room temperature ready for use.
Film thicknesses were measured by surface profilometry (Dektak, Veeco) on films of the different compositions that had been dropcast onto microscope cover slips. Film thicknesses were estimated by obtaining the surface profile of the probe tip scanned across scratches made in the film with a needle tip, profiles of the edges of dropcast films gave an indication of film homogeneity and of drying behaviours.
Results and discussion
Electrode preparation
The initial goal of this work was to obtain direct electron transfer between the heme centers and the electrode when the heme was incorporated into a silk film cast on the electrode. Achieving direct electron transfer in protein film voltammetry is not a trivial matter and a large number of electrode modifications have been employed to assist this process.34,35We decided to use CNTs to modify a glassy carbon electrode given the former's good electrical conductivities, large surface area and commercial availability.36 Following modification of the glassy carbon electrode with nanotubes, a silk film was drop-cast on top of the electrode and heme was leached into the film (Fig. 1A). Through this process the heme becomes incorporated into the protein and an amino acid from the silk coordinates to the heme iron center, thereby producing a silk material which functions as a mimic of naturally occurring heme proteins.19,20
 | ||
| Fig. 1 (A) Cartoon representation of the heme–silk film cast on a glassy carbon electrode modified with carbon nanotubes. (B) SEM images of a multiwalled carbon nanotube (MWCNT) film. (C) SEM image of a MWCNT film covered with a heme–silk film. | ||
The drop-cast films were found to be approximately 5 μm thick with significant variation between films, ranging from 5–8 μm. In addition, the films had a thicker outer region due to the coffee ring effect,37 similar to what we observed previously for drop-cast films.21 Scanning electron microscopy (SEM) was used to analyze both the CNT layer used to modify the glassy carbon electrode and the heme–silk film covering the carbon nanotube layer. SEM imaging showed that the silk film completely covers the CNT layer (Fig. 1B and C) and that no heme is observed on the surface of the silk (ESI Fig. 1†).
To determine the amount of heme incorporated into the film, quartz crystal microbalance with dissipation monitoring (QCM-D) was employed (see ESI†). QCM-D showed that overnight soaking and subsequent washing led to an approximately 20% increase in film mass. In addition, leaching of heme into the films stiffened the adlayer and increased its ellipsometric thickness by about 7%. The stiffening is similar to that observed upon crosslinking38,39 and is consistent with the irreversible incorporation of heme into the silk protein using 70% methanol.19,40
Non-catalytic voltammetry
To determine if electron transfer was occurring from the heme center to the glassy carbon electrode, cyclic voltammetry was used. Oxygen was removed from the electrochemical buffer solution by purging with argon gas to avoid interference from the oxygen reduction reaction. Under these anaerobic conditions, a pronounced reversible peak was observed with a formal potential (E1/2) of −165 mV, ΔE −62 mV (Fig. 2A). No peaks were observed in control experiments without heme (Fig. 2A). The redox potential of this voltammetric response is similar to that noted for other heme protein systems and is consistent with the Fe3+/Fe2+ redox couple.8,41 No redox peaks were observed when a heme–silk film was cast on an unmodified electrode. This confirmed that in the presence of CNT, we had achieved direct electron transfer from the heme iron to the electrode. | ||
| Fig. 2 Voltammetric response from heme–silk films in a degassed 50 mM phosphate buffer (pH 7.4). (A) Cyclic voltammograms of a silk film electrode with heme (solid line) compared to without heme (dashed line); scan rate, 20 mV s−1. (B) The effect of scan rate (mV s−1) on the voltammetric response from heme–silk films. Inset, plot of peak current (ip) vs. scan rate ip - peak current, ic - capacitance. | ||
The scan rate in voltammetry is a useful experimental parameter that can be varied to investigate the nature and kinetics of electron transfer.34 The peak current (ip) was found to be proportional to the scan rate and not to the square root of the scan rate (Fig. 2B). This result is consistent with a redox couple that is adsorbed on the electrode42 and demonstrates that the heme was bound within the silk film and electron transfer was achieved within the silk–heme matrix, despite the insulating nature of the silk protein.
The surface area of the heme–silk films was determined from the capacitance of the heme–silk electrode (ic – Fig. 2A) compared to the capacitance of an unmodified glassy carbon electrode.43 The surface area of the films was found to be an average of 14 ± 9 cm2, the variation in surface areas is primarily attributed to the changes in film thickness obtained with drop casting.
Catalytic voltammetry
When oxygen was present in the buffer solution of the electrochemical cell, there was a pronounced increase in the reduction current (Fig. 3A). Such an increase in current is typical of catalysis where the catalytic redox center is continuously regenerated leading to an amplification in the current.22 | ||
| Fig. 3 Comparison of oxygen reduction by heme–silk films (A) and silk films without heme (B). Solid lines show cyclic voltammogram in an aerated 50 mM phosphate buffer (pH 7.4). Dashed lines show the cyclic voltammogram in a degassed buffer. Scan rate, 100 mV s−1. | ||
Silk films without heme also showed a reduction reaction in the presence of oxygen (Fig. 3B). However, the reaction occurred at a more negative potential (∼250 mV relative overpotential, Fig. 3Avs.Fig. 3B). The potential of this reaction is similar to that reported for the oxygen reduction reaction at a bare glassy carbon electrode.1 In addition, the reductive current recorded for the silk film was approximately three times lower than that obtained for the heme–silk film, again indicating that this current was due to non-catalytic reduction of oxygen at the glassy carbon electrode.
Given that CNTs are used as oxygen reduction catalysts,2,3 we compared the oxygen reduction by heme–silk films on a CNT modified glassy carbon electrode to a CNT film without any further modification. Similar to that noted for silk films without any heme, oxygen reduction occurred at a relative overpotential of 250 mV (ESI Fig. 4†). This result confirms the importance of heme for the catalytic reduction of oxygen.
To determine whether the oxygen reduction reaction obtained using heme–silk films is diffusion-limited, the effect of scan rate on oxygen reduction was measured (Fig. 4A). A linear relationship between peak current and the square root of the scan rate was noted, which is expected for the reversible reduction of a reactant that is under diffusion control (Fig. 4A, inset).34,42
 | ||
| Fig. 4 The effect of scan rate for a stationary electrode (A) and electrode rotation rate at a scan rate of 100 mV s−1 (B) on the voltammetric response from heme–silk films in 50 mM phosphate buffer (pH 7.4) in the presence of oxygen. Inset, the peak current as a function of the square root of the scan rate. | ||
Given that the drop-cast heme–silk films are thick and not monolayers,21 two diffusion processes are required. Firstly, oxygen is diffusing from the solution to the silk film surface. Secondly, oxygen needs to diffuse through the silk film to the heme center where catalysis occurs.42
Rotating disk voltammetry (RDV) forces convection of the solution and enhances the transport of reactant to the outer surface of the silk film.34 The shape and magnitude of the reduction current recorded from a heme–silk film in aerated buffer was found to be dependent on the rate of rotation (Fig. 4B). At rotation rates greater than 1000 rpm, a sigmoidal voltammetric response was obtained in which the forward and the reverse scans are almost identical. This result confirms that diffusion of oxygen to the outer surface of the heme–silk film is the rate-limiting step rather than diffusion through the silk film.
Using rotation rates greater than 1000 rpm, the catalytic potential (Ecat) of the heme–silk films was determined to be 25 mV vs. NHE. This Ecat is significantly lower than that obtained using platinum electrodes (290 mV vs. NHE, pH 7.4 – ESI Fig. 4†), and biocatalysis using bilirubin oxidase30 and other multicopper oxidases.31 We44–46 and others47–49 have previously used metal-ion-substituted porphyrins to vary the chemical properties of heme proteins. The engineering approach that we have adopted in this work using honeybee silk proteins presents multiple ways to reduce the over-potential required for oxygen reduction, such as varying the protein scaffold and changing the metal cofactors incorporated.19,20 This is an avenue for future research which may lead to an increase in the catalytic potential of these heme–silk films.
Mechanism of oxygen reduction
Oxygen can be either partially reduced to hydrogen peroxide (eqn (1)) or fully reduced to water (eqn (2)). The full reduction of oxygen is desirable in fuel cells as it maximizes the power output and avoids the production of peroxide which damages the cell electrodes.1| O2 + 2H+ + 2e− → H2O2 | (1) |
| O2 + 4H+ + 4e− → 2H2O | (2) |
The number of electrons involved in the reduction of oxygen can be determined by monitoring the change in the catalytic current with increasing rotation rate following the Koutecký–Levich equation:
| icat−1 = iK(E)−1 + (0.62nFA[O2]0D2/3ω1/2ν−1/6)−1 | (3) |
485 C mol−1), A is the macroscopic area of the disc (0.125 cm2), [O2]0 is the initial concentration of O2 in an air-saturated buffer (0.26 mM) at 25 °C, D is the diffusion coefficient of oxygen (2.0 × 10−5 cm2 s−1), ν is the kinematic viscosity of the solution (0.01 cm2 s−1), ω is the angular velocity of the disc, icat is the catalytic current and iK(E) is the potential-dependent current.
The catalytic response was measured at different rotation rates (Fig. 5A) and from the slope of the Koutecky–Levich plot (Fig. 5B), the average number of electrons was determined to be 3.74.
 | ||
| Fig. 5 Determining the mechanism of oxygen reduction by heme–silk films. (A) The effect of rotation on oxygen reduction by a heme–silk film in 100 mM phosphate buffer, pH 7. (B) Koutecký–Levich plot of the catalytic current of heme–silk films in the presence of oxygen to determine the number of electrons transferred to the substrate. Solid line, fit of the experimental data; dashed lines, theoretical fit for 2- and 4-electron reductions. | ||
In addition to calculating the number of electrons transferred to oxygen in the reduction reaction, the rate of the oxygen reduction reaction (kORR) can be determined from the intercept of the Koutecky–Levich plot (eqn (3) and (4)).
| iK(E) = kORRnFA[O2]Γcat | (4) |
485 C mol−1), A is the macroscopic area of the disc (0.125 cm2), [O2] is the concentration of dissolved oxygen (0.26 mM) and Γcat is the electroactive area (determined from an anaerobic CV) to be 895 pmol cm−2.
Using eqn (4) and the experimental data (Fig. 5B), kORR was calculated to be 1.07 × 105 M−1 s−1. This rate constant is similar to those reported for synthetic and biosynthetic models of cytochrome c oxidase, 1.2 × 105 M−1 s−1 and 1.98 × 107 M−1 s−1, respectively.8,11
The number of electrons transferred, calculated using the Koutecký–Levich plot, and indicated that while water was the major product of the reaction, some hydrogen peroxide was also produced. The amount of hydrogen peroxide produced was determined using rotating ring disk voltammetry (RRDV) and is shown in Fig. 6.
 | ||
| Fig. 6 Determining the amount of peroxide produced by a heme–silk film in 100 mM phosphate buffer, pH 7. (A) Rotating ring disk voltammetry (RRDV) of heme–silk film. Rotation rate, 2500 rpm; scan rate, 100 mV s−1. Black line, disk electrode; grey line, ring electrode held at +1.5 V vs. NHE. The ring current is expanded five-fold for clarity. (B) Plot of the ratio of disk current to ring current against rotation rate, the solid lines indicate the least-squared linear fits to the experimental data. Ratio determined at four different potential points of the disk electrode linear sweep. | ||
In RRDV, the oxidative current recorded on the platinum ring is due to the oxidation of hydrogen peroxide produced by the heme–silk film during the oxygen reduction reaction (Fig. 6A). At potentials above +20 mV, an oxidative current is observed, indicating the production of hydrogen peroxide, while at more reducing potentials below +20 mV, the amount of hydrogen peroxide produced decreases (Fig. 6A, grey line). A similar potential-dependent selectivity of oxygen reduction has been observed in other iron porphyrin systems such as iron protoporphyrin IX (heme b) on a graphite electrode.6,9
If the collection efficiency of the rotating ring disk electrode is known, then both the number of electrons transferred to oxygen (eqn (5)) and the amount of hydrogen peroxide produced (eqn (6)) can be quantified.50
 | (5) |
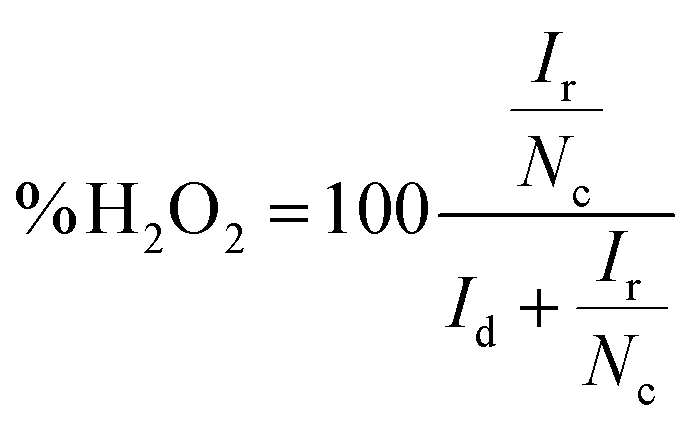 | (6) |
The collection efficiency of the rotating ring disk electrode was calculated to be 1.42 (ESI†). From the disk current (id) and ring current (ir) at −345 mV vs. NHE at 2500 rpm (Fig. 5), n was determined to be 3.71 and the amount of hydrogen peroxide produced was calculated to be 14% (eqn (6)).
Calculations of hydrogen peroxide production using RRDV can be artificially low as the hydrogen peroxide produced at the disk electrode can react further at the disk, thereby reducing the peroxide to water (eqn (7)). Such a two-step process with H2O2 as an intermediate occurs in many simple iron porphyrin catalysts.6,51,52
| H2O2 + 2e− + 2H+ → 2H2O | (7) |
To determine if hydrogen peroxide was being reduced by the heme–silk film on the disk electrode, RRDV was carried out at rotation rates ranging from 500 to 5000 rpm (Fig. S2†). The ratio of the disk-to-ring current at different rotations was plotted at four different potentials, at +100 mV (which is close to the onset of catalysis) at +40 mV (the point of maximum ring current) and at −100 mV and −260 mV (the plateau region of the catalysis).
Both the slope and intercept of the plots were found to be dependent on the potential of the disk electrode (Fig. 6B). This is typical of a mechanism in which intermediates are produced in parallel but do not react further53 and suggests that the heme–silk film does not reduce H2O2. The quantities detected at the ring electrode are indicative of the total amount of peroxide produced by the heme–silk electrocatalyst.
Given that hydrogen peroxide is produced as a parallel product that does not react further, the potential-dependent production of peroxide can be rationalized using a mechanism proposed by Boulatov and co-workers6 involving two possible pathways (ESI Scheme 1†). The pathway followed is based on kinetic competition between the release of H2O2 (Pathway A, ESI Scheme 1†) and its reduction to a ferrous-hydroperoxide species (Pathway B, ESI Scheme 1†).
We proposed that in our system, at potentials greater than 0 mV, Pathway A is followed, leading to the production of H2O2. As the potential of the electrode is lowered, the driving force for the reduction of ferric-hydroperoxo to ferrous-hydroperoxo increases, promoting Pathway B over Pathway A.
The percentage of H2O2 produced by heme–silk films is lower than that reported for other immobilized heme systems (Table 1) and close to the lowest H2O2 production levels reported for binuclear copper–heme mimics of cytochrome c oxidase. Both the axial ligand trans to bound oxygen and the secondary coordination sphere of iron porphyrins have been shown to promote selective and efficient oxygen reduction.6,54 Therefore, varying the silk protein, such as the use of a histidine coordinating residue rather than tyrosine, may further decrease the amount of hydrogen peroxide produced by heme–silk films.
| % H2O2 produced | |||
|---|---|---|---|
| Homogenous | Immobilized | Reference | |
| a Carried out in organic solvents. For further details see Rigsby et al.15 b Data obtained from Mayer and co-workers.15 Complexes were immobilized in Nafion, Nafion/carbon or on edge plane pyrolytic graphite – the range from lowest to highest is reported here. RRDE was carried out in 0.1 M HClO4, except heme–silk, which was carried out in phosphate buffer pH 7. While not a direct comparison, the apparent redox stoichiometry is known to be pH-independent.6n.r., not reported; n.d., not determined. | |||
| Heme–silk | n.d. | 14 | This work |
| Tetraphenyl iron porphyrin | 15a | 27–58b | 15 |
| 2-Carboxylphenyl iron porphyrin | 0a | 20–5b | 15 |
| 4-Carboxyphenyl iron porphyrin | 15a | 28–56b | 15 |
| 2-Pyridyl iron porphyrin | 2–6a | 18–35b | 15 |
| 4-Pyridyl iron porphyrin | 9–11a | 36–56b | 15 |
| Cytochrome c oxidase synthetic model | n.r. | 10 | 11 |
| Cytochrome c oxidase | |||
| Biosynthetic model | n.r. | 6 | 8 |
Electrode stability
One of the principal factors that prevents biologically derived materials from being used in commercial fuel cells is their poor stability. Simple iron porphyrins are notoriously unstable oxygen reduction catalysts and rapidly degrade after a few turnovers.6,17 The rapid degradation of metalloporphyrin-based catalysts is attributed to the production of highly destructive hydroxyl radicals from O–O bond homolysis (dotted lines, ESI Scheme 1†) that rapidly oxidize the porphyrin macrocycle.9,11 The development of a thermally-stable protein material is the main force driving our research on heme–silk materials.20One method which has been used to improve the stability of iron porphyrins is to immobilize them in polymers such as Nafion55,56 and lipid films.10 Consequently, we were interested to determine whether incorporating the heme within a silk material would improve the stability of the heme catalyst under oxygen reduction.
We determined the stability of our bioelectrocatalyst under representative operating conditions. Fig. 7A shows the amperometric plot of a freshly prepared heme–silk electrode used for continuous oxygen reduction (−200 mV vs. NHE) overnight at pH 7.
 | ||
| Fig. 7 Stability testing of heme–silk film. (A) Stability at pH 7.4 – amperometric measurements at – 200 mV vs. NHE 50 mM phosphate (pH 7.4) with stirring 800 rpm. (B) Stability at pH 3. The electrode was operated at 0 mV vs. NHE over two hours to determine the stability in acidic medium. Cyclic voltammetry was carried out before testing (0 hours) and after each hour to monitor changes in catalytic activity. 100 mM citrate buffer (pH 3). Scan rate, 100 mV s−1 and rotation rates of 1500 rpm. | ||
In addition to testing stability at pH 7, the stability of the heme–silk electrodes was tested under acidic conditions. Fig. 7B shows that the heme–silk electrode maintained 85% of its catalytic activity after operating at pH 3 for two hours.
This stability of the heme–silk electrocatalyst suggests that immobilizing the heme within a protein environment, which provides both an axial ligand and protein distal environment, has increased the selectivity toward heterolysis of the O–O bond rather than homolysis (ESI Scheme 1†).9 An alternative possibility is that any hydroxyl radical produced reacts with the silk protein rather than the heme center. Regardless of the exact production and fate of radicals produced, the stability of heme–silk films highlights the advantages of employing a silk protein which provides axial ligation and a robust protein scaffold for immobilization.
Conclusions
The results presented above demonstrate that heme–silk materials are a promising new avenue for the development of oxygen reduction electrocatalysts. Recombinant honeybee silk (AmelF3) films with heme incorporated can perform efficient reduction of oxygen, with low levels of peroxide produced. The reaction is limited by the rate of oxygen diffusion to the silk film. The silk film provides both a coordinating axial ligand and a distal environment to the heme center which facilitates heterolytic cleavage of the O–O bond, thereby producing heme-based electrocatalysts with significantly improved stability. The ability of heme–silk to reduce oxygen efficiently at neutral buffers makes them ideal for use in enzymatic fuel cells or microbial fuel cells. Furthermore, the stability of the electrocatalysts at mild acidic conditions would allow these materials to be used as the cathode in hybrid fuel cells with an inorganic anode catalyst.Acknowledgements
TDR thanks the Churchill Trust for a travel fellowship enabling him to build new international collaborations. The authors thank Dr Stephen Edmonson (U. Manchester) for ellipsometry measurements and analyses. J. B. and C. F. B. acknowledge the support of the UK's Engineering and Physical Sciences Research Council (EPSRC) CDT in Science and Applications of Graphene and Related Nanomaterials (EP/L01548X/1). We acknowledge funding from CSIRO.References
- K. Shimizu, L. Sepunaru and R. G. Compton, Chem. Sci., 2016, 7, 3364–3369 RSC.
- J. Yang, H. Sun, H. Liang, H. Ji, L. Song, C. Gao and H. Xu, Adv. Mater., 2016, 28, 4606–4613 CrossRef CAS PubMed.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- C. Costentin, H. Dridi and J.-M. Savéant, J. Am. Chem. Soc., 2015, 137, 13535–13544 CrossRef CAS PubMed.
- Y. Xu, M. Shao, M. Mavrikakis and R. R. Adzic, in Fuel Cell Catalysis: A Surface Science Approach, ed. M. T. M. Koper, 2009, pp. 271–315 Search PubMed.
- R. Boulatov, in Fuel Cell Catalysis, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008, pp. 637–693 Search PubMed.
- J. P. Collman and R. A. Decréau, Chem. Commun., 2008, 356, 5065–5076 RSC.
- S. Mukherjee, A. Mukherjee, A. Bhagi-Damodaran, M. Mukherjee, Y. Lu and A. Dey, Nat. Commun., 2015, 6, 8467 CrossRef CAS PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- J. P. Collman and R. Boulatov, Angew. Chem., Int. Ed., 2002, 41, 3487–3489 CrossRef CAS PubMed.
- R. Boulatov, J. P. Collman, I. M. Shiryaeva and C. J. Sunderland, J. Am. Chem. Soc., 2002, 124, 11923–11935 CrossRef CAS PubMed.
- S. Safarian, C. Rajendran, H. Müller, J. Preu, J. D. Langer, S. Ovchinnikov, T. Hirose, T. Kusumoto, J. Sakamoto and H. Michel, Science, 2016, 352, 583–586 CrossRef CAS PubMed.
- K. Mittra, S. Chatterjee, S. Samanta and A. Dey, Inorg. Chem., 2013, 52, 14317–14325 CrossRef CAS PubMed.
- B. D. Matson, C. T. Carver, A. Von Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Chem. Commun., 2012, 48, 11100 RSC.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Rodríguez, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2014, 136, 12544–12547 CrossRef CAS PubMed.
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS.
- J. P. Collman, M. Kaplun and R. A. Decréau, Dalton Trans., 2006, 7, 554–559 RSC.
- T. D. Rapson, T. D. Sutherland, J. S. Church, H. E. Trueman, H. Dacres and S. C. Trowell, ACS Biomater. Sci. Eng., 2015, 1, 1114–1120 CrossRef CAS.
- T. Rapson, Molecules, 2016, 21, 919 CrossRef PubMed.
- T. D. Rapson, J. S. Church, H. E. Trueman, H. Dacres, T. D. Sutherland and S. C. Trowell, Biosens. Bioelectron., 2014, 62, 214–220 CrossRef CAS PubMed.
- F. A. Armstrong and G. S. Wilson, Electrochim. Acta, 2000, 45, 2623–2645 CrossRef CAS.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- T. D. Rapson, U. Kappler and P. V. Bernhardt, Biochim. Biophys. Acta, 2008, 1777, 1319–1325 CrossRef CAS PubMed.
- T. D. Rapson, U. Kappler, G. R. Hanson and P. V. Bernhardt, Biochim. Biophys. Acta, 2011, 1807, 108–118 CrossRef CAS PubMed.
- C. F. Blanford, Chem. Commun., 2013, 49, 11130–11132 RSC.
- L. Xu and F. A. Armstrong, RSC Adv., 2015, 5, 3649–3656 RSC.
- T. W. Woolerton, S. Sheard, Y. S. Chaudhary and F. A. Armstrong, Energy Environ. Sci., 2012, 5, 7470–74790 CAS.
- P. V. Bernhardt, Aust. J. Chem., 2006, 59, 233–256 CrossRef CAS.
- K. Singh, T. McArdle, P. R. Sullivan and C. F. Blanford, Energy Environ. Sci., 2013, 6, 2460–2464 CAS.
- J. A. Cracknell and C. F. Blanford, Chem. Sci., 2012, 3, 1567–1581 RSC.
- S. Weisman, V. S. Haritos, J. S. Church, M. G. Huson, S. T. Mudie, A. J. W. Rodgers, G. J. Dumsday and T. D. Sutherland, Biomaterials, 2010, 31, 2695–2700 CrossRef CAS PubMed.
- T. D. Sutherland, S. Weisman, A. A. Walker and S. T. Mudie, Biopolymers, 2012, 97, 446–454 CrossRef CAS PubMed.
- V. Fourmond and C. Léger, in Adv Biochem Eng Biotechnol, Springer, Berlin Heidelberg, 2016, pp. 1–43 Search PubMed.
- C. Léger, S. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- P. J. Yunker, T. Still, M. A. Lohr and A. G. Yodh, Nature, 2011, 476, 308–311 CrossRef CAS PubMed.
- T. McArdle, T. P. McNamara, F. Fei, K. Singh and C. F. Blanford, ACS Appl. Mater. Interfaces, 2015, 7, 25270–25280 CAS.
- L. Richert, F. Boulmedais, P. Lavalle, J. Mutterer, E. Ferreux, G. Decher, P. Schaaf, J.-C. Voegel and C. Picart, Biomacromolecules, 2004, 5, 284–294 CrossRef CAS PubMed.
- M. G. Huson, J. S. Church, J. M. Poole, S. Weisman, A. Sriskantha, A. C. Warden, P. M. Campbell, J. A. M. Ramshaw and T. D. Sutherland, PLoS One, 2012, 7, e52308 CAS.
- S. Mukherjee, K. Sengupta, M. R. Das, S. S. Jana and A. Dey, J. Biol. Inorg Chem., 2012, 17, 1009–1023 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley-VCH Verlag, 2nd edn., 2001 Search PubMed.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS.
- T. D. Rapson, H. Dacres and S. C. Trowell, RSC Adv., 2014, 4, 10269–10272 RSC.
- T. D. Rapson, S. Warneke, M. M. Musameh, H. Dacres, B. C. T. Macdonald and S. C. Trowell, RSC Adv., 2015, 5, 89003–89008 RSC.
- C. C. Horgan, Y.-S. Han, H. Trueman, C. J. Jackson, T. D. Sutherland and T. D. Rapson, RSC Adv., 2016, 6, 39530–39533 RSC.
- M. B. Winter, E. J. McLaurin, S. Y. Reece, C. Olea, D. G. Nocera and M. A. Marletta, J. Am. Chem. Soc., 2010, 132, 5582–5583 CrossRef CAS PubMed.
- M. B. Winter, P. J. Klemm, C. M. Phillips-Piro, K. N. Raymond and M. A. Marletta, Inorg. Chem., 2013, 52, 2277–2279 CrossRef CAS PubMed.
- P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1018 CrossRef CAS PubMed.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- J. P. Collman, P. Denisevich, Y. Konai, M. Marrocco, C. Koval and F. C. Anson, J. Am. Chem. Soc., 1980, 102, 6027–6036 CrossRef CAS.
- K. Shigehara and F. C. Anson, J. Phys. Chem., 1982, 86, 2776–2783 CrossRef CAS.
- A. Damjanovic, M. A. Genshaw and J. O. Bockris, J. Chem. Phys., 1966, 45, 4057–4059 CrossRef CAS.
- T. Ohta, P. Nagaraju, J. G. Liu, T. Ogura and Y. Naruta, J. Biol. Inorg Chem., 2016, 21, 745–755 CrossRef CAS PubMed.
- D. A. Buttry and F. C. Anson, J. Am. Chem. Soc., 1984, 106, 59–64 CrossRef CAS.
- F. C. Anson, C. L. Ni and J. M. Saveant, J. Am. Chem. Soc., 1985, 107, 3442–3450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta02322g |
| This journal is © The Royal Society of Chemistry 2017 |