
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper on carbon materials: stabilization by nitrogen doping†
Dmitri A.
Bulushev
 *abc,
Andrey L.
Chuvilin
de,
Vladimir I.
Sobolev
ab,
Svetlana G.
Stolyarova
b,
Yury V.
Shubin
bc,
Igor P.
Asanov
bc,
Arcady V.
Ishchenko
ac,
Giacomo
Magnani
f,
Mauro
Riccò
f,
Alexander V.
Okotrub
bc and
Lyubov G.
Bulusheva
*bc
*abc,
Andrey L.
Chuvilin
de,
Vladimir I.
Sobolev
ab,
Svetlana G.
Stolyarova
b,
Yury V.
Shubin
bc,
Igor P.
Asanov
bc,
Arcady V.
Ishchenko
ac,
Giacomo
Magnani
f,
Mauro
Riccò
f,
Alexander V.
Okotrub
bc and
Lyubov G.
Bulusheva
*bc
aBoreskov Institute of Catalysis, SB RAS, Novosibirsk, 630090, Russia. E-mail: dmitri.bulushev@catalysis.ru
bNikolaev Institute of Inorganic Chemistry, SB RAS, Novosibirsk, 630090, Russia. E-mail: bul@niic.nsc.ru
cNovosibirsk State University, Novosibirsk, 630090, Russia
dCIC nanoGUNE Consolider, San Sebastian, 20018, Spain
eIKERBASQUE, Basque Foundation for Science, Bilbao, 20013, Spain
fDipartimento di Scienze Matematiche, Fisiche e Informatiche, Università di Parma, 43124 Parma, Italy
First published on 26th April 2017
Abstract
The applicability of Cu/C catalysts is limited by sintering of Cu leading to deactivation in catalytic reactions. We show that the problem of sintering could be resolved by N-doping of the carbon support. Cu nanocatalysts with 1 at% of metal were synthesized by Cu acetate decomposition on N-free and N-doped (5.7 at% N) mesoporous carbon supports as well as on thermally expanded graphite oxide. Catalytic properties of these samples were compared in hydrogen production from formic acid decomposition. The N-doping leads to a strong interaction of the Cu species with the support providing stabilization of Cu in the form of clusters of less than 5 nm in size and single Cu atoms, which were observed in a significant ratio by atomic resolution HAADF/STEM even after testing the catalyst under harsh conditions of the reaction at 600 K. The mean size of the obtained Cu clusters was by a factor of 7 smaller than that of the particles in the N-free catalyst. The N-doped Cu catalyst possessed good stability in the formic acid decomposition at 478 K for at least 7 h on-stream and a significantly higher catalytic activity than the N-free Cu catalysts. The nature of the strongly interacting Cu species was studied by XPS, XRD and other methods as well as by DFT calculations. The presence of single Cu atoms in the N-doped catalysts should be attributed to their strong coordination by pyridinic nitrogen atoms at the edge of the graphene sheets of the support. We believe that the N-doping of the carbon support will allow expanding the use of Cu/C materials for different applications avoiding sintering and deactivation.
Introduction
Over the last decade, interest in the conversion of biomass-derived acids to valuable products has increased. The simplest organic acid is formic acid, which can be produced from biomass cellulose as well as from fats and oils.1 This acid can be used for storage of hydrogen for different applications. Supported Pt-group metal catalysts are often applied for formic acid decomposition. Hydrogen production from formic acid decomposition over bulk Cu and Cu supported on oxide supports attracted a lot of attention long time ago and the results were discussed by Iglesia and Boudart.2 These authors showed that the reaction is structure-insensitive at least in the range of particle sizes between 5 and 40 nm. However, recently Li et al.3 showed with the help of density functional theory (DFT) calculations that the reaction is structure-sensitive as it occurred differently on different Cu planes. They predicted that the pathway via a formate intermediate is preferable as compared to the pathway via a carboxyl intermediate, and the reaction proceeds more readily on the terrace Cu sites. Cu catalysts are much cheaper than Pt-group metals, but they are normally less active and prone to aggregation at elevated reaction temperatures causing deactivation. Since stabilization of Cu in the active state poses a problem, this seriously limits the applicability of the Cu-based catalysts.Although gas-phase formic acid decomposition over Cu takes place at higher temperatures than that over noble metals, Cu catalysts may find applications for hydrogen transfer hydrogenation/deoxygenation reactions, in which formic acid is used as a hydrogen donor.4 This provides benefits in transportation and storage as compared to molecular hydrogen. When the hydrogenation/deoxygenation of a substrate by hydrogen takes place at high temperatures, there is no necessity for catalysts for hydrogen production from formic acid that operate at very low temperatures. This catalyst can work at temperatures slightly lower than the temperatures of substrate conversion. A higher resistance of Cu with respect to CO formed in traces can be an additional advantage for certain applications as compared to Pt-group metals.
To the best of our knowledge formic acid decomposition has not been studied yet over Cu catalysts supported on carbon. Carbon materials, especially those with a planar sp2 structure, are often considered as weakly interacting supports with Cu particles.5,6 The stability could be improved by utilization of the supports with a significant fraction of edge sites like carbon nanofibers, but still the mean Cu particle size proved to be relatively big (20–40 nm) and the particle size distribution was broad.5 N-Doping of carbon materials provides new interesting opportunities for utilization of the carbon materials as catalyst supports.7,8 There is a reason to deposit Cu on the surface of a nitrogen-doped carbon taking advantage of the known high affinity of Cu ions to some nitrogen species.9 In this case, we could expect a stronger interaction of Cu with the support than in the case of N-free carbon, thus decreasing the tendency to aggregate. A study of interaction of Cu with the N-doped carbon is a key point of the present investigation. Earlier, a few catalytic reactions have been studied on Cu catalysts supported on N-doped carbon nanotubes10 and microporous carbon,11 with high concentrations of Cu (10 wt%).
Recently, we have demonstrated that Pt-group metals can be dispersed on N-doped carbon in the form of single atoms and small clusters.1,12–14 Interestingly, the catalytic activity in the formic acid decomposition of single Pt atoms stabilized by N-doped carbon was by 1 order of magnitude higher than that of Pt atoms on the surfaces of unsupported Pt powder or supported Pt particles.7,12 The adsorption and catalytic properties of Cu are very much different from the properties of Pt-group metals.15 However, at small concentrations of Cu (1 and 3 wt%), Yamada et al.16 have also shown that N-doping of exfoliated graphene leads to stabilization of atomically dispersed Cu. They proposed that this may take place because of the strong coordination of Cu2+ ions by N-doped graphene sheets. At the same time, small clusters were also present in their samples. The authors did not provide any application of these materials. The objectives of the present work are to determine the character of interaction of Cu species with an N-doped carbon, and to elucidate the role of this interaction in hydrogen production from formic acid decomposition.
Experimental
Materials
Three carbon materials have been used as catalyst supports in this work. A thermally expanded graphite oxide (TEGO), which was obtained by exfoliation of graphite oxide at 1423 K, has been described earlier.17 N-Free and N-doped porous carbon networks (PCN and N-PCN) were synthesized by chemical vapor deposition from ethanol and acetonitrile, respectively, at 1073 K using iron-doped (1.4 wt%) calcium tartrate as a template.1,13,18 Cu (1 at% or 4.5 wt%) was deposited on carbon supports by their impregnation with anhydrous Cu(II) acetate (Sigma-Aldrich) dissolved in anhydrous tetrahydrofuran (99.9% purity) at 348 K for 17 h. Then, the solvent was evaporated and the adsorbed Cu acetate was decomposed by placing the material under dynamic vacuum conditions (10−3 Pa) into a hot furnace at 573 K, and keeping there for 15 min.Characterization
Nitrogen adsorption–desorption measurements of the supports were carried out at 77 K using an ASAP 2400 (Micromeritics) instrument. The surface area was calculated using the Brunauer–Emmett–Teller (BET) equation and the average pore diameter was estimated from this measurement.The powder X-ray diffraction analysis (XRD) of the samples was performed at room temperature on a Shimadzu XRD-7000 diffractometer (CuKα radiation, Ni filter on the reflected beam). In order to decrease the background contribution to the diffraction patterns, the samples were deposited on a zero diffraction plate of SiO2 single crystal. The data were collected with a 0.1° step size in the 2Θ range of 10–80°. The volume-averaged crystallite sizes for the carbon materials were determined from the integral broadening of the peaks using the Scherrer equation19 after exclusion of the instrumental contribution. Deconvolution and fitting of the XRD lines was based on the Pearson (PVII) function and was performed using WinFit 1.2.1 software.20
N 1s spectra of the samples were taken using X-ray photoelectron spectroscopy (XPS) at the Berliner Elektronen-Speicherring für Synchrotronstrahlung (BESSY) using radiation from the Russian–German beamline. The N 1s spectra were measured at the energy of monochromatized synchrotron radiation of 830 eV with a resolution of 0.1 eV. The Au 4f7/2 line at a binding energy of 84.0 eV was used for reference energy calibration.
In addition, XPS measurements of the samples were performed on a SPECS Phoibos 150 laboratory spectrometer using a monochromatized AlKα radiation (1486.6 eV). In situ reduction of the samples in a preparation chamber of the XPS unit was performed in 1 Pa of hydrogen at 533 K for 30 or 60 min. After this pretreatment, the sample was transferred to an analytical chamber without contact with air.
The morphology of the samples was studied using a FEI Titan 60-300 TEM/STEM image-side Cs-corrected microscope at an acceleration voltage of 80 kV for TEM and 300 kV for Scanning Transmission Electron Microscopy (STEM). Size distributions of Cu particles were obtained using a JEOL JEM-2010 microscope at an acceleration voltage of 200 kV in TEM mode. The samples for analysis were prepared by ultrasound-assisted deposition of acetone or ethanol suspension of the material on lacey carbon film grids. The macrostructure of supports was studied by scanning electron microscopy (SEM) on a JEOL JSM-6700F microscope.
Catalytic experiments
Vapor-phase formic acid decomposition was carried out in a fixed-bed tubular glass reactor. Activity tests were performed at atmospheric pressure with 20 mg of catalyst. The catalysts were pretreated while heating in a flow of 5 vol% formic acid in He (20 cm3 min−1) up to 600 K with a heating rate of 2 K min−1 and cooling in the same mixture to the reaction temperature (first heating cycle). A second heating cycle was performed in order to study the temperature dependence of the formic acid conversion for comparison with other samples. Characterization after the reaction implied that it was performed after two heating cycles in formic acid.The reactant and products were analyzed using a gas chromatograph (Tsvet-500). For determination of CO and CO2, they were separated on a 1.5 m × 3 mm steel column filled with Porapak-Q at 293 K followed by methanation and analysis with a flame-ionization detector. As no other carbon containing products were detected, the conversion of formic acid was determined as a sum of the concentrations of CO and CO2 formed relative to the initial concentration of formic acid.
Computational details
Theoretical modeling was carried out using a gradient-corrected functional of Perdew–Burke–Ernzerhof21 (PBE) with local and nonlocal exchange and correlation within the quantum-chemical program package Jaguar (Jaguar, version 9.2, Schrödinger, LLC, New York, NY, 2016). Atomic orbitals were described by an LACVP basis set, where the outermost core orbitals were included for Cu and the 6-31G set was applied for light elements. An N-doped carbon substrate was modeled using a nitrogen-containing graphene fragment with hydrogen-terminated edges. The geometry of the models with a Cu atom located at different sites of the fragment was optimized by an analytical gradient method until the energy was converged to be better than 5 × 10−5 Hartree/Bohr. The interaction energy of a Cu atom with the graphene fragment was calculated as: Eint = Etot(fragment) + Etot(Cu) − Etot(model), where Etot(fragment), Etot(Cu) and Etot(model) are the total energy of the isolated initial N-doped graphene fragment, the energy of a single Cu atom, and the energy of the model, respectively.Results and discussion
Characterization of catalyst supports
Since we have already described the carbon supports used in this work,1,13,17,18 only some new or most important features will be mentioned below. The major idea of the present work was to elucidate the effect of nitrogen-doping on the stability of Cu species in the formic acid decomposition reaction. Hence, N-free and N-doped porous carbon networks (PCN and N-PCN)1,13,18 with a similar morphology as is seen from SEM images (Fig. S1†) have been prepared. XRD patterns of these materials (Fig. 1) show broad lines for graphite C(101) and C(002) indicating a low degree of graphitization. As we reported earlier, Raman spectra of the similarly prepared carbon materials were also almost the same.1 They allowed estimation of an average size of graphite crystallites in the materials used,17 which was equal to about 10 nm for both. The Brunauer–Emmet–Teller (BET) surface area (Table 1) and the average pore diameter for the N-free material were the following: 873 m2 g−1 and 9.6 nm, while those for the N-doped material – 474 m2 g−1 and 8.7 nm. The average pore diameters corresponded to the range of mesopores and the carbon materials used could be related to mesoporous catalyst supports.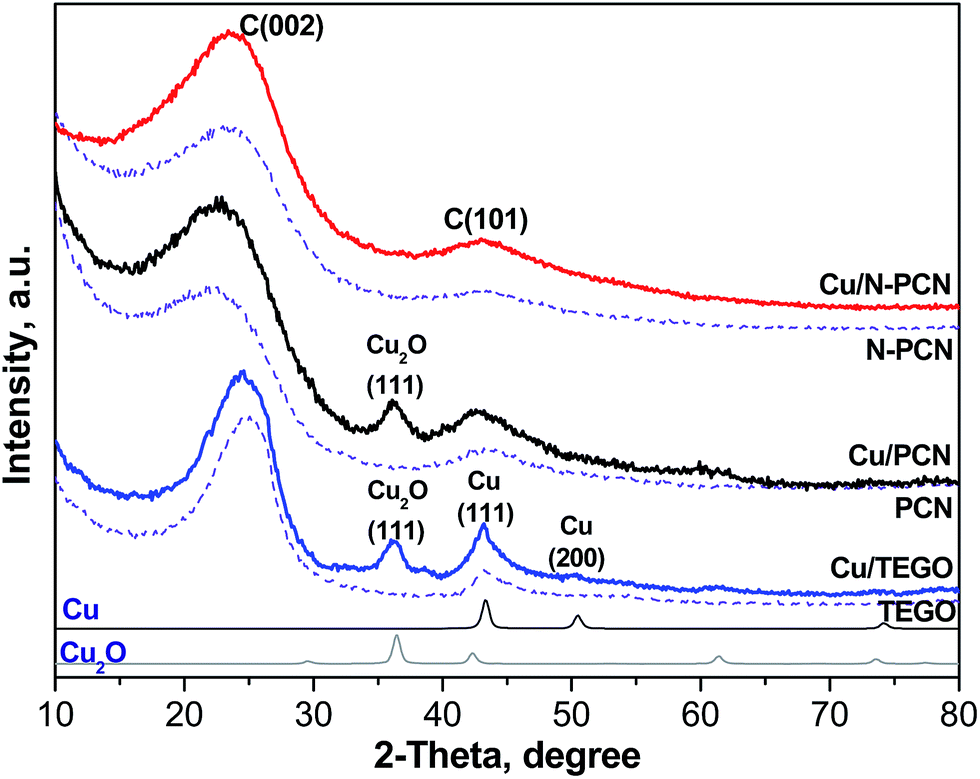 | ||
| Fig. 1 X-ray powder diffraction patterns of the catalyst supports (dashed lines) and 1 at% Cu catalysts before the reaction (solid lines). The patterns of Cu (PDF2 card 000-04-0836) and Cu2O (PDF2 card 000-05-0667) are shown for comparison. | ||
N-free thermally expanded graphite oxide (TEGO)17 with an average crystallite size of about 12 nm was used for comparison. Its BET surface area was 505 m2 g−1.
XRD study
The earlier obtained thermogravimetric analysis data demonstrated that the decomposition of Cu acetate in a nitrogen flow takes place at temperatures lower than 573 K with the formation of metallic Cu.22 Hence, the formation of metallic Cu is expected after thermal treatment of supported Cu acetate under our conditions (vacuum, 573 K, 15 min). An XRD study of the samples after their contact with ambient air was performed. The contact with air must lead to oxidation of the metallic Cu. Fig. 1 shows the XRD patterns of all the samples before the reaction. In the Cu/TEGO sample, the presence of two Cu phases is seen. These are Cu2O (PDF2 card 000-05-0667) and metallic Cu (PDF2 card 000-04-0836) with a mass ratio of 1.5. Only the Cu2O phase was clearly seen in the Cu/PCN sample. In contrast to the results for the N-free samples, no diffraction pattern for Cu species was found for the N-doped material (Cu/N-PCN, Fig. 1). This shows that Cu is present in the amorphous state, probably, in a highly dispersed state and that the interaction of Cu species with support sites is stronger than the mutual interaction between Cu species leading to aggregation and crystallization.Electron microscopy study
In order to elucidate the size and morphology of Cu species an electron microscopy study of the samples has been performed. As seen from Fig. 2 and 3, the N-free catalysts Cu/TEGO and Cu/PCN before the reaction contain Cu particles with the mean size of 23 and 16 nm, respectively. The particle size distributions for the samples presented in Fig. 3 demonstrate substantial differences in the samples. In the case of the Cu/TEGO catalyst (Fig. 3a), the size distribution is broad, while for the Cu/PCN sample (Fig. 3b), it is narrow. This difference could be related to the difference in the porous structures of the substrates. It is also interesting that the particles are often hollow inside (Fig. 2b and its inset). The formation of such particles is known for Cu2O/Cu systems23,24 and is normally attributed to the Kirkendall effect.25,26 | ||
| Fig. 2 TEM images of the N-free (a) Cu/TEGO and (b) Cu/PCN catalysts before the reaction. | ||
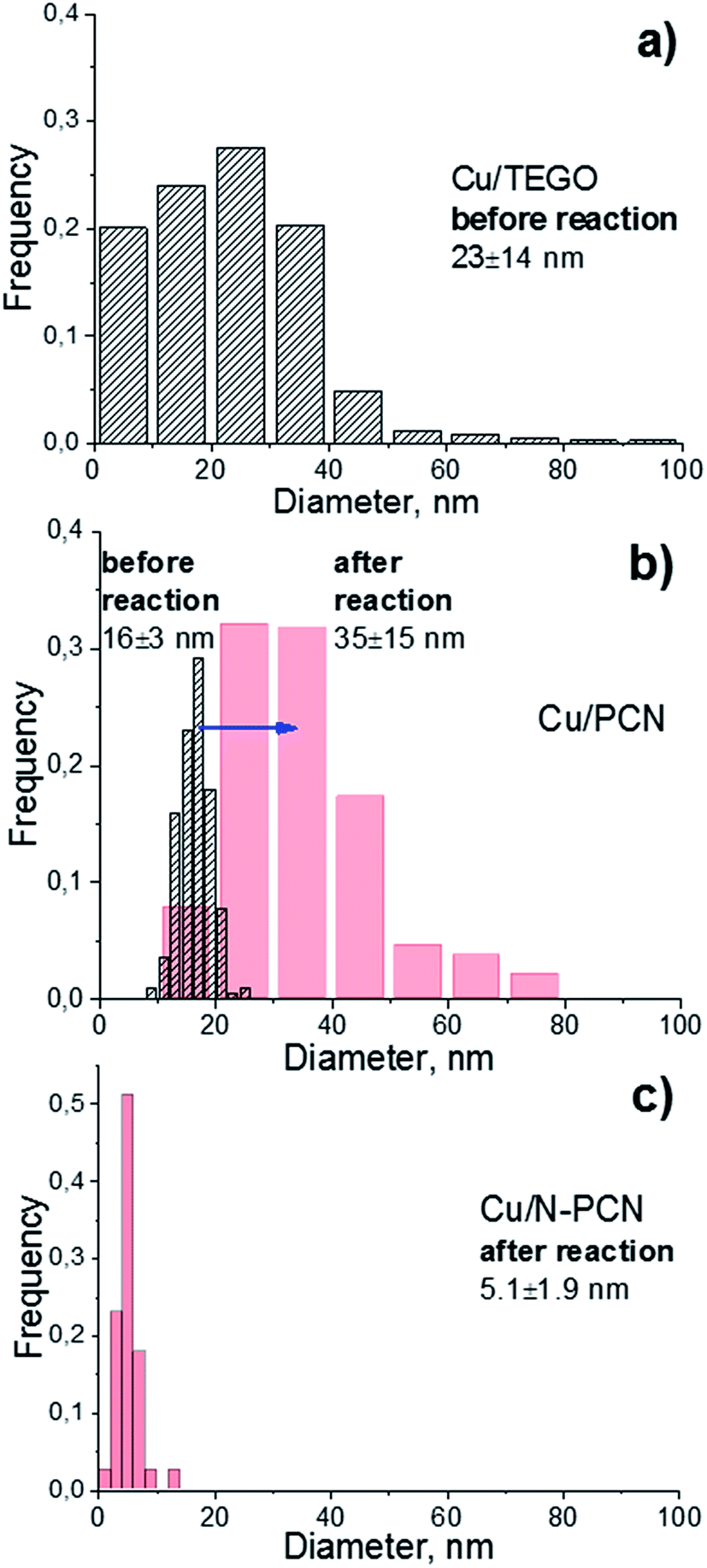 | ||
| Fig. 3 Particle size distributions in the (a) Cu/TEGO before the reaction, (b) Cu/PCN before (dashed) and after (red) the reaction, and (c) Cu/N-PCN catalysts after the reaction. | ||
Treatment of the Cu/PCN sample under the harsh conditions of formic acid reaction at 600 K leads to significant broadening of the particle size distribution (Fig. 3b) and an increase of the mean particle size from 16 (Fig. 2b) to 35 nm (Fig. 4a), but the hollow structure of the particles is mainly preserved. Electron diffraction patterns taken for some hollow particles (not shown here) corresponded to the Cu2O phase, which is consistent with the XRD data (Fig. 1). Single Cu atoms were not clearly seen in the Cu/PCN sample by atomic resolution HAADF/STEM, but a few nanoclusters (<1 nm) were seen (Fig. S2†). These nanoclusters could be stabilized by oxygen containing functional groups. In this sample, the content of single Cu atoms and nanoclusters was very small and copper was mainly present in the form of nanoparticles with a size of about 35 nm.
 | ||
| Fig. 4 (a, b) TEM and (c) atomic resolution HAADF/STEM (Titan 60-300) images of the (a) Cu/PCN and (b), (c) Cu/N-PCN catalysts after the reaction. | ||
In contrast to the N-free samples, the number of Cu particles in the Cu/N-PCN sample even after the reaction at 600 K was negligible as seen in TEM (Fig. S3a†) and HAADF/STEM (Fig. S3b†) images. In this sample, copper was mainly present in the form of poorly crystallized clusters of less than 5 nm in size, seen by TEM as weak gray spots (Fig. 4b), and in the form of nanoclusters and single Cu atoms, as revealed by atomic resolution HAADF/STEM (Fig. 4c). This high dispersion, narrow size distribution (Fig. 3c) and the stability under the aggressive acidic conditions cannot be attributed to the differences in the BET surface area (Table 1) or porous structure of the support; it rather indicates that Cu species interact much stronger with the support as compared to analogous N-free supports.
XPS study
The content of nitrogen in the N-PCN support determined by XPS was 5.7 at%. It did not change after the deposition of Cu. The content of oxygen in all samples with copper was in the range of 3.4–5.2 at%. This oxygen should be assigned to the oxygen functional groups of the support, since the content of oxygen in Cu2O is low relative to the substrate. We did not find a noticeable difference in the oxygen nature and content for the PCN and N-PCN supports, hence, the difference in Cu dispersion in the samples cannot be attributed to the oxygen species, but is rather determined by the presence of N on the support. Therefore, we studied in detail the electronic state of Cu and N species by XPS to understand their interaction.As mentioned, all the supports were loaded with the same content of Cu. However, the content of Cu in the Cu/TEGO, Cu/PCN, and Cu/N-PCN samples determined by XPS was different and equal to 0.14, 0.26 and 0.57 at%, respectively (Table 1). This difference is explained by the different degrees of Cu dispersion, in accordance with the electron microscopy data. The in situ reduction of the Cu samples in hydrogen causes some decrease in the Cu content (Table 1), probably due to the transformation of oxidized Cu particles to the reduced ones and different degrees of wetting of the support by these particles leading to a low content of surface atoms, as seen by XPS.
Fig. 5 and S4† show the results of the XPS study of the same samples that were studied by XRD (Fig. 1). It is seen that the spectra of the N-free samples (Cu/PCN and Cu/TEGO) before the reduction are the same (Fig. 5a and S4†). The binding energy at 932.5 eV corresponds to copper in the Cu0 and Cu+ states, which cannot be discriminated from each other. Their presence was in agreement with the XRD study revealing Cu2O and metallic Cu phases (Fig. 1). The intensive broad lines at 934.1 eV for the Cu/PCN sample and at 934.5 eV for the Cu/TEGO sample accompanied by satellites at 939–946 eV correspond to the Cu2+ state (Fig. 5a and S4†). The presence of CuO or any other crystalline Cu2+ compound was not found in sufficient concentrations by XRD and was also not in accordance with the XPS data, as the Cu 2p3/2 binding energy for CuO is expected at about 933.5 eV,27 but was not observed. Hence, the appearance of the Cu2+ species could be attributed to further oxidation of the surface of Cu2O particles by oxygen from ambient air with the formation of an amorphous layer of hydroxycarbonates. Accordingly, the literature data for hydroxides and carbonates are in the range of 934.4–934.8 eV.28,29 For the N-doped Cu catalyst, some significant differences were found in the XPS spectra as compared to the N-free Cu catalysts. Instead of one broad line for the Cu2+ species seen for the N-free samples (Fig. 5a and S4†), two distinct lines, narrow and broad, with binding energies at 934.6 and 935.2 eV (Fig. 5b) were observed, respectively. The assignment of these lines to Cu species will be discussed below.
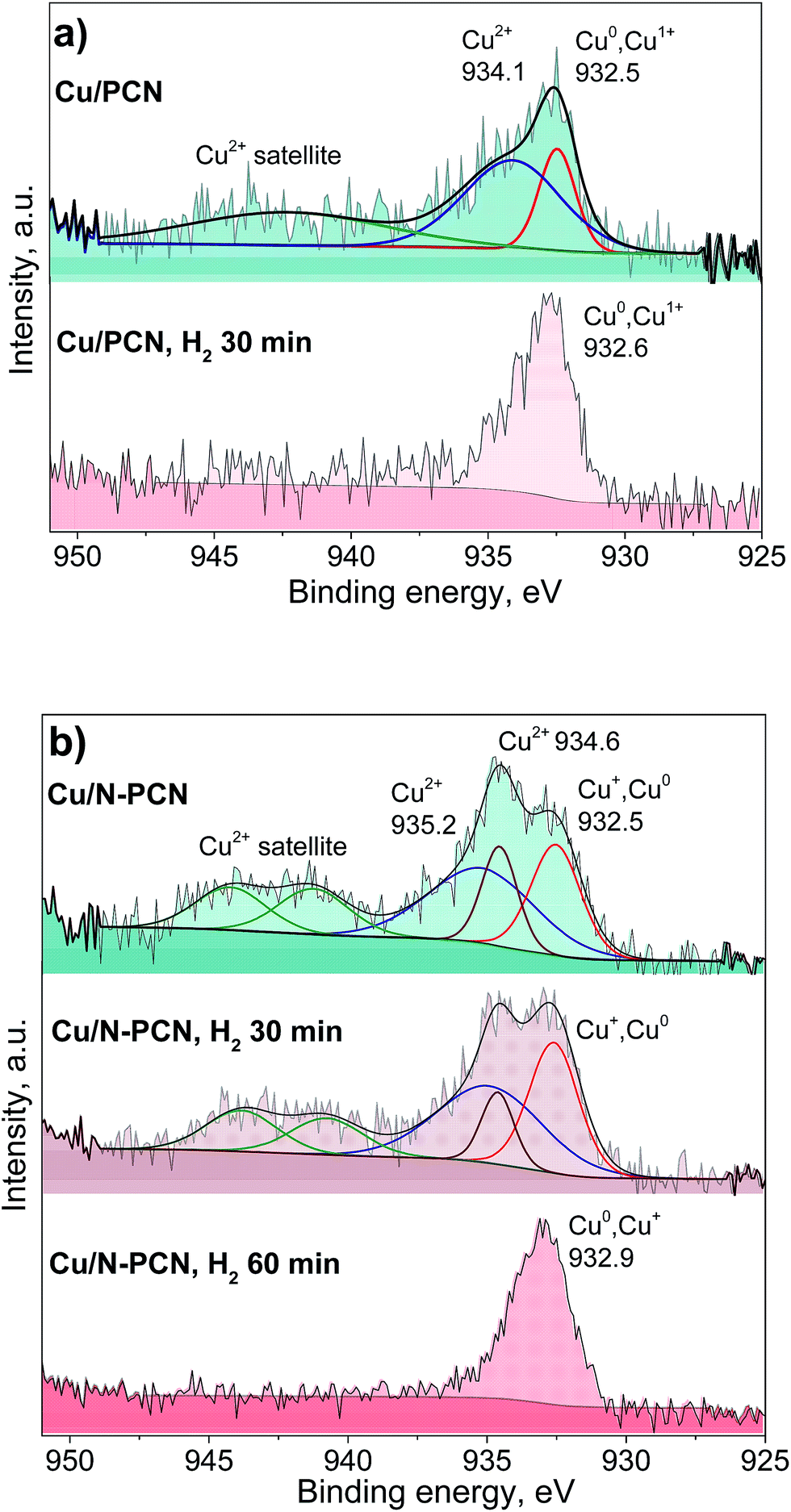 | ||
| Fig. 5 Cu 2p3/2 XPS spectra of the (a) Cu/PCN and (b) Cu/N-PCN catalysts before the reaction and after the reduction in H2 (1 Pa, 533 K) in the XPS preparation chamber. | ||
Additionally, we studied the Cu/PCN and Cu/N-PCN catalysts after the in situ reduction in hydrogen. The in situ reduction was performed in order to reproduce and preserve the electronic state of Cu similar to the one existing during the catalytic reaction. As seen in Fig. 5b, the reduction of Cu2+ species in the hydroxycarbonate layer of the Cu/PCN sample takes place easily in H2 within 30 min at 533 K, since no Cu2+ species were observed afterwards and only a single line at 932.6 eV corresponding to Cu0 and Cu+ species was present.
In contrast to the N-free Cu sample, it was not possible to completely reduce the Cu2+ ions in the Cu/N-PCN sample within 30 min. The reduction is complete only after 60 min. Interestingly, the two types of Cu2+ species existing in this sample showed different abilities to be reduced in H2. The Cu2+ species responsible for the binding energy at 934.6 eV are reduced faster than the Cu2+ species giving a broad line at 935.2 eV. The reduction of the Cu2+ species in this sample takes place, probably, with an induction period, explained by the necessity for vacancy/metallic Cu sites for H2 activation. The presence of an induction period in the reduction of unsupported Cu oxides was reported earlier.30 The size of Cu oxide particles may also affect the reduction rate leading to easier reduction of bigger particles.31
The reduction data assist the assignment of the lines to certain Cu species in the N-doped sample. As the position of the binding energy for the narrow peak (934.6 eV) is quite the same as that observed for the Cu2+ species in the N-free samples and these species are reduced quicker than the species characterized by the binding energy at 935.2 eV, we assign this line similar to the N-free samples to amorphous hydroxycarbonate species formed after contact of the sample with ambient air, while we assign the line with the binding energy at 935.2 eV to Cu2+ species directly interacting with the nitrogen species of the support. This is in accordance with the data of XPS studies of Cu(II) complexes with 1,10-phenanthroline ligand found in the literature,32,33 which provide the binding energy for the Cu 2p3/2 line at 935.0–935.4 eV corresponding to Cu2+ ions coordinated by N-species of the ligand. The difference in the Cu2+ binding energies for the N-doped sample as compared to the N-free sample and the slower reduction of the former species also correspond to a stronger interaction of Cu2+ species with the support surface, which could be provided by the N species of the support.
The position of the Cu 2p3/2 lines attributed to Cu0 and Cu+ species obtained after the reduction of the samples is also important (Fig. 5a and b). It is seen that the binding energy for the N-doped sample is higher by 0.3 eV than that for the N-free sample. This could be related to a significantly smaller size of the Cu particles obtained after the reduction in accordance with the results of Peters et al.34 Alternatively, this could be attributed to a stronger interaction of the obtained reduced Cu species with the support owing to coordination with nitrogen species, which gives rise to their electron-deficient nature. Earlier, for Pt catalysts, we have observed a significant shift of the Pt 4f7/2 binding energy by 0.6 eV for N-doped catalysts to a higher region as compared to N-free catalysts, despite the same mean particle size at about 1.1 nm for these catalysts.7 Hence, we attribute the observed shift for the N-doped Cu catalyst to the formation of electron-deficient Cu species coordinated by the nitrogen species of the support.
In order to understand the nature of nitrogen species strongly interacting with Cu species, we performed an XPS study based on synchrotron radiation. This approach provides a higher resolution than that obtained with conventional XPS and, additionally, the depth of analysis is smaller giving information only from a few layers of surface atoms. The spectra of the N 1s region of the N-PCN support and Cu/N-PCN catalyst after the reaction are compared in Fig. 6a. It is seen that the spectrum of the N 1s line of the support is represented by four species. These species are attributed to pyridinic (N1, 398.2 eV), pyrrolic (N2, 399.8 eV), graphitic or in plane substitutional (N3, 400.9 eV) and oxidized (402.4 eV) nitrogen species. Cu deposition on the support leads to significant changes in the spectra. Thus, a new line at 399.0 eV appears. The line corresponding to the position of graphitic nitrogen becomes stronger. At the same time, the lines of pyridinic and pyrrolic nitrogen species become weaker. The appearance of the new line at 399.0 eV and diminishing of the line corresponding to pyridinic species must be attributed to the interaction of Cu species with pyridinic nitrogen. We reported earlier the same feature for a Pd catalyst on N-doped carbon.13 It is important that Clarke et al.35 demonstrated a shift of the N 1s line by 0.8–1.2 eV to a higher binding energy region after the coordination of Cu ions by 1,10-phenanthroline.
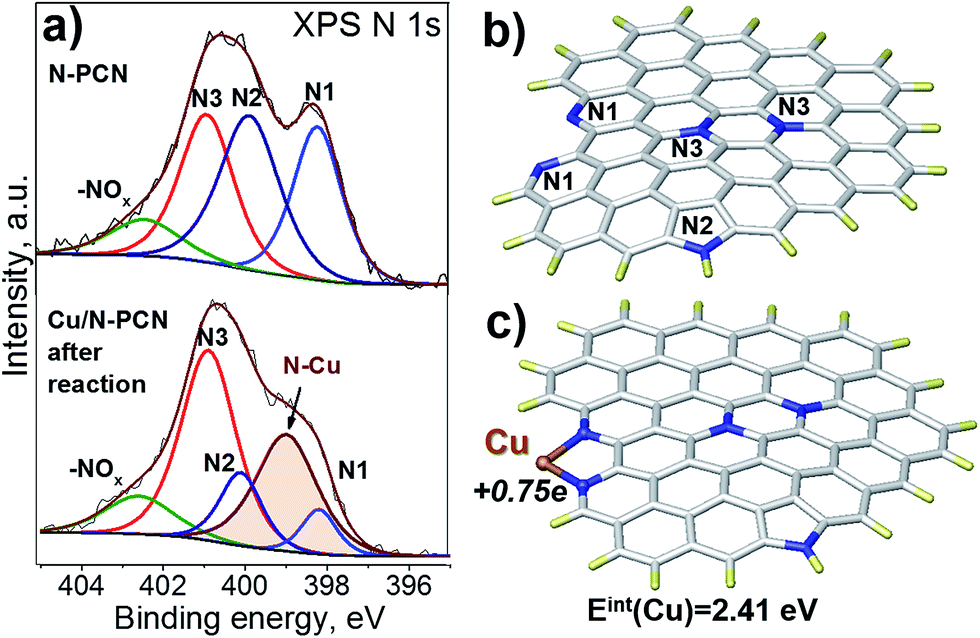 | ||
| Fig. 6 (a) N 1s XPS spectra of the N-PCN support and Cu/N-PCN catalyst after the reaction, (b) an N-doped graphene fragment model, and (c) a model of binding of a Cu atom to the N-doped graphene fragment obtained after optimization at the PBE/LACVP level. The white color indicates C, dark blue – N, yellow – H, and brown – Cu. The interaction energy of the Cu atom and NBO charge on it are indicated. | ||
The data on the reversed change in the intensity of the pyrrolic and graphitic nitrogen lines could also be explained by the formation of a Cu complex with involvement of pyrrolic nitrogen causing a shift of the pyrrolic line toward the position of the graphitic line. This feature was not seen for the Pd catalyst on a similar support13 indicating the different character of interaction of Cu and Pd species with N species of the support.
DFT study
To confirm that the interaction of a Cu atom with nitrogen species is sufficiently strong, we performed a DFT study. As our carbon is a graphitic material, we modeled it using a graphene fragment. The calculation of the location of a Cu atom on the non-doped graphene basal plane did not point to a stable configuration indicating that defects, vacancies or edge sites together with functional groups should be considered. For this reason, we introduced a pair of pyridinic species on the armchair edge (1,10-phenanthroline-like site), pyrrolic and graphitic N species (Fig. 6b) in accordance with the XPS N 1s spectrum (Fig. 6a). The interest towards the 1,10-phenanthroline-like site is explained by the results of earlier studies, where we showed that such nitrogen species strongly interact with single Pt-group metal atoms, and the attached metal atom may also lead to dissociation of the bonds in the incoming formic acid molecule, which leads to the further formation of H2 and CO2 products.1,12,13The present calculations found that the interaction energy of a Cu atom with a pair of pyridinic species (Fig. 6c) is equal to 2.41 eV, which is higher than that obtained earlier for the interaction of a Pd atom (1.9 eV), but lower than that for a Pt atom (2.97 eV) attached to a similar graphene fragment. The distance between the Cu atom and nitrogen atom in this configuration is 0.19 nm and the natural bond orbital (NBO) charge on the Cu atom is equal to +0.75e. The latter value is significantly higher than those for the Pt (+0.24e) and Pd (+0.32e) atoms and may affect further interaction with the formic acid molecule. The high charge is in accordance with the electron-deficient nature of Cu atoms evidenced by XPS data after the reduction (Fig. 5b). Additionally, the calculations showed that the coordination of the Cu atom to the pair of pyridinic atoms should result in the shift of the binding energy of N 1s to a higher region by 1.47 eV. This value is higher than that obtained in the XPS experiments (0.8 eV, Fig. 6a), but the direction of the shift well fits the results of our measurements and the literature data.35
We have also considered a few configurations with single pyridinic nitrogen species to stabilize a Cu atom (Fig. S5†). The Cu atom interacts even stronger with an armchair site consisting of a carbon–nitrogen pair (Fig. S5a†). In this case, the interaction energy is equal to 3.57 eV and the NBO charge is equal to +0.58e. There is a question, however, whether unterminated carbon may exist under our conditions. The bonding of the Cu atom with the pyridinic atom is weaker, when this atom is located at an atomic vacancy (Fig. S5b†). The interaction energy is equal to 2.12 eV and the NBO charge is +0.93e. Interestingly, the C–C bond in the carbon pentagon breaks because of geometry optimization and the Cu atom links to the pyridinic nitrogen atom and two sp2-carbon atoms of the vacancy. Finally, we showed that a pyridinic atom surrounded by hydrogen-terminated carbon atoms on the graphene edge does not interact with a Cu atom as strongly as in the previous cases, providing an interaction energy of only 1.06 eV and NBO charge of +0.42e (Fig. S5c†).
In addition, high energy of interaction (1.92 eV) is obtained for the interaction of Cu atom with the deprotonated pyrrolic nitrogen atom (Fig. S5d†). The NBO charge on the Cu atom is also high (+0.60e). Here, the properties of the nitrogen atom are close to the properties of pyridinic nitrogen. An upshift of the N 1s core level by 0.97 eV should be expected in this case, which is close to the experimental data for the upshift of the pyridinic line (Fig. 6a).
For the normal pyrrolic nitrogen, the NBO charge and interaction energy of a Cu atom were very low and equal to +0.18e and 0.13 eV, respectively (Fig. S6a†). These data indicate that the interaction of a single Cu atom with the pyrrolic nitrogen atom is hardly possible despite that the N 1s binding energy upshift for this nitrogen species was equal to 1.07 eV, which is close to the shift of the pyrrolic nitrogen line towards the graphitic nitrogen line observed by XPS (Fig. 6a). Hence, we can suppose that pyrrolic sites seen by XPS could be more complex being located in proximity to pyridinic sites, which attach a Cu atom directly. We have considered such a site involving both pyridinic and pyrrolic nitrogen atoms located in a zigzag configuration (7-azaindole-like site) in Fig. S6b.† It turned out that the Cu atom interacted with the pyridinic nitrogen atom with the interaction energy of 1.1 eV acquiring an NBO charge of +0.38e. In this case, upshifts of the binding energies of the pyridinic and pyrrolic nitrogen atoms by 1.24 eV and 0.4 eV are expected, respectively. These data indicate that the real site could involve both nitrogen species in proximity providing a sufficiently strong energy of interaction with the Cu atom and the shifts of the binding energy of the N 1s lines in correct directions.
The Cu atom does not interact directly with the graphitic nitrogen, but prefers bonding to the neighboring carbon atom (Fig. S6c and d†). When there is a single graphitic nitrogen atom, the interaction is even endothermic (−0.14 eV). The interaction energy is low (0.78 eV), when two graphitic nitrogen atoms are present in proximity. This should lead to a shift of the N 1s binding energy to a lower region by only 0.14 eV. Hence, the situation is quite similar to that observed for the graphene plane without nitrogen species. The NBO charges are also low and equal to +0.17e and +0.19e, respectively. These data indicate that the graphitic nitrogen atoms may not stabilize the Cu atom on the graphene fragment. In contrast, for a cluster of 20 atoms, Zhang et al.11 found the strongest interaction with the graphitic nitrogen (2.14 eV) as compared to pyrrolic and pyridinic nitrogen atoms. This attachment led to a very small negative shift of the binding energy related to the graphitic nitrogen while no other changes were observed by these authors. However, in contrast to our study the authors used microporous carbon, a higher concentration of Cu and considered only a vacancy site for location of the pyrrolic and pyridinic species. The trend could change, if the attachment would be considered on the edge site.
Therefore, the formation of the atomic Cu species attached to pyridinic nitrogen species of the support on the edge of the graphene sheet provides the strongest interaction as compared to interaction with graphitic and pyrrolic nitrogen species. Hence, single Cu atoms observed by atomic resolution HAADF/STEM (Fig. 4c) are most probably attached to these pyridinic species of the support.
Catalytic study
The reaction of formic acid was performed while heating every catalyst up to 600 K with a heating rate of 2 K min−1 in two heating cycles, during which the conversion was measured. The first heating cycle was considered as a necessary pretreatment. This pretreatment was performed in order to reduce Cu oxides and stabilize the catalyst before the second heating cycle, which was used for comparison of the activity of the samples (Fig. 7). The first and second heating cycles are also compared in Fig. S7.† A hysteresis was always observed. Interestingly, for the N-free Cu samples, the formic acid conversion was lower in the second heating cycle than in the first one, while the opposite was observed for the N-doped catalyst. Hence, activation of the N-doped Cu catalyst and deactivation of the N-free Cu catalysts in formic acid took place. The decrease of the activity for the N-free samples in the second heating cycle as compared to the first one is attributed to sintering of Cu particles, as this was in accordance with the increase of the mean particle size in the Cu/PCN catalyst after the reaction (Fig. 3b). In contrast, the activation of the N-doped catalyst in formic acid could be related to further formation of active Cu sites by reduction of oxidized Cu species by formic acid. We suppose that after the first heating cycle some Cu species are still in an inactive oxidized state and they could be further reduced during a longer time of treatment in formic acid. This was in accordance with the XPS – H2 reduction experiments (Fig. 5a and b), which demonstrated slower reduction of the Cu/N-PCN catalyst as compared to the Cu/PCN catalyst.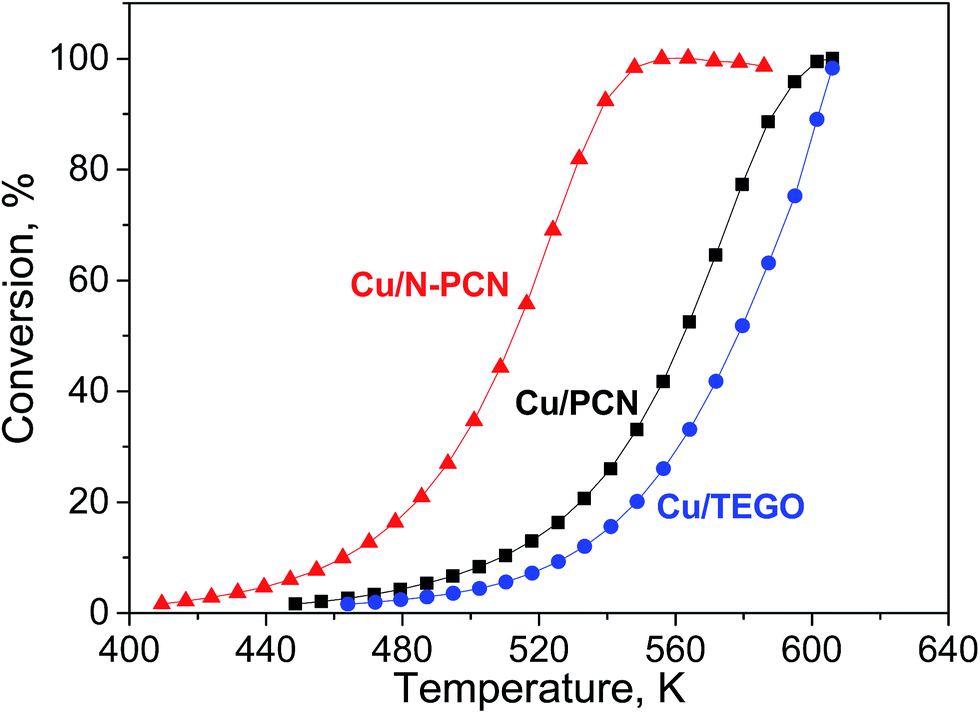 | ||
| Fig. 7 Formic acid conversion – temperature dependences for the studied catalysts in second heating cycles. | ||
Formic acid conversions obtained in the second heating cycles are shown in Fig. 7. At the same temperature, the conversions for the N-doped catalyst were significantly higher than those for the both N-free samples. The reaction rates determined per total number of Cu atoms at low conversions for the Cu/N-PCN and Cu/PCN catalysts differed by a factor of 4. The difference of the temperature for 50% conversion was about 50 K.
The selectivity of hydrogen formation at 50% conversion was also higher for the N-doped sample (97.4 vs. 94.4%). The stability of the Cu/N-PCN catalysts in formic acid decomposition was studied for 2 h at 513 and for 7 h at 478 K (Fig. 8). It is seen that the conversion of formic acid was very stable. This result is in accordance with the data of Yamada et al.,16 who reported that single Cu atoms and small clusters on N-doped graphene sheets are thermally stable until 773 K. Also, Kondo et al.36 compared the stability of monolayer Pt clusters on N-doped and N-free graphite surfaces under conditions of the H2–D2 exchange reaction and found a much better stability of Pt clusters on the N-doped graphite.
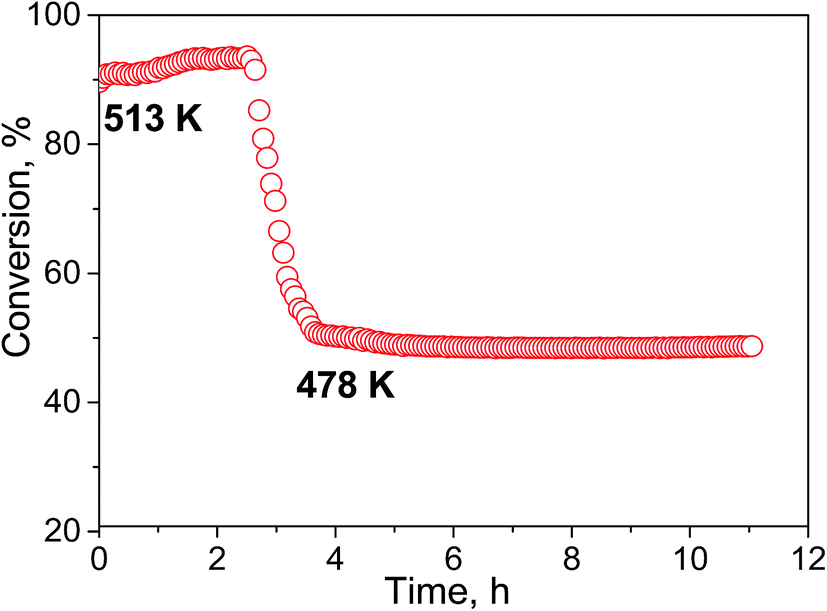 | ||
| Fig. 8 Long-term stability experiment with the Cu/N-PCN catalyst for formic acid decomposition. | ||
Therefore, the catalytic data indicate a substantially better performance of the N-doped Cu catalyst in the reaction of formic acid decomposition as compared to the N-free catalysts. This improvement in performance is attributed to the presence of single Cu atoms strongly interacting with the N species of the support, which could participate in the reaction directly or participate as anchoring sites for the formation of active nanoclusters or clusters. We showed that the character of interaction of Cu species with the N-doped and N-free supports is different. The mobility of Cu species is decreased significantly by a strong interaction with the N containing support preventing sintering. The oxygen functional groups do not provide the necessary strong interaction with Cu species, as Cu particles of a much bigger size were formed on the N-free support and increased significantly in size during the reaction (Fig. 3b).
The performed XPS study found some important differences in the state of Cu in the initial and reduced samples for the N-doped and N-free carbon materials. In agreement with the concept of strong interaction, it was difficult to reduce the oxidized Cu species in the N-doped sample in contrast to those in the N-free sample. Moreover, electron-deficient Cu species were formed on the N-doped sample because of the reduction in hydrogen. According to XPS and DFT studies the single Cu atoms were stabilized by pyridinic nitrogen species located at the edge of the graphene sheet (Fig. 6c and S5a†). This interaction is chelate binding of Cu ions, similar to that taking place in Cu(II) complexes with 1,10-phenanthroline, providing a stronger interaction with transition metal ions than in the case of coordination by single pyridinic nitrogen atoms. The high activity of the N-doped Cu catalyst was in accordance with the data of Lisitsyn37 who reported a strong increase of the rate of decarboxylation of aromatic acids by doping of Cu catalysts with a chelating promoter (2,2′-bipyridine or 1,10-phenanthroline).
Therefore, in the present work we have provided an understanding of the Cu interaction with the N-doped carbon support. This opens new ways for further studies and utilization of Cu catalysts on N-doped carbon materials for different applications. As a variety of Cu species, strongly interacting with the N-doped support, even not limited to those considered in the present work, could be formed in the studied systems, there is a question: which Cu species are catalytically active? This could be a subject of further research leading to synthesis of new Cu-based materials with these particular Cu species providing the highest activity, selectivity and stability.
Conclusions
By a combination of instrumental methods (atomic resolution HAADF/STEM, XPS, XRD, etc.) we have shown that incorporation of 5.7 at% of nitrogen into a mesoporous carbon support leads to a dramatic increase in dispersion and activity in formic acid decomposition and stability of Cu based catalysts prepared on such supports. In accordance with the results of the DFT study, we attribute these effects to a strong interaction of Cu with the edge pyridinic nitrogen atoms, which stabilize metal species in the form of strongly bound electron-deficient single atoms, which could be the anchoring sites for the growth of nanoclusters and clusters. The clusters on the N-doped carbon were by a factor of 7 smaller than those particles obtained on the N-free carbon after the reaction.Acknowledgements
This study was conducted with the financial support of the Russian Science Foundation (Grant 16-13-00016). The authors highly appreciate the assistance of Mr E. Lobiak and Dr M. Kanygin.References
- M. Zacharska, L. G. Bulusheva, A. S. Lisitsyn, S. Beloshapkin, Y. Guo, A. L. Chuvilin, E. V. Shlyakhova, O. Y. Podyacheva, J. J. Leahy, A. V. Okotrub and D. A. Bulushev, ChemSusChem, 2017, 10, 720–730 CrossRef CAS PubMed.
- E. Iglesia and M. Boudart, J. Catal., 1983, 81, 214–223 CrossRef CAS.
- S. Li, J. Scaranto and M. Mavrikakis, Top. Catal., 2016, 59, 1580–1588 CrossRef CAS.
- D. A. Bulushev and J. R. H. Ross, Catal. Today, 2011, 163, 42–46 CrossRef CAS.
- J. Ma, C. Park, N. M. Rodriguez and R. T. K. Baker, J. Phys. Chem. B, 2001, 105, 11994–12002 CrossRef CAS.
- A. Dandekar, R. T. K. Baker and M. A. Vannice, J. Catal., 1999, 183, 131–154 CrossRef CAS.
- L. J. Jia, D. A. Bulushev, O. Y. Podyacheva, A. I. Boronin, L. S. Kibis, E. Y. Gerasimov, S. Beloshapkin, I. A. Seryak, Z. R. Ismagilov and J. R. H. Ross, J. Catal., 2013, 307, 94–102 CrossRef CAS.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- The Chemistry of Organocopper Compounds, ed. Z. Rappoport and I. Marek, Wiley, Chichester, 2009, vol. 2 Search PubMed.
- V. G. Ramu, A. Bordoloi, T. C. Nagaiah, W. Schuhmann, M. Muhler and C. Cabrele, Appl. Catal., A, 2012, 431, 88–94 CrossRef.
- P. Zhang, Q. N. Wang, X. Yang, D. Wang, W. C. Li, Y. Zheng, M. Chen and A. H. Lu, ChemCatChem, 2017, 9, 505–510 CrossRef CAS.
- D. A. Bulushev, M. Zacharska, A. S. Lisitsyn, O. Y. Podyacheva, F. S. Hage, Q. M. Ramasse, U. Bangert and L. G. Bulusheva, ACS Catal., 2016, 6, 3442–3451 CrossRef CAS.
- D. A. Bulushev, M. Zacharska, E. V. Shlyakhova, A. L. Chuvilin, Y. Guo, S. Beloshapkin, A. V. Okotrub and L. G. Bulusheva, ACS Catal., 2016, 6, 681–691 CrossRef CAS.
- M. Zacharska, O. Y. Podyacheva, L. S. Kibis, A. I. Boronin, B. V. Senkovskiy, E. Y. Gerasimov, O. P. Taran, A. B. Ayusheev, V. N. Parmon, J. J. Leahy and D. A. Bulushev, ChemCatChem, 2015, 7, 2910–2917 CrossRef CAS.
- J. A. Herron, J. Scaranto, P. Ferrin, S. Li and M. Mavrikakis, ACS Catal., 2014, 4, 4434–4445 CrossRef CAS.
- Y. Yamada, M. Miyauchi, J. Kim, K. Hirose-Takai, Y. Sato, K. Suenaga, T. Ohba, T. Sodesawa and S. Sato, Carbon, 2011, 49, 3375–3378 CrossRef CAS.
- M. Gaboardi, A. Bliersbach, G. Bertoni, M. Aramini, G. Vlahopoulou, D. Pontiroli, P. Mauron, G. Magnani, G. Salviati, A. Zuttel and M. Riccò, J. Mater. Chem. A, 2014, 2, 1039–1046 CAS.
- E. V. Shlyakhova, L. G. Bulusheva, M. A. Kanygin, P. E. Plyusnin, K. A. Kovalenko, B. V. Senkovskiy and A. V. Okotrub, Phys. Status Solidi B, 2014, 251, 2607–2612 CrossRef CAS.
- B. D. Cullity, Elements of X-Ray Diffraction, Addison-Wesley Publishing Company, Reading, Massachusetts, USA, 1978 Search PubMed.
- S. Krumm, in European Powder Diffraction: Epdic Iv, Pts 1 and 2, ed. R. J. Cernik, R. Delhez and E. J. Mittemeijer, 1996, vol. 228, pp. 183–188 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. Y. Obaid, A. O. Alyoubi, A. A. Samarkandy, S. A. Al-Thabaiti, S. S. Al-Juaid, A. A. El-Bellihi and E. M. Deifallah, J. Therm. Anal. Calorim., 2000, 61, 985–994 CrossRef CAS.
- J. Ma, N. M. Rodriguez, M. A. Vannice and R. T. K. Baker, J. Catal., 1999, 183, 32–44 CrossRef CAS.
- D. Tokozakura, R. Nakamura, H. Nakajima, J. G. Lee and H. Mori, J. Mater. Res., 2007, 22, 2930–2935 CrossRef CAS.
- A. A. El Mel, R. Nakamura and C. Bittencourt, Beilstein J. Nanotechnol., 2015, 6, 1348–1361 CrossRef CAS PubMed.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- S. Poulston, E. Rowbotham, P. Stone, P. Parlett and M. Bowker, Catal. Lett., 1998, 52, 63–67 CrossRef CAS.
- C. Q. Zhu, A. Osherov and M. J. Panzer, Electrochim. Acta, 2013, 111, 771–778 CrossRef CAS.
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- J. Y. Kim, J. A. Rodriguez, J. C. Hanson, A. I. Frenkel and P. L. Lee, J. Am. Chem. Soc., 2003, 125, 10684–10692 CrossRef CAS PubMed.
- J. Pike, S. W. Chan, F. Zhang, X. Q. Wang and J. Hanson, Appl. Catal., A, 2006, 303, 273–277 CrossRef CAS.
- Y. Yano, M. Nakano and D. Takakura, Anal. Sci., 1997, 13, 355–358 CrossRef CAS.
- J. Wang, K. Wang, F. B. Wang and X. H. Xia, Nat. Commun., 2014, 5, 5285 CrossRef CAS PubMed.
- S. Peters, S. Peredkov, M. Neeb, W. Eberhardt and M. Al-Hada, Phys. Chem. Chem. Phys., 2013, 15, 9575–9580 RSC.
- R. Clarke, K. Latham, C. Rix, M. Hobday and J. White, CrystEngComm, 2005, 7, 28–36 RSC.
- T. Kondo, T. Suzuki and J. Nakamura, J. Phys. Chem. Lett., 2011, 2, 577–580 CrossRef CAS.
- A. S. Lisitsyn, Appl. Catal., A, 2007, 332, 166–170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta02282d |
| This journal is © The Royal Society of Chemistry 2017 |