
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted deep eutectic-solvothermal preparation of iron oxide nanoparticles for photoelectrochemical solar water splitting†
Oliver S.
Hammond
 a,
Salvador
Eslava
*b,
Andrew J.
Smith
c,
Jifang
Zhang
b and
Karen J.
Edler
*a
a,
Salvador
Eslava
*b,
Andrew J.
Smith
c,
Jifang
Zhang
b and
Karen J.
Edler
*a
aCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: k.edler@bath.ac.uk
bDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: S.Eslava@bath.ac.uk
cDiamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot OX11 0DE, UK
First published on 5th June 2017
Abstract
Here, we present a new microwave-solvothermal method for the preparation of iron oxide nanostructures using deep eutectic solvents as a more sustainable reaction medium. By varying the synthesis temperature and solvent water fraction, the methodology offers control over iron oxide phase, size, and morphology, using efficient, rapid (10 minute) microwave heating. Synthesis with pure DES gives small (<5 nm) superparamagnetic samples of γ-Fe2O3 or α-Fe2O3, whereas hydrated DES yielded either nanoshards or large rhombohedral nanoparticles without the superparamagnetic response. Nanostructures were solution-cast onto F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SnO2 films. The photoelectrochemical response of the prepared photoanodes was assessed, with a maximum measured photocurrent response of 0.7 mA cm−2 at 1.23 V vs. RHE. We measured the solvent structure using synchrotron WAXS, demonstrating the differences between the dry and hydrated solvent before and after heat-treatment, and showing that the hydrated solvent is remarkably resilient to extensive degradation.
:SnO2 films. The photoelectrochemical response of the prepared photoanodes was assessed, with a maximum measured photocurrent response of 0.7 mA cm−2 at 1.23 V vs. RHE. We measured the solvent structure using synchrotron WAXS, demonstrating the differences between the dry and hydrated solvent before and after heat-treatment, and showing that the hydrated solvent is remarkably resilient to extensive degradation.
Introduction
Solution processes, whereby liquid solvents are used to facilitate chemical reactions and processes, dominate chemistry because of the unmatched convenience that they offer.1 This fact is problematic in the face of increasing levels of environmental regulation that curtail the application of volatile organic solvents, which make up the majority of solvents that are used today.2 Therefore, there is an increasing drive towards replacing traditional solvent systems with more environmentally-friendly ones. Important developments have included systems such as supercritical fluids,3 greener alternative molecular solvents,4 and ionic liquids (ILs).5 Of these alternative solvents, ionic liquids are particularly fascinating because of their intrinsically ‘designer’ nature that allows them to be optimised and tuned to suit certain processes.6 Deep Eutectic Solvents (DESs) represent another, newer, class of alternative mixed solvent system, formed upon the complexation of various hydrogen-bonding salts and charge-neutral species, to create a low-melting liquid from the ensuing melting point depression.7 Originally described as a sub-category of ILs, an increasing bank of evidence suggests that they are more akin to an ionic mixture,8 with high entropy arising from a relatively disordered nanostructure9 comprising hundreds of intermolecular bonding interactions with similar strengths.10 Regardless, this broad definition allows DES to be made from a wide array of components, providing even more ways to tune and tailor the solvent around the matter of interest.11 As a result, DES offer new and unprecedented opportunities to improve upon the sustainability of important and industrially-relevant chemical processes.12 Research interest is therefore increasing rapidly in these solvents,7,13 particularly for the preparation of nanomaterials,14 where new structure-directing effects are becoming apparent.15Iron is the most common element on Earth. Because of their inherent sustainability and combination of interesting physicochemical properties, iron oxides are used for a wide variety of applications. For example, magnetic iron oxide nanoparticles such as Fe3O4 (magnetite) have useful applications in medicine (i.e. magnetic resonance imaging),16 ferrofluids, and biosensors.17 Haematite (α-Fe2O3) nanomaterials have attracted significant research interest as a vector for the sustainable storage of solar energy,18 much like many other metal oxides, because it can split water into H2 and O2 in a photoelectrochemical (PEC) process.19–28 This is facilitated by the 2.2 eV bandgap of haematite, allowing it to absorb a meaningful component of solar radiation. However, the PEC performance of haematite has been limited because of various factors; charge transport is often poor, the photon penetration depth produces carriers far from the active liquid junction, and the carriers that are produced subsequently recombine on the picosecond timescale.22 Therefore, a significant level of effort has gone towards nanostructuring haematite films to improve their PEC performance,21 and has resulted in structures such as nanowires,18 nanorods,29 and extremely active highly fractal structures,23 all offering various levels of activity. A common theme amongst newly-developed nanostructuring techniques for haematite thin film photoelectrodes is spiralling levels of complexity. Many nanostructures require increasingly exotic reagents, and experimental techniques with expensive instruments such as chemical vapour deposition,23 or spray pyrolysis and plasma-based methods.18 Here, we aim to use DESs as an alternative green solvent medium towards the goal of active haematite photoelectrodes. We present a synthesis of nanostructured iron oxides using a highly simple, fast, efficient and low-toxicity solvothermal process based on the biodegradable choline chloride–urea DES and its aqueous mixtures.30 To do this, we utilise a simple, energy-efficient microwave-solvothermal methodology.31
Experimental
DES preparation
The DES components choline chloride (Acros, 99%), and urea (Sigma-Aldrich, ≥99.5%) were used as provided. The deep eutectic solvent reline (1 choline chloride:2 urea) was prepared by mixing the two components in a sealed container and heating to 60 °C with regular agitation. After the formation of a homogeneous phase, the mixture was dried using a lyophiliser for one week, yielding the pure DES with a water content of 100.6 ppm, as assessed by 1H NMR. Hydrated DESs were subsequently prepared from dry reline by the addition of several mole equivalents of water, giving a choline chloride:urea:water ratio of 1:2:10, referred to here as reline-10w.
Solvothermal synthesis
In a polytetrafluoroethylene (PTFE) autoclave liner (CEM OMNI XP-1500), 5 g of Fe(NO3)3·9H2O (Acros, ≥99%) was added to 50 g of the chosen DES composition (either pure reline or reline-10w) to obtain a constant molal concentration of 0.25 mol kg−1. A standard (solvent only) sample was simultaneously prepared in a separate vessel, using the same quantity of the same DES composition but with no inorganic precursor. The two vessels were sealed, jacketed, and placed into a CEM Corporation MARS-5 microwave reactor equipped with temperature and pressure sensors. Using a maximum of 300 W of microwave power, the vessels were heated, and regulated to a maximum pressure of 5.5 MPa up to the desired temperature (100 °C, 150 °C or 200 °C) for a duration of 10 minutes. Following reaction, the samples were left to cool to below 60 °C before isolation. The reacted mixture containing inorganic materials was decanted into cellulose dialysis tubing (Sigma-Aldrich, 99.99% retention, MWCO 12.4 kDa) and dialysed against purified water (Elga, 18.2 MΩ) to remove ionic impurities. The products were then washed with ethanol to remove organic impurities prior to drying in an oven, yielding a series of fine powders of varying hue, from amber to crimson.Synchrotron small- and wide-angle X-ray scattering
Synchrotron SWAXS measurements of the as-prepared and heat-treated DES were made using the I22 instrument at Diamond Light Source, Harwell, UK. The incoming beam was monochromated at 18 keV, giving effective q-ranges for the two Dectris Pilatus-2M detectors of 0.007 Å−1 ≤ q ≤ 0.6 Å−1 at small-angle, and 0.4 Å−1 ≤ q ≤ 8 Å−1 for the wide-angle array. Samples were placed into soda-glass X-ray capillaries with a path-length of 1.5 mm and wall diameter of 10 μm. The capillaries were measured in a water-recirculating brass block sample holder with the temperature regulated to 30 °C. The instrumental background was removed and the data were reduced using the standard procedures of the DAWN software suite.32Nanomaterial characterisation
Powder X-ray diffraction of the prepared inorganic samples was carried out using a Bruker D8-ADVANCE instrument equipped with a Bruker VÅNTEC-1 CCD X-ray detector. The monochromated Cu-Kα radiation from the X-ray source has a wavelength of 1.5418 Å, and is slit-collimated to coincide upon the powder sample under Bragg–Brentano geometry. Diffraction data was collected across a scattering vector range of 1.42 Å−1 ≤ q ≤ 5.76 Å−1, corresponding with an angular range of 20°–90° 2θ in this configuration. Laboratory small- and wide-angle X-ray scattering (SWAXS) measurements of the prepared powders, films, and colloids were made using an Anton Paar SAXSess instrument, using a slit-collimated beam of Cu-Kα radiation and a static phosphorescent image plate as a SWAXS detector, to achieve a q-range of 0.01 Å−1 ≤ q ≤ 2.80 Å−1. The desmearing procedure for the slit geometry and subsequent SWAXS data processing was completed using Anton Paar SAXSQuant software. 1D 1H NMR experiments were carried out using d6-DMSO as a solvent, with data collected using an Agilent ProPulse 500 MHz NMR system, and processed using Bruker TopSpin software. Preliminary SEM and EDX measurements were made using a JEOL SEM6480LV instrument. FE-SEM images of the prepared photoanodes were collected using a JEOL JSM6301F instrument using 10 nm of conductive chromium coating. TEM imaging of the prepared nanoparticles was done using a JEOL JEM-2100 Plus instrument operating with 200 kV of accelerating potential.The magnetic properties of the prepared nanoparticles were determined using instrumentation at the Materials Characterisation Lab of the STFC ISIS Pulsed Neutron and Muon Source. Analogous procedures were carried out using either a Quantum Design PPMS Vibrating Sample Magnetometer, or a Quantum Design MPMS-XL SQUID Magnetometer. Samples (∼100 mg) were weighed into a gelatin capsule and packed with PTFE tape. The M(T) measurement of the magnetic moment as a function of temperature was carried out by first cooling the sample under zero-field conditions. After charging the instrument to a field of 100 oersted, measurements were periodically made as the sample chamber was heated to 300 K at a ramp rate of 10 K min−1. The magnetic moment of the samples was measured as a function of applied field at 300 K. The M(H) scan began under zero-field, before ramping the applied field to 5000 Oe at a rate of 100 Oe min−1. After settling at the desired field strength, the hysteresis behaviour was determined by ramping the field to −5000 Oe and back again to 5000 Oe at the same rate. Finally, the measured magnetic moment data were normalised to absolute units of emu g−1.
Photoanode fabrication
Photoanodes were prepared by adapting a previously-described solution-processed colloidal method from Sivula et al.22 A paste was prepared from 100 mg of the desired iron oxide powder and 0.1 mL of a 10% solution of acetylacetone (acac) in octanol. This paste was subsequently diluted by the addition of aliquots of a 10% solution of acac in isopropanol (IPA), until 2.5 mL of the acac/IPA solution had been added. The dispersions were then sonicated using an ultrasonic bath for 10 minutes. At this stage, a small (∼200 μL) aliquot of each colloid was sampled in case further (SWAXS) analysis was necessary. 1 mL of a 10% solution of hydroxypropylcellulose in IPA was then added to the colloidal dispersions as a porogen and viscosity-modifier, before a final sonication step of ten minutes. Aluminoborosilicate glass slides (Solaronix) coated with F:SnO2 (FTO) as a transparent conducting layer were used as a substrate, with a spacer of 40 μm invisible Scotch tape (3M). The final colloidal iron solution was doctor-bladed onto the substrate and allowed to air-dry. The dried films were pre-treated to ensure the removal of organics by heating in a tubular furnace up to 400 °C, with a temperature ramp rate of 1.5 °C min−1 and holding at temperature for 12 hours. After allowing to cool, the final treatment step was performed by placing samples directly into a tubular furnace at 800 °C for 20 minutes.
Photoelectrochemical testing
The solar photoelectrochemical water splitting performance of the prepared films was estimated using in-house photocurrent measurements. The Fe2O3-coated substrate was connected as the working electrode of a three-electrode cell, with an electrolyte solution of 1 M NaOH, a platinum counter electrode and a Ag/AgCl (3.5 M KCl) reference electrode. A PTFE circular mask (radius 0.3 cm) was placed on the reverse (uncoated) side of the sample, and this slide side was subsequently illuminated. Back illumination was used because the entire surface area of the film is wetted due to the high porosity, and therefore photogenerated hole sites are able to reach the liquid junction at any point within the film structure, and an optimal photocurrent value is subsequently obtained. A representative comparison of front and back illumination is given in the ESI.† Linear sweep voltammetry (LSV) measurements were made of the prepared photoanodes, under dark conditions and under simulated sunlight, and finally using a chopped shutter oscillating between light and dark conditions with a periodicity of 0.5 s−1. The prepared Fe2O3 electrodes were scanned between −300 mV and 800 mV against the Ag/AgCl reference using a scan rate of 20 mV s−1, and data reported herein are stated against the potential of the reversible hydrogen electrode (RHE). Simulated sunlight was calibrated to standard solar conditions (100 mW cm−2) at the sample position, derived from a 300 W Xe lamp and an AM1.5 filter. The electrode stability was assessed by holding the electrode at a constant potential of 0.22 V vs. Ag/AgCl (1.25 V vs. RHE), cycling between illuminated and dark conditions every 3 minutes for one hour.Results and discussion
Deep eutectic-solvothermal microwave synthesis
As far as was possible, elements of the synthesis described herein were specifically designed with the principles of green chemistry in mind.33 For example, reactive mixtures were prepared using a relatively high constant molal concentration of the iron precursor (0.25 mol kg−1), which takes advantage of the high mutual compatibility of many metal salts in DESs,34 thereby maximising the atom efficiency of the process whilst minimising solvent requirements. The choline chloride–urea DES (reline) was chosen for various positive attributes; it is simple to prepare, it is derived from cheap, abundant and natural precursors, and is biodegradable.35 Furthermore, we have shown previously that in inorganic syntheses based around metal nitrate precursors containing a highly-charged cation, the reline solvent effectively acts as a supramolecular catalyst for the reaction, with a pre-structuring effect bringing the reactants together to enable milder synthesis conditions.15 A microwave-solvothermal methodology was chosen because of obvious rate and efficiency improvements from microwave heating, which has already been shown to work synergistically with the benefits offered by using DESs as reaction media in the synthesis of organic molecules.36 Anhydrous procedures using reline and FeCl3 were discarded because HCl was liberated during the strongly exothermic mixing, which formed an intractable and corrosive brown mixture. Hydrated iron (III) nitrate (Fe(NO3)3·9H2O) was favoured, due to mixing safely, spontaneously and endothermically with the DES, and with the additional benefit of increasing tractability relative to the pure DES due to the presence of small hydrogen-bonding molecules (water and nitrate). A further viscosity improvement was achieved by dosing the DES with a known quantity of water to produce a DES–water mixture with hydration level of 10w (∼41 wt% H2O).37 This additional water eases the processing of the products,38 and simultaneously modifies the solvent environment, whilst remaining below the hydration level where DESs become aqueous solutions.30 Although different reaction times were trialled the results presented here are derived solely from the shortest reaction time (10 minutes) in the interests of energy efficiency; although in some cases relatively high (100, 150, and 200 °C) temperatures are used for the solvothermal reaction, in every case the power input was limited to a maximum of 300 W of microwave irradiation. Products are purified by dialysis against deionised water, such that a final ethanol rinse is the only time that a ‘traditional’ molecular volatile organic solvent is used. Hereafter, inorganic products are described as Fe-x-y, where x refers to the synthesis temperature and y refers to the chosen DES–water molar hydration ratio (w).15,30Characterisation of nanomaterials
Characterisation of the as-prepared iron oxide nanoparticles using powder X-ray diffraction (Fig. 1) demonstrated an unusual variation of phase as a function of the chosen synthesis temperature, occurring independently of the solvent composition. Samples synthesised at 200 °C demonstrated reflections corresponding with the rhombohedral α-Fe2O3 structure (haematite; JCPDS 85-0987). Conversely, those synthesised at 150 °C yielded Bragg peaks corresponding with one of either of the indistinguishable phases Fe3O4 (magnetite; JCPDS 85-1436) or γ-Fe2O3 (maghemite; JCPDS 25-1402). With γ-Fe2O3 representing a defect structure of Fe3O4, other techniques are required to distinguish between these two spinel ferrites. Efforts to characterise the different phases using Raman spectroscopy (shown in the ESI†) were confounded by their tendency to oxidise to α-Fe2O3 under even weak and brief laser illumination.39 However, as these conditions do not contain a Fe2+ ion source and are performed under air at a reasonably high temperature, γ-Fe2O3 is the most likely product.17 The XRD data also highlighted differences in the particle sizes between the products synthesised in hydrated and pure DESs, which is evident from the greater Bragg peak height and width for the hydrated syntheses. For the Fe-150-0 product no scattering features are observed, and the sample is essentially X-ray amorphous, due to peak broadening arising from small particle size. Scherrer analysis of the peak FWHM for the dominant (104) and (110) reflections of the haematite-type Fe-200-y products shows a mean crystallite size of 15.9 nm for Fe-200-0, as compared to 49.1 nm for the Fe-200-10 product. This suggests a greater degree of crystallite growth in the hydrated case, likely to reflect the faster kinetics of the less viscous aqueous mixture.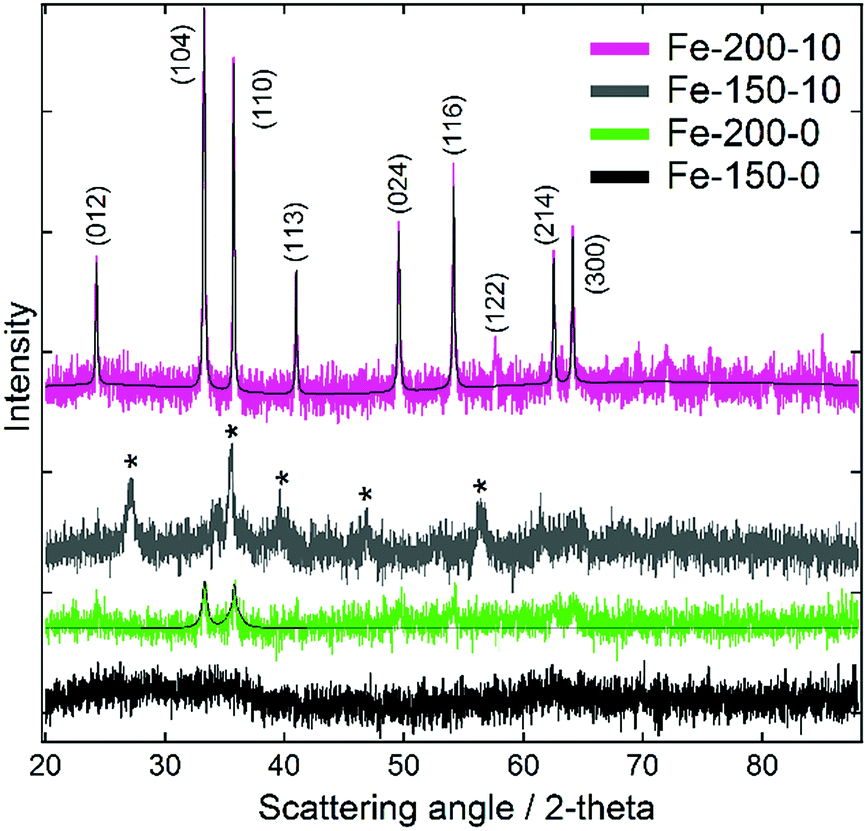 | ||
| Fig. 1 Powder X-ray diffraction data for the iron oxide nanoparticles prepared using pure reline and hydrated reline. Pseudo-Voigt fits calculated using Fityk software are shown for the haematite-type iron oxide products.40 Bragg peaks suspected to correspond with the γ-Fe2O3 phase are denoted with an asterisk. | ||
TEM imaging of the iron oxide nanoparticles showed a variety of nanostructures, with size and morphology determined by the synthesis conditions. Representative images are displayed in Fig. 2. In all cases, the prepared nanoparticles showed a strong tendency to aggregate, which is not surprising since no stabilising species were used. We observe that in the case of the reline-0w synthesis at 150 °C, aggregated spongelike composite spheres are formed with an average diameter of 50 nm. Interestingly, these spheres are composed of many extremely small spherical nanoparticles, which have an individual size of 2–3 nm and are relatively homogeneous. Upon adding water and reacting at the same temperature, a completely different 1D nanoshard morphology is formed, rather than the extremely small nanoparticles. These nanostructures are not as homogeneous as the particles prepared in the water-free case, and accurately determining a mean size is difficult because of the bundle formation. However, the nanoshards generally display a width ranging from 10–20 nm, with lengths of 80–100 nm. Interestingly, the lattice planes of the γ-Fe2O3 nanoshards can be seen in the TEM imaging without extremely high levels of magnification, and were found universally to run parallel with the major axis of extension. The TEM data therefore suggest that the main reason for the diffuse X-ray scattering of the Fe-150-0 product is small particle size and the accompanying peak broadening rather than any amorphous nature, and we believe that this is the same γ-Fe2O3 phase as the Fe-150-10 product, which is clearly larger and evidently crystalline. The nanoshard morphology may be driven by the selective capping of certain crystal planes by the DES components during crystal growth.
 | ||
| Fig. 2 TEM images of the prepared iron oxide nanoparticles as a function of synthesis temperature, and DES hydration level. | ||
Increasing the synthesis temperature, the Fe-200-0 product also yields a system of relatively monodisperse spheroid nanoparticles with a tendency to aggregate, much like Fe-150-0. In this case, the morphology is similar to the Fe-150-0 particles but with slightly larger sub-particles, with an apparent size of around 5 nm. This suggests once again that the diffuse α-Fe2O3 Bragg peaks in the XRD data are indicative not of an amorphous nature, but a small particle size. Increasing the water content for the 200 °C synthesis yields again a completely different nanostructure. Very large nanoparticles are formed, displaying a rhombohedral morphology that is evocative of the crystal structure of α-Fe2O3. The prepared particles are not homogeneous, and range from 100–200 nm in length, with a width of around 100 nm. These data therefore make it clear that the reaction temperature and reaction water content are two independent variables that both have an impact upon the nanoparticle growth rate. Additionally, there appears to be a subtle structure-directing effect exerted by the DES, because a similar morphological relation of the prepared nanoparticles was observed for a DES-solvothermal preparation of ceria, with smaller, less crystalline materials when the pure DES is used relative to the hydrated system, and 1D nanostructures formed in hydrated DES at low reaction temperatures.15 This is particularly interesting because ceria has a cubic fluorite structure that is entirely different from either the α-Fe2O3 or γ-Fe2O3 unit cell. The Fe-200-10 synthesis combines the most extreme reaction conditions of the highest water content and highest temperature, to give the highest in situ autoclave pressure and the most rapid kinetics of reaction and growth. The DES-solvothermal methodology therefore offers tunability of the size, shape, and phase of the prepared iron oxide nanoparticles, and does so whilst being rapid, simple, and environmentally friendly.
The magnetic properties of iron oxide nanoparticles are known to have a strong dependence upon the morphology, phase, and size. This has given rise to a number of important medical applications, such as their usage in bioseparations or as MRI contrast agents.16 The magnetisation of the prepared materials was therefore measured firstly as a function of temperature under 100 Oe of applied field, having been cooled under zero-field conditions (ZFC). Following this, M(H) measurements were made on the samples at 300 K to determine the hysteresis behaviour as the field was varied from 5000 Oe to −5000 Oe. The results of the M(T) scans are shown in Fig. 3. The observed ZFC magnetisation curves show completely different behaviours that can be related to the nanoparticle phase and size, which are themselves a function of the synthesis temperature and water content. The Fe-150-0 and Fe-200-0 nanoparticles show analogous behaviour, with a sharp rise in magnetisation as the temperature is increased, with a relatively strong peak of 0.16 emu g−1 for Fe-150-0 and 0.23 emu g−1 for Fe-200-0, before the magnetisation falls with increasing temperature. γ-Fe2O3 generally displays ferrimagnetic behaviour in the bulk phase, whereas α-Fe2O3 is typically a weak ferromagnet or canted antiferromagnet.41 This kind of magnetisation response is characteristic of superparamagnetic iron oxide particles, which are formed with hyperfine nanoparticles that are below 10 nm, such that each nanoparticle acts as a single-domain paramagnet, not large enough to have multi-domain ordering.41 This is confirmed by the observations made during TEM experiments. The superparamagnetic blocking temperatures for the prepared Fe-150-0 and Fe-200-0, respectively, are 69 K and 62 K.
 | ||
| Fig. 3 Magnetometry measurements showing variations in the magnetic moment of the samples as a function of the sample temperature, for the Fe-x-0 materials (upper panel) and the Fe-x-10 materials (lower panel). Measurements were made under a constant external field of 100 Oe. | ||
Conversely, the nanoparticles synthesised in hydrated DES show an entirely different and far weaker magnetic response. The Fe-150-10 product shows a minor fluctuation in the degree of magnetisation as a function of temperature, with two ‘fine’ transitions occurring at 110 K and 250 K, which is a more typical response for a nanoparticulate maghemite phase. The Fe-200-10 product shows classic α-Fe2O3 characteristics, with minimal magnetic response until a temperature of 250 K, at which point it undergoes the Morin transition to either a canted antiferromagnetic or ferromagnetic state.17 This is in accordance with the large, highly crystalline nanoparticles that can be observed using TEM. The suspected magnetic phases are confirmed by M(H) measurements at room temperature, which are shown in Fig. 4. The Fe-150-0 and Fe-200-0 products both show a completely straight line with negligible hysteresis, characteristic of a paramagnetic, or superparamagnetic state, in this case. The Fe-200-10 product shows a classic ferromagnetic behaviour, with a weak hysteresis of approximately 0.06 emu g−1. The Fe-150-10 material shows a weak ferrimagnetic response at room temperature with very minor hysteresis and curvature, and so this sample was also measured at 220 K, below the fine transition observed at 250 K. An increase in the overall magnetic moment and a slightly stronger hysteresis behaviour was observed, consistent with an increase in magnetic ordering occurring as the nanoparticles are cooled.42 Additionally, these magnetisation measurements offer definitive proof that the Fe-150-y nanoparticles are composed of a γ-Fe2O3 phase rather than Fe3O4, because the measured magnitude of the magnetisation is significantly lower than would be seen for magnetite.16
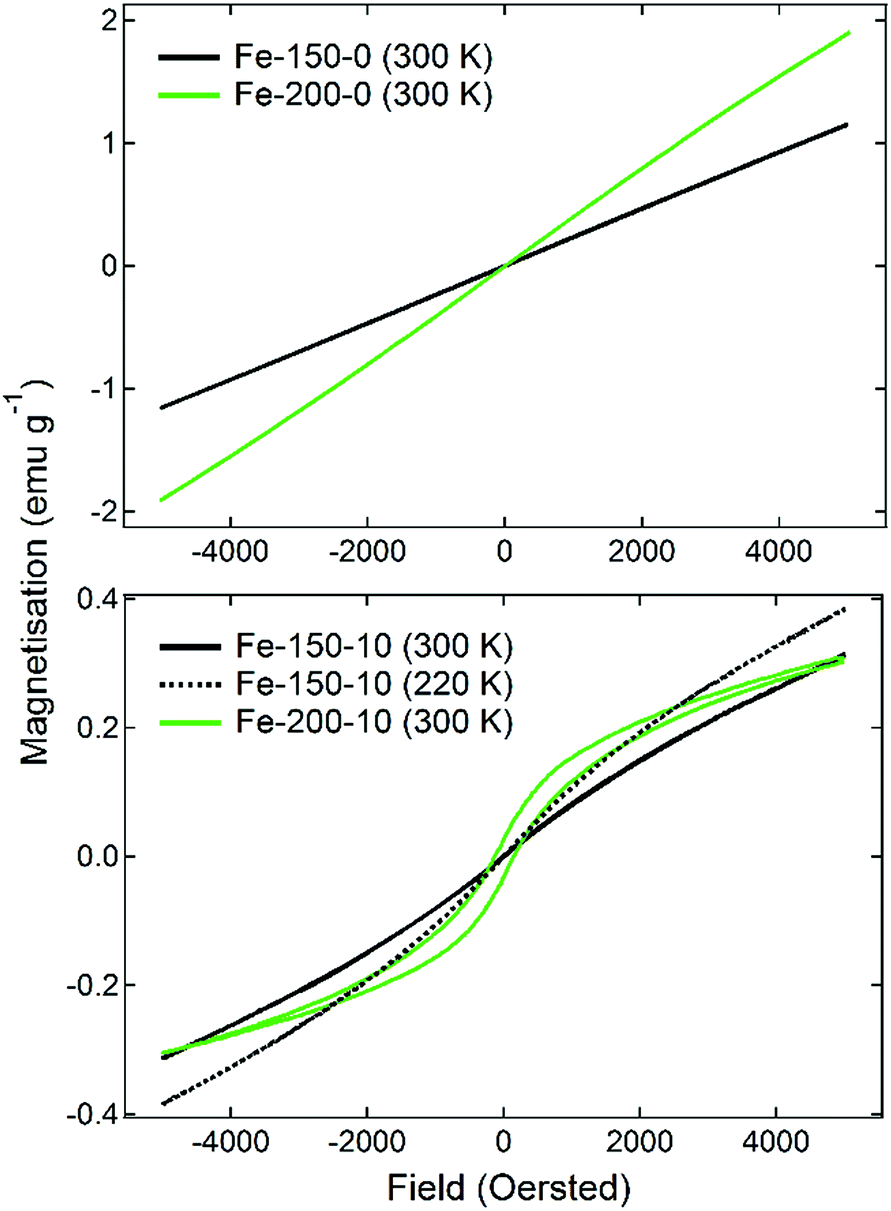 | ||
| Fig. 4 Magnetometry measurements showing the variations in the magnetic moment as a function of applied field, for the Fe-x-0 materials (upper panel) and Fe-x-10 materials (lower panel). Measurements were made using a field sweep rate of 100 Oe minute−1. | ||
Solvothermal reaction mechanism and solvent degradation structural studies
Efforts were made to determine the mechanism of solvothermal reaction. We note that this is not the first time that DES have been used for the preparation of iron oxides; Chen et al. used a co-precipitation route to prepare magnetic Fe3O4 nanoparticles,43 and Xiong et al. prepared haematite nanospindles using a precipitation method from heated, dry DES upon the addition of water.44 Despite the promising properties of the prepared materials, these precipitation methods cannot truly be described as a solvothermal process, but there are obvious similarities in the conditions because they propose the same urea-hydrolytic pathway that has been seen in numerous other preparations.45 The materials characterisation detailed above provided no evidence that the novel methodology reported here represents a significant deviation from other solvothermal protocols in terms of the urea hydrolysis and subsequent iron-oxide forming mechanisms, which have been previously and exhaustively addressed.15,46–48The reaction temperature and water fraction have an important effect upon the urea hydrolysis rate in DESs. The decomposition of DES at elevated temperature was noted previously by Parnham et al. in their studies of DESs as alternative solvent media for the synthesis of hybrid inorganic materials.46,47 Interestingly, they observed that the DES predominantly plays a templating function, with the controlled degradation of the labile species such as urea and its functionalised analogues delivering structuring agents for the synthesis. In our studies of the more closely related metal oxide ceria, we directly observed a pre-structuring of the reactive components within the reline mixture, effectively decreasing the activation energy for the reaction, whilst also noting hydrolysis and subsequent reaction of urea with solvated cerium ions.15 Based on the morphological dependence upon synthesis conditions, there may be some similar processes occurring in the iron oxide synthesis. Samples of the pure reline-0w and reline-10w solvents (50 g) without iron precursor were therefore placed into separate vessels and exposed to the microwave heating treatment alongside the reacting samples. The urea decomposition was monitored using 1H NMR spectroscopy, the results of which are shown in Table 1. It was observed that the pure reline DES did not undergo any significant urea hydrolysis after heat treatment at either 100 °C or 150 °C. The pure DES had to be heated to 200 °C before there was any quantifiable degradation of urea, with the choline chloride–urea ratio diminished to around 1:1.8 after this treatment. Interestingly, the iron-forming reactions for the pure DES were found to form a water-soluble crust around the autoclave lid. Analysis of this product using 1H NMR and powder X-ray diffraction suggests it contained a highly crystalline combination of the various likely DES degradation products, such as biuret, cyanuric acid, and ammonium carbonate (see ESI†). For the hydrated DES, significantly more urea hydrolysis was seen. Treatment at 100 °C and 150 °C again yielded almost identical results of a 1:1.8 choline chloride–urea final ratio, and in the most extreme circumstances, the 200 °C hydrated synthesis resulted in a 1:1, distinctly off-eutectic mixture of choline chloride–urea following the thermal treatment. Unlike the study of Querejeta-Fernández et al., we do not observe any significant signal corresponding with the formation of NH4OH in the solvent.48 Generally, when contrasting with previous syntheses, we observe significantly less solvent degradation, which is almost certainly a product of the rapid reaction times facilitated by the efficiency of microwave heating. This can be visualised by the relative simplicity of the NMR spectra collected after treatment (see ESI†), which can be contrasted with the extensive degradation observed when a DES is heat-treated in a conventional oven for a week.46
| DES composition | [Urea/ChCl] 100 °C | [Urea/ChCl] 150 °C | [Urea/ChCl] 200 °C |
|---|---|---|---|
| a It should be noted that the measured hydrolysis in the pure DES may not necessarily be representative of the reacting system, which contains additional low-level water from the iron precursor, and may experience some further effect from the paramagnetic iron content. Errors are stated assuming a standard 5% deviation in the veracity of the NMR integrals. | |||
| Reline-0w | 2.086 ± 0.104 | 1.939 ± 0.097 | 1.781 ± 0.089 |
| Reline-10w | 1.773 ± 0.089 | 1.804 ± 0.092 | 1.014 ± 0.051 |
We subsequently aimed to determine the degradative effects of urea hydrolysis upon the nanostructure of the DES and the hydrated DES. It is unclear what effect that both the loss of urea, and the subsequent integration of the hydrolysis products themselves, have on the overall solvent structure in the case of both the pure and hydrated systems.49 To understand this, measurements of the primary structure factor S(q) of the solvents were made using synchrotron wide-angle X-ray scattering (WAXS) at beamline I22 of Diamond Light Source, UK. Data were collected before and after heat-treatment, with background-corrected and normalised scattering patterns shown in Fig. 5. In pure d17-choline chloride:urea, there are two primary constructive scattering interferences, giving peaks at 1.45 and 2.15 Å−1, which represent the two most common interaction lengths in the disordered liquid, and respectively describing real-space separations of 4.3 and 2.9 Å (d = 2π/q). The measured X-ray structure factor S(q) for reline therefore matches accurately with the scattering form factor and primary correlation lengths observed in the pure DES by wide q-range neutron diffraction.9 Upon adding water, the data show that the system becomes more disordered, signified here by the additional peak broadening.50 The intensity of the secondary ‘shoulder’ peak at 2.15 Å−1 is increased noticeably in the hydrated system, signifying the partial loss of the DES–DES (i.e. the choline–choline, choline–chloride, choline–urea, urea–urea and urea–chloride) interactions.8 Despite this, the position of the main scattering feature (at q = 1.45 Å−1; d = 4.3 Å) remains in the hydrated system. This demonstrates that the hydrated DES still contains a significant portion of the pure DES hydrogen-bonding nanostructure,30 and validates the approach of adding water as a processing enhancement option for DES.37
 | ||
| Fig. 5 WAXS data from beamline I22, Diamond Light Source, UK, showing untreated and microwave-treated samples of the pure reline-0w DES (a), and the hydrated reline-10w DES (b). Data are background-corrected and the intensity of the major correlation peak scaled to unity, to facilitate comparison and account for manufacturing variations in the X-ray capillaries used for measurement. | ||
In the case of the 100 °C and 150 °C heat treatments, there is very little variation in the structure between both the pure solvent and the hydrated solvent before and after microwave treatments. The pure DES is confirmed to degrade slightly because of the minor peak broadening observed when the solvent is treated at 150 or 200 °C, whereas the reline-10w DES sees some broadening alongside a significant extension to the satellite correlation at 2.15 Å−1, signifying that the hydrated DES is somewhat more affected by the heat treatment, as was suggested by the NMR analysis. However, in both instances the DES display remarkable nanostructural resilience with regard to shifting to an off-eutectic composition.8 This is likely to be a product of the short reaction time that is facilitated by the usage of microwave irradiation, the hydrogen-bonding contribution of certain likely degradation product molecules such as isocyanuric acid and biuret, and the hydrogen-bonding contribution from water in the hydrated system. These findings raise the possibility that the DES could even be recovered and recycled after such syntheses, further improving the efficiency.
Attempts to further reduce the reaction time and temperature met kinetic limitations; unlike the 150 °C or 200 °C preparations, syntheses performed for 10 minutes at 100 °C had only fractional Fe2O3 yields of 40% (Fe-100-0) or 73% (Fe-100-10). EDX measurements suggested that the prepared Fe-100-0 product had only a surface coating of the desired Fe2O3, with the particulate bulk composed of crystallised FeCl3. Because this salt was not used, this must represent the dominant dynamically-solvated iron species in the reline DES, which is necessarily chloride-rich.51 In spite of any preferential solvent-reactant structuring, it seems likely that in this case the kinetic limitation of the pure DESs lies in their relatively high viscosity, which represents a diffusion-limited regime. In the case of the aqueous DES this kinetic limitation is mitigated, as the additional water has the effect of dramatically reducing the solvent viscosity and hence, increasing the solvent self-diffusion coefficient relative to the pure DES.49 Therefore, there are clearly some synthetic advantages to be had by using hydrated DESs over the pure form. The optimal conditions for a hydrated deep eutectic-solvothermal reaction can be found by tailoring the DES hydration level to obtain the desired combination of solvent diffusion and pre-structuring effects, whilst remaining below an aqueous regime.37
Application as photoelectrodes
Photoanodes were prepared by adapting a previously-described solution-processed colloidal methodology, rather than developing a DES-based method because of the likely introduction of impurities such as chloride and other organics from the incomplete calcination of the low vapour pressure ionic mixture.52 In this process, a stable colloid of the iron oxide nanoparticles is prepared,22 using acetylacetone to stabilise the nanoparticles by acting as a ‘capping’ hydrotrope within the isopropanol dispersant phase. A structure-directing agent (hydroxypropylcellulose) is then added to ensure the produced electrodes are porous after the thermal treatment.21 The films were cast onto F:SnO2-coated aluminoborosilicate glass and heat-treated in two steps, with a preliminary 400 °C, 12 hour treatment to remove the organic residues and convert the γ-Fe2O3 phase to the photoactive and stable α-Fe2O3 phase. The final step is a fast 20 minute treatment at 800 °C. This latter treatment is unfortunately intensive, but is necessary to produce sufficiently active haematite photoelectrodes without precious metal dopants, because the Sn4+ diffuses into the iron oxide during sintering, modifying the lattice parameters and resulting in electronic modification.22 The optical properties were found to vary according to the nanoparticle size, with the most transparent films prepared from the smallest nanoparticles (Fe-150-0) and the most opaque films derived from the largest nanoparticles (Fe-200-10). FE-SEM measurements of the prepared photoanodes after the dual heat treatments yielded a similar morphology to those prepared previously from the related casting method, with iron oxide from the thermal decomposition of Fe(CO)5,22 and these are shown in Fig. 6. These images show the partial sintering and growth of the nanoparticles, which form necked aggregates reaching a diameter of around 50 nm in the Fe-150-0 film and 100 nm in the Fe-150-10 film. The larger feature size of the Fe-150-10 film can be related to the larger size of the nanoshards that are used for the preparation, as compared to the miniscule Fe-150-0 nanoparticles. This is additionally reflected in the clearly more tightly-packed Fe-150-0 film, and the very large feature size of the Fe-200-10 film. The porous nature of the photoelectrode is confirmed by the imaging, suggesting a high accessible surface area, ideal for the photoelectrochemical splitting of water.21
 | ||
| Fig. 6 FE-SEM images of the Fe-150-0 (a), Fe-150-10 (b), Fe-200-0 (c) and Fe-200-10 (d) films after calcination, measured using a thin-film coating (10 nm) of chromium. Scale bars depict 1 μm. | ||
The prepared photoanodes were measured under standard solar conditions (100 mW cm−2 at the sample position) in a three-electrode configuration, using a 1 M NaOH electrolyte, platinum counter-electrode and 3.5 M KCl reference electrode. The reverse (uncoated) side of the electrode was found to give the maximum photocurrent response, because despite the complete wetting of the electrode nanostructures by the electrolyte, more photoinduced electrons are generated closer to the FTO substrate than with front illumination, with more electrons then moving to the cathode. Additionally, there is inevitable absorption of light with a corresponding decay in intensity when films are greater than a threshold thickness.53 An example of front vs. back illumination performance is given in the ESI.† Linear sweep voltammetry data for the electrodes under both light and dark conditions are shown in Fig. 7, and the calculated values of the photocurrent density at 1.23 V versus the RHE are shown in Table 2. The majority of the prepared systems deliver a photocurrent density competitive with other examples in the literature, which can be related to the properties of the iron oxides used to prepare the respective photoelectrodes. The Fe-150-0 electrode has a photocurrent of 0.53 mA cm−2 at 1.23 V vs. RHE, identical to the value obtained for the Fe-200-0 electrode. This is representative of the very similar nanoparticle size and morphology of the two systems, with the minor differences between the two systems negated after the high-temperature sintering treatment. The anode derived from the Fe-200-10 rhombohedral nanoparticles gave the weakest measured photocurrent response of 0.19 mA cm−2. This can be related to the particularly large and more difficult to coalesce, low-surface area nanoparticles that this anode is derived from. Conversely, the strongest observed photocurrent response was for the Fe-150-10 film, derived from the nanoshards prepared in the hydrated DES at lower temperature. This anode gave a photocurrent response of 0.7 mA cm−2 at 1.23 V vs. RHE, which is competitive with other literature preparations, and notably higher than photoanodes prepared using the thermal decomposition method from Fe(CO)5, despite the possibility of our products passing through a hydroxyl-containing goethite phase due to the synthesis mechanism.54 The FE-SEM data (Fig. 6) reflect this, in the high solvent-accessible surface area of this electrode and the high quantity of elongated, necked arrays that are derived from the sintered shard nanostructures. We note that the average width of the features is commensurate with the photon penetration depth in haematite.22 This level of photocurrent response does not match extremely high performance benchmarks such as the 2.2 mA cm−2 that can be achieved by chemical vapour deposition of high-surface-area, porous cauliflower-type fractal haematite.23 However, the simple solvothermal process reported here is comparatively rapid, facile, and more environmentally benign, whilst not requiring CVD equipment, or volatile and harmful reagents such as tetraethylorthosilicate (TEOS).21
 | ||
| Fig. 7 Photoelectrochemical water oxidation data showing the normalised photocurrent density as a function of potential for the Fe-x-0 products (upper panel) and the Fe-x-10 products (lower panel), measured either in the dark (dashed lines) or under illumination (solid lines). Analogous data for the Fe-150-y samples are shown in the inset, measured using a chopping light shutter whilst a standard sweep of current as a function of voltage is performed. | ||
| DES composition | J 150 °C (mA cm−2) | J 200 °C (mA cm−2) |
|---|---|---|
| a Data are calculated as the mean photocurrent density from data measured during a sweep with a rate of 20 mV s−1, across the potential range 1.22–1.24 V versus the RHE, corresponding with a measurement time of 1 s. Errors are reported as the standard deviation of this value. | ||
| Reline-0w | 0.528 ± 0.015 | 0.532 ± 0.012 |
| Reline-10w | 0.701 ± 0.008 | 0.187 ± 0.002 |
Some interesting differences were observed in the photocurrent response between those products isolated from dry and hydrated DESs. Firstly, the different DES resulted in a slight shift in the position of the dark current onset potential. The dark current occurs at approximately 1.6 V for the Fe-x-0 materials, but consistently occurs at a lower potential of 1.55 V for the Fe-x-10 products. Moreover, the Fe-150-0 measurements do not show any significant transient in the dark current alongside the photocurrent onset potential, whereas there is a slight dark current at lower potentials for the Fe-x-10 films, likely due to trace contamination. Finally, an interesting feature is noted in the insets of Fig. 7. The prepared samples were also measured using sweeping current-potentiometry scans with a chopped shutter in order to determine the presence of photocurrent transients as the incident light is periodically cycled between on and off. The Fe-x-10 photoanodes show sharper characteristic transient spikes upon cycling, corresponding with a high concentration of photo-generated hole sites being generated within 100 nm of the semiconductor liquid junction (SCLJ).55 This suggests that the recombination step is likely to be limiting for both sets of materials,29 which can occur either in the bulk or at grain boundaries.18
Finally, the stability of the prepared photoanodes was measured in order to demonstrate the resilience of the prepared films. This was done by measuring at a constant potential, cycling between illuminated and dark with a periodicity of 0.33 min−1 for one hour, and representative data for the Fe-200-y electrodes are shown in Fig. 8. In each case, the fabricated photoanodes show good resistance to the repeated cycling. For the Fe-200-0 thin film, the average first photocurrent response of 0.532 ± 0.005 mA cm−2 drops to 0.526 ± 0.004 mA cm−2 after one hour of this treatment. This corresponds with an activity reduction of 5.3 μA cm−2, or just 1% of the total activity. Similarly, the Fe-200-10 photoanode response falls from the first average of 0.114 ± 0.001 mA cm−2 to the final measurement of 0.108 ± 0.001 mA cm−2, corresponding with a comparable absolute loss in photocurrent of 5.5 μA cm−2, but a 5% loss in this case due to the lower total photocurrent density of this anode. Therefore, the prepared electrodes are remarkably stable, with only minimal loss in photocurrent density after repetitive on-off cycling.
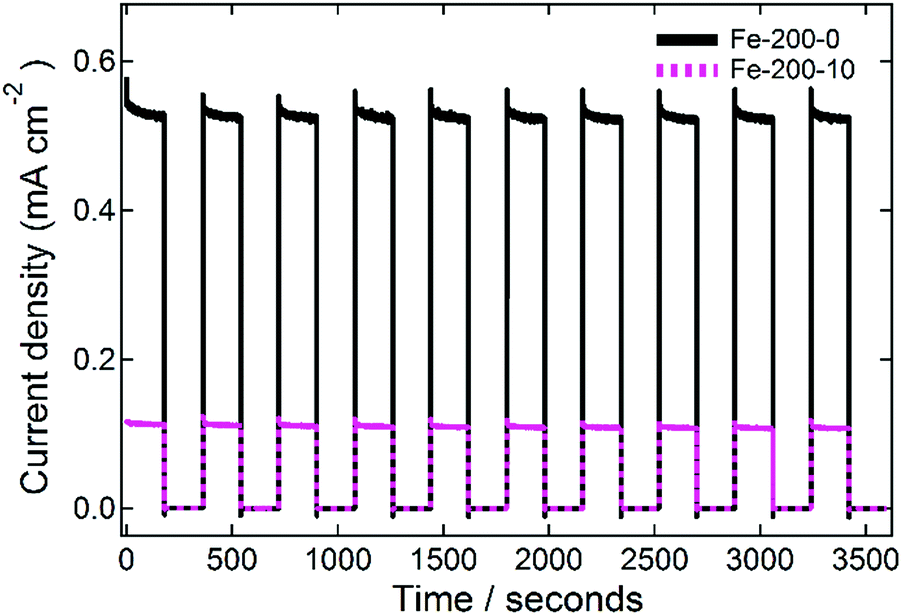 | ||
| Fig. 8 Stability testing data for the fabricated Fe-200-y photoanodes. The incident simulated-solar radiation was chopped on or off every 3 minutes for a total of one hour, whilst the photoanodes were held at a constant potential of 0.22 V vs. Ag/AgCl (1.25 V vs. RHE). | ||
Conclusions
We have demonstrated that the DES reline and its hydrated mixtures can be used as viable sustainable alternative solvents in the manufacturing of green materials for the photoelectrochemical splitting of water to hydrogen. The microwave-assisted solvothermal methodology is rapid, malleable, and represents a significantly more environmentally-friendly route towards this goal due to lower energy usage than traditional solvothermal methods and the avoidance of volatile organic solvents. We have found that the reaction mechanism is similar to previous preparations following a urea-hydrolysis pathway. The solvent degradation was monitored, showing that following the reaction the mixtures are in an off-eutectic composition. Synchrotron WAXS measurements showed that this has surprisingly little effect on the major correlation structure in the solvent, signifying their resistance to this change. We find additionally using WAXS that the hydrated solvent has a different structure from the pure system, with a more disordered structure, but one that retains some of the DES intermolecular bonding.The prepared iron oxide nanostructures vary in phase, size, and morphology as the synthesis conditions are varied. The DES was found to have some inherent structuring effect, in line with previous studies. Preparations using the pure DES always yielded very small nanoparticulates, whereas synthesis using hydrated DESs gave either 1D nanoshards at 150 °C, or large rhombohedral nanoparticles at 200 °C. Samples prepared at 150 °C were the γ-Fe2O3 phase, whereas the syntheses conducted at 200 °C yielded the α-Fe2O3 phase. Investigations into the magnetic properties of these nanoparticles showed that the small γ-Fe2O3 and α-Fe2O3 nanoparticles were sufficiently small that they exhibited superparamagnetic behaviour. The large, more crystalline samples synthesised with hydrated DESs showed ferrimagnetic or ferromagnetic hysteresis. Photoanodes were prepared from the nanoparticles using a previously-developed solution-processed colloidal method, and photoelectrochemical measurements of these showed a competitive photocurrent density, with a maximum measured photocurrent of 0.7 mA cm−2 at 1.23 V vs. RHE. Whilst short of the most extreme reported values, this is a strong response when considering the environmental credentials of the process that was used to prepare them. We additionally demonstrate that the measured photocurrent is remarkably stable under repeated cycling.
We therefore present here a new route towards functional and highly active iron oxide nanomaterials to be used in photocatalytic water splitting applications, based around greener DESs as a structure-directing solvent medium. These new fundamental insights into the DES role in nanomaterials synthesis, and in particular the solvent structure information from synchrotron WAXS studies will aid with the development of future, greener processes towards other nanomaterials using DESs and hydrated DESs.
Acknowledgements
Data supporting this paper have been made openly available, using the University of Bath Research Data Archive system (DOI: 10.15125/BATH-00376). We acknowledge the STFC ISIS Pulsed Neutron and Muon source (Rutherford Appleton Laboratory) and the UK Engineering and Physical Sciences Research Council (EPSRC) for co-funding the PhD studentship of OSH in the Centre for Doctoral Training in Sustainable Chemical Technologies, at the University of Bath (EP/L016354/1; STFC Studentship Agreement 3578). SE acknowledges EPSRC funding EP/P008097/1. We thank Diamond Light Source for access to the I22 SWAXS instrument under beamtime allocation SM15194. We thank the ISIS Pulsed Neutron and Muon source for access to VSM and SQUID magnetometers in the Materials Characterisation Laboratory (MCL), and Dr Gavin Stenning for aid in magnetometer operation. We acknowledge the University of Bath Chemical Characterisation and Analysis Facility (CCAF) for providing access to NMR spectroscopy. We thank the University of Bath Microscopy and Analysis Suite (MAS) for access to Raman spectroscopy, SEM, FE-SEM and TEM imaging, and aid with collecting data using the instrumentation.References
- C. Reichardt, Solvents and solvent effects in organic chemistry, Wiley-VCH, Weinheim, 4th edn, 2011 Search PubMed.
- F. M. Kerton, Alternative Solvents for Green Chemistry, Royal Society of Chemistry, Cambridge, 2009 Search PubMed.
- J. Peach and J. Eastoe, Beilstein J. Org. Chem., 2014, 10, 1878–1895 CrossRef PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 6358–6405 Search PubMed.
- N. V. Plechkova and K. R. Seddon, in Methods and Reagents for Green Chemistry: An Introduction, 2007, pp. 103–130 Search PubMed.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- R. Stefanovic, M. Ludwig, G. B. Webber, R. Atkin and A. J. Page, Phys. Chem. Chem. Phys., 2017, 114, 11060–11082 Search PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- M. Francisco, A. Van Den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Eur. J. Org. Chem., 2016, 612–632 CrossRef CAS.
- D. V Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef PubMed.
- O. S. Hammond, K. J. Edler, D. T. Bowron and L. Torrente-Murciano, Nat. Commun., 2017, 8, 14150 CrossRef CAS PubMed.
- M. Mohapatra and S. Anand, Int. Trans. J. Eng., Manage., Appl. Sci. Technol., 2010, 2, 127–146 Search PubMed.
- G. Sharma and P. Jeevanandam, RSC Adv., 2013, 3, 189–200 RSC.
- B. D. Chernomordik, H. B. Russell, U. Cvelbar, J. B. Jasinski, V. Kumar, T. Deutsch and M. K. Sunkara, Nanotechnology, 2012, 23, 194009 CrossRef PubMed.
- C. Ray and T. Pal, J. Mater. Chem. A, 2017, 5, 9465–9487 CAS.
- J. Gu, Y. Yan, J. W. Krizan, Q. D. Gibson, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Am. Chem. Soc., 2014, 136, 830–833 CrossRef CAS PubMed.
- J. Brillet, M. Grätzel and K. Sivula, Nano Lett., 2010, 10, 4155–4160 CrossRef CAS PubMed.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 15714–15721 CrossRef CAS PubMed.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2009, 113, 772–782 CAS.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS PubMed.
- I. Cesar, A. Kay, J. A. G. Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- N. Beermann, L. Vayssieres, S.-E. Lindquist and A. Hagfeldt, J. Electrochem. Soc., 2000, 147, 2456 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201702486.
- L. Pan, X. Liu, Z. Sun and C. Q. Sun, J. Mater. Chem. A, 2013, 1, 8299 CAS.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Náray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- A. P. Abbott, G. Capper, D. L. Davies and R. K. Rasheed, Chem.–Eur. J., 2004, 10, 3769–3774 CrossRef CAS PubMed.
- Q. Wen, J.-X. Chen, Y.-L. Tang, J. Wang and Z. Yang, Chemosphere, 2015, 132, 63–69 CrossRef CAS PubMed.
- K. M. Taylor, Z. E. Taylor and S. T. Handy, Tetrahedron Lett., 2017, 58, 240–241 CrossRef CAS.
- Y. Dai, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Food Chem., 2015, 187, 14–19 CrossRef CAS PubMed.
- N. Fechler, N. P. Zussblatt, R. Rothe, R. Schlögl, M.-G. Willinger, B. F. Chmelka and M. Antonietti, Adv. Mater., 2015, 28, 1287–1294 CrossRef PubMed.
- M. Hanesch, Geophys. J. Int., 2009, 177, 941–948 CrossRef CAS.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- F. Bødker, M. Hansen, C. Koch, K. Lefmann and S. Mørup, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 6826–6838 CrossRef.
- C. Pascal, J. L. Pascal, F. Favier, M. L. E. Moubtassim and C. Payen, Chem. Mater., 1999, 11, 141–147 CrossRef CAS.
- F. Chen, S. Xie, J. Zhang and R. Liu, Mater. Lett., 2013, 112, 177–179 CrossRef CAS.
- Q. Q. Xiong, J. P. Tu, X. Ge, X. L. Wang and C. D. Gu, J. Power Sources, 2015, 274, 1–7 CrossRef CAS.
- F. N. Sayed and V. Polshettiwar, Sci. Rep., 2015, 5, 9733 CrossRef CAS PubMed.
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Z. Slawin and R. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 4962–4966 CrossRef CAS PubMed.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS PubMed.
- A. Querejeta-Fernández, J. C. Hernàndez-Garrido, H. Yang, Y. Zhou, A. Varela, M. Parras, J. J. Calvino-Gàmez, J. M. Gonzàlez-Calbet, P. F. Green and N. A. Kotov, ACS Nano, 2012, 6, 3800–3812 CrossRef PubMed.
- C. D'Agostino, L. F. Gladden, M. D. Mantle, A. P. Abbott, E. I. Ahmed, A. Y. M. Al-Murshedi and R. C. Harris, Phys. Chem. Chem. Phys., 2015, 15297–15304 RSC.
- T. Zhekenov, N. Toksanbayev, Z. Kazakbayeva, D. Shah and F. S. Mjalli, Fluid Phase Equilib., 2017, 1–6 Search PubMed.
- J. M. Hartley, C. M. Ip, G. C. H. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed.
- O. Aschenbrenner, S. Supasitmongkol, M. Taylor and P. Styring, Green Chem., 2009, 1217–1221 RSC.
- S. Eslava, G. Eymery, P. Marsik, F. Iacopi, C. E. A. Kirschhock, K. Maex, J. A. Martens and M. R. Baklanov, J. Electrochem. Soc., 2008, 155(5), G115–G120 CrossRef CAS.
- D. Maiti, V. Aravindan, S. Madhavi and P. Sujatha Devi, J. Power Sources, 2015, 276, 291–298 CrossRef CAS.
- J. Krysa, M. Zlamal, S. Kment, M. Brunclikova and Z. Hubicka, Molecules, 2015, 20, 1046–1058 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Solvent degradation experiments, Raman spectra and residual solid in the reactors. See DOI: 10.1039/c7ta02078c |
| This journal is © The Royal Society of Chemistry 2017 |