
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH dependent photocatalytic hydrogen evolution by self-assembled perylene bisimides†
Michael C.
Nolan
 abc,
James J.
Walsh
cd,
Laura L. E.
Mears
bc,
Emily R.
Draper
ab,
Matthew
Wallace
b,
Michael
Barrow
b,
Bart
Dietrich
ab,
Stephen M.
King
e,
Alexander J.
Cowan
*c and
Dave J.
Adams
*ab
abc,
James J.
Walsh
cd,
Laura L. E.
Mears
bc,
Emily R.
Draper
ab,
Matthew
Wallace
b,
Michael
Barrow
b,
Bart
Dietrich
ab,
Stephen M.
King
e,
Alexander J.
Cowan
*c and
Dave J.
Adams
*ab
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK
cStephenson Institute for Renewable Energy, University of Liverpool, Liverpool L69 7ZF, UK. E-mail: a.j.cowan@liverpool.ac.uk
dSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland
eSTFC Pulsed Neutron and Muon Source, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
First published on 29th March 2017
Abstract
There is growing interest in the design of supramolecular structures that are photocatalytically active. Perylene bisimides can be self-assembled to produce structures for photocatalytic hydrogen evolution. Herein we explore the role of pH in controlling self-assembly and photocatalysis. It is shown that self-assembly, which occurs as the pH of the system is decreased, is required for hydrogen evolution to occur.
Introduction
Photoconductive low molecular weight gelators (LMWGs) are becoming increasingly important due to their tuneable mechanical, electrical and optical properties.1 In particular perylene diimide, or perylene bisimide (PBI) derivatives are of interest. In addition to being strongly absorbing in the visible region, many possess n-type semiconductor properties when aggregated1–3 which means they have potential use as light harvesting materials in photovoltaics and photocatalysis.2,4–7 Interestingly, some PBIs are used as LMWGs as they can self-assemble into a wide range of structures as a result of non-covalent interactions including hydrogen bonding, van der Waals interactions and π–π stacking.8,9 PBIs have also been successfully used as photocatalysts for a range of reactions including hydrogen evolution,10–16 water oxidation17 and the reduction of aryl halides.18,19Proton reduction, or the Hydrogen Evolution Reaction (HER), is a widely studied half reaction from the water-splitting process.20 The HER provides potentially a direct route to storing solar energy in the stable chemical bonds found in H2.21 The key to any emerging technology is to provide a cheap and competitive system; LMWG can be assembled easily at low cost and are comprised only of abundant elements – this warrants their investigation as an alternative to the inorganic materials usually investigated.21,22 Weingarten et al. reported the first work on photocatalytic π-conjugated gels for proton reduction, where the gel phase showed superior activity to that of the solid powder of the same compound,13,16,23 showing how structuring is key. It is important to understand how a self-assembled network can affect the photocatalytic activity of a LMWG and how subtle changes in molecular packing can affect their photocatalytic ability.24
The self-assembly of LMWGs in water can be triggered using several methods, including a solvent switch or a pH switch.15–17,25,26 Recently, PBIs substituted with amino acids in the imide position, for example as shown in Fig. 1a, have been studied for their photoconductive properties as thin films.27–30 The carboxylic acid functionality on the amino acid side groups allows control over the solubility of the gelator in aqueous solutions at high pH (Fig. 1b).27,31 A subsequent decrease in the pH results in the consecutive protonation of the carboxylic acid groups, reducing the solubility and allowing for further self-assembly in solution (Fig. 1c).
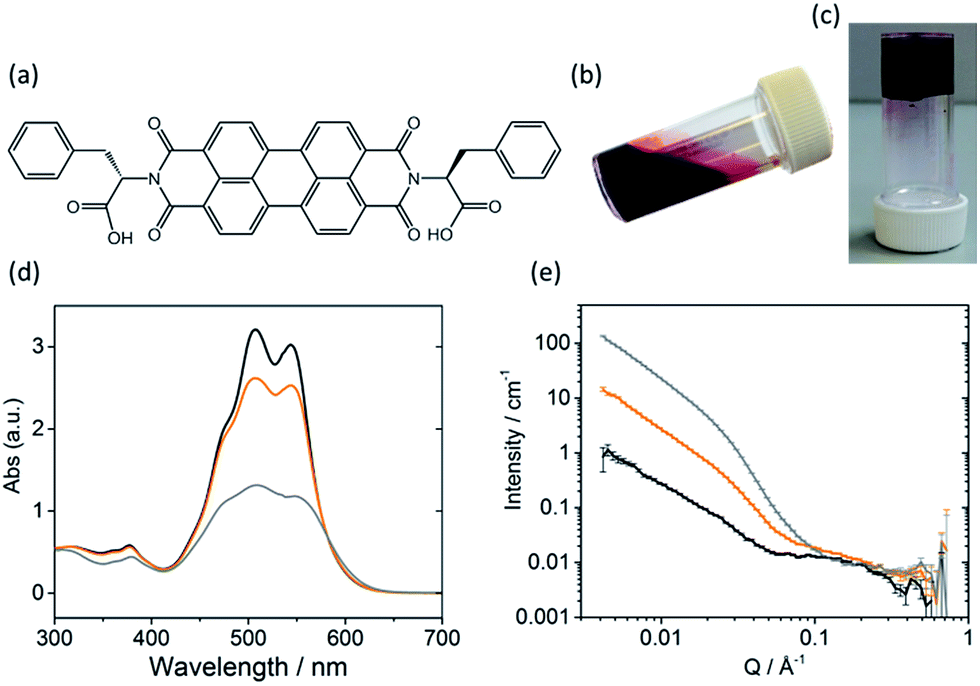 | ||
| Fig. 1 (a) Molecular structure of PBI-F; (b) high pH solution (10 mg mL−1 PBI-F, 2 eq. NaOH, 20 v/v% methanol); (c) low pH gel (10 mg mL−1 PBI-F, 2 eq. NaOH, 8 mg mL−1 GdL, 24 hours); (d) UV-Vis absorption spectra and (e) SANS data at pH 11 (black), 6 (orange), and 2 (grey). pH was lowered in (d) by HCl addition and in (e) by DCl addition. | ||
In this paper, we show that pH is crucial for controlling the self-assembly of amino-acid functionalised PBIs and that subtle changes in structure can also have a profound effect on the photocatalytic properties of the material. We demonstrate the role of aggregation by showing that we can optimise the rate of hydrogen evolution in water by fine-tuning the pH of the perylene bisimide assemblies.
Results and discussion
Self-assembly
Sufficient π–π stacking between perylene cores allows for the formation of PBI fibres and electron transport along these fibres.17,32 PBIs have characteristic absorption bands at visible wavelengths related to the vibrational transitions of the S0–S1 absorption; the intensity, resolution and shift of the absorbance bands are dependent upon the environment around the aromatic core. This allows UV-Vis absorption spectroscopy to be used as a tool for studying aggregation processes such as the formation of H- or J-type aggregates.33 The HOMO/LUMOs of the perylene core have a small electronic coupling to the substituents on the peripheral imide positions and the orbital energy can be manipulated by adding substituents onto the core or by π-stacking with adjacent molecules.34–36The PBI used here (PBI-F) is substituted with L-phenylalanine at the imide positions (Fig. 1a). Aqueous solutions of PBI-F at a concentration of 10 mg mL−1 with 20 v/v% methanol and 2 equivalents of NaOH were prepared, resulting in deep red solutions with a pH between 11 and 12 (Fig. 1b). Methanol was included as a sacrificial electron donor for photocatalysis, as discussed below. The pH of solutions was then lowered, while stirring, by the addition of 10 μL aliquots of 0.1 M HCl to form aggregates in solution. Initial studies showed that PBI-F has two apparent pKa values (Fig. S1†) at 8.6 and 5.7. These are related to the consecutive protonations of the carboxylic acid groups. Whilst it might be expected that there is only one pKa since the two carboxylic acids are apparently identical, these elevated pKa values can be associated with the pKa of self-assembled structures.37 It is therefore expected that structural changes will occur within the solutions at each apparent pKa.37,38 For ease, we will describe solutions at pH > 8.6, at pH 5.7–8.6, and at pH < 5.7 as high, medium, and low pH respectively. The PBI-F solutions can also be gelled by a slow acidification using glucono-δ-lactone (GdL) (Fig. 1c and S2†).27,39 The stable structures that PBI-F forms in aqueous solutions of 20 v/v% methanol were probed using UV-Vis absorption spectroscopy, viscometry and small angle neutron scattering (SANS) (Fig. 1d–e and S3–S7†).
The higher intensity of absorbance of the 0–1 vibronic transition of the S0–S1 band at 506 nm compared to the 0–0 transition at 543 nm is an indication of H-type aggregation and is observed in the PBI-F samples at 10 mg mL−1 (Fig. 1d and S3†) at all pH values.40–45 H-type aggregation describes a face-to-face stacking of the perylene cores of adjacent molecules and coincides with a bathochromic shift in the UV-Vis absorption spectra. In the case of PBI-F, we expect a slight rotational displacement to account for the steric hindrance and directional H-bonding of the peripheral phenylalanine groups.46 Upon decreasing the solution pH, the absorption peaks broaden indicating increased intermolecular interactions and increased aggregation.47
SANS is well suited to study LMWGs as neutrons are non-destructive and have the ability to probe the bulk material. The fitted data provide details of the size and shape of structures in the sample.48–50 For the SANS experiments, deuterated solvents were used to enhance the contrast; UV-Vis absorption spectroscopy and viscometry studies with deuterated solvents showed no changes in structures as compared with the protonated solvents (Fig. S4a and b†). The pD of a deuterated solution of PBI-F in 20 v/v% d4-methanol(aq) was reduced by adding 0.1 M DCl. Aliquots were taken from the solution spanning the whole pD range and a scattering pattern was obtained from each sample (Fig. S4c and d†). All pD measurements were converted to pH to allow effective comparison with data collected in pH. The SANS data were fitted by combining flexible cylinder, power law and spherical models (Fig. S5†).51–53 The flexible cylinder model depicts a worm-like micellar structure which is expected for this class of PBIs.27,54,55 The parameters reported here are derived by model-fitting the scattering data to analytic expressions describing different shapes. In some instances this has resulted in flexible cylinder lengths that are beyond the actual measurement limit of the instrument (>150 nm) in the configuration used. This does not invalidate the analysis, it just means that affected values are associated with a greater uncertainty.
Fitting the SANS curves to the flexible cylinder and power law models revealed that anisometric structures are present in solutions at both high and low pH (Fig. S6 and S7†), although as will be shown below the relative concentration of these structures is strongly pH dependent. These structures may be described as worm-like micelles at high and medium pH where the PBI still holds a negative charge. However, at low pH, below the second apparent pKa, there is expected to be little charge on the PBI molecules and we describe these as fibres. The fitting parameters from the flexible cylinder model (Fig. S7†) depict the worm-like micelles with radii of 6.2 (±1.0) nm and a length of 414 (−110, +86.0) nm (where the minimum and maximum errors are stated in brackets and plotted in Fig. S7†). The fibres at low pH have radii of 5.1 (±1.0) nm and a length parameter >500 nm. This shows that at low pH the extent of stacking in a single structure is increased due to the absence of strong coulombic interactions and the dominance of the weaker intermolecular interactions that promote self-assembly, including π-stacking, H-bonding, van der Waals and hydrophobic interactions. The low pH fibres also have a Kuhn length parameter of 7.8 (−1.0, +7.0) nm compared to 26.3 (−3.00, +4.00) nm for the high pH worm-like micelles; the Kuhn length is a segment of the flexible cylinder model (Fig. S5†) and a smaller Kuhn length indicates a more flexible structure.52,53 For the fibres at low pH, the lack of a supporting hydrated shell of water molecules around the fibre may give rise to the higher flexibility of the structures. Additionally, the reduced coulombic interactions at low pH may open up additional interactions between PBI-F molecules via the peripheral phenyl rings on the imide groups.
The solutions at high and medium pH also showed an additional type of structure dominating the scattering curve, which exhibits a hump-like feature at around Q = 0.1 Å−1 correlating to structures a few nanometres in size (Fig. S5–S7†). We hypothesised that free molecules were present in solution above the second apparent pKa at pH 5.7; in order to test this, diffusion 1H-NMR was carried out on a high pH solution. Broad NMR signals, reduced relaxation times and negative NOEs clearly indicated the presence of aggregated species in solution (Fig. S8 and S9†). However, the observed species were found to possess hydrodynamic radii of only 1.7 (±0.03) nm. The hump-like feature in the scattering pattern at Q = 0.1 Å−1 was found to fit well to the spherical model with a sphere radius of 0.9 (±0.1) nm. The SANS and NMR data thus strongly suggest the presence of free molecules at high pH, which co-exist with worm-like micelles. The difference between the radii calculated from the two techniques can be related to the uncertainties associated with the fitting and calculation56,57 as well as solvation effects.
The combination of the spherical model with the flexible cylinder and power law models provided a convincing fit for all data (Fig. S6†). We can also further our understanding by addressing the contributions of each of the models to the fits (Fig. S7†). The power law scale factor increases as the pH is lowered showing a corresponding increase in long-range structures. Interestingly, at high pH the spherical model dominates ca. 99%, which is in general agreement with 1H-NMR measurements which show ca. 90% of PBI-F exists as free molecules in solution at high pH. At low pH, the SANS data fits to 100% flexible cylinder model indicating that the PBI-F molecules are assembled into fibres.
At pH 6.5, the power law scale contribution along with its exponent m begins to significantly increase, showcasing the increase in assembly of the molecules into structures which scatter. An increase in the contribution of the flexible cylinder model is also observed at pH 6.0. The transition from free molecules to aggregated species at ∼pH 6.5 proves to be vital for photocatalysis, as discussed below.
The optical density (O.D.) of the PBI-F samples at 506 nm and the SANS intensity at Q = 0.01 Å−1 help to reveal how aggregation increases as the pH decreases (Fig. 2a). As the pH is lowered below the first apparent pKa at pH 8.6, the O.D. begins to decrease quickly and the scattering intensity increases slowly. At the second pKa at pH 5.7 the scattering intensity increases at a notably greater rate with not much change thereafter. It is clear that the greatest change in aggregation occurs as the pH is decreased below pH 5.7; this is where gelation of these materials occurs and where the formation of longer fibrous structures is observed in SANS. It is also interesting to note that UV-Vis absorption data shows that aggregation begins to occur below pH 8.6 when the mono-protonated PBI-F begins to form and that SANS indicates the structures begin to change significantly below pH 5.7.
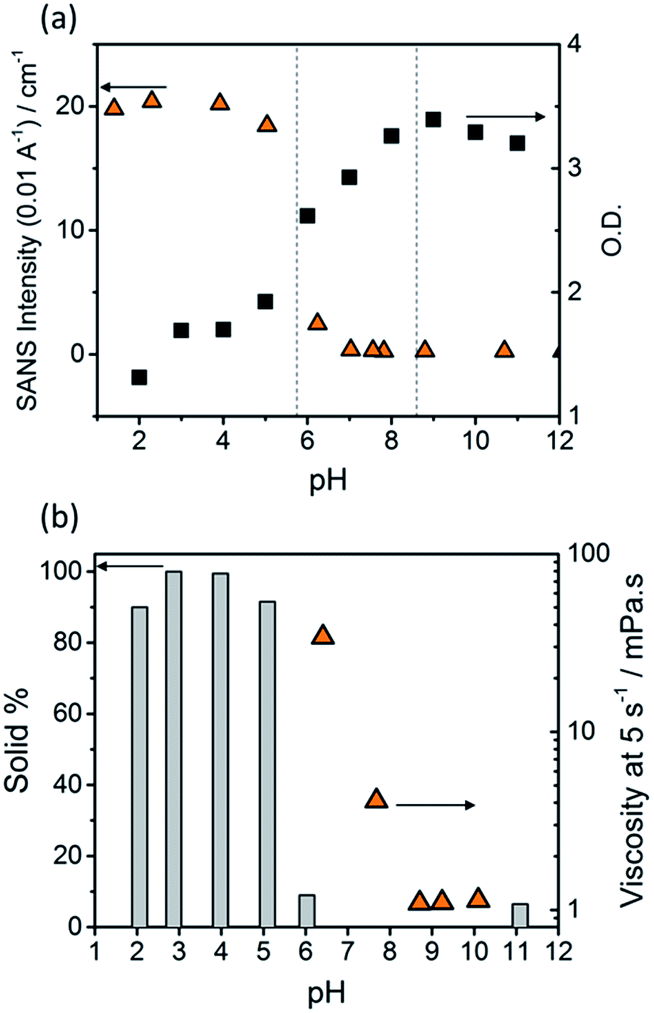 | ||
| Fig. 2 (a) SANS intensity (triangles) from scattering patterns and optical density (O.D., squares) from UV-Vis absorption spectra of PBI-F (10 mg mL−1) solutions in 20 v/v% d4-methanol(aq). SANS intensity taken at 0.01 Å−1 and O.D. at 506 nm; vertical dotted lines indicate pKa1 (pH 8.6) and pKa2 (pH 5.7) (b) percentage of PBI-F collected from a 2 mL solution by filtration (column) and viscosity of solutions at a shear rate of 5 s−1 (triangles). Arrows indicate which axis data correspond to. | ||
Viscometry was used to identify the extent of networks in the solutions (Fig. 2b and S10†). Samples were filtered, using filter paper, to collect any solid aggregates present in solution. Fig. 2b shows the percentage of solid collected after filtering 2 mL of a sample compared to the viscosity intensity at a shear rate of 5 s−1. Greater than 95% solid, compared to the amount of PBI-F added into solution, was collected from the fully protonated solutions below the second apparent pKa. This shows we have aggregated species suspended in solution. The dried solutions at high pH and collected solids from low pH were imaged by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Fig. S11†). The microscopic data shows aggregates much larger than those observed from SANS. Drying artefacts are highly prevalent in these systems as drying leads to further aggregation and structural changes.58 Therefore, the SANS data, which probes the bulk sample, along with UV-Vis spectroscopy and viscometry are better representations of the system used.
The viscosity of solutions (Fig. 2b, and S10†) increases greatly between pH 8.6 and 5.7 showing that the singly protonated species promotes the formation of a self-assembled network; this correlates with the broadening of absorption bands in the UV-Vis spectra and changes in the SANS parameters towards longer fibres. It is likely that the increasing viscosity of the solutions down to pH 5.7 is due to the presence of a higher concentration of worm-like micelles in solution.
Hence we conclude from the UV-Vis, SANS and viscometry data that a low concentration of short, worm-like micelles and a high concentration of free solvated molecules are present in the PBI-F solutions at high pH. The free molecules aggregate to form more worm-like micellar structures below pH 8.6. As the pH approaches 5.7 and the molecules become fully protonated, all the molecules are aggregated into long fibres. The protonation steps lead to intermolecular interactions prevailing over solvating forces, allowing self-assembly on a larger scale to occur.
Photocatalysis
Photocatalytic hydrogen evolution was tested at a range of pHs using PBI-F (10 mg mL−1) in the presence of polyvinylpyrrolidone (PVP)-capped platinum nanoparticles (1 mol%, ca. 8 nm diameter) and methanol (20 v/v%). Methanol is a widely used sacrificial electron donor that induces a minimal pH change upon addition and operates across a range of pH values. Platinum nanoparticles have been widely studied as proton reduction co-catalysts and have a relatively simple synthesis and characterisation (Fig. S12–S15†). Samples were irradiated for 4 hours (90 mW cm−2, 300–795 nm (KG1 filter)) while stirring as described in the Experimental (Fig. S16 and S17†). The amount of hydrogen evolved was measured using gas chromatography (GC) via a gas injection from the quartz cuvette headspace.Initial control experiments in the absence of platinum yielded minimal hydrogen. In the absence of methanol a large amount of CO evolved during photocatalysis indicating that PBI-F oxidation occurs without the sacrificial donor.30 The amount of CO evolved was significantly reduced when methanol was present. However, no photodegraded product could be detected by NMR or FTIR; we note that this would only be present in very small amounts and so might be below the limit of detection.
After 4 hours of irradiation, no hydrogen was evolved at pH values above 8, Fig. 3. Between pH 7–5.4 a low rate of hydrogen was produced, but repeat experiments to yield error estimates clearly demonstrate the photocatalytic formation of hydrogen. The solution noticeably begins to form a gel at pH 5 which results in a reduced rate of hydrogen production, likely due to the increased viscosity. Below pH 5 the structures aggregate and are present as a suspension in solution at which point the fibres still hold a residual charge.59 At pH 4.75 a sharp increase in the rate of H2 evolution is observed to occur. The hydrogen evolution reaches a maximum at pH 4.5 with a decline at lower pHs. At pH 4.5 our structural characterisation studies show that PBI-F has self assembled into long-fibres and it is proposed that these large aggregates are the photocatalytically active species. The PBI-F/PVP–Pt/methanol solution was stable for up to 13 days (307 hours) of irradiation (Fig. S18†); the solution became inactive after this time due to the complete evaporation of methanol from the system as a result of the daily purging of the cell. After 307 hours, a turnover number of 158 per PVP–Pt NP, or 1.32 × 10−2 per Pt atom (see ESI† for calculations), was achieved. The resulting solution was freeze dried and analysed to try and characterise any products of PBI photodegradation (Fig. S19†). Again, no product of degradation was detected by NMR or FTIR. This could be due to the amount produced being below the detectable limit, a significantly lower solubility of the degraded product, or simply that no degradation had occurred. Therefore it is apparent that PBI-F supramolecular structures show a reasonable degree of photocatalytic stability.
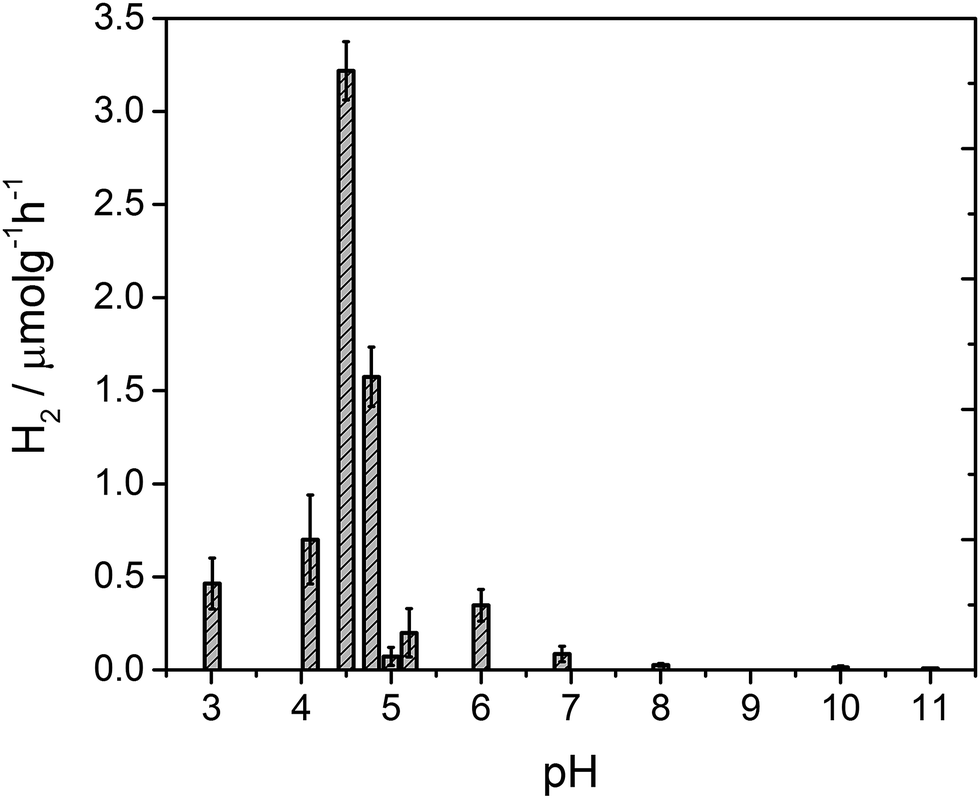 | ||
| Fig. 3 Amount of hydrogen evolved from 4 hour photocatalysis of PBI-F/PVP–Pt/methanol mixtures at different pH values (column with error bars). pH measurements between pH 4–5 are grouped together with ±0.2 pH units, all other measurements are grouped up to ±0.3 pH units. | ||
Electrochemical characterisation
We have explored the mechanism of photocatalytic hydrogen evolution and its pH dependence further through electrochemical measurements. PBIs can be chemically, electrochemically and photochemically reduced to form radical anion (PBI˙−) and dianion (PBI2−) species.9,27,30,35,60,61 Square wave voltammetry (SWV) (10 mg mL−1, 20 v/v% methanol, 0.1 M NaCl, 1 Hz) was carried out at a range of different pHs, Fig. 4. A complex voltammogram was obtained with three close lying reductions (−0.285, −0.411 and −0.472 VAg/AgCl). The characteristic absorptions in the UV-Vis absorption spectra of the PBI˙− at 725 nm and PBI2− at 610 nm allow for these reduced species to be identified by UV-Vis absorption spectroscopy.60 Therefore, spectroelectrochemical measurements of PBI-F at a lower concentration (1 mg mL−1, 20 v/v% methanol, 0.1 M NaCl, vs. Ag/Ag+ pseudo-reference, pH 11) were carried out (Fig. 5). Lower PBI-F concentrations are required here due to the high extinction coefficient of the material. Prolonged reduction (1 min) at potentials close to the first reduction potential gives rise to partial conversion of the PBI to PBI˙− with an increase in the 725 nm absorption. As the potential is moved negative, we observe PBI2− forming with a UV-Vis absorption maximum at 610 nm as the PBI˙− band at 725 nm decreases. This begins to occur at the 2nd reduction potential, but does not complete until the most negative reduction potential is reached. It is therefore proposed that the first and third reduction features at −0.285 and −0.472 VAg/AgCl can be assigned to the PBI/PBI˙− and PBI˙−/PBI2− couples. The spectroelectrochemical measurements indicate that the reduction at −0.411 VAg/AgCl also appears to be related to the PBI˙−/PBI2− couple. The presence of this additional reduction peak is tentatively ascribed to the presence of multiple PBI-F environments in the assembled structures or may also be due to the formation of states with charge delocalised across the aggregated structure.62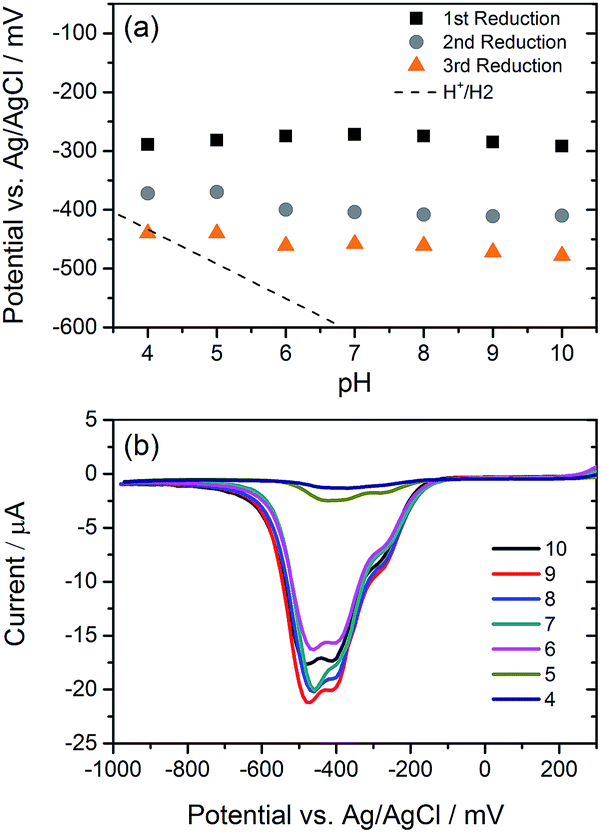 | ||
| Fig. 4 (a) Reduction potentials of PBI-F (10 mg mL−1) in 20 v/v% methanol(aq) at various pHs. Reduction peaks were picked from SWV measurements shown in (b) which shows three reductions. The dotted line indicates the energy required for hydrogen evolution–reduction potentials which lie underneath this line can feasibly drive hydrogen evolution. (b) SWVs of PBI-F (10 mg mL−1) at pH 10–4 in 20 v/v% methanol(aq). Measured using Ag/AgCl electrode with a scan rate of 1 Hz. The SWVs were scaled in the y-axis (μA) to roughly the same size. | ||
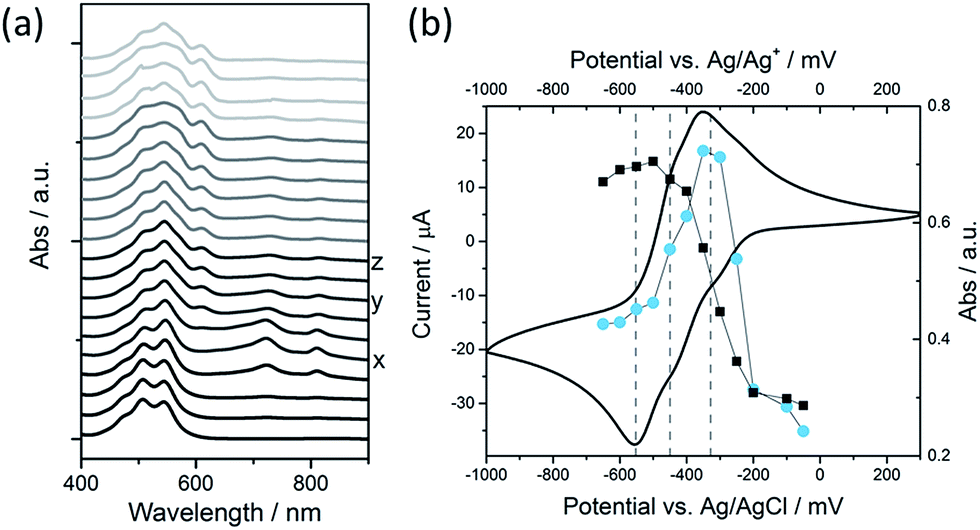 | ||
| Fig. 5 (a) UV-Vis absorption spectra of PBI-F (1 mg mL−1, 20 v/v% methanol(aq)) from thin layer spectroelectrochemical (SEC) measurements. The voltage applied through the cell during data collection was (from bottom to top): −0.05, −0.1, −0.2, −0.25, −0.3 (x), −0.35, −0.4, −0.45 (y), −0.5, −0.55 (z), −0.6, −0.65, −0.7, −0.8, −0.9, −0.95, −1.1, −1.15, −1.2, −1.25 V vs. Ag wire. x = −328; y = −450; z = −552 mV vs. Ag wire. (b) Cyclic Voltammogram (CV) of PBI-F (line, mV vs. Ag/AgCl) overlaid with the UV-Vis absorption intensity at 725 nm (PBI˙−, circles, mV vs. Ag/Ag+) and 610 nm (PBI2−, squares, mV vs. Ag/Ag+). Vertical dotted lines from right to left correspond to x, y, z in (a). | ||
All three reductions are found to show minimal pH dependence between pH 10–4 with a <50 mV difference between pH 6 and 4 for all reductions, Fig. 4. Notably only the 3rd reduction of the solution PBI (PBI˙−/PBI2−) at a pH of approximately 5 and lower is predicted to be sufficiently reducing for hydrogen evolution to occur (Fig. 4a). However, it should be noted that this electrochemical analysis focuses on soluble species, as indicated by the decrease in current response at lower pHs where the soluble species are present in lower concentrations (Fig. 4b). At low pH, the increasing viscosity of the solution (Fig. 2) and the gradual formation of insoluble aggregates hampers studies and attempts to electrochemically characterise the collected solid are to date unsuccessful. As we measure photocatalytic hydrogen production at pH 7, a pH where we would not anticipate PBI˙−/PBI2− in solution to be active for H2 production, it is likely that the active species are the large aggregated structures which begin to be present in significant levels at these pHs.
To explore if PBI˙− or PBI2− generation is required for hydrogen evolution, UV-Vis absorption spectroscopy was used to probe the formation of radical anion and dianion species after visible (470 nm) and UV (365 nm) irradiation (Fig. S20†). For both pH 9.5, pH 6, and pH 4.5 solutions, the radical anion is formed under 470 nm irradiation. Under 365 nm irradiation a decreased yield of PBI˙− is observed and a new UV-Vis absorption at ∼610 nm is present, assigned to PBI2−. Nonetheless the findings at pH 9.5 and 6 are in agreement with the wavelength dependence of a pH 4.5 sample for hydrogen evolution activity (Fig. S21 and S22†) where activity is only observed during UV irradiation. Here, the irradiation wavelength overlaps with the UV absorption band of the S0–S2 transition and this may suggest that PBI2− is generated during photocatalysis, however further studies are required to explore the aggregated structures. An apparent quantum efficiency of 0.018% was obtained from 365 nm irradiation, calculated for a two electron reduction of H+ to H2. These findings are in agreement with other work where <400 nm irradiation induces conductivity and photocatalytic activity.12,27,30 Although this value is low compared to many inorganic semiconductors, it is inline with comparable perylene-based systems.12,13,16 Here, it is important to note that the aim of this work is to understand the relationship between supramolecular structure and photocatalytic activity and not to optimise activity.
We have therefore demonstrated that PBI-F solutions become photochemically active for H2 production at low pH with a maximum hydrogen evolution rate being observed at pH 4.5. Characterisation of the PBI-F samples demonstrates that at the pH where hydrogen evolution occurs long self-assembled fibres are present. Notably, we begin to observe a level of hydrogen evolution at around pH 7, the point at which the SANS experiment indicates that an increased amount of assembled structures begin to form. Strikingly we also find that at this pH the potential of the PBI˙−/PBI2− couple in solution is not sufficiently reducing for proton reduction to occur. All of these observations strongly suggest that hydrogen evolution occurs from the self-assembled structures and not from free molecules in solution. The large increase in hydrogen evolution rate is in line with the structural changes and the point at which we observe the highest concentration of the self-assembled fibrous structures. It is likely that the formation of these fibrous structures, which are known to be effective for long-range electron transport,27 are vital in enabling photoelectron transfer to the sites of the platinum co-catalysts. The rationale for the subsequent decrease in hydrogen evolution rate at lower pH is not yet clear. It is likely that activity is a product of a delicate balance between aggregate size, self-assembled structure, charge state and availability of edges in the material and is still to be fully understood.
Conclusions
Through a combination of pH-dependent electrochemical and photocatalytic studies, we have shown the importance of the self-assembly of a perylene bisimide on its photocatalytic activity. Currently, there are very few reported examples of self-assembled materials for H2 evolution and our focus on understanding the pH-induced structural changes of perylene bisimides provides important new insights for the field. This work provides a route to open up a wealth of opportunities for the optimisation of self-assembled PBIs for photocatalytic applications.Experimental
Materials
Perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA), L-phenylalanine and imidazole were obtained from Sigma Aldrich for the synthesis of PBI-F. K2PtCl6, polyvinylpyrrolidone (PVP, MW = 40k) and potassium L-tartrate monobasic were obtained from Sigma Aldrich for the preparation of PVP-Pt nanoparticles. Photometric grade methanol, glucono-δ-lactone, HCl, DCl, NaOH, NaOD and D2O were obtained from Sigma Aldrich and used as received. d4-Methanol was obtained from Apollo Scientific. MilliQ water was used throughout.Procedure for preparation of PVP-capped Pt nanoparticles
The preparation of PVP-Pt NPs was carried out using a previously described method.63 A 20 mL aqueous solution of potassium L-tartrate monobasic (0.5 wt%) was brought to reflux (ca. 100 °C). Then, a 20 mL aqueous solution of H2PtCl6 (1 mM Pt) and PVP (1.0 wt%) was added into the vortex of the stirring reflux solution and left to reflux for 60 minutes. A dark brown solution formed after 5 minutes, with no further visible change thereafter. The solution was cooled to room temperature and then spin filtered by distributing across 3 × 20 mL Corning® Spin-X® UF concentrators containing a polyethersulfone membrane with a 50k molecular weight filter. The solutions were centrifuged for 3 × 30 minutes at 5000 rpm and then redispersed in ultra-pure water to a total volume of 20 mL and 1.0 mM Pt. The PVP-Pt NPs were characterised by dynamic light scattering (DLS), TEM, thermogravic analysis (TGA) and UV-Vis absorption spectroscopy (Fig. S12–S15†).DLS measurements were performed on a Malvern Zetasizer Nano ZS using non-invasive backscatter optics with a He–Ne laser source at 633 nm. Measurements were collected at room temperature using 3 runs of 25 scans.
TGA measurements were carried out on a TA Instruments SDT Q600 TGA machine using a constant air flow of 100 mL min−1. Samples were heated up to 120 °C at a heating rate 10 °C min−1. The samples were kept at 120 °C for 20 minutes to remove any water, then ramped to 200 °C at a heating rate of 10 °C min−1.
Synthesis of PBI-F
The synthesis of PBI-F was scaled up from a previously described synthesis.27 In a 100 mL Schlenk flask, PTCDA (3.0 g, 7.62 mmol), imidazole (10.42 g, 152.94 mmol) and L-phenylalanine (2.43 g, 15.3 mmol) were mixed and purged with nitrogen for 10 minutes. Once purged, the mixture was heated up to 120 °C and the resulting molten solution was stirred for 5 hours at 120 °C under nitrogen. The reaction was then cooled to 90 °C and 5 mL of deionised water was added. The reaction was stirred at 90 °C for 1 hour and then cooled to room temperature before filtering to remove unreacted PTCDA. The pH of the filtrate was then adjusted to 2–3 using 2 M HCl (ca. 100 mL). The resulting mixture was stirred at 60 °C for 8 hours. The precipitate was collected via vacuum filtration and washed thoroughly with acidified H2O. The final compound was analysed by NMR spectroscopy, mass spectroscopy and FTIR spectroscopy (Fig. S23–S25†).Preparation of LMWG solutions
7 mL solutions of PBI-F were prepared by weighing out 70 mg PBI-F (10 mg mL−1) into 14 mL vials then, while stirring, adding 4.98 mL deionised H2O, 1.02 mL 0.2 M NaOH(aq) (2 eq.) and 1.4 mL methanol (20 v/v%). The solutions were stirred overnight and pH was adjusted the next day by adding 0.1 M HCl dropwise to the solution while stirring and measuring the pH. The solutions were stirred for another 30 minutes and the pH was checked and adjusted if needed before use. Any aggregates of PBI in solution were present as a suspension. The solutions were not stirred during UV-Vis absorption spectroscopy, electrochemistry, and SANS measurements.pH measurements
A FC200 pH probe from HANNA instruments with a 6 mm × 10 mm conical tip was used for pH measurements. pD measurements were collected with the same probe and corrected with a constant offset of pH = pD − 0.4.64UV-Vis absorption spectroscopy
UV-Vis absorption spectra were taken using a Shimadzu UV-2600 spectrometer and a 0.1 mm demountable quartz cuvette (Starna). For irradiation experiments an LED (RS Electronics) was pointed at the sample and the spectrometer covered with a black cloth.Small angle neutron scattering
SANS measurements of the gelator solutions were performed using the SANS2D instrument (STFC ISIS Pulsed Neutron Source, Oxfordshire, UK). A simultaneous Q-range [Q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ/2)/λ, where θ is the scattering angle] of 0.005 to 0.7 Å−1 was achieved using an incident wavelength (λ) range of 1.75 to 16.5 Å and employing a sample-to-detector distance of 4 m, with the 1 m2 detector offset vertically 60 mm and sideways 100 mm. Samples were housed in 5 mm quartz cuvettes and measured for ∼30 minutes. Scattering data were normalized for the sample transmission and background corrected using a quartz cell with 20 v/v% d4-methanol in D2O and also corrected for the linearity and efficiency of the detector response using the Mantid framework.65 The scattering data were then fitted in the SasView software (version 3.1.1)66 to a customised model comprising of a (Kratky-Porod) flexible cylinder, a sphere and an absolute power law.51–53 The power law (Q−m) accounts for the mass fractal contribution to the scattering intensity which is combined with the flexible cylinder and sphere models (Fig. S5†).
sin(θ/2)/λ, where θ is the scattering angle] of 0.005 to 0.7 Å−1 was achieved using an incident wavelength (λ) range of 1.75 to 16.5 Å and employing a sample-to-detector distance of 4 m, with the 1 m2 detector offset vertically 60 mm and sideways 100 mm. Samples were housed in 5 mm quartz cuvettes and measured for ∼30 minutes. Scattering data were normalized for the sample transmission and background corrected using a quartz cell with 20 v/v% d4-methanol in D2O and also corrected for the linearity and efficiency of the detector response using the Mantid framework.65 The scattering data were then fitted in the SasView software (version 3.1.1)66 to a customised model comprising of a (Kratky-Porod) flexible cylinder, a sphere and an absolute power law.51–53 The power law (Q−m) accounts for the mass fractal contribution to the scattering intensity which is combined with the flexible cylinder and sphere models (Fig. S5†).
Viscometry
Viscosity measurements were performed on an Anton Paar Physica MCR301 rheometer utilising a 75 mm cone and plate geometry. 1.0 mL of solutions were pipetted onto the plate and left to stabilise for 2 minutes before measurements. Experiments were run at 25 °C. The viscosity of each solution was recorded under the rotation, with the shear rate varying from 1 to 100 s−1.Electrochemistry
Cyclic voltammetry (CV) measurements were collected using a three-electrode system and a PalmSens1 potentiostat. All systems were measured in aqueous conditions using 5 mL of sample. 0.1 M sodium chloride (NaCl) was used as the electrolyte, glassy carbon as the working electrode (d = 3 mm, BASi), a Pt mesh as a counter electrode and an Ag/AgCl electrode with a double junction as the reference electrode. CVs were measured from 1.0 V to −1.0 V then back to 1.0 V and repeated over a range of scan rates: the most successful in observing the desired peaks was 0.01 V s−1. All samples were purged with argon for 30 minutes before a measurement was run.SWV measurements were collected using a three-electrode system and a Dropsens potentiostat with a glassy carbon working electrode, a Pt wire counter electrode and a Ag/AgCl reference electrode. The supporting electrolyte was 0.1 M NaCl. The measurements were scanned from 0.3 V to −1.0 V at a rate of 1 Hz (0.01 V s−1).
Spectroelectrochemical experiments were carried out on 1 mg mL−1 samples of PBI-F using a thin layer SEC cell containing a Pt counter electrode and an Ag wire pseudo-reference electrode. The cell was placed inside a Shimadzu UV-2600 spectrometer. The current through the sample was monitored while a chosen potential was applied across the cell and a UV-Vis absorption spectrum was recorded when the current reached a plateau. This took approximately 1 minute due to the slow diffusion through the cell.
Photocatalytic experiments
Photocatalytic experiments were carried out using a 75 W Xe lamp, a lens and 2× KG1 filters to achieve an output of 100 mW cm−2 onto the sample (Fig. S16 and S17†). Solutions tested for photocatalysis were stirred in a 1 cm pathlength quartz cuvette with a 25 mL headspace sealed with a rubber septum. Solutions were pre-purged for 30 minutes with N2, and H2 was measured via gas chromatography at t = 0 and after 4 hours of irradiation.For wavelength dependence studies a similar setup was used and the lamp exchanged for an LED. A photodiode was used to monitor the power output of the LEDs onto the sample and the power was altered accordingly to ensure all samples received a matching irradiance as 5 mW cm−2 at 365 nm (Fig. S21†).
GC headspace analysis was performed using an Agilent 6890N employing N6 helium as the carrier gas (5 mL min−1). A 5 Å molecular sieve column (ValcoPLOT, 30 m length, 0.53 mm ID) and a pulsed discharge detector (D-3-I-HP, Valco Vici) were employed. H2 peak areas were quantified with multiple calibrant gas injections.
Acknowledgements
We thank the EPSRC and the University of Liverpool for funding a DTA (MCN). We thank the STFC for funding our time at the ISIS facility (Experiment Number RB1610240). We would also like to thank Ana Castilla-Manjon, Mark Forster, Jonathan Lee and Tom McDonald for their time and help throughout the project. Matthew Wallace would like to thank the EPSRC and Unilever for a Case Award. AJC and DJA thank the EPSRC for Fellowships (EP/K006851/1 and EP/L021978/1 respectively). LLEM, ERD, and BD thank the EPSRC for funding (EP/L021978/1). MB thanks UKRMP for funding (MR/K026739/1). The NMR spectrometers used for this work were funded by the EPSRC (EP/K039687/1 and EP/C005643/1). This work benefited from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.Notes and references
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- J. Choi, H. Song, N. Kim and F. S. Kim, Semicond. Sci. Technol., 2015, 30, 64002 CrossRef.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377 CAS.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- F. Fernández-Lázaro, N. Zink-Lorre and Á. Sastre-Santos, J. Mater. Chem. A, 2016, 4, 9336–9346 Search PubMed.
- J. López-Andarias, M. J. Rodriguez, C. Atienza, J. L. López, T. Mikie, S. Casado, S. Seki, J. L. Carrascosa and N. Martín, J. Am. Chem. Soc., 2015, 137, 893–897 CrossRef PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- F. Würthner, Chem. Commun., 2004, 14, 1564–1579 RSC.
- S. Chen, C. Wang, B. R. Bunes, Y. Li, C. Wang and L. Zang, Appl. Catal., A, 2015, 498, 63–68 CrossRef CAS.
- F. S. Liu, R. Ji, M. Wu and Y. M. Sun, Acta Phys.-Chim. Sin., 2007, 23, 1899–1904 CAS.
- S. Chen, Y. X. Li and C. Y. Wang, RSC Adv., 2015, 5, 15880–15885 RSC.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- S. Chen, D. L. Jacobs, J. Xu, Y. Li, C. Wang and L. Zang, RSC Adv., 2014, 4, 48486–48491 RSC.
- T. Abe, Y. Tanno, N. Taira and K. Nagai, RSC Adv., 2015, 5, 46325–46329 RSC.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, J. Am. Chem. Soc., 2015, 137, 15241–15246 CrossRef CAS PubMed.
- J.-X. Li, Z.-J. Li, C. Ye, X.-B. Li, F. Zhan, X.-B. Fan, J. Li, B. Chen, Y. Tao, C.-H. Tung and L.-Z. Wu, Catal. Sci. Technol., 2016, 6, 672–676 CAS.
- B. K. Indrajit Ghosh, T. Ghosh and J. I. Bardagi, Science, 2014, 346, 725–728 CrossRef PubMed.
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and C. Duan, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed.
- H. Ahmad, S. K. Kamarudin, L. J. Minggu and M. Kassim, Renewable Sustainable Energy Rev., 2015, 43, 599–610 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, J. Am. Chem. Soc., 2016, 138, 11762–11774 CrossRef CAS PubMed.
- D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Adv. Mater., 2016, 1–7 Search PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Zamith Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jackel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC.
- E. R. Draper, O. O. Mykhaylyk and D. J. Adams, Chem. Commun., 2016, 52, 6934–6937 RSC.
- J. J. Walsh, J. R. Lee, E. R. Draper, S. M. King, F. Jäckel, M. A. Zwijnenburg, D. J. Adams and A. J. Cowan, J. Phys. Chem. C, 2016, 120, 18479–18486 CAS.
- E. Kozma, G. Grisci, W. Mróz, M. Catellani, A. Eckstein-Andicsovà, K. Pagano and F. Galeotti, Dyes Pigm., 2016, 125, 201–209 CrossRef CAS.
- A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H.-W. Schmidt and R. Hildner, Nature, 2015, 523, 196–199 CrossRef CAS PubMed.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS PubMed.
- F. Würthner and A. Sautter, Chem. Commun., 2000, 2, 445–446 RSC.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- H. Langhals, S. Demmig and H. Huber, Spectrochim. Acta, Part A, 1988, 44, 1189–1193 CrossRef.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- J. Sung, P. Kim, B. Fimmel, F. Würthner and D. Kim, Nat. Commun., 2015, 6, 8646 CrossRef CAS PubMed.
- U. Rosch, S. Yao, R. Wortmann and F. Wurthner, Angew. Chem., Int. Ed., 2006, 45, 7026–7030 CrossRef PubMed.
- X.-Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 37, 3871–3873 RSC.
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. Van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. Van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudholter, J. Am. Chem. Soc., 2000, 122, 11057–11066 CrossRef CAS.
- F. Meinardi, M. Cerminara, A. Sassella, R. Bonifacio and R. Tubino, Phys. Rev. Lett., 2003, 91, 247401 CrossRef CAS PubMed.
- Y. Che, A. Datar, K. Balakrishnan and L. Zang, J. Am. Chem. Soc., 2007, 129, 7234–7235 CrossRef CAS PubMed.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- P.-A. Plötz, S. Polyutov, S. D. Ivanov, F. Fennel, S. Wolter, T. A. Niehaus, Z. Xie, S. Lochbrunner, F. Würthner and O. Kuehn, Phys. Chem. Chem. Phys., 2016, 53, 1689–1699 Search PubMed.
- J.-B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- V. Croce, T. Cosgrove, C. A. Dreiss, S. King, G. Maitland and T. Hughes, Langmuir, 2005, 21, 6762–6768 CrossRef CAS PubMed.
- C. A. Dreiss, Soft Matter, 2007, 3, 956–970 RSC.
- A. Guinier and G. Fournet, Small angle scattering of X-rays, John Wiley Sons, Inc., New York, 1955,18, pp. 1–19 Search PubMed.
- W. R. Chen, P. D. Butler and L. J. Magid, Langmuir, 2006, 22, 6539–6548 CrossRef CAS PubMed.
- J. S. Pedersen and P. Schurtenberger, Macromolecules, 1996, 29, 7602–7612 CrossRef CAS.
- R. van der Weegen, P. A. Korevaar, P. Voudouris, I. K. Voets, T. F. A. de Greef, J. A. J. M. Vekemans and E. W. Meijer, Chem. Commun., 2013, 49, 5532–5534 RSC.
- A. Arnaud, J. Belleney, F. Boué, L. Bouteiller, G. Carrot and V. Wintgens, Angew. Chem., Int. Ed., 2004, 43, 1718–1721 CrossRef CAS PubMed.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- D. K. Wilkins, S. B. Grimshaw, V. Receveur, C. M. Dobson, J. A. Jones and L. J. Smith, Biochemistry, 1999, 38, 16424–16431 CrossRef CAS PubMed.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Soft Matter, 2016, 12, 3612–3621 RSC.
- M. Wallace, J. A. Iggo and D. J. Adams, Soft Matter, 2015, 11, 7739–7747 RSC.
- R. O. Marcon and S. Brochsztain, J. Phys. Chem. A, 2009, 113, 1747–1752 CrossRef CAS PubMed.
- T. H. Reilly, A. W. Hains, H. Y. Chen and B. A. Gregg, Adv. Energy Mater., 2012, 2, 455–460 CrossRef CAS.
- F. Schlosser, M. Moos, C. Lambert and F. Würthner, Adv. Mater., 2013, 25, 410–414 CrossRef CAS PubMed.
- Y. Tan, X. Dai, Y. Li and D. Zhu, J. Mater. Chem., 2003, 13, 1069–1075 RSC.
- A. Krezel and W. Bal, J. Inorg. Biochem., 2004, 98, 161–166 CrossRef CAS PubMed.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- http://www.sasview.org .
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S25. See DOI: 10.1039/c7ta01845b |
| This journal is © The Royal Society of Chemistry 2017 |