
Translationally invariant colloidal crystal templates
Pankaj
Popli
 a,
Saswati
Ganguly
b and
Surajit
Sengupta
*a
a,
Saswati
Ganguly
b and
Surajit
Sengupta
*a
aTIFR Centre for Interdisciplinary Sciences, 36/P Gopanapally, Hyderabad 500107, India. E-mail: pankajp@tifrh.res.in; surajit@tifrh.res.in
bInstitut für Theoretische Physik II: Weiche Materie, Heinrich Heine-Universität Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: saswati@thphy.uni-duesseldorf.de
First published on 29th November 2017
Abstract
We show that dynamic, feed-back controlled optical traps, whose positions depend on the instantaneous local configuration of particles in a pre-determined way, can stabilise colloidal particles in finite lattices of any given symmetry. Unlike in a static template, the crystal so formed is invariant under uniform translations and retains all possible zero energy modes. We demonstrate this in silico by stabilising the unstable two-dimensional square lattice in a model soft solid with isotropic interactions.
1 Introduction
The discovery and development of techniques to form stable, complex assemblies of colloidal particles has emerged as a rather vibrant sub-field of soft condensed matter physics and materials science.1–4 Colloidal particles, synthesized using a variety of routes into an array of shapes and sizes from nanometers to microns, are now readily available for use.4 Apart from many technological applications, the assembly of such colloids into ordered crystals offers us unique insights into the properties of ordinary solids and their behaviour. This is facilitated by the relatively large size and consequent slow timescales5–7 of a colloidal particle, enabling one to use simple optical means to observe and manipulate them.8–10 One encounters, mainly, two paradigms related to the assembly of colloids into complex structures.In the first case, interactions between colloidal particles are tuned. This may be done either by controlling the shape,11 by alloying12,13 or by adding specially reactive patches or tethers.18,19 Apart from this, confinement, either between glass plates14,15 or at an interface,16,17 induces effective interactions between colloidal particles influencing their structure. While one obtains a degree of control over the symmetry and properties of the colloidal crystals so produced, producing complex structures is difficult and needs careful synthesis and/or cumbersome fine tuning of many parameters.
On the other hand, one may also produce ordered colloidal crystals using templates.20 These templates may be either permanent, such as etched onto a surface, or reconfigurable, if produced by optical means.21–23 The latter technique has the advantage that a large variety of crystals,21 quasi-crystals22 and even random structures23 may be produced. Nevertheless, all periodic crystals induced by static templates necessarily suffer from a fundamental flaw.
While crystallisation due to inter-particle interactions necessarily breaks the continuous translational and orientational symmetry of the liquid spontaneously, uniform translations of the crystal as a whole in any direction or global rotations about any axis still do not cost energy. This gives rise to Goldstone modes viz. acoustic phonons whose frequencies disappear with increasing wavelength (or decreasing wavenumber).24 On the other hand, templates break translational invariance explicitly by destroying spatial homogeneity.25 Uniform translations of the whole crystal in one or more directions now cost energy and some or all vibrational modes become massive, i.e. their frequencies do not disappear with decreasing wavenumber. In the extreme case of deep periodic traps, particles do not interact with each other at all and oscillate independently in isolation, making the spectrum of vibrational frequencies resemble that of a trivial Einstein crystal.26 This has a profound influence on scattering processes at small wavenumbers since fewer low energy vibrational modes remain. While for many applications, such as colloidal epitaxy,4,20 this may not be a problem, for many others it is an issue to be addressed. For example, constructing colloidal models of solid–solid transformations27 and interfaces,28 the mechanical behaviour of crystals,29 crystal–glass transition,30etc. requires such spurious effects to be avoided.
In this paper, we propose a means to stabilise finite-sized colloidal crystals in any desired lattice symmetry. The particles may, in principle, have almost any kind of pairwise or many-body interactions. For example, one may now stabilise open lattices such as a square or honeycomb in two dimensions (2d) and a simple cube or diamond cube in three dimensions (3d), which are unstable for most colloids interacting with central pair potentials. As a specific example of the general principle, we stabilise a square lattice in such a system of colloidal particles using computer simulations. Our proposed method is able to generate additional interactions which stabilise such a lattice “on the fly” and requires tuning of only a single parameter. The lattices so formed are invariant under uniform translations and retain acoustic phonon modes.
The rest of the paper is organised in the following way. In the next section we introduce our proposal, which uses a feedback mechanism to stabilise target structures for any kind of colloidal particle. This is followed by our specific results for first a harmonic network of particles as a test case and then a colloidal solid modelled by an isotropic pair potential. This is followed by a three part discussion section where we first explain the mechanism behind our proposal, then indicate how our scheme may be generalised to more complex structures and to higher dimensions and end by discussing caveats for its experimental realisation, comparing our strategy with other existing methods. Finally we conclude with a summary and outlook for future work.
2 The feedback loop
First, the target lattice, ST, is “read in” as a set of reference coordinates {Ri} (see Fig. 1). We assume for simplicity that ST is a Bravais lattice with a single particle basis. Later we show how this procedure may be generalised to more complex structures. Unlike a physical template, the target lattice or structure, which we wish to stabilise, remains in the memory of a computer coupled to an optical setup. Note that there is no restriction on the choice of {Ri}. Next, around each particle i, we fix a neighbourhood Ω containing a fixed set of tagged particles whose instantaneous coordinates ri are recorded. Particle positions undergo thermal fluctuations due to the presence of a solvent. Using a well-defined projection formalism, which we have established in previous publications,31,32 we separate these thermal particle displacements into affine and non-affine subspaces. A laser tweezer is used to exert additional forces,![[scr F, script letter F]](https://www.rsc.org/images/entities/char_e141.gif) χ(ri,Ri) to particle i which bias displacement fluctuations so that the non-affine component of the displacement is suppressed. Since χ depends on instantaneous particle positions, they need to be continuously updated by tracking particle trajectories in real time. Since the intrinsic timescales of colloids are large, this is achievable using the current video microscopic and spatial light modulation technology.33 We derive below χ for any given ST.
χ(ri,Ri) to particle i which bias displacement fluctuations so that the non-affine component of the displacement is suppressed. Since χ depends on instantaneous particle positions, they need to be continuously updated by tracking particle trajectories in real time. Since the intrinsic timescales of colloids are large, this is achievable using the current video microscopic and spatial light modulation technology.33 We derive below χ for any given ST.
 | ||
| Fig. 1 The schematic diagram showing the steps involved in generating feed-back controlled optical traps. Particle coordinates are obtained from video microscopy. The neighbourhood of each particle is recorded and the force χ(ri,Ri) (see text) is calculated from the Hamiltonian 1. Next, the positions of optical traps needed for applying these forces are calculated and the traps are deployed. If this procedure is continuously repeated for all particles at a rate comparable to typical vibrational frequencies of the colloidal particles, a uniform stabilising field is obtained. | ||
Additional forces are computed from the following extended Hamiltonian,32,34 ![[script letter H]](https://www.rsc.org/images/entities/char_e142.gif) = 0 + X. Here 0 represents any Hamiltonian for the interacting particles and,
= 0 + X. Here 0 represents any Hamiltonian for the interacting particles and,
 | (1) |
X involves the “projection operator”  and the particle displacements ui = ri − Ri. The projection operator is a function only of the reference lattice and for any particle, i is given by
and the particle displacements ui = ri − Ri. The projection operator is a function only of the reference lattice and for any particle, i is given by 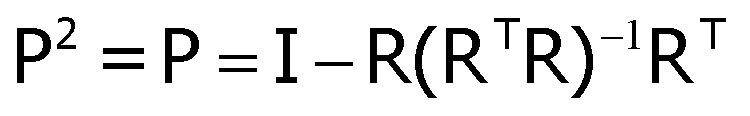 and
and  , where Greek indices go over spatial components and j ∈ Ω is a neighbour of i.
, where Greek indices go over spatial components and j ∈ Ω is a neighbour of i.
Note that (1) preserves translational invariance viz.ui → ui + constant. One can also show32 that  where χi is the least square error made while replacing particle displacements in Ω by the “best fit” affine strains.35 The quantity χi = ΔTPΔ where Δ is the column vector of displacement differences31 with components Δj = uj − ui between particles i and all its neighbours j within Ω. The projection operator therefore projects out of the non-affine part of Δ and χi is the local non-affine parameter. Finally, the forces iχ = − ∂X/∂ui. We show below that suppressing non-affine fluctuations using negative values of hX is sufficient to stabilise ST. The Hamiltonian X introduces new interactions among particles, which guarantee that the target lattice ST is stabilised as long as |hX| is larger than a system size dependent threshold.
where χi is the least square error made while replacing particle displacements in Ω by the “best fit” affine strains.35 The quantity χi = ΔTPΔ where Δ is the column vector of displacement differences31 with components Δj = uj − ui between particles i and all its neighbours j within Ω. The projection operator therefore projects out of the non-affine part of Δ and χi is the local non-affine parameter. Finally, the forces iχ = − ∂X/∂ui. We show below that suppressing non-affine fluctuations using negative values of hX is sufficient to stabilise ST. The Hamiltonian X introduces new interactions among particles, which guarantee that the target lattice ST is stabilised as long as |hX| is larger than a system size dependent threshold.
3 Results
3.1 The harmonic network
It is instructive to demonstrate our procedure first for the case of N particles arranged as a 2d square lattice (see Fig. 2) interacting according to the Hamiltonian: | (2) |
 | ||
| Fig. 2 (a) The phonon dispersion curve ω(q) plotted along high symmetry directions for a square lattice network of vertices connected by springs. Both the nearest neighbour and next nearest neighbour springs with spring constants K1, K2 > 0 are needed for stability. The longitudinal (transverse) modes are shown as solid (dashed) lines. The colours denote K2 = 0 (black), 0.1 (red) and 0.5 (blue). (b) Phonon dispersion for K2 = 0 but now for various values of hX = 0 (black), 0.003 (red), 0.03 (blue). Insets show the interaction volume Ω in the square lattice with bonds (left) and the high symmetry points of the corresponding Brillouin zone (right) | ||
Here pi is the momentum of the particle i, m is the mass, ri are the instantaneous positions, and Ri is the reference position in the target lattice. In this system, the length and energy scales are set by the lattice parameter l and Kl2 respectively. The timescale is set by  . We may choose l = m = K = 1 without the loss of generality. The dimensionless inverse temperature is given by β = Kl2/kBT, with kB as the Boltzmann constant. The interactions have a range equal to the size of the coordination volume Ω. The nearest and next nearest neighbour vertices are connected by harmonic springs of strengths K1 = 1 and K2 respectively. The square lattice is mechanically unstable in the limit K2 → 0 due to softening of the transverse phonon modes.36 This is illustrated in Fig. 2a where we have plotted the phonon dispersion ω(q) for this harmonic square network for three different values of K2. For K2 = 0 the transverse mode disappears. We now add the term proportional to hX in (1) to the harmonic Hamiltonian 0 using the interaction volume, namely, the first and second neighbour shells of the square lattice as our choice of Ω (see Fig. 2 inset). The dynamical matrix,24,26 corresponding to the full Hamiltonian, can be written as
. We may choose l = m = K = 1 without the loss of generality. The dimensionless inverse temperature is given by β = Kl2/kBT, with kB as the Boltzmann constant. The interactions have a range equal to the size of the coordination volume Ω. The nearest and next nearest neighbour vertices are connected by harmonic springs of strengths K1 = 1 and K2 respectively. The square lattice is mechanically unstable in the limit K2 → 0 due to softening of the transverse phonon modes.36 This is illustrated in Fig. 2a where we have plotted the phonon dispersion ω(q) for this harmonic square network for three different values of K2. For K2 = 0 the transverse mode disappears. We now add the term proportional to hX in (1) to the harmonic Hamiltonian 0 using the interaction volume, namely, the first and second neighbour shells of the square lattice as our choice of Ω (see Fig. 2 inset). The dynamical matrix,24,26 corresponding to the full Hamiltonian, can be written as ![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) μν = μν0 + μνX, where μν0 is the dynamical matrix from 0 and
μν = μν0 + μνX, where μν0 is the dynamical matrix from 0 and
 | (3) |
μνX(q) = 2hX![[scr A, script letter A]](https://www.rsc.org/images/entities/char_e520.gif) X(q)δμν. The function X(q) ∼ q4 for small wavenumbers37 so that X does not affect the elastic properties, e.g. the speed of sound, of a mechanically stable lattice. Furthermore, these results do not depend on the nature of 0.
X(q)δμν. The function X(q) ∼ q4 for small wavenumbers37 so that X does not affect the elastic properties, e.g. the speed of sound, of a mechanically stable lattice. Furthermore, these results do not depend on the nature of 0.
In Fig. 2b we plot the resulting phonon dispersion curves all at K2 = 0 but for three different values of hX. As hX is made more negative, the transverse phonon mode appears to revive. One must note however that as q → 0 the nature of AX(q) dictates that the speed of transverse sound vanishes in the hydrodynamic limit. Nevertheless, for finite lattices, this limit is never reached since wavenumbers are cut off at q = 2π/L where L ∝ N1/d is the linear system size.
3.2 The Gaussian core solid
Consider, next, a system of particles interacting with a soft, purely repulsive, “Gaussian core” model potential (GCM)38,39 where the harmonic interaction between particles i and j is replaced by the functionϕij = ε![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(−rij2/σ2). exp(−rij2/σ2). |
When particles interacting with the GCM potential are arranged in a square lattice, one obtains a mechanically unstable solid. Small displacement fluctuations from the ideal square lattice positions (where forces still disappear due to symmetry) make the solid deform into a stable triangular structure via soft transverse modes.36 This is clear from the calculated dispersion curve shown in Fig. 3a for hX = 0.
 | ||
| Fig. 3 (a) Phonon dispersion curve ω2(q) plotted along high symmetry directions for a square lattice of particles interacting with the GCM potential at ρ = 0.5 and T = 1 × 10−3. The small q region where ω2 < 0 is shown in the inset. We follow the same convention as in Fig. 2. There are two sets of curves for hX = 0 (red curves) and hX = 0.5 (blue curves). (b) A stability diagram for finite size square lattices constructed from the dispersion curve in (a). The green shaded region represents −hX values below which the square lattice becomes unstable (ω2(q) < 0). We have also marked six points on the graph such that black (white) circles denote stable (unstable) square lattices. | ||
We now turn on hX defined exactly as in the case of the network. As hX is decreased below zero, again, we see a revival of the transverse phonon mode. Unlike the network however, for all hX, the transverse mode now has ω2 < 0 in a small region of q → 0. Making hX more negative can, nevertheless, restrict this region to extremely small q values which are not accessed by a solid of finite size due to the infrared cut-off discussed earlier. Since within harmonic approximation temperature enters only as a pre-factor, this leads to a T independent “stability diagram” as shown in Fig. 3b.
To verify the stability diagram presented in Fig. 3b we perform Monte Carlo simulations with standard Metropolis updates42 of a GCM solid with various N and hX values keeping ρ and T the same as before, equilibrating the system for a minimum of 106 Monte Carlo steps (MCS) starting from the initial square lattice. To check our results we obtained the probability distribution of the non-affine parameter P(χ) and compared it with the predictions from the harmonic theory using the calculated dynamical matrix μν as the input.31,43
Equilibrated configurations from our simulations are presented in Fig. 4a. The hX and N values for these configurations are marked in Fig. 3b. It is clear that these results follow the expectations from our stability criterion. Once the stability threshold is breached, the square lattices destabilise due to q → 0 modulations. The local non-affine parameter χ rapidly rises as the crystal becomes unstable and is shown as a colour map. For all the stable square solids studied, our results were indistinguishable from the theoretical prediction though they deviate, as they should, if instability sets in (Fig. 4b). If ρ is increased, harmonic approximation becomes more accurate and the magnitude of hX needed to stabilise the square lattices becomes even smaller.
 | ||
| Fig. 4 (a) Configurations obtained from Monte Carlo simulations after 6 × 106 MCS arranged according to the corresponding points marked in Fig. 3b verifying the stability condition. Colours correspond to the local value of χ. (b) P(χ) obtained from the equilibrated configurations where the square lattice is stable (top) and unstable (bottom). Data points are computed from the results of our simulations and lines are predictions of the harmonic theory.43 The configurations and curves are labelled by the corresponding hX and N values. | ||
4 Discussion
4.1 Stabilisation mechanism
We begin by commenting on the mechanism by which the ST structure is stabilised. Consider two crystal lattices as the target, ST, and the equilibrium structure that the solid prefers without hX, viz. SE. The lowest energy path from ST to SE (a) may involve non-affine displacements of particles within Ω, or “atomic shuffles” or (b) may be purely affine as is the case of structures connected by a group–subgroup relation.44 Possibility (a) is easy to analyse since, in this case, the term involving hX in (1) increases the energy of SE relative to that of ST, stabilising the latter; any small fluctuation taking ST → SE always costs energy. Possibility (b) is more subtle.For example, both square and triangular crystals may be considered as special cases of a general oblique lattice and one can be obtained from the other by a purely affine transformation. Along this transformation path, which involves a bulk homogeneous strain, hX will not contribute. However, such an event is statistically unlikely except for extremely small systems. What is more likely is that a small patch of particles locally transforms to the SE structure, creating χ at the interface. Within the classical nucleation theory24 the free energy cost of such a patch of size Lp is p = AΔLp2 + BhXLp, where we ignore interfacial terms independent of hX. Here Δ is the bulk free energy difference per unit area between the two lattices and A and B are constants. This patch is stable only if it is larger than a critical size Lp* ∼ −hX/Δ and costs interfacial energy ∼hX2/Δ. Lp* can be large if Δ is small and can be made even larger by tuning hX. If Lp* > L, again, a finite ST crystal will be stable due to the inability of the system to create a sufficiently low energy SE|ST interface.
It is possible to demonstrate both these mechanisms for the square to triangular transition. It is known that a mechanically unstable square lattice may decay at nonzero temperatures into a stable triangular structure in many different ways.36 For example, (a) alternate rows of particles in the square lattice may shift by half-a-lattice spacing, hence producing a distorted triangular lattice that subsequently equilibrates. On the other hand, a patch of particles inside the square matrix may undergo distortion to a triangular structure (b). Our stabilisation strategy should be able to recover the target square lattice ST from both (a & b) of these distortions.
In Fig. 5a and b we demonstrate this explicitly by starting from initial (square) configurations of an N = 2500 GCM solid incorporating these two kinds of distortions (a) and (b) and equilibrating with hX = −2.0, where the square lattice is stable. In the first case (Fig. 5a), the local shuffles of particles cost energy and are quickly removed from the solid. In the second case (Fig. 5b(i)), we first create a large patch of particles with a local triangular order using an inhomogeneous affine strain. Next, we equilibrate the surrounding matrix keeping the particles within the patch immobile. This produces a mechanically relaxed (but high energy with large local χ) interface between a square solid and a triangular inclusion. The constraint is then removed and the whole system is equilibrated. Fig. 5b(iii) shows that the sub-critical patch thus created disappears. As a check (Fig. 6) we ensure that P(χ) obtained from the equilibrated configurations again matches theoretical predictions.31,43
 | ||
| Fig. 5 Configurations showing the recovery of the square lattice after an initial distortion: (a) The square lattice of 2500 GCM particles at ρ = 0.5 and T = 1 × 10−3 was distorted by displacing alternate rows of particles by half a lattice spacing (see schematic inset), producing a triangular lattice close to the SE structure (i). Under a stabilising field of hX = −2.0, the target lattice quickly recovers as seen in (ii) (4.8 × 104 MCS) and (iii) (24 × 104 MCS). (b) Here we produce a patch of triangular lattice within a square matrix (i) and again let the solid recover the square structure: (ii and iii as above) under the influence of the same field as in (a). The particles are coloured according to the value of the local χ field. | ||
 | ||
| Fig. 6 The resulting P(χ) at the end of the runs in Fig. 5a (red circles) and Fig. 5b (blue squares); the solid line is the harmonic theory prediction.43 | ||
In 2d, long wavelength displacement fluctuations are known to destroy the crystalline order24 and therefore a discussion of this effect is germane for the 2d square solid discussed here. Crystals in 2d posses only “quasi”, rather than true, long-ranged order since the mean squared displacement 〈u2〉 diverges as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) L. This is an effect of dimensionality and appears in the thermodynamic limit for any 2d crystal, even those with non-pathological phonon spectra. These fluctuations have also recently been observed even for amorphous solids,45–47 considerably increasing the scope of its general applicability. How do these fluctuations affect our results?
L. This is an effect of dimensionality and appears in the thermodynamic limit for any 2d crystal, even those with non-pathological phonon spectra. These fluctuations have also recently been observed even for amorphous solids,45–47 considerably increasing the scope of its general applicability. How do these fluctuations affect our results?
Our proposal is designed to stabilise finite-sized crystals. Indeed, for any |hX|, as the system size L is increased, one reaches a threshold above which the solid is no longer stable (see Fig. 3b). In order to stabilise larger systems, |hX| has to be increased as ∼L2 = N. To obtain stable solids with large L requires large and probably experimentally un-realisable optical field strengths. The weak logarithmic divergence of displacements discussed above is therefore unobservable at the system sizes obtainable with any realistic |hX| and does not alter our results.
4.2 Complex open lattices
In this paper, we present a “proof of principle” demonstration for a relatively simple case of the 2d square lattice. We now indicate how our procedure may be generalised in a straight-forward manner to periodic crystalline structures of greater complexity. These lattices are “open” in the sense that their packing density is much lower than the “close-packing” density for the dimensionality considered and are usually metastable or unstable. The only closely packed structure in 2d is the triangular lattice, while in three dimensions (3d), there are a large number of ordered closely packed structures possible which are obtained by stacking 2d triangular planes in specific sequences. The face-centered cubic and hexagonal closely packed lattices are the most common ones.24,26Open lattices may be of two kinds. Firstly they may be sparsely coordinated primitive lattices. Maxwell's condition for stability48,49 requires that the coordination number, z, must be strictly >2 × d, which amounts to z > 4 in 2d and z > 6 in 3d. The square lattice in 2d and the simple cubic lattice in 3d are marginal having 4 and 6 nearest neighbours respectively. Including the second coordination shell while constructing the coarse grain volume Ω is essential in these cases. This ensures that phonon modes which are soft, such as the shear modes encountered in the 2d square lattice, involve particles within Ω. These modes need to be suppressed in order to ensure stability of the lattice.
Open lattices may also result from decorating a primitive lattice with a basis i.e. a motif consisting of more than one inequivalent particle. For example, in 2d one has a planar honeycomb lattice where a dimer replaces every site of the triangular lattice.50 Similarly, in the Kagomé lattice, the equivalent motif contains five particles arranged as a pair of equilateral triangles, which share a vertex.49 In 3d, the diamond cubic structure contains repeated vertex sharing tetrahedra.
Such lattices also contain “floppy” modes which involve relative twists of the basis with respect to the rest of the particles in the structure.43,49 In the planar honeycomb structure localised modes representing a twist of the dimer results in the creation of a Stone–Wales defect.43 The coarse grain volume needs to be large enough to be able to describe such modes and Ω needs to contain all symmetry dictated particles belonging to copies of the basis, centred on each site of the primitive lattice. The single parameter hX may then be used to suppress these modes. In this case, however, our formulae for relative displacement Δ and projection  matrices need to be generalised as follows.
matrices need to be generalised as follows.
To obtain Δ we arrange the entries ΔJmn = uJm − uIn as a column vector. The first (capitalized) index numbers the basis and the second index numbers the ![[scr N, script letter N]](https://www.rsc.org/images/entities/char_e52d.gif) Ω particles within the basis. The elements of the matrix of reference coordinates are similarly given by
Ω particles within the basis. The elements of the matrix of reference coordinates are similarly given by  . These elements are arranged so that the matrix
. These elements are arranged so that the matrix  has the dimensions of d
has the dimensions of d![[scr B, script letter B]](https://www.rsc.org/images/entities/char_e13f.gif) Ω × d2, where Ω is the total number of distinct lattice vectors, {R}, in Ω. In terms of
Ω × d2, where Ω is the total number of distinct lattice vectors, {R}, in Ω. In terms of  the dΩ × dΩ dimensional projection matrix remains,
the dΩ × dΩ dimensional projection matrix remains,  . Once a suitable Ω is chosen, Ω, Ω,
. Once a suitable Ω is chosen, Ω, Ω,  and
and  are known and the feedback loop can be implemented exactly as discussed earlier. As the complexity of the lattices increases, the calculation of the forces becomes more involved but always scales linearly with Ω. Real time feedback is therefore still limited by the response time of the optical system and not computation.
are known and the feedback loop can be implemented exactly as discussed earlier. As the complexity of the lattices increases, the calculation of the forces becomes more involved but always scales linearly with Ω. Real time feedback is therefore still limited by the response time of the optical system and not computation.
4.3 Experimental realisation of hX
We now comment briefly on the foreseeable technical challenges which may need to be overcome to implement our proposal in the laboratory. Colloidal particles have sizes in the range from 1 nm to a few μm, with the larger sized particles being easier to trap and manipulate.4 For colloids at the larger end of the size range, typical timescales are of the order of a second.7 These particles need to be tracked in real time, and forces need to be calculated and applied using laser traps.Particle tracking at a frame rate of 10 kHz is possible using the current technology51 so this is not a rate limiting step. Since the computations needed to calculate forces even for the most complex lattices are simple, these are also quite fast. The crucial experimental step therefore is the deployment of traps to generate forces.
For this purpose one may use a spatial light modulator and holographic tweezer technology33 which produce a reconfigurable optical surface based on inputs. Typical spatial resolutions of these devices are about 1–3 μm which is appropriate for our purpose. The input frame rate can theoretically go from 500 up to 1 kHz, although the actual frame rates can be much slower. In any case, considering that large colloids are slow, one has a reasonable margin of exploration.
Are the values of hX for reasonably sized lattices too high to be realised in the lab? The quantitative estimation of energy due to the hX term shows that it is comparable to the energy of interactions which is of the order of a few kBT. This is the regime where all previous optical manipulations of colloidal crystals have been traditionally performed.21–23 However, spatial light modulators also decrease the light intensity which depends on the make of the device; so higher laser powers are needed limiting the number of particles which may be trapped without causing excessive heating. An alternative procedure is to use time shared traps52 in a dense enough array fixed in space with their intensities modified intermittently to approximate the optical potential surface.
Feedback controlled traps of somewhat different nature have already been employed to study the crystallisation of colloids.53 The principle behind this implementation is quite different from ours. Instead of coupling to a local configuration-dependent parameter such as χ, the trap responds to a global order parameter for crystallisation. The symmetry of the crystal is still determined by inter-particle interactions and very limited control over the crystal structure is possible.
5 Conclusion
In conclusion, we present a proposal for producing crystalline templates for colloidal particles using dynamic, feedback controlled laser traps. These templates are reconfigurable and can, in principle, stabilise any structure in any dimension. Unlike static templates, our method does not merely provide a restoring force to the target structure. All possible affine fluctuations around ST are allowed, preserving all symmetries of the crystal. Only non-affine excursions away from ST are selectively suppressed.One may view the stabilising forces that we use as originating from an effective three-body potential viz., eqn (1). The form of this potential depends on the reference configuration, guaranteeing that these forces necessarily stabilise the target structure. Our method therefore automatically determines the potential which stabilises ST. The advantage of our method is that no further assumption about particle interactions is necessary. While it may be possible, for the case of simple target structures, to guess interactions that give rise to them and perhaps design colloids which produce these structures,18 this becomes progressively difficult as the complexity of ST increases. This is especially important when we realise that using a similar strategy even inhomogeneous structures such as surfaces and interfaces of any specified orientation as well as random glassy configurations may be stabilised.
Finally, we emphasise again that fast particle tracking and the realisation of fast response times of light fields required for our method to work is feasible for colloidal solids because of their slow timescales. Similarly heating problems associated with these light fields should also be minimal especially since the magnitude of the forces needed is small (≪kBT). Of course, only actual experiments can finally decide upon the feasibility of our proposal. We hope that our work motivates experimental work in this direction in the near future. Extension of our method to stabilise complex open lattices in 2d and 3d, in collaboration with experiments, is an attractive future goal.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank J. Horbach, P. Nath, A. K. Sood, R. Ganapathy and S. Egelhaaf for discussion. SS acknowledges travel support from the FP7-PEOPLE-2013-IRSES grant no: 612707, DIONICOS.References
- A. K. Sood, Solid State Phys., 1991, 45, 1 CAS.
- A. K. Arora and B. V. R. Tata, Ordering and Phase Transitions in Charged Colloids, VCH, New York, 1995 Search PubMed.
- H. Löwen, Phys. Rep., 1994, 237, 76 CrossRef.
- A. Ivlev, H. Löwen, G. Morfill and C. P. Royall, Complex Plasmas and Colloidal Dispersions: Particle-resolved Studies of Classical Liquids and Solids, World Scientific, Singapore, 2012 Search PubMed.
- A. J. Hurd, N. A. Clark, R. C. Mockler and W. J. O'Sullivan, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2869 CrossRef CAS.
- Z. Cheng, J. Zhu, W. B. Russel and P. M. Chaikin, Phys. Rev. Lett., 2000, 85, 1460 CrossRef CAS PubMed.
- P. Keim, G. Maret, U. Herz and H. H. von Grünberg, Phys. Rev. Lett., 2004, 92, 215504 CrossRef CAS PubMed.
- M. Bowick and P. M. Chaikin, Nat. Mater., 2016, 15, 1151 CrossRef CAS PubMed.
- K. Zahn, A. Wille, G. Maret, S. Sengupta and P. Nielaba, Phys. Rev. Lett., 2003, 90, 155506 CrossRef CAS PubMed.
- W. T. M. Irvine, A. D. Hollingsworth, D. G. Grier and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15544 CrossRef CAS PubMed.
- A. van Blaaderen, Nature, 2006, 439, 545 CrossRef CAS PubMed.
- A.-P. Hynninen, J. H. J. Thijssen, E. C. M. Vermolen, M. Dijkstra and A. van Blaaderen, Nat. Mater., 2007, 6, 202 CrossRef CAS PubMed.
- A. D. Law, D. M. A. Buzza and T. S. Horozov, Phys. Rev. Lett., 2011, 106, 128302 CrossRef PubMed.
- S. Neser, C. Bechinger, P. Leiderer and T. Palberg, Phys. Rev. Lett., 1997, 79, 2348 CrossRef CAS.
- R. P. A. Dullens and W. K. Kegel, Phys. Rev. Lett., 2004, 92, 195702 CrossRef PubMed.
- P. Pieranski, Phys. Rev. Lett., 1980, 45, 569 CrossRef CAS.
- D. Ershov, J. Sprakel, J. Appel, M. A. Cohen Stuart and J. van der Gucht, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9220 CrossRef CAS PubMed.
- S. Glotzer and M. Solomon, Nat. Mater., 2007, 6, 557 CrossRef PubMed.
- J. Russo, P. Tartaglia and F. Sciortino, Soft Matter, 2010, 6, 4229 RSC.
- A. van Blaaderen, R. Ruel and P. Wiltzius, Nature, 1997, 385, 321 CrossRef CAS.
- A. Chowdhury, B. J. Ackerson and N. A. Clark, Phys. Rev. Lett., 1985, 55, 833 CrossRef CAS PubMed.
- J. Mikhael, G. Gera, T. Bohlein and C. Bechinger, Soft Matter, 2011, 7, 1352 RSC.
- J. Bewerunge, A. Sengupta, R. F. Capellmann, F. Platten, S. Sengupta and S. U. Egelhaaf, J. Chem. Phys., 2016, 145, 044905 CrossRef PubMed.
- P. Chaikin and T. Lubensky, Principles of Condensed Matter Physics, Cambridge Press, Cambridge, 1995 Search PubMed.
- E. Frey, D. R. Nelson and L. Radzihovsky, Phys. Rev. Lett., 1999, 83, 2977 CrossRef CAS.
- N. W. Ashcroft and N. D. Mermin, Solid State Physics, Holt, Rinehart, and Winston, New York, 1976 Search PubMed.
- Y. Peng, F. Wang, Z. Wang, A. M. Alsayed, Z. Zhang, A. G. Yodh and Y. Han, Nat. Mater., 2014, 14, 101 CrossRef PubMed.
- D. Meyers, Surfaces, Interfaces, and Colloids: Principles and Applications, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- Rob Phillips Crystals, defects and microstructures: Modeling across scales, Cambridge Press, Cambridge, 2004.
- N. B. Simeonova, R. P. A. Dullens, D. G. A. L. Aarts, V. W. A. de Villeneuve, H. N. W. Lekkerkerker and W. K. Kegel, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73, 041401 CrossRef PubMed.
- S. Ganguly, S. Sengupta, P. Sollich and M. Rao, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 87, 042801 CrossRef PubMed.
- S. Ganguly, S. Sengupta and P. Sollich, Soft Matter, 2015, 11, 4517 RSC.
- G. C. Spalding, J. Courtial and R. D. Leonardo, in Structured Light and its Applications, ed. D. L. Andrews, Elsevier, Oxford, 2008 Search PubMed.
- S. Ganguly, P. Nath, J. Horbach, P. Sollich, S. Karmakar and S. Sengupta, J. Chem. Phys., 2017, 146, 124501 CrossRef PubMed.
- M. L. Falk and J. S. Langer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1998, 57, 7192 CrossRef CAS.
- X. Mao, A. Souslov, C. I. Mendoza and T. C. Lubensky, Nat. Commun., 2015, 6, 5968 CrossRef CAS PubMed.
- S. Ganguly and S. Sengupta, J. Chem. Sci., 2017, 129, 891 CrossRef CAS.
- F. H. Stillinger, J. Chem. Phys., 1976, 65, 3968 CrossRef CAS.
- F. H. Stillinger and T. A. Weber, J. Chem. Phys., 1981, 74, 4015 CrossRef CAS.
- S. Prestipino, F. Saija and P. V. Giaquinta, Phys. Rev. Lett., 2011, 106, 235701 CrossRef PubMed.
- M. Zu, J. Liu, H. Tong and N. Xu, Phys. Rev. Lett., 2016, 117, 085702 CrossRef PubMed.
- D. Frenkel and B. Smit, Understanding Molecular Simulations, Academic Press, San Diego, 2002 Search PubMed.
- S. Ganguly, P. S. Mohanty, P. Schurtenberger, S. Sengupta and A. Yethiraj, Soft Matter, 2017, 13, 4689 RSC.
- K. Bhattacharya, G. Conti, G. Zanzotto and J. Zimmer, Nature, 2004, 428, 55 CrossRef CAS PubMed.
- H. Shiba, Y. Yamada, T. Kawasaki and K. Kim, Phys. Rev. Lett., 2016, 117, 245701 CrossRef PubMed.
- B. Illing, S. Fritschi, H. Kaiser, C. L. Klix, G. Maret and P. Keim, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1856 CrossRef CAS PubMed.
- S. Vivek, C. P. Kelleher, P. M. Chaikin and E. R. Weeks, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1850 CrossRef CAS PubMed.
- J. C. Maxwell, Philos. Mag., 1864, 27, 294 Search PubMed.
- T. C. Lubensky, C. L. Kane, X. Mao, A. Souslov and K. Sun, Rep. Prog. Phys., 2015, 78, 073901 CrossRef CAS PubMed.
- A. Mitra, S. Ganguly, S. Sengupta and P. Sollich, J. Stat. Mech.: Theory Exp., 2015, P06025 CrossRef.
- O. Otto, F. Czerwinski, J. L. Gornall, G. Stober, L. B. Oddershede, R. Seidel and U. F. Keyser, Opt. Express, 2010, 18, 22722 CrossRef CAS PubMed.
- Y. Tanaka and S. Wakida, Biomed. Opt. Express, 2015, 6, 3670 CAS.
- J. J. Juárez and M. A. Bevan, Adv. Funct. Mater., 2012, 22, 3833 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |