
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of aggregation temperatures in mixed and blended cytocompatible thermoresponsive block co-polymer nanoparticles†
Ruggero
Foralosso
a,
Lee
Moir
a,
Francesca
Mastrotto
ab,
Luana
Sasso
a,
Aleksandra
Tchoryk
a,
Amjad
Selo
a,
Anna
Grabowska
c,
Marianne B.
Ashford
d,
Jonathan
Aylott
a,
Paul R.
Gellert
d,
Sebastian G.
Spain
 ae and
Cameron
Alexander
*a
ae and
Cameron
Alexander
*a
aSchool of Pharmacy, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: Cameron.alexander@nottingham.ac.uk
bDivision of Cancer Biology, School of Medicine, University of Nottingham, NG7 2RD, UK
cDipartimento di Scienze del Farmaco, via Marzolo, 5, 35131 Padova, Italy
dAstraZeneca, Silk Road, Macclesfield, SK10 4TF, UK
eDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK
First published on 2nd October 2017
Abstract
A small library of thermoresponsive amphiphilic copolymers based on polylactide-block-poly((2-(2-methoxyethoxy)ethyl methacrylate)-co-(oligoethylene glycol methacrylate)) (PLA-b-P(DEGMA)-co-(OEGMA)), was synthesised by copper-mediated controlled radical polymerisation (CRP) with increasing ratios of OEGMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEGMA. These polymers were combined in two ways to form nanoparticles with controllable thermal transition temperatures as measured by particle aggregation. The first technique involved the blending of two (PLA-b-P(DEGMA)-co-(OEGMA)) polymers together prior to assembling nanoparticles (NPs). The second method involved mixing pre-formed nanoparticles of single (PLA-b-P(DEGMA)-co-(OEGMA)) polymers. The observed critical aggregation temperature Tt did not change in a linear relationship with the ratios of each copolymer either in the nanoparticles blended from different copolymers or in the mixtures of pre-formed nanoparticles. However, where co-polymer mixtures were based on (OEG)9MA ratios within 5–10 mole%, a linear relationship between (OEG)9MA composition in the blends and Tt was obtained. The data suggest that OEGMA-based copolymers are tunable over a wide temperature range given suitable co-monomer content in the linear polymers or nanoparticles. Moreover, the thermal transitions of the nanoparticles were reversible and repeatable, with the cloud point curves being essentially invariant across at least three heating and cooling cycles, and a selected nanoparticle formulation was found to be readily endocytosed in representative cancer cells and fibroblasts.
:DEGMA. These polymers were combined in two ways to form nanoparticles with controllable thermal transition temperatures as measured by particle aggregation. The first technique involved the blending of two (PLA-b-P(DEGMA)-co-(OEGMA)) polymers together prior to assembling nanoparticles (NPs). The second method involved mixing pre-formed nanoparticles of single (PLA-b-P(DEGMA)-co-(OEGMA)) polymers. The observed critical aggregation temperature Tt did not change in a linear relationship with the ratios of each copolymer either in the nanoparticles blended from different copolymers or in the mixtures of pre-formed nanoparticles. However, where co-polymer mixtures were based on (OEG)9MA ratios within 5–10 mole%, a linear relationship between (OEG)9MA composition in the blends and Tt was obtained. The data suggest that OEGMA-based copolymers are tunable over a wide temperature range given suitable co-monomer content in the linear polymers or nanoparticles. Moreover, the thermal transitions of the nanoparticles were reversible and repeatable, with the cloud point curves being essentially invariant across at least three heating and cooling cycles, and a selected nanoparticle formulation was found to be readily endocytosed in representative cancer cells and fibroblasts.
Introduction
Thermoresponsive nanoparticles are of interest for a range of applications including sensing, diagnostics, adaptive optics and therapeutics.1–6 The most widely studied nanoparticles of this type have been those intended for drug delivery, as the thermal triggering of a response in a carrier vehicle potentially allows exogenous control of particle location or drug release at a specific site.6–10 The thermoresponsive behaviour of these nanoparticles is usually bestowed by a surface corona of polymer chains which undergo a phase transition at a specified temperature in solution/suspension. The thermoresponsive polymers can be attached to a pre-formed nanoparticle, or the particles themselves can be composed of self-assembling block co-polymers in which the inner block is solvophobic. In both cases the outer block or corona polymers exhibit changes in their solvation properties according to a change in temperature.The thermoresponsive polymers forming the corona are characterised by their Lower or Upper Critical Solution Temperature (LCST11 or UCST12) behaviour. For the LCST polymers, which have been most widely investigated for thermoresponsive nanoparticles, there is a critical temperature above which the polymer is only partially miscible in its solvent. Below the LCST, the polymer and the solvent are in the same phase, stabilized for example by hydrogen bonds between the solvent and polymeric chain. Above the LCST, the process is entropy-driven and leads to phase separation, resulting in collapse of the polymer chain.5 In the case of nanoparticles with a corona of polymers displaying an LCST in water, the result of the polymer phase change is a decrease in surface solvation and an enhancement in apparent particle hydrophobicity. In pure aqueous suspensions, the LCST change of surface-displayed polymers results in a loss in colloidal stability, which then leads to particle precipitation or aggregation, dependent on concentration.1 In more complex environments, for example in biological fluids or in tissue, the phase changes and increase in surface hydrophobicity can result in differential protein adsorption or cell membrane interaction.13 These variations in bio-interfacial behaviour14 have been the most widely explored, as nanoparticles with these properties have been shown to exhibit controllable cell attachment and endocytosis.6,15
As a result, there have been many efforts to tune nanoparticle–surface interactions by varying the temperatures at which phase changes occur. However, while there have been many papers showing that the LCST (and UCST) of linear polymers in solution can be controlled very precisely by co-monomer content, molar mass and end-group content,16–19 the control of nanoparticle thermoresponsive properties has been more elusive.1,20 In part, this is because many studies have had a strong application focus, particularly in drug delivery, where interpretations of data are often confounded by alterations in polymer LCST by electrolytes21 and biopolymers, which in turn lead to highly variable protein adsorption or cell membrane association dependent on the specific environment.22 There are also the well-known issues in characterising polymers grown from the surface of nanoparticles, or estimating polymer surface coverage when the polymers are grafted to pre-formed nanoparticles. Very recently, work from the Gibson group has offered important insight into the thermoresponsive behaviour of poly(N-isopropylacrylamide) (pNIPAM) coated gold NPs.23 Mixtures of thermoresponsive particles with different core sizes and chain lengths were shown to display controllable aggregation behaviour, with co-operativity in association between particles demonstrated convincingly in spectroscopic and transmission electron microscopy studies.
In parallel, we have been investigating routes by which precise and reliable thermoresponsive behaviour can be introduced into ‘polymer-only’ nanoparticles, ultimately to develop predictable and constant release systems for drug delivery. We are also interested in developing simple formulation rules, through which temperature response can be ‘dialled in’ to nanoparticles in a manner analogous to that in linear responsive co-polymers. Here we describe how mixtures of different thermoresponsive polymers, based on oligo(ethyleneglycol)methacrylate (OEGMA) and 2-(2-methoxyethoxy)ethyl methacrylate (also known as diethyleneglycolmethacrylate or DEGMA) monomers, and their resulting nanoparticles can be mixed to yield a range of temperature responses. We base this work on the hypothesis that aggregation temperature of nanoparticles can be controlled through 2 ways: (a) mixing of pre-formed nanoparticles from constituent individual PLA-b-p-OEGMA-stat-DEGMAs of varying critical aggregation temperature (single polymer corona nanoparticles) and (b) mixing of pre-formed co-polymers of poly(lactic acid) (PLA)-block-poly(oligo(ethyleneglycol)methacrylate) (p(OEGMA))-co-poly(diethyleneglycomethacrylate) (pDEGMA) with varying LCST prior to nanoparticle formation through nanoprecipitation (binary polymer corona nanoparticles). Accordingly, we report copolymer and nanoparticle mixtures at different constituent ratios, and show the results of dynamic light scattering and turbidimetry experiments to characterise size and transition temperatures of the resulting nanoparticles (Fig. 1).
 | ||
| Fig. 1 Structure of polymers and cartoon representation of different PLA–p(OEGMA) copolymers used to form thermoresponsive nanoparticles. Individual polymers with varying ratios of (OEG)9MA and DEGMA were formed into nanoparticles (“Single polymer corona nanoparticles”) or mixtures of co-polymers were prepared prior to nanoparticle formation (“binary polymer corona nanoparticles”). The resulting nanoparticles displayed variable aggregation temperatures dependent on the relative concentrations of the copolymers in the blends. | ||
Materials and methods
Materials
D,L-Lactide (Sigma Aldrich), (2-(2-methoxyethoxy)ethyl methacrylate) (DEGMA, average Mnca. 188, Sigma Aldrich), oligo(ethylene glycol) methyl ether methacrylate (OEGMA, average Mnca. 500, Sigma Aldrich), copper(II) bromide (Alfa Aesar, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) (Sigma Aldrich, 99%), tin(II)2-ethylhexanoate (Sn(Oct)2) (Sigma Aldrich, 95%), 2-butanone (Sigma Aldrich, 99%) were used as received.Synthesis of polymers
| Polymer | PLA Mn (kDa) | Total Mn, GPC (kDa) | Đ GPC | M n (kDa) NMR | (OEG)9MA, theor. (mol%) | (OEG)9MA content (mol%) NMR | NP size (DLS)a (nm) | NP Tt (°C) |
|---|---|---|---|---|---|---|---|---|
| a Mean radius. | ||||||||
| PLA–pDEGMA 100 | 23.4 | 25.1 | 1.48 | 47.7 | 0 | 0 | 118 | 28.8 |
| PLA–pOEGMA 5 | 23.4 | 23 | 1.39 | 39.4 | 5 | 5.2 | 121 | 40.7 |
| PLA–pOEGMA 10 | 23.4 | 27.7 | 1.48 | 52.7 | 10 | 8.9 | 111 | 43.9 |
| PLA–pOEGMA 15 | 23.4 | 27.4 | 1.53 | 50.9 | 20 | 14.5 | 111 | 51.1 |
| PLA–pOEGMA 20 | 23.4 | 32.8 | 1.56 | 69.0 | 35 | 21.2 | 139 | 55.2 |
Characterisation of polymers – NMR
1H NMR spectra were recorded at 20 °C on a Bruker instrument operating at 400 MHz. Chemical shifts (δ) are referenced to CDCl3 (δ 7.26 ppm). The data were processed using MestReNova (v. 6.0.2) software. To determine DP of PLA macroinitiator the proton peak for lactide (δ 5.3–5.1 ppm) was integrated in relation to terminal methyl of isobutyryl bromide group (δ 1.96 ppm, 6H). The DP of copolymers were measured from DP of PLA (δ 5.3–5.1 ppm, set to 1H) then integrating terminal PEGMA methyl (δ 3.46–3.36 ppm, yH), y × (PLA DP) = copolymer DP.Gel permeation chromatography (GPC)
GPC was performed on a Polymer Laboratories GPC 50 system equipped with a refractive index detector. Separations were achieved with a pair of PLgel Mixed-D (5 μm bead, 7.8 × 300 mm) columns with a matching guard (7.8 × 50 mm) and chloroform as eluent at a flow rate of 1 mL min−1. Calibration was performed using narrow molar mass range polystyrene standards (Polymer Labs) in the molecular weight range 0.13–210 kDa. Molar masses and dispersity values were calculated using Cirrus GPC 3.0 software.Full details of the characterisation data for these polymers are provided in the ESI.†
Nanoparticle preparation
The copolymers were dissolved in acetone at a concentration of 5 mg mL−1. They were then mixed together at different ratios. Each blend was prepared using a double syringe pump. One syringe was used to load the polymeric solution; the other one was loaded with an equal amount of water. The two solutions were loaded at the same time and mixed together in a T-junction, using the same flow rate of 1 mL min−1 and loading the same volume for both syringes. Copolymers rapidly formed nanoparticle suspensions through solvent exchange between water and acetone. The final suspension was then left at room temperature in order to reach complete acetone evaporation.Transition temperature determination by cloud point analysis
The transition temperature (Tt) was determined by measuring the change in absorbance of light using UV-vis spectrophotometry at 550 nm. This allowed for the increase in scattered light upon sample aggregation to be measured (cloud point). Cloud point evaluations were performed using a DU-800 UV/vis Spectrophotometer from Beckman Coulter, US, working at concentrations ranging from 2 to 5 mg mL−1 in deionised water. Samples were heated at a rate of 0.5 °C min−1. Measurements were taken every 0.5 °C. The absorbance was converted to % transmittance (%transmittance = 100 × (10−Abs)). Transmittance data were plotted versus temperature and Tt taken to be the point at which there was a 50% drop in the normalised transmittance (normalised to 100%).Heating–cooling cycles
Polymeric blends were analysed working at a concentration of 5 mg mL−1. The blends were heated and cooled consecutively for 3 times using a rate of 0.5 °C min−1, taking measurements every 0.5 °C. The heating and cooling curves were recorded for each blend for each cycle.DLS measurements
Dynamic light scattering was performed using Nano-ZS Zetasizer and Viscotek Model 802 instruments. Samples were taken from nanoparticle suspension in ultrapure water (1:20 dilution), typically at 0.25 mg mL−1 and 25 °C measurements. Analyses were repeated 3 times for each sample, and a minimum of 10 measurements was performed for each analysis.
Cell culture and nanoparticle internalisation studies
Human colon cancer HCT116 cells and 3T3 fibroblasts were seeded at a concentration of 3 × 105cells per cm2 on rounded glass coverslips in 6 well plates and incubated at 37 °C and 5% v/v CO2 to allow cells to attach overnight. Subsequently, full media from the cells were aspirated and replaced, and the cells were incubated with 250 μg mL−1 of nanoparticles formulated from a sample of PLA-b-PLA–pOEGMA 5 polymer which had been labelled via incorporation of rhodamine B methacrylate dye ([dye]:[OEGMA/DEGMA], 1:1000). These nanoparticles exhibited a thermal transition temperature in cell culture media (HBSS/HEPES 20 mM) of 28 °C, which was lower than that recorded in deionized water (41 °C, Table 1). The incubation step was carried out overnight at 37 °C prior to confocal microscopy analysis. Cells were stained with Hoechst 33342 1 μg mL−1 and/or CellMask deep red plasma membrane staining 1 μg mL−1 for 30 minutes and the staining solution removed prior to confocal microscopy on a Zeiss Confocal microscope 710.
Results and discussion
Nanoparticle synthesis and characterisation
The prerequisite for the study was the preparation of co-polymers with a hydrophobic block to provide a driving force for self-assembly in aqueous media and a thermoresponsive segment to provide temperature-switchable behaviour. A small library of copolymers was synthesised (Scheme 1) by varying the feed ratio of (2-(2-methoxyethoxy)ethyl methacrylate), (DEGMA, average Mn: 188 Da) and oligo(ethylene glycol)methacrylate ((OEG)9MA, average Mn 500 Da). | ||
| Scheme 1 Synthesis of thermoresponsive PLA-b-P((DEGMA)-stat-(OEGMA)) polymers used in this study. | ||
A range of polymers was produced with systematically varied mol:mol ratios of OEGMA and DEGMA (Scheme 1) via the AGET-ATRP route.25 The synthesised polymers varied in calculated molar mass (Mn 38–68 kDa, NMR) and in polydispersity (∼1.3–1.5). We refer throughout the manuscript to measured co-monomer content in these polymers rather than feed ratios: full details are shown in Table 1.
The synthesised polymers were then formulated into kinetically-trapped core–shell nanoparticles by two different methods. In the first method, individual solutions of each block co-polymer in a good solvent for both blocks, were added into water, thus producing a range of nanoparticles each with a thermal transition temperature dependent on the specific co-polymer forming the outer corona. We termed these as ‘single component’ nanoparticles, as only one block co-polymer type was used to form each nanoparticle. In the second method, solutions of two block co-polymers with different DEGMA:(OEG)9MA ratios were prepared in the good solvent, then added to water, producing co-precipitated i.e. ‘blended’ polymer nanoparticles. Previous studies have showed that the sizes of nanoparticles with thermoresponsive exteriors can significantly affect observed cloud points, owing to crowding effects which are strongly influenced by surface curvature.19,26 We therefore aimed to produce NPs of ≥100 nm diameter: the sizes of the resultant nanoparticles obtained by light scattering measurements are shown in Table 1 and in ESI.† Subsequently, the thermal transition temperatures (Tt) were measured for the single component NPs (Table 1), the blended polymer NPs, and mixtures thereof.
Temperature–turbidity studies of single component polymer nanoparticles (NPs) (“Single polymer corona nanoparticles”)
The measured solution cloud points (Tt from turbidimetry) of the block co-polymers in this study ranged from 29 to 55 °C with an increase in Tt with OEGMA content (Fig. 2). These values were in accord with those reported previously for similar block co-polymers of poly(poly(propyleneglycolmethacrylate)-co-OEGMA-b-PLGA).5,24 and also for statistical poly(DEGMA-co-OEGMA) thermoresponsive polymers.21,27,28 It has previously been shown that the transition temperatures of POEGMA-based polymers are not dependent on the number average molar mass,21 and thus it is unlikely that the changes in particle transition temperature observed were a function of variations in molar mass in the constituent polymers. | ||
| Fig. 2 (a–d) Turbidimetric analysis of formulation series 1 nanoparticles resulting from blends between PLA–pDEGMA 100 and PLA–pOEGMA 5, 10, 15 and 20. Cloud point measurements were performed at 5 mg mL−1. In each set of panels (a–d) the measured absorbance is shown on the left, with the transmittance shown on the right normalised to compensate for the lower overall absorbance at intermediate pOEGMA:pDEGMA ratios. | ||
Temperature–turbidity studies of nanoparticles formed by simultaneous co-precipitation of polymers with different individual cloud points (“Binary polymer corona nanoparticles”)
In order to investigate possible control of cloud point by nanoparticle composition, mixtures of PLA-b-P(DEGMA-stat-OEGMA) polymers each with different Tt were precipitated from acetone into water to obtain the blended NPs. The ratios of the individual polymers in the formulated nanoparticles were systematically varied from 0% pOEGMA (i.e. PLA–pDEGMA 100), 25% (i.e. PLA–pDEGMA 75% and PLA–pDEGMA-stat-OEGMA 25%), 50% (PLA–pDEGMA 50% and PLA–pDEGMA-stat-OEGMA 50%), 75% (PLA–pDEGMA 25% and PLA–pDEGMA-stat-OEGMA 75%) and 100 weight percent (PLA–pDEGMA-stat-OEGMA 100%) such that any relative enrichment of one co-polymer over another in the surface corona might be detected. In addition, the PLA–pDEGMA-stat-OEGMA co-polymers in the mixtures were varied, employing co-polymers with increasing OEGMA content (i.e. PLA–pDEGMA 95-stat-OEGMA 5%, 10%, 15% and 20% mole% and thus higher individual Tt), in the blend. The compositions and the temperature vs. turbidity (data expressed in absorbance and transmittance) of the resulting copolymers are reported in Fig. 2 and 3. | ||
| Fig. 3 Turbidimetric analysis of nanoparticles resulting from blends between PLA–pOEGMA 10 and PLA–pOEGMA 15, performed at 5 mg mL−1. (a) The absorbance, (b) the normalised transmittance. | ||
As is apparent from Fig. 2, the change in absorbance of nanoparticle suspensions with temperature varied with the different compositions of nanoparticle blends. However, for most of the formulations, the cloud points of the blends were close to that of PLA–pDEGMA 100, even if the nanoparticle formulation contained up to 50% of PLA–pOEGMA-co-DEGMA co-polymer with a higher original cloud point (Tt). Only where the PLA–pOEGMA-co-DEGMA content reached 75% relative to PLA–pDEGMA 100 polymer did the observed Tt become similar to the Tt of the introduced PLA–pOEGMA-co-DEGMA, irrespective of which PLA–pOEGMA-co-DEGMA (i.e. PLA–pOEGMA 10, 15, 20) had been added. The only exception to this occurred in the case of PLA–pDEGMA 100–PLA–pOEGMA 5 blends, where the Tt was almost exactly at the mid-point between the Tt for pure PLA–pDEGMA 100 and PLA–pOEGMA 5 when the PLA–pOEGMA 5 content was 75%. In addition, the turbidities of the solutions above the cloud points were lower for the blends at the same concentration of particles compared to the turbidities of the ‘single-co-polymer’ NPs. In particular, absorbance was strongly reduced for NPs containing PLA–pDEGMA 100 weight fractions of 50% and 25%. This behaviour was observed for all polymers that were mixed with PLA–pDEGMA 100 (see Fig. 2). These results suggested that there was limited ‘cooperativity’ between co-polymers of different Tt undergoing coil-to-globule collapse at the surfaces of the particles as such behaviour would have been manifest by bulk aggregation. This also indicated that the most important factor in the aggregation was the proportion of the PLA–pDEGMA 100 component in the blend, as when this was progressively diluted in composition in the blended nanoparticles, the overall turbidity decreased. In turn, this suggested that either there was insufficient pDEGMA exposed at the surface of the lower PLA–pDEGMA 100-content NPs to render the surface sufficiently hydrophobic on chain collapse to aggregate, or that blending of co-polymers during nanoprecipitation generated nanoparticles with heterogeneous surfaces.
In experiments to test these individual hypotheses, we then turned to NPs formulated from copolymers in which the co-monomer ratios in the constituent individual co-polymers were different by no more than 5 mole% OEGMA:DEGMA, as we reasoned that these polymers would be most likely to mix. In addition, we selected PLA–pOEGMA-10 and PLA–pOEGMA-15, as these polymers displayed cloud points at temperatures achievable clinically by local hyperthermia.29 The results of the temperature–turbidity measurements for blends of these terpolymers in nanoparticles are shown in Fig. 3.
The data show that single transition temperatures were observed for the blends of the PLA–pOEGMA-10 and PLA–pOEGMA-15 across all compositions tested, and that cloud point values ranged from 45 °C for pure PLA–OEGMA 10 increasing linearly to 50 °C of pure PLA–OEGMA 15 according to (OEG)9MA percentage (Fig. 3 and Fig. S2, ESI†).
Heating–cooling cycles
For many practical applications, there is a need for particle self-association and aggregation to be reversible, thus a number of heating–cooling cycle experiments were conducted. Binary polymer corona nanoparticles as shown in Fig. 2 and 3 were subjected to multiple heat–cool cycles and the temperature–turbidity curves were compared (Fig. 4). As exemplified for nanoparticles prepared from blends of PLA–pOEGMA 10 with PLA–pOEGMA 15, which showed a linear variation of Tt with increased PLA–OEGMA content (Fig. S2, ESI†), there were minimal differences in the temperature–turbidity curves over 3 consecutive heat–cool cycles (Fig. 4b). The same heat–cool cycle experiments with nanoparticles formed from blends of PLA–pDEGMA 100–PLA–pOEGMA 10 also showed complete reversibility of the temperature turbidity curves, suggesting that any changes in nanoparticle structure over the temperature ranges were transient (see Fig. 4). | ||
| Fig. 4 Turbidimetric heating–cooling cycles. (a) PLA–pOEGMA 10–PLA–pOEGMA 15 blends. (b) PLA–pDEGMA 100–PLA–pOEGMA 10 blends. The analysis was performed at 5 mg mL−1 for both experiments. Cooling curves (dotted lines) are similar to the heating curves over all three measurements. | ||
These experiments demonstrated ‘formulation stability’ of the particles formed from blends of two thermoresponsive polymers. However, the cloud points in the cooling processes were slightly lower when compared to those obtained during the heating cycles: this hysteresis can be explained by the interactions that are generated among polymer chains once the Tt is passed. Rehydration and chain extension of the collapsed polymer chains as the temperature drops below the polymer phase transition can be slower than initial polymer collapse above the thermal transition temperature: similar effects have been observed with pOEGMA-based polymers previously.22
Temperature–turbidity studies to compare nanoparticles formed from co-polymer blends with mixtures of single polymer corona nanoparticles
Subsequent experiments were designed to test the hypothesis that tuning of thermal transition temperature might be obtained through mixing preformed nanoparticles with varying individual Tt, rather than by mixing polymers of different thermal transition temperatures prior to nanoparticle formation. Single polymer corona nanoparticles prepared from PLA–pDEGMA 100, PLA–pOEGMA 10 and PLA–pOEGMA 15 were mixed to give particle blends at the same ratios that were used to prepare nanoparticles from the mixtures of PLA–DEGMA 100–PLA–pOEGMA 10 and PLA–pOEGMA 10–PLA–pOEGMA 15 co-polymers. As apparent from Fig. 5 (and compared with Fig. 2 and 3), the resulting temperature–turbidity graphs for single polymer corona nanoparticles resembled closely the curves for the binary polymer corona nanoparticles obtained by mixing co-polymers in different ratios prior to nanoparticle formation. | ||
| Fig. 5 Turbidimetric analysis of: (a) PLA–pDEGMA 100–PLA–pOEGMA 5, (b) PLA–pDEGMA 100–PLA–pOEGMA 10, (c) PLA–pDEGMA 100–PLA–pOEGMA 15, (d) PLA–pDEGMA 100–PLA–pOEGMA 20 and (e) PLA–pOEGMA 10–PLA–pOEGMA 15 blends. On the left are temperature–turbidity curves for the nanoparticles prepared from binary polymer blends, on the right are the corresponding curves for mixtures of pre-formed single polymer corona nanoparticles. | ||
Preliminary analysis of nanoparticle cytocompatibility
The potential for these materials to be used in an exemplar biomedical setting was assessed by evaluating the endocytosis of a selected sub-set of the nanoparticles. As our previous data with similar polymers had indicated uptake of nanoparticles in MCF7 breast cancer cells,5 we decided to investigate a cell line, HCT116, representing an additional solid cancer, colon cancer, and a fibroblast cell-line, 3T3, as an example of a stromal cell. We chose the PLA-b-P((DEGMA)-stat-(OEGMA)) polymer (PLA–pOEGMA 5) polymer as our investigational polymer, owing to its thermal transition temperature of 40 °C, and found that when prepared with ∼1% rhodamine methacrylate to provide a fluorescent label, the thermal transition temperature for these polymers in cell culture media was 28 °C. Accordingly, at 37 °C (optimal for cell culture), we expected that these nanoparticles would be readily endocytosed by both cell lines. Initial Alamar Blue assays indicated that the polymers had little or no effect on metabolic activity up to a concentration of at least 1 mg mL−1 (Fig. S45, ESI†). As apparent from Fig. 6, the nanoparticles were apparent in the cytoplasm of both the fibroblast and cancer cell lines following the selected incubation time period.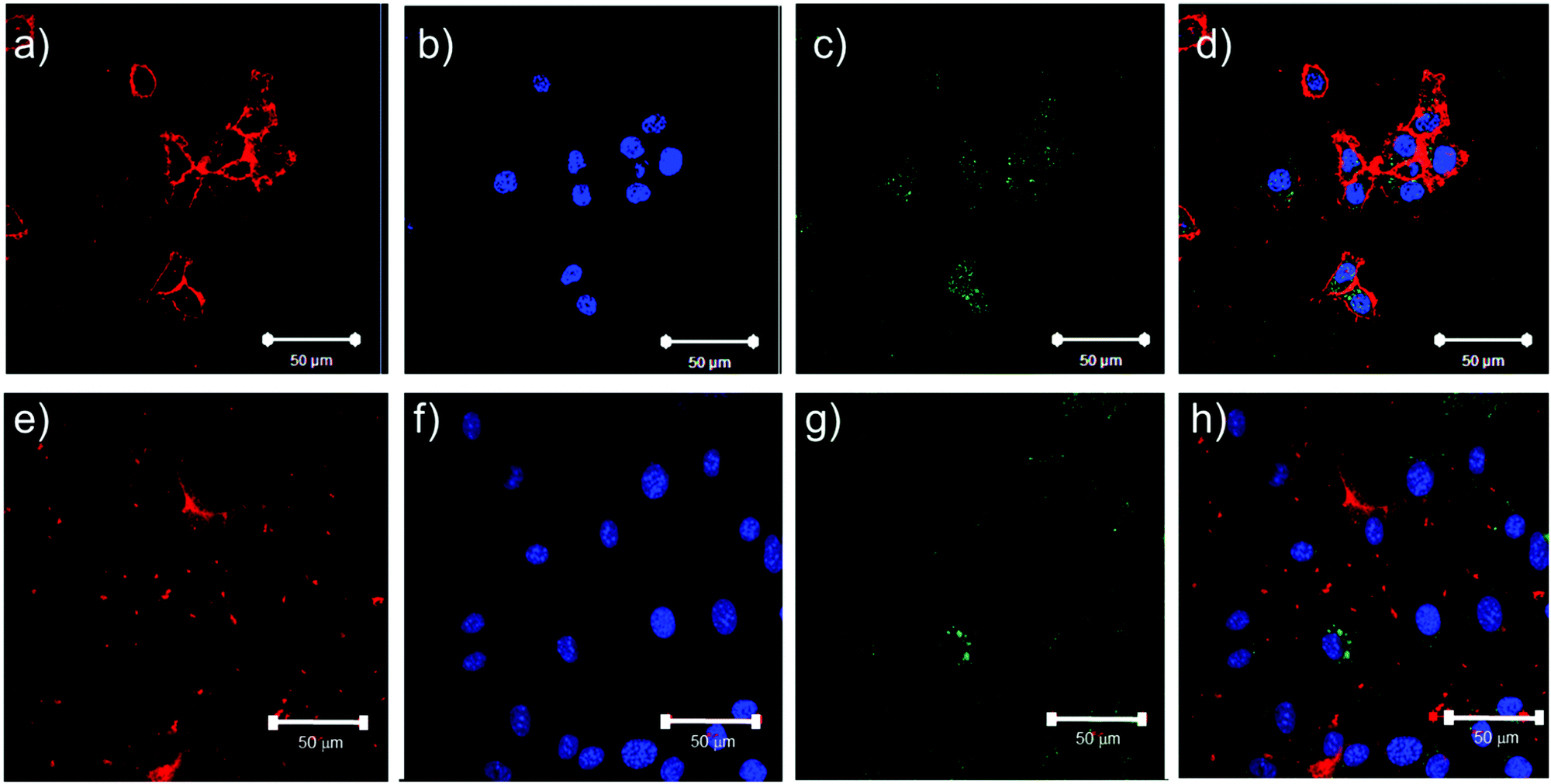 | ||
| Fig. 6 Confocal microscopy images of HCT116 and 3T3 cells incubated with PLA-b-P((DEGMA)-stat-(OEGMA)) polymer (PLA–pOEGMA 5 nanoparticles) at a concentration of 250 μg mL−1. Plates a–d show HCT116 cells incubated with polymers overnight at 37 °C then imaged. Plates e–h show 3T3 cells incubated with polymers overnight at 37 °C then imaged. Cells were labelled with CellMask deep red membrane stain (red) and Hoechst nuclear stain (blue). Polymers were labelled with Rhodamine B which has been re-coloured in green for easy distinction of the micelles from the Deep Red CellMask dye. Scale bars represent 50 μm. | ||
Discussion
The data obtained for both the ‘binary polymer corona’ and ‘single polymer corona’ nanoparticles showed that tuning of the thermoresponsive properties and the aggregation temperature of the poly(oligoethylene glycol methacrylate)-co-poly(diethylene glycol methyl ether methacrylate) (p(OEGMA-stat-DEGMA)) was possible by systematic variation of shell co-monomer content. However, unlike the case for linear co-polymers which display Lower Critical Solution Temperatures (LCST) dependent on the ratios of constituent co-monomers,21,27 there was no simple and general rule linking composition of these nanoparticles to aggregation temperature. It is important to emphasise that observed aggregation of particles by turbidimetry is concentration-dependent30 and cannot provide detailed mechanistic information on the thermal transitions of individual co-polymers at the surfaces of nanoparticles. However, as an easily accessible measurement technique, solution turbidimetry provides valuable practical information concerning the effects of polymer transitions on nanoparticle self-association and solution stability. In addition, the temperature–turbidity curves obtained for the different families of nanoparticles enable some insights to be gained into the mechanisms by which different nanoparticles associate and aggregate.Temperature-induced nanoparticle aggregation
The key factor controlling aggregation in nanoparticle formulations is colloidal stability. For the PLA-b-P((DEGMA)-stat-(OEGMA)) polymer nanoparticles in this study, all but two of the zeta potentials (Table S1, ESI†) were close to zero indicating a limited extent of charge-mediated colloidal stabilisation. While zeta potentials did become more negative over time due most likely to partial hydrolysis of lactide blocks (data not shown), the primary stabilising contribution at the start of the study was expected to be the presence of the chain-extended hydrophilic pOEGMA at the nanoparticle surface. When co-polymerised with DEGMA the resultant polymers displayed predictable phase transition temperatures in water, but when grown from a hydrophobic PLA block and assembled into kinetically-trapped nanoparticles, the coil-to-globule collapse of the corona co-polymers was expected to be constrained. The constraints arise from surface geometry, i.e. the extent to which polymers can collapse without sterically hindering neighbouring chains, and also concentration/conformation effects, for example if the polymers are in ‘brush’ or ‘mushroom’ domains. The nanoparticles in this study were varied in diameters between 110–140 nm and thus while surface geometry cannot be ignored, the effects were likely to be very similar across the set of nanoparticles tested. This is because the sizes of the particles were in all cases much greater than the expected end-to-end distances of the individual polymer chains. Based on degrees of polymerisation of 75–150 in the pDEGMA and pOEGMA segments, even for fully extended ‘bottle brush’ chains the maximum geometric end-to-end distance would be ∼18 nm per pOEGMA chain. A more realistic block length assuming some conformational flexibility would be ∼7 nm, which is rather less than any of the radii of nanoparticles. The method of assembly of the nanoparticles was also constant across the set of formulations, and thus was likely to generate polymer coronae which were similar in conformation and effective surface concentration irrespective of the individual polymers used. The most observable effects of size on aggregation phenomena were thus likely to have been on efficiency of particle packing above the aggregation temperature rather than the likelihood of different sized NPs having different surface compositions and architectures.Temperature–turbidity responses of ‘binary corona’ nanoparticles
For the first set of binary mixtures selected (Fig. 2), the nanoparticles were formed from mixtures of a PLA–pDEGMA di-block and a PLA–pDEGMA-co-pOEGMA block, with variations in pOEGMA content of the PLA–pDEGMA-co-pOEGMA block. Thus, all the nanoparticles shown in Fig. 2 were prepared from a di-block co-polymer and a di-block terpolymer, with the corona expected to be formed from a pDEGMA block and/or a pDEGMA-co-OEGMA block. The extent of mixing of different polymers is complex, with a strong thermodynamic driving force towards phase separation of dissimilar segments, but also changes in polymer association over time even in polymer blends.31 Thus, while the co-polymers in this study were similar in overall composition and molar mass, it is likely that some enrichment of one of the components of the blends may have occurred, leading to heterogeneity in nanoscale domains.32,33 This may have resulted in corresponding variations in self-associative and aggregative behaviour. As apparent from Fig. 2 and 3, the cloud points for these “binary polymer corona” nanoparticles were most strongly influenced by the most abundant copolymer. When PLA–pDEGMA 100 was the predominant component, transition temperatures closely matched those of nanoparticles made of PLA–pDEGMA 100 alone. Above 50% incorporation of PLA–pOEGMA-co-DEGMA, the Tt values for the particles were closer to those made of the PLA–pOEGMA-co-DEGMA exclusively, suggesting that either the increased pOEGMA content in the corona prevented collapse of pDEGMA chains or provided sufficient steric colloidal stabilisation as to make collapse of pDEGMA chains negligible.The overall turbidities of the solutions above the transition temperatures were informative also, as the progressive drop in turbidity with increased pOEGMA content up to the 50% w/w suggested only a proportion of the nanoparticles were aggregating or the overall aggregate size was smaller and therefore scattering less light. In two cases at the higher pOEGMA compositions, this phenomenon was manifest in difficulties in detecting the transition temperatures of the nanoparticles with high precision, with apparently the same, or marginally higher, Tt values for 75% PLA–pOEGMA 15 and PLA–pOEGMA 20 nanoparticles than those with 100% PLA–pOEGMA 15 and PLA–pOEGMA 20.
However, by changing the coronae from mixtures of a ‘homopolymer’ (i.e. the pDEGMA block of PLA–pDEGMA 100) and statistical co-polymers of pDEGMA-co-OEGMA, to mixtures of different pDEGMA-co-OEGMA co-polymers, a more predictable tuning of aggregation temperature was obtained (Fig. 3). This may be attributed to the similarity in the PLA–pDEGMA90-co-OEGMA10 and PLA–pDEGMA85-co-OEGMA15 (“PLA–pOEGMA 10 and PLA–pOEGMA 15”) co-polymers in the outer layer, and the possibility of better mixing during nanoparticle formation. This effect was also observed for PLA–pDEGMA-co-OEGMA co-polymers with higher overall OEGMA content as long as the two co-polymers used in the mixture did not vary in OEGMA:DEGMA mole fraction by more than 10% (data not shown). The overall turbidities declined to a lesser extent with changes in OEGMA content in these ‘all OEGMA-co-DEGMA’ copolymer particles, compared to the nanoparticles containing the PLA–pDEGMA mixed with PLA–pDEGMA-co-pOEGMA polymers.
In all cases, the transition temperatures of the nanoparticles were essentially the same over multiple heating/cooling cycles (Fig. 4). These data indicated that the gross surface structures and morphologies of the nanoparticles formed during the nanoprecipitation process were invariant across the solution phase transition temperatures of the individual polymers. Experiments were carried out to probe any changes in nanoparticle structure and morphology by TEM following these heat–cool cycles. No obvious variations in the nanoparticle sizes or shapes were apparent in TEM after heating and cooling (Fig. S47, ESI†), and DLS also indicated no significant change in size.
Temperature–turbidity responses for mixtures of ‘single corona’ nanoparticles
The final approach to tune aggregation temperature was to mix nanoparticles prepared from specific individual co-polymers. The temperature–turbidity curves for these mixtures of pre-formed co-polymer nanoparticles were in general very similar to those of the nanoparticles prepared from the polymer blends, as can be discerned by comparison of the graphs on the left and right hand side of Fig. 5. Thus for blends of 25% PLA–pDEGMA 100 and 75% PLA–pOEGMA 10, PLA–pOEGMA 15 and PLA–pOEGMA 20, the observed transition temperatures were similar to those of mixtures containing pre-formed NPs of PLA–pDEGMA 100 and PLA–pOEGMA 10, PLA–pOEGMA 15 and PLA–pOEGMA 20 in the same % compositions. The same pattern of transition temperature variation was apparent for blends of 75% PLA–pDEGMA 100 and 25% PLA–pOEGMA 10, PLA–pOEGMA 15 and PLA–pOEGMA 20 and the corresponding compositional mixtures of pre-formed nanoparticles. Intriguingly however, variations in the observed Tt values for the pre-formed co-polymer nanoparticles containing 50% PLA–pDEGMA 100 and 50% of PLA–pOEGMA 10, PLA–pOEGMA 15 or PLA–pOEGMA 20, compared to their blend co-polymer counterparts were apparent. For example, in the nanoparticles containing a blend of 50% PLA–pDEGMA 100 and PLA–pOEGMA 10, the observed Tt was 30 °C, whereas for the mixture of pre-formed nanoparticles in the same ratio the Tt was hard to discern. The reason for the difference in aggregation temperature between blended polymer nanoparticles and mixtures of pre-formed nanoparticles at this specific compositional ratio is not obvious, however interaction between the particle types, e.g. surface adsorption, may prevent bulk aggregation even when particles with lower Tt have collapsed coronas. DLS and zeta potential data (Table S1, ESI†) for these materials were not very different across the whole set, except for the PLA–pDEGMA 100 (50% w/w)–PLA–pOEGMA 10 (50% w/w) blends and PLA–pDEGMA 100 (50% w/w)–PLA–pOEGMA 10 (50% w/w) nanoparticles and even in this case the difference in diameter was less than 30 nm and ζ-potential variation of ∼5 mV. In addition, for the most highly negatively charged samples, the particles still aggregated above a critical temperature, indicating that the loss of steric stabilisation above this point overrode any charge-mediated stabilisation. Thus the most likely explanation of the observed aggregation behaviour in the mixtures compared to the blends is that there were lower associative forces between the mixtures of pre-formed nanoparticles. This may have arisen because the different constituent polymers in the blended nanoparticles may have generated regions where one polymer was enriched, leading to surface heterogeneities and the greater possibility of inter-chain penetration on contact with another heterogeneous nanoparticle. The single-component nanoparticles probably had less heterogeneity in the outer layer, leading to less inter-chain association. Thus the subtle heterogeneities in surface structures in the 50% mixtures may have been more dominant in overall aggregative behaviour than for nanoparticles containing greater proportions of one co-monomer.It has been noted many times before that the aggregation behaviour of thermoresponsive nanoparticles is highly complex,23,34,35 and there are many interlinked variables which can complicate attempts to define structure–property relationships.36,37 For the nanoparticles in this study, we chose to keep concentrations and solution conditions constant, in order that factors such as aggregation kinetics and ionic strength were minimised.21,36 We also used poly(ethyleneglycol)methacrylate-derived monomers such that the resulting polymers could act only as lone-pair donors and thus not form inter-chain hydrogen bonds as reported for poly(N-isopropylacrylamide)-based materials.38 The data obtained for these nanoparticles, even in the absence of competing salts, proteins or other components present in environments where responsive materials are likely to find use, suggest that particle aggregation behaviour is not easily predictable based purely on polymer composition. Indeed, the preliminary cell culture studies showed that components in the cell growth media contributed to a drop in thermal transition temperature of more than 10 °C (from ∼40 to ∼28 °C) for the PLA-b-P((DEGMA)-stat-(OEGMA)) polymer (PLA–pOEGMA 5) nanoparticles. In addition, for hydrolytically degradable polymers, such as the PLA cores used in this study, there are possible further variations in polymer physical properties dependent on sample age. The thermal transition temperature changes we observed suggest that formulations intended for specific biomedical applications need to be pre-screened in environments close to which they would be used in practice, i.e. in the presence of salts, metabolites etc., as data are not directly transferable from model studies in highly controlled environments. We are currently collating data from cell culture and drug delivery studies of other nanoparticle formulations we have introduced in this manuscript, as the preliminary experiments with the PLA–pOEGMA 5 nanoparticles indicated rapid endocytosis above the thermal transition temperature in 2 cell lines. The full endocytosis evaluations for all 36 formulations prepared in this study (to be reported in future) may require bespoke optimisations in the specific culture media for individual cell lines. Nevertheless, for co-polymers which are similar in structure, such as the pOEGMA-based materials used here, and for nanoparticles blended from chemically similar components, it should still be possible to tune thermal response temperature in their respective nanoparticle formulations based on the strategies we have described.
Conclusions
In summation, this study has shown that critical aggregation temperatures of thermoresponsive nanoparticles can be tuned by two complementary methods. These involve the simple blending of co-polymers with different co-monomer ratios prior to nanoprecipitation, and by the mixing of pre-formed nanoparticles derived from nanoprecipitated copolymers of different monomer ratios. However, the relationship between aggregation temperature and co-polymer or nanoparticle composition is only linearly variable over specific formulation regions. This ‘temperature tuning region’ is predictable if the copolymers used to form the nanoparticles are similar in their OEGMA substitution ratio and chain length. It was also found that the nanoparticles were stable to repeated heat–cool cycles and that the thermal aggregation behaviours were fully reversible over multiple heating–cooling experiments. These data suggest that it is possible to design nanoparticles with specific aggregation temperatures but over a wide range via simple mixing of small families of chemically similar pre-cursor copolymers. The data also imply that any variations in surface structure of responsive nanoparticles, such as those possible from polymer demixing, may have marked effects on associative or adsorptive properties, and these will need careful evaluation prior to the use of these materials in biomedical environments.Data access statement
All raw data created during this research are openly available from the corresponding author (http://Cameron.alexander@nottingham.ac.uk) and at the University of Nottingham Research Data Management Repository (http://https://rdmc.nottingham.ac.uk/) and all analysed data supporting this study are provided as ESI,† accompanying this paper.Author contributions
R. F., L. M., F. M., L. S., A. T., and A. S., performed the experiments. A. G., M. A., P. G., J. A., S. G. S., and C. A. conceived the project and supervised the research. All authors contributed data or text for the manuscript and C. A. wrote the paper.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/H005625/1, EP/I01375X/1, EP/L01646X/1]; the Royal Society [Wolfson Research Merit Award WM150086]; and Astra Zeneca. We also thank Christine Grainger-Boultby, Tom Booth and Paul Cooling for excellent technical support.References
- M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC.
- D. J. Phillips, J. P. Patterson, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2014, 5, 126–131 RSC.
- J. R. McDaniel, S. R. MacEwan, X. Li, D. C. Radford, C. D. Landon, M. Dewhirst and A. Chilkoti, Nano Lett., 2014, 14, 2890–2895 CrossRef CAS PubMed.
- A. Barhoumi, W. Wang, D. Zurakowski, R. S. Langer and D. S. Kohane, Nano Lett., 2014, 14, 3697–3701 CrossRef CAS PubMed.
- S. R. Abulateefeh, S. G. Spain, K. J. Thurecht, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Biomater. Sci., 2013, 1, 434–442 RSC.
- F. Mastrotto, P. Caliceti, V. Amendola, S. Bersani, J. P. Magnusson, M. Meneghetti, G. Mantovani, C. Alexander and S. Salmaso, Chem. Commun., 2011, 47, 9846–9848 RSC.
- H. Wang, J. Yang, Y. Li, L. Sun and W. Liu, J. Mater. Chem. B, 2013, 1, 43–51 RSC.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- S. Louguet, B. Rousseau, R. Epherre, N. Guidolin, G. Goglio, S. Mornet, E. Duguet, S. Lecommandoux and C. Schatz, Polym. Chem., 2012, 3, 1408–1417 RSC.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- H. Ringsdorf, J. Simon and F. M. Winnik, Macromolecules, 1992, 25, 7306–7312 CrossRef CAS.
- L. P. N. Rebelo, Z. P. Visak, H. C. De Sousa, J. Szydlowski, R. G. De Azevedo, A. M. Ramos, V. Najdanovic-Visak, M. N. Da Ponte and J. Klein, Macromolecules, 2002, 35, 1887–1895 CrossRef CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- W. Hassouneh, K. Fischer, S. R. MacEwan, R. Branscheid, C. L. Fu, R. Liu, M. Schmidt and A. Chilkoti, Biomacromolecules, 2012, 13, 1598–1605 CrossRef CAS PubMed.
- J. R. McDaniel, S. R. MacEwan, M. Dewhirst and A. Chilkoti, J. Controlled Release, 2012, 159, 362–367 CrossRef CAS PubMed.
- F. Fernandez-Trillo, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Chem. Commun., 2008, 2230–2232 RSC.
- F. Fernandez-Trillo, A. Dureault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef CAS.
- J. Buller, A. Laschewsky, J.-F. Lutz and E. Wischerhoff, Polym. Chem., 2011, 2, 1486–1489 RSC.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- D. J. Phillips and M. I. Gibson, Polym. Chem., 2015, 6, 1033–1043 RSC.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Aberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- S. Won, D. J. Phillips, M. Walker and M. I. Gibson, J. Mater. Chem. B, 2016, 4, 5673–5682 RSC.
- S. R. Abulateefeh, A. O. Saeed, J. W. Aylott, W. C. Chan, M. C. Garnett, B. R. Saunders and C. Alexander, Chem. Commun., 2009, 6068–6070 RSC.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139–4146 CrossRef CAS.
- N. S. Ieong, K. Brebis, L. E. Daniel, R. K. O'Reilly and M. I. Gibson, Chem. Commun., 2011, 47, 11627–11629 RSC.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- T. M. Zagar, J. R. Oleson, Z. Vujaskovic, M. W. Dewhirst, O. I. Craciunescu, K. L. Blackwell, L. R. Prosnitz and E. L. Jones, Int. J. Hyperthermia, 2010, 26, 618–624 CrossRef PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- R. Koningsveld, Macromol. Symp., 1994, 78, 1–13 CrossRef CAS.
- T. P. T. Dao, F. Fernandes, M. Er-Rafik, R. Salva, M. Schmutz, A. Brûlet, M. Prieto, O. Sandre and J. F. Le Meins, ACS Macro Lett., 2015, 4, 182–186 CrossRef CAS.
- C. LoPresti, M. Massignani, C. Fernyhough, A. Blanazs, A. J. Ryan, J. Madsen, N. J. Warren, S. P. Armes, A. L. Lewis, S. Chirasatitsin, A. J. Engler and G. Battaglia, ACS Nano, 2011, 5, 1775–1784 CrossRef CAS PubMed.
- L. Wang, X. Zhao, Y. Zhang, W. Zhang, T. Ren, Z. Chen, F. Wang and H. Yang, RSC Adv., 2015, 5, 40437–40443 RSC.
- C. I. Biggs, S. Edmondson and M. I. Gibson, Biomater. Sci., 2015, 3, 175–181 RSC.
- L. Luo, J. Tam, D. Maysinger and A. Eisenberg, Bioconjugate Chem., 2002, 13, 1259 CrossRef CAS PubMed.
- L. D. Blackman, D. B. Wright, M. P. Robin, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2015, 4, 1210–1214 CrossRef CAS.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: This includes DLS measurements for all blends tested. See DOI: 10.1039/c7sm00920h |
| This journal is © The Royal Society of Chemistry 2017 |