
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of dextran(ethylene glycol) hydrogel films for giant unilamellar lipid vesicle production and their application for the encapsulation of polymersomes†
Nestor Lopez
Mora‡
 *a,
Yue
Gao
a,
M. Gertrude
Gutierrez
b,
Justin
Peruzzi
c,
Ivan
Bakker
a,
Ruud J. R. W.
Peters
d,
Bianka
Siewert
e,
Sylvestre
Bonnet
e,
Roxanne E.
Kieltyka
*a,
Jan C. M.
van Hest
d,
Noah
Malmstadt
b and
Alexander
Kros
a
*a,
Yue
Gao
a,
M. Gertrude
Gutierrez
b,
Justin
Peruzzi
c,
Ivan
Bakker
a,
Ruud J. R. W.
Peters
d,
Bianka
Siewert
e,
Sylvestre
Bonnet
e,
Roxanne E.
Kieltyka
*a,
Jan C. M.
van Hest
d,
Noah
Malmstadt
b and
Alexander
Kros
a
aLeiden Institute of Chemistry, Leiden University, Supramolecular & Biomaterials Chemistry, Einsteinweg 55, 2333 CC, Leiden, The Netherlands. E-mail: r.e.kieltyka@chem.leidenuniv.nl
bDepartments of Chemical Engineering & Materials Science, Biomedical Engineering, and Chemistry, University of Southern California, 925 Bloom Walk, 90089, Los Angeles, CA, USA
cDepartment of Chemical Engineering, University of Virginia, 102 Engineer's Way, 400741, Charlottesville, VA, USA
dRadboud University Nijmegen, Department of Organic Chemistry, Heyendaalseweg 135 6525 AJ, Nijmegen, The Netherlands
eLeiden Institute of Chemistry, Leiden University, Metals in Catalysis, Biomimetics & Inorganic Materials, Einsteinweg 55, 2333 CC, Leiden, The Netherlands
First published on 21st July 2017
Abstract
Giant Unilamellar Vesicles (GUVs) prepared from phospholipids are becoming popular membrane model systems for use in biophysical studies. The quality, size and yield of GUVs depend on the preparation method used to obtain them. In this study, hydrogels consisting of dextran polymers crosslinked by poly(ethylene glycol) (DexPEG) were used as hydrophilic frameworks for the preparation of vesicle suspensions under physiological ionic strength conditions. A comparative study was conducted using hydrogels with varied physicochemical properties to evaluate their performance for GUV production. The prepared GUVs were quantified by flow cytometry using the Coulter Principle to determine the yield and size distribution. We find that hydrogels of lower mechanical strength, increased swellability and decreased lipid interaction favour GUV production, while their resulting size is determined by the surface roughness of the hydrogel film. Moreover, we embedded polymersomes into the crosslinked hydrogel network, creating a DexPEG – polymersome hybrid film. The re-hydration of lipids on those hybrid substrates led to the production of GUVs and the efficient encapsulation of polymersomes in the lumen of GUVs.
1 Introduction
The cellular membrane defines the boundary between the cytoplasm and the cell exterior regulating important intra- and intercellular processes in the biological milieu.1 Therefore, the development of minimal cell models that allow in vitro studies become highly relevant to simplify investigations into these phenomena.2 Giant Unilamellar Vesicles (GUVs) are excellent in vitro biomembrane models because of their membrane curvature and similar size to cells, being readily observable by optical microscopy.3,4 The GUV model has been used in the study of biomembrane properties,5–8 membrane–protein,9–13 and membrane–peptide interactions,14 channel formation in membranes15,16 or transmembrane transport of ions.17,18 Thus, the efficient formation of high quality GUVs under relevant physiological conditions is highly desirable.The use of hydrogels as substrates for GUV growth is attractive due to their potential to circumvent disadvantages encountered in traditionally used preparation methods such as natural swelling19 and electroformation.20 Covalent hydrogels are insoluble, chemically crosslinked polymer networks consisting of hydrophilic precursors that swell rapidly upon the addition of water to create a three-dimensional network whereas physical hydrogels rely on non-covalent interactions to form such materials.21 Most often, these networks have been applied in the areas of controlled drug delivery22–28 and tissue engineering,29,30 but rarely in the growth of cell-sized vesicles (GUVs). Only recently, it has been shown that hydrogel films can enable GUV formation under physiological conditions. Horger et al.31 proposed the use of physical gel substrates based on non-crosslinked agarose for the preparation of GUVs under near physiological conditions using several lipid compositions. However, traces of agarose remain in the vesicle inner volume and the lipid membrane affecting their mechanical properties. To tackle these drawbacks Lira et al.32 used thermal post-treatment of agarose-GUVs to release the encapsulated agarose thereby recovering GUV responses in electro-deformation studies. Alternatively, films of hydrogels based on poly(vinyl alcohol)33 and crosslinked polyacrylamide,31 have also been employed in the preparation of GUVs. While these polymers are not detected in the lipid bilayer of GUVs, they afford minimal control over their production and size.34 We recently presented a facile method to form GUVs under physiological ionic strength conditions using a neutral, chemically crosslinked hydrogel substrate (DexPEG) consisting of a dextran polymer crosslinked by polyethylene glycol (PEG).35 Maleimide–thiol coupling chemistry was used to simultaneously crosslink the biocompatible polymer chains and a glass surface forming an immobilized hydrogel material. Chemically anchoring the hydrogel to a glass surface prevented its detachment during the hydration of the lipid-DexPEG hydrogel film when forming GUVs. Moreover, the DexPEG system demonstrated the potential to form defect-free vesicle structures when rehydrated from the dry state using physiologically relevant buffers.
To further understand GUV production on such chemically crosslinked substrates, we herein examine the effect of modulating the physicochemical properties of the DexPEG network by controlling its crosslink density. We synthesized various DexPEG hydrogels by reacting a dextran polymer with varying degrees of maleimide substitution and poly(ethylene glycol) (PEG) polymers with distinct molecular weights, a number of thiol functional groups and architectures (Scheme 1 and Table 1), and examined their physicochemical properties. GUV production on various DexPEG hydrogel substrates was compared with respect to their size and yield by flow cytometry using the same lipid composition, ionic strength and growth times. Additionally, GUV formation was imaged over time during hydrogel swelling to better understand the GUV growth process from the DexPEG chemically crosslinked hydrogel and calorimetry studies were performed to probe lipid–polymer interactions. Moreover, we demonstrate the potential of this method to encapsulate biological and colloidal cargo, such as polymersomes, in a facile manner.
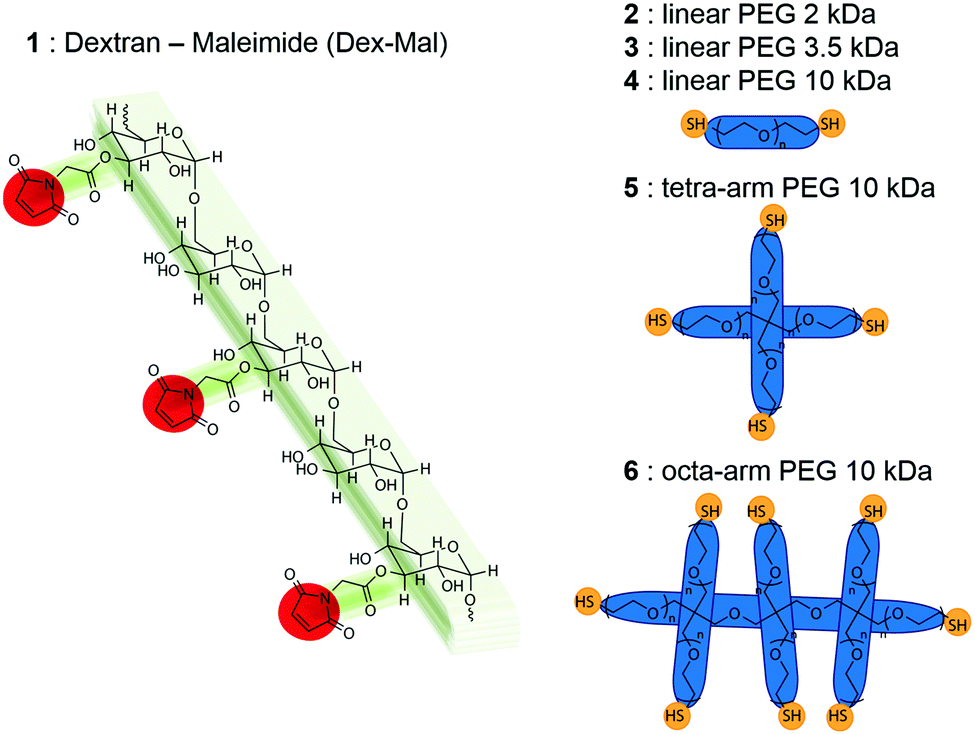 | ||
| Scheme 1 Chemical structures of DexPEG hydrogel precursors. | ||
| PEG crosslinker | 1 DS = 2 | 1 DS = 4 | 1 DS = 6 | 1 DS = 9 | 1 DS = 12 |
|---|---|---|---|---|---|
| n.p.: not prepared. | |||||
| 2 | ✓ | ✓ | ✓ | ✓ | ✓ |
| 3 | ✓ | ✓ | ✓ | n.p. | ✓ |
| 4 | n.p. | ✓ | n.p. | n.p. | n.p. |
| 5 | ✓ | ✓ | ✓ | n.p. | ✓ |
| 6 | ✓ | ✓ | ✓ | n.p. | ✓ |
2 Experimental
2.1 Chemicals and materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (PEG2000-PE) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NBD-PE) were purchased from Avanti Polar Lipids. Dextran from Leuconostoc (Mn = 70000 Da, dried in vacuum oven for several days before use), cholesterol (CH), β-alanine, 4-(dimethylamino)pyridine (DMAP), magnesium sulfate (MgSO4), poly(ethylene glycol)dithiol (MW = 3400 Da) (3), N,N′-diisopropylcarbodiimide (DIC), acetic acid, ethyl acetate, toluene, dimethyl sulfoxide (DMSO) and 2-propanol were purchased from Sigma-Aldrich. Maleic anhydride and p-toluene sulfonic acid monohydrate (PTSA) were purchased from Fluka. Poly(ethylene glycol) dithiols (MW = 2000 Da and 10000 Da) (2 and 4) were purchased from Iris Technologies GmbH. 4-arm poly(ethylene glycol) thiol (pentaerythritol core) (5) and 8-arm poly(ethylene glycol) thiol (tripentaerythritol core) (6) were purchased from JenKem Technology, USA. NIST traceable latex particles standard with nominal sizes 10 μm and 20 μm were purchased from Beckman Coulter. The salt of 4-(dimethylamino)pyridinium 4-toluenesulfonate (DPTS) was prepared from DMAP and PTSA in equimolar quantities.36N-Maleoyl-β-alanine was prepared following the previously reported procedure for obtaining maleimido alkanoic acids.37 DexMal (1) was synthesized by DIC mediated esterification of the hydroxyl groups of dextran with N-maleoyl-β-alanine as previously reported.35 PS300-b-PEG44-OMe and PS280-b-PEG44-Atto488 (5 wt%) block copolymers were used for the synthesis of 500 nm size polystyrene-block-polyethylene glycol polymersomes following a previously reported procedure (synthetic details can be found in the ESI†).38,39
:1 NBD-PE) were purchased from Avanti Polar Lipids. Dextran from Leuconostoc (Mn = 70000 Da, dried in vacuum oven for several days before use), cholesterol (CH), β-alanine, 4-(dimethylamino)pyridine (DMAP), magnesium sulfate (MgSO4), poly(ethylene glycol)dithiol (MW = 3400 Da) (3), N,N′-diisopropylcarbodiimide (DIC), acetic acid, ethyl acetate, toluene, dimethyl sulfoxide (DMSO) and 2-propanol were purchased from Sigma-Aldrich. Maleic anhydride and p-toluene sulfonic acid monohydrate (PTSA) were purchased from Fluka. Poly(ethylene glycol) dithiols (MW = 2000 Da and 10000 Da) (2 and 4) were purchased from Iris Technologies GmbH. 4-arm poly(ethylene glycol) thiol (pentaerythritol core) (5) and 8-arm poly(ethylene glycol) thiol (tripentaerythritol core) (6) were purchased from JenKem Technology, USA. NIST traceable latex particles standard with nominal sizes 10 μm and 20 μm were purchased from Beckman Coulter. The salt of 4-(dimethylamino)pyridinium 4-toluenesulfonate (DPTS) was prepared from DMAP and PTSA in equimolar quantities.36N-Maleoyl-β-alanine was prepared following the previously reported procedure for obtaining maleimido alkanoic acids.37 DexMal (1) was synthesized by DIC mediated esterification of the hydroxyl groups of dextran with N-maleoyl-β-alanine as previously reported.35 PS300-b-PEG44-OMe and PS280-b-PEG44-Atto488 (5 wt%) block copolymers were used for the synthesis of 500 nm size polystyrene-block-polyethylene glycol polymersomes following a previously reported procedure (synthetic details can be found in the ESI†).38,39
2.2 Preparation of DexPEG and PEG hydrogel substrates for GUV growth
2.3 Formation of GUVs
Giant Unilamellar Vesicles (GUVs) were grown on various DexPEG and hybrid DexPEG-polymersome hydrogel coated microscope glass slide substrates. A lipid solution (10 μL) composed of POPC:cholesterol (80:20 molar ratio, 14 mM) and DOPE-NBD (0.7 mol%) or POPC:cholesterol:PEG 2000-PE (75:20:5 molar ratio, 14 mM) and DOPE-NBD (0.7 mol%) were drop-casted on a hydrogel-coated glass slide. The drop-casted lipid solution was evaporated by using a gentle stream of nitrogen and the coated glass slide was placed in a vacuum oven overnight at room temperature. A liquid chamber was made by placing a 15 mm (OD) glass O-ring on top of the hydrogel using high vacuum silicon grease to generate a seal. GUV growth was initiated by hydrating the lipid-DexPEG hydrogel film with 400 μL of PBS. The hydrated substrates were left to stand overnight at room temperature.
3 Results and discussion
The degree of substitution (DS) of 1 and the architecture of the polymer crosslinkers (2–6) were systematically varied to modulate the physicochemical properties of the DexPEG hydrogel materials (Scheme 1). Esterification of the dextran polymer with various equivalents of N-maleoyl-β-alanine at room temperature resulted in functionalized polymers of 1 with various degrees of substitution. The DS of the maleimide-modified dextran polymer (DexMal) is defined as the number of maleimide groups per 100 glucopyranose residues of dextran from 1H NMR measurements. A proportional increase in the DS was observed up to DS = 6 with the number of equivalents of N-maleoyl-β-alanine added. For DS > 6, less N-maleoyl-β-alanine was required to achieve a greater increase in the DS value (Fig. S1, ESI†). Additionally, the successful reaction of N-maleoyl-β-alanine with the dextran polymer was confirmed by IR spectroscopy through the growth of the band 1700 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1650 and 700 (vinyl) cm−1 increasing sharply with the substitution degree (Fig. S2, ESI†). In order to form hydrogels, polymers of 1 were reacted with thiolated linear (molecules 2, 3 and 4), tetra-arm or octa-arm PEGs (molecules 5 and 6) in an equimolar ratio (maleimide:thiol) resulting in their crosslinking by Michael addition. The polymer mixture (DexPEG) was drop-casted on thiolated glass slides as previously described.35 The quality of the hydrogel film formed on the glass surface varied with DS by visual inspection. DexPEG hydrogels with a DS from 2 to 4 on polymer 1 produced clear and homogeneous films on the glass surface regardless of the architecture of the PEG crosslinker used, whereas higher DS polymers of 1 resulted in inhomogeneous hydrogel films.
O), 1650 and 700 (vinyl) cm−1 increasing sharply with the substitution degree (Fig. S2, ESI†). In order to form hydrogels, polymers of 1 were reacted with thiolated linear (molecules 2, 3 and 4), tetra-arm or octa-arm PEGs (molecules 5 and 6) in an equimolar ratio (maleimide:thiol) resulting in their crosslinking by Michael addition. The polymer mixture (DexPEG) was drop-casted on thiolated glass slides as previously described.35 The quality of the hydrogel film formed on the glass surface varied with DS by visual inspection. DexPEG hydrogels with a DS from 2 to 4 on polymer 1 produced clear and homogeneous films on the glass surface regardless of the architecture of the PEG crosslinker used, whereas higher DS polymers of 1 resulted in inhomogeneous hydrogel films.
Oscillatory rheology time sweeps were performed to provide insight into the gelation time and mechanical properties of the various DexPEG hydrogels. Their mechanical stiffness was examined as a function of the DS of 1, the molecular weight, the number of thiols, and architecture of the crosslinker precursor (2, 3, 4, 5 and 6). Samples consisting of 1 with a highest DS (DS = 9) produced weaker network structures with a slower gelation rate (Fig. 1A) in comparison to those with a lower DS (DS = 4, 6). Conversely, fast gelation and increased mechanical strength were observed when higher molecular weight PEG crosslinkers (4, 5, 6) (Fig. 1B) were compared to those with a lower molecular weight (2, 3). Interestingly, increasing the amount of thiol reactive groups 2-fold in the crosslinker led to a higher storage modulus of 5 over 6. Most likely, this unexpected decrease in mechanical properties arises due to the amount of unreacted groups and intramolecular loops formed during the coupling reaction between 1 and the various PEG polymer chains when the number of thiols are increased 4-fold in the crosslinker precursor 6.30
 | ||
| Fig. 1 Mechanical and equilibrium swelling data of various DexPEG hydrogels with varied DS of 1 and crosslinkers 2–6. Top: Oscillatory time sweeps for DexPEG hydrogels: (A) 1 DS = 4, 6 and 9 (3.5 wt%) with 2 (1.2 wt%). (B) 1 DS = 4 with molecules 2–6. Bottom: Equilibrium mass swelling ratio for DexPEG hydrogels with (C) 1 DS = 2, 4, 6, 9 and 12 with molecule 2. (D) 1 DS = 9 with molecules 2–6. Error bars in C and D represent the standard deviation of three independent mass swelling determinations. | ||
The swelling properties of the various DexPEG hydrogels were examined to better understand the differences in their network architecture. We studied the equilibrium mass swelling ratio of DexPEG hydrogels in glass vials excluding lipids. The equilibrium mass swelling ratio is defined as Ws/Wd, where Ws is the swollen weight of the gel after equilibration in buffer and Wd is the dry weight of the lyophilized gel. With respect to the degree of substitution of the dextran polymer, the swelling ratio immediately decreases when the DS of 1 increases (from DS = 2 to 4), due to the increasing number of crosslinks. The swelling ratio then gradually increases (from DS = 4 to 9) with higher substitution degrees until reaching a plateau after DS = 9 (Fig. 1C). The increase in the swelling ratio revealed that polymers of 1 with the lowest and highest DS provided a loose network structure. At lower DS this is possibly due to the lack of crosslinking, while at higher DS the insolubility of the maleimide-substituted polymer becomes important. The opposite effect was observed, in comparison to the substitution degree in polymers of 1, for PEG crosslinkers with different molecular weights. The mass swelling ratio was decreased in PEG crosslinkers with a higher molecular weight (5, 6) with respect to PEG crosslinkers with a lower molecular weight (2, 3) in the swelling of the hydrogel with a higher DS (DS = 9, Fig. 1D). Increasing the amount of thiol reactive groups in the polymer architecture produced lower swelling ratios relative to the linear crosslinkers with a less amount of thiol reactive groups. This trend is consistent with mechanical measurements, suggesting that increased crosslinking and higher molecular weight reduce the swelling of the hydrogel material. Additionally, the DexPEG hydrogel containing 6 displayed a higher swelling ratio as compared to hydrogels crosslinked with 5, further supporting the formation of fewer effective crosslinks as observed in rheological measurements and a more swellable network in the former.
Next, we examined the microstructure of selected DexPEG hydrogels in the swollen state by cryo-scanning electron microscopy (cryo-SEM) (Fig. 2). With increasing substitution degree of 1, larger pore sizes or discontinuities in the network were observed in the hydrogel material. DexPEG hydrogels of 1 with a DS = 4 (Fig. 2B) displayed a porous network morphology with pores around 200 nm in size, whereas a DS = 6 (Fig. 2E) showed larger pores (∼1 μm). In contrast, DexPEG hydrogels with a DS > 6 on 1 lacked the ability to form a three-dimensional network structure (i.e.Fig. 2F). Furthermore, increasing the number of thiol groups in the PEG crosslinker 2- and 4-fold, yielded DexPEG hydrogels with a highly porous honeycomb-like structure. Thus modulation of the PEG architecture by varying the crosslink density had a clear effect on the hydrogel microstructure as observed by a direct comparison of linear PEG precursors 2 and 3 (Fig. 2A and B) with tetra-arm PEG 5 (Fig. 2D) and octa-arm PEG 6 (Fig. 2C).
 | ||
| Fig. 2 Cryo-SEM of DexPEG hydrogels with different DS values and the PEG crosslinker. (A) 1 DS = 4 and crosslinker 2; (B) 1 DS = 4 and crosslinker 3; (C) 1 DS = 4 and crosslinker 5; (D) 1 DS = 4 and crosslinker 6, (E) 1 DS = 6 and crosslinker 3, (F) 1 DS = 12 and crosslinker 3. | ||
We previously found that DexPEG hydrogels with a decreased crosslink density grow GUVs with a larger mean diameter.35 Therefore, the effect of the maleimide/thiol ratio on the morphology of these hydrogels was also examined by cryo-SEM (Fig. S3, ESI†). When the PEG crosslinker concentration was decreased, an inhomogeneous network structure was observed due to fewer crosslinks formed. These results are in good agreement with the obtained mechanical data (Fig. S4, ESI†) indicating the formation of a weak and loose network.
The correlation between the hydrogel crosslink density and its effect on GUV yield was quantified by flow cytometric (FC) experiments. Stock solutions of the lipid mixtures POPC:cholesterol (80:20 mol%) or POPC:cholesterol:PEG2000-PE (75:20:5 mol%) were deposited on the DexPEG hydrogel coated microscope slides with varied DS and crosslinkers. Both lipid compositions also contained 0.7 mol% of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3 benzoxadiazol-4-yl) (ammonium salt) (DOPE-NBD) to allow fluorescence detection of the formed GUVs. Subsequently, the solvent was evaporated under a gentle stream of nitrogen and placed in a vacuum oven overnight to prepare the lipid film on the hydrogel surface. The lipid-coated hydrogel films were then hydrated in phosphate-buffered saline (PBS, pH 7.4) and the resulting free-floating GUVs were collected and characterized by FC. PEGylated and non-PEGylated GUVs were formed on all hydrogels and detected in both fluorescence and side scattering channels, producing typical populations of GUVs (Fig. S5 and S6, ESI†). The use of PEGylated lipids in the lipid composition decreased GUV aggregation, resulting in an increased frequency of single events (i.e. a more precise counting of individual GUVs) for all DexPEG combinations (Fig. 3). Firstly, increasing the DS from 2 to 12 in 1 resulted in a higher GUV yield (Fig. 3A). In contrast, increasing 2-fold the number of thiol groups on the PEG crosslinkers decreased the GUV yield (Fig. 3B). Only hydrogels synthesized with 6 did not follow this trend due to higher swelling of this DexPEG combination (Fig. 3B). These experiments suggest that the main driving force for GUV production is the swelling of the dextran hydrogel network during the rehydration step of the lipid-coated hydrogel films with the degree of substitution of the polymer and crosslink density affecting this process.
 | ||
| Fig. 3 Yield of fluorescently-labelled GUVs produced on the various DexPEG substrates as determined by FC. Cross-hatched bars represent GUVs with the lipid composition POPC:cholesterol (80:20 mol%) and line patterned bars represent PEGylated GUVs with the lipid composition POPC:cholesterol:PEG2000-PE (75:20:5 mol%). (A) Effect of the DS on the production of GUVs for DexPEG hydrogel films with 1 (DS from 2–12) and crosslinker 3. (B) Effect of the PEG crosslinker architecture on the production of GUVs for DexPEG hydrogel films with 1 (DS = 2) and crosslinkers 2, 3, 5 and 6. Error bars represent the standard deviation of three analysed data sets (see details in the ESI†). | ||
The size distribution of the produced GUVs was estimated using the Coulter Principle in a Quanta SC FC instrument based on electrical impedance.34,35 The electric volume (EV) parameter is proportional to the electrical impedance and does not depend on the laser wavelength, geometry or refractive index of the sample, overcoming limitations of forward and side scattering monotonic measurements for the determination of particle size by flow cytometry.36 We transformed the EV parameter measured to the GUV diameter (μm) (see eqn (S1) and (S2) in the ESI†). Validation of this method was made with microsphere standards with nominal sizes of 10 μm and 20 μm (Fig. S7, ESI†). These measurements yielded a size distribution profile for a large set of PEGylated GUVs. The advantage of this method is the possibility of counting large populations of GUVs, in comparison to microscopy-based methods that only consider smaller population sizes due to the small focal volume. All tested DexPEG hydrogel substrates, independent of the DS of 1 or the PEG crosslinker architecture, produced GUV populations with sizes ranging between 5 μm and 20 μm in diameter (Fig. 4). DexPEG hydrogels prepared from multi-arm PEG crosslinkers 5 and 6 produced GUVs with a similar size distribution as compared to linear PEG crosslinker hydrogels composed of 2 or 3, but with lower yields (Fig. 4B).
 | ||
| Fig. 4 Determination of the size distribution by the Coulter principle for GUV populations with the lipid composition POPC:cholesterol:PEG2000-PE (75:20:5 mol%) produced on various DexPEG substrates. (A) Effect of the DS on the size of GUVs for DexPEG hydrogel films with 1 (DS from 2–12) and crosslinker 3. (B) Effect of the polymer architecture of the PEG crosslinker on the size of GUVs for DexPEG hydrogel films with 1 (DS = 2) and crosslinkers 2, 3, 5 and 6. Error bars represent the standard deviation of three analysed data sets (see details in the ESI†). | ||
These size distributions are in line with those reported previously by optical microscopy using a lipid composition of POPC:cholesterol (80:20 mol%) on DexPEG substrates with an equimolar ratio of precursors (diameter = 10 ± 5 μm, N = 55 GUVs).35 We previously found that GUV size could be tuned by decreasing the crosslink density within the hydrogel by reducing the molar ratio of 2 with respect to 1 DS = 4. However in this work, we find that further increasing the crosslink density in these networks modulates their ability to swell, and therefore, affects the GUV yield.
The GUV formation and growth process from DexPEG hydrogels was further studied by differential interference contrast (DIC) microscopy. Lipids (POPC:cholesterol, 80:20 mol%) were spread on DexPEG hydrogels composed of dextran 1 (DS = 2) and crosslinker 2 (linear) or 5 (tetra-arm). The hydrogel/lipid film was swollen with 200 mM sucrose in PBS (pH 7.4) and images were taken every second for 10 minutes. The initial GUV diameter was a few microns and became larger due to coalescence from crowding. These GUV coalescence events are quantified as a function of time in Fig. 5. The average size of GUVs increases while the number of GUVs decreases upon GUV coalescence. Interestingly, variation in the initial GUV sizes was observed as a function of the varying surface roughness within the DexPEG hydrogel using crosslinker 2 or 5. Qualitative inspection of hybrid lipid/DexPEG hydrogel films under a microscope showed varying surface roughnesses; representative examples are shown in Fig. 5B (relatively rough) and Fig. 5C (relatively smooth). Varying surface roughness could also be observed by a simple direct visual inspection of the prepared samples; rough areas appeared as an opaque film (see Fig. S9, ESI†). On rougher surfaces (Fig. 5B) GUVs coalesced less and maintained a smaller size (∼5 μm diameter), while on smoother surfaces (Fig. 5C), the formed GUVs were larger (∼14 μm diameter). This phenomenon was consistent across samples from crosslinker 2 or 5, measured each in triplicate. Therefore, the thickness and roughness of the DexPEG hydrogel film needs to be considered for controlling polydispersity of GUVs. Additionally, the growth of fluorescently labelled GUVs was followed using confocal microscopy imaging reconstruction (Fig. 6). GUVs were found to swell from the surface of the DexPEG hydrogel matrix as depicted. The orthogonal reconstruction of the confocal image allowed the imaging of the budding of vesicles during the first minutes of lipid hydration on the hydrogel scaffold, clearly showing the self-assembly of small vesicles on the hydrogel surface and the mechanism of growth by coalescence.
 | ||
| Fig. 5 GUV swelling observed on the DexPEG surface. (A) DIC time lapse series of micrographs of POPC:cholesterol (80:20 mol%) on the DexPEG hydrogel with 1 (DS = 2) and crosslinker 5. The scale bars are 10 μm. The first image shows a dried lipid film on top of the hydrogel. The subsequent images are indicated according to time of swelling. GUVs form off of the DexPEG hydrogel network and coalesce to form larger GUVs. (B) Left micrographs show areas of rough DexPEG with lipid before hydration (time = 0 s) and after hydration (600 s). The right plot shows the change in the GUV number and average size with respect to time. (C) Micrographs are examples of areas of smooth DexPEG with lipid and the associated plot of swelling kinetics is on the right. As coalescence occurs, GUV size increases and their amount decreases. Smooth areas form larger GUVs as compared with rougher areas on the DexPEG hydrogels. | ||
 | ||
| Fig. 6 Confocal images GUVs formed from a DexPEG surface. (A) A single confocal slice of an image stack of GUVs swollen on DexPEG. Lipid composition is POPC:cholesterol (80:20 mol%) fluorescently labelled with ATTO-488-DPPE (0.4 mol%). The yellow lines indicate the orthogonal slices in B and C. (B) Horizontal orthogonal slice of GUVs in A reconstructed from confocal image stack. (C) Vertical orthogonal slice of GUVs in A. (B and C) Show GUVs that are not yet fully formed closed spheres. Rather, they are still attached by budding necks to the surface of the lipid film. | ||
To better understand the effect of the various hydrogel components on GUV growth, the interaction of lipids with the various hydrogel components was probed by differential scanning calorimetry (DSC). The melting temperature (Tm) of DOPC lipids in the presence and absence of the DexPEG film was determined by differential scanning calorimetry (DSC). A single, sharp endothermic transition arising from the gel to the liquid-crystalline phase was found for pure DOPC at −6 °C. This transition is higher than the reported value of the hydrated lipid indicating that the water/lipid ratio was ∼3.37 In the presence of the DexPEG hydrogel, the Tm for DOPC shifted to −18 °C and was broadened (Fig. S8A, ESI†), indicative of a moderate lipid–DexPEG interaction. As a comparison, we also determined the Tm of DOPC in the presence of a gel consisting solely of disulphide crosslinked PEG (2 wt% of 5). Surprisingly, the intensity of gel-to-liquid crystalline phase transition of DOPC was suppressed (Fig. S8B, ESI†) suggestive of a stronger interaction of the lipids with the PEG network over DexPEG. In addition, the growth of GUVs on hydrogel films composed only of 5 was not observed. While the strong interaction of lipids with the PEG crosslinker decreases GUV formation, their lack of interaction with the dextran scaffold may have an important role in the vesicle self-assembly. These results suggest the potential to control this process by using polymers that show minimal lipid interactions for their construction.
Once the effect of the physicochemical properties of DexPEG hydrogels on GUV formation was examined using various methods, we sought to explore the potential of this method to create compartmentalized GUVs. Compartmentalization is a fundamental characteristic of a cellular structure and it is desired in the formation of minimal cell models for the understanding of multi-step biological processes. However, the formation of compartmentalized GUVs using rehydration methods is still challenging, unlike the encapsulation of a biomolecular cargo that is achievable using gentle hydration and microfluidic methods.40–42 Firstly, a 10 bp fluorescent oligonucleotide (5′-FAM-TGGAGAAGGC-3′) was encapsulated by direct rehydration of a dried lipid film with the composition of DOPC:DOPE:cholesterol (40:40:20 mol%) on the DexPEG hydrogel substrate (1 DS = 4, crosslinker 2) using the nucleic acid solution. The free-floating GUVs were collected from the growth chamber and a non-encapsulated fluorescent oligonucleotide was removed with a washing step in PBS prior to imaging by confocal microscopy (Fig. 7A). Secondly, the same method was applied in the encapsulation of colloidal particles in the form of fluorescently labelled polymersomes synthesised from PS300-b-PEG44-OMe and labelled with PS280-b-PEG44-Atto488 (5 wt%) block copolymers (polymersome diameter of 500 nm and zeta potential ca. −20 mV in PBS pH 7.4). Fluorescence imaging of free-floating GUVs showed that polymersomes were present only in the outside of GUVs without being encapsulated (Fig. S10, ESI†). Finally, to successfully encapsulate GUVs we followed a different approach by embedding the polymersomes into the DexPEG hydrogel scaffold, forming a hybrid DexPEG-polymersome hydrogel film. The DexMal precursor (1 DS = 4) was mixed with a solution of polymersomes, prior to the addition of the reactive PEG crosslinker 2. Subsequently, the DexPEG solution containing polymersomes was drop-casted on thiolated glass slides to form an anchored hybrid DexPEG-polymersome hydrogel film. A lipid film was drop-casted on the surface of the dried hydrogel film as described previously and its direct re-hydration using PBS resulted in GUVs with encapsulated polymersomes in the inner aqueous volume as imaged by dual colour confocal microscopy (Fig. 7B). Polymersomes were not observed either trapped or aggregated in the lipid membrane of GUVs and the localization of polymersomes in the inner volume of GUVs was confirmed by cryo-SEM imaging (Fig. 7C and D). In these images, the wavy GUV surface, most likely arising from the freezing process during sample preparation, helped in localizing GUVs and identifying polymersomes within the vesicle lumen. Moreover, the effect of the lipid composition for the encapsulation of polymersomes was probed with the lipid mixtures DOPC:DOPE:cholesterol:PEG2000-PE (40:39.5:20:0.5 mol%) to promote steric repulsion and POPC:POPG (50:50 and 90:10 mol%) for increasing the electrostatic repulsion between lipids and polymersomes during the vesicle budding process on the hybrid DexPEG – polymersome substrate. The encapsulation of polymersomes in the lumen of GUVs was observed by fluorescence microscopy for all lipid combinations (Fig. S11, ESI†). During the fluorescence imaging of polymersomes loaded GUVs, we always observed super-filled GUVs (∼5–10% of the total GUV population), which contained a significantly higher number of polymersomes per GUV as one could expect from the initial polymersome concentration in the buffer solution (Fig. 7B). This phenomenon has been previously reported by Luisi et al. in a small liposomal subpopulation due to particular environmental local conditions that might lead to the perturbation of the vesicle formation mechanism.43–45 Therefore, further physicochemical studies of the formed super-filled GUVs from DexPEG substrates are required to better understand the overcrowding process in cell-sized GUVs. All these data together show that DexPEG chemically crosslinked hydrogel films can be easily adapted for the facile encapsulation of biomolecular cargos and colloidal particles without the use of any sophisticated equipment. Importantly, if the polymersomes were not embedded within the hydrogel during the synthesis step, polymersomes were not found within the GUV interior.
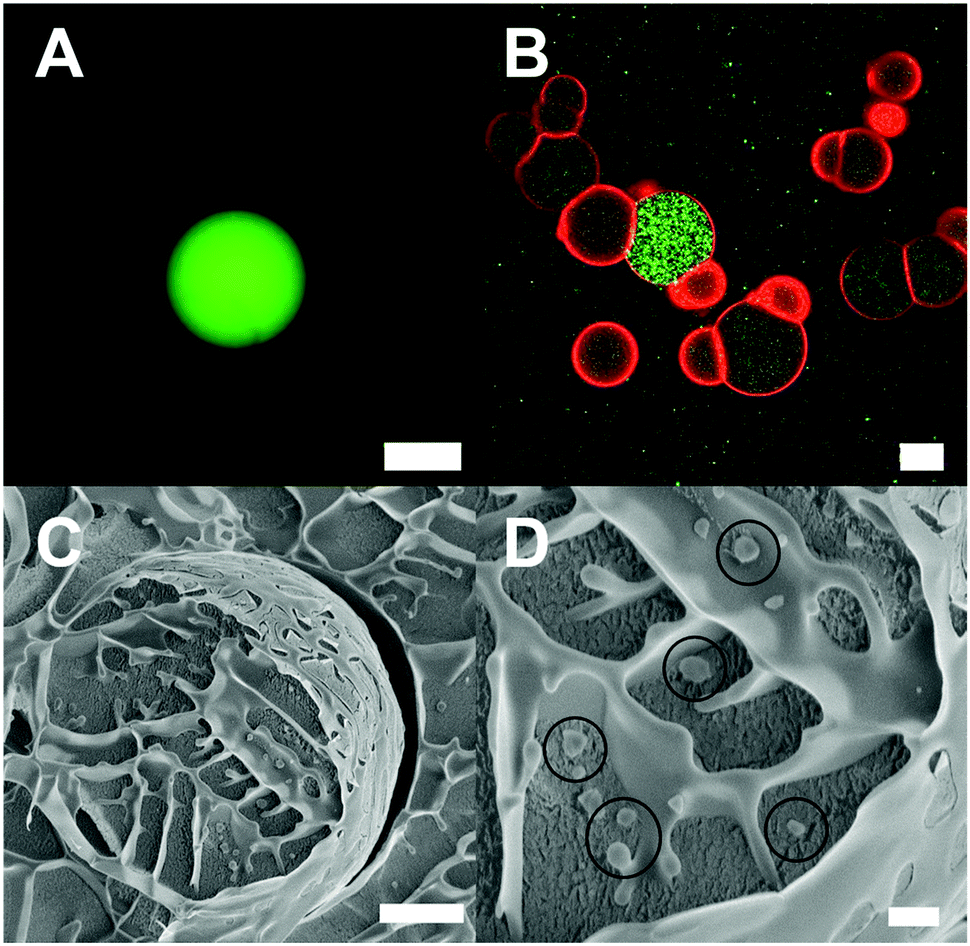 | ||
| Fig. 7 Encapsulation of cargo in the lumen of GUVs with the lipid composition DOPC:DOPE:cholesterol (40:40:20 mol%). (A) Encapsulation of biomolecular cargo, namely 10 bp fluorescently labelled oligonucleotide (5-FAM/TGGAGAAGGC). The scale bar is 50 μm. (B) Encapsulation of colloidal cargo, namely 500 nm polymersomes labelled with ATTO 488 (green). The GUV membrane was labelled with DOPE-lissamine rhodamine. The non-encapsulated polymersomes are not removed by a washing step in order to show the contrast between the external global concentration, the filled GUVs and the super-filled GUV. The scale bar is 50 μm. (C) Cryo-SEM of a single GUV containing 500 nm polymersomes. The scale bar is 5 μm. (D) Zoom in of C. The 500 nm polymersomes are highlighted in circles. The scale bar is 1 μm. | ||
4 Conclusions
Collectively, oscillatory rheology, equilibrium swelling, cryo-SEM and FC experiments show that the physicochemical properties of the polymer network strongly affect GUV production in terms of size and yield under physiologically relevant conditions. Polymer scaffolds of lower mechanical strength and increased swelling ratios favour the budding of vesicles and GUV growth, whereas an increased crosslink density negatively impacts the GUV yield. By imaging using various microscopes, we observed that the GUV size is significantly affected by the homogeneity of the network and surface roughness of the film. We find that lipid interactions with the hydrogel film should be considered in the design of these scaffolds as strong lipid–polymer interactions preclude the formation of GUVs. Hence, we look to further understand the impact of lipid–polymer interactions and the effect of surface roughness on GUV formation such that monodisperse populations of GUVs can be made. We demonstrate, for the first time, that a chemically crosslinked hydrogel film, consisting of DexPEG, can be used as a suitable scaffold for the GUV encapsulation of a colloidal cargo, such as polymersomes. We believe that the formation of multicompartmentalized GUVs using this method can be an extremely useful tool to create minimal cell models that can provide insight into cellular structure and function to support research in the areas such as the origin of life.Acknowledgements
Dr Sander van Kasteren from the bio-organic synthesis group at Leiden University is acknowledged for the access to FlowJo software. We thank Wim Jesse and Viorica Tudor for technical assistance. NLM acknowledges the National Council for Science and Technology (CONACyT) for scholarship 181940. Y. G. would like to thank the China Scholarship Council (CSC) for their support. NM acknowledges the National Institutes of Health (1R01GM093279). RK acknowledges the Netherlands Organisation for Scientific Research (NWO) for a VENI grant. AK acknowledges the Netherlands Organisation for Scientific Research (NWO) for a VICI grant.References
- S. F. Fenz and K. Sengupta, Integr. Biol., 2012, 4, 982–995 RSC.
- F. C. Tsai, B. Stuhrmann and G. H. Koenderink, Langmuir, 2011, 27, 10061–10071 CrossRef CAS PubMed.
- F. M. Menger and J. S. Keiper, Curr. Opin. Chem. Biol., 1998, 2, 726–732 CrossRef CAS PubMed.
- P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef CAS PubMed.
- R. Lipowsky, Curr. Opin. Struct. Biol., 1995, 5, 531–540 CrossRef CAS PubMed.
- S. Pautot, B. J. Frisken and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10718–10721 CrossRef CAS PubMed.
- D. Needham and E. Evans, Biochemistry, 1988, 27, 8261–8269 CrossRef CAS PubMed.
- A. Kato, A. Tsuji, M. Yanagisawa, D. Saeki, K. Juni, Y. Morimoto and K. Yoshikawa, J. Phys. Chem. Lett., 2010, 1, 3391–3395 CrossRef CAS.
- J. S. Hansen, J. R. Thompson, C. Helix-Nielsen and N. Malmstadt, J. Am. Chem. Soc., 2013, 135, 17294–17297 CrossRef CAS PubMed.
- M. G. Gutierrez and N. Malmstadt, J. Am. Chem. Soc., 2014, 136, 13530–13533 CrossRef CAS PubMed.
- K. S. Horger, H. Y. Liu, D. K. Rao, S. Shulda, D. Sept, S. V. Ambudkar and M. Mayer, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 643–653 CrossRef CAS PubMed.
- I. Motta, A. Gohlke, V. Adrien, F. Li, H. Gardavot, J. E. Rothman and F. Pincet, Langmuir, 2015, 31, 7091–7099 CrossRef CAS PubMed.
- N. Kahya, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1392–1398 CrossRef CAS PubMed.
- D. Koller and K. Lohner, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2250–2259 CrossRef CAS PubMed.
- Y. Tamba and M. Yamazaki, J. Phys. Chem. B, 2009, 113, 4846–4852 CrossRef CAS PubMed.
- Y. Tamba, H. Ariyama, V. Levadny and M. Yamazaki, J. Phys. Chem. B, 2010, 114, 12018–12026 CrossRef CAS PubMed.
- K. Akashi, H. Miyata, H. Itoh and K. Kinosita, Biophys. J., 1996, 71, 3242–3250 CrossRef CAS PubMed.
- H. Valkenier, N. L. Mora, A. Kros and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 2137–2141 CrossRef CAS PubMed.
- J. P. Reeves and R. M. Dowben, J. Cell. Physiol., 1969, 73, 49–60 CrossRef CAS PubMed.
- M. I. Angelova and D. S. Dimitrov, Faraday Discuss., 1986, 81, 303–311 RSC.
- J. Chen, H. Park and K. Park, J. Biomed. Mater. Res., 1999, 44, 53–62 CrossRef CAS PubMed.
- W. S. W. Shalaby, W. E. Blevins and K. Park, Biomaterials, 1992, 13, 289–296 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- J. Henise, B. R. Hearn, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 270–278 CrossRef CAS PubMed.
- K. Peng, I. Tomatsu, B. van den Broek, C. Cui, A. V. Korobko, J. van Noort, A. H. Meijer, H. P. Spaink and A. Kros, Soft Matter, 2011, 7, 4881–4887 RSC.
- K. Peng, I. Tomatsu and A. Kros, Chem. Commun., 2010, 46, 4094–4096 RSC.
- K. Peng, I. Tomatsu, A. V. Korobko and A. Kros, Soft Matter, 2010, 6, 85–87 RSC.
- K. Peng, C. Cui, I. Tomatsu, F. Porta, A. H. Meijer, H. P. Spaink and A. Kros, Soft Matter, 2010, 6, 3778–3783 RSC.
- T. G. Kim, H. Shin and D. W. Lim, Adv. Funct. Mater., 2012, 22, 2446–2468 CrossRef CAS.
- M. A. Daniele, A. A. Adams, J. Naciri, S. H. North and F. S. Ligler, Biomaterials, 2014, 35, 1845–1856 CrossRef CAS PubMed.
- K. S. Horger, D. J. Estes, R. Capone and M. Mayer, J. Am. Chem. Soc., 2009, 131, 1810–1819 CrossRef CAS PubMed.
- R. B. Lira, R. Dimova and K. A. Rieke, Biophys. J., 2014, 107, 1609–1619 CrossRef CAS PubMed.
- A. Weinberger, F. C. Tsai, G. H. Koenderink, T. F. Schmidt, R. Itri, W. Meier, T. Schmatko, A. Schroder and C. Marques, Biophys. J., 2013, 105, 154–164 CrossRef CAS PubMed.
- M. Garten, C. Prevost, C. Cadart, R. Gautier, L. Bousset, R. Melki, P. Bassereau and S. Vanni, Phys. Chem. Chem. Phys., 2015, 17, 15589–15597 RSC.
- N. L. Mora, J. S. Hansen, Y. Gao, A. A. Ronald, R. Kieltyka, N. Malmstadt and A. Kros, Chem. Commun., 2014, 50, 1953–1955 RSC.
- J. S. Moore and S. I. Stupp, Macromolecules, 1990, 23, 65–70 CrossRef CAS.
- R. M. de Figueiredo, P. Oczipka, R. Frohlich and M. Christmann, Synthesis, 2008, 1316–1318 CAS.
- S. F. M. van Dongen, W. P. R. Verdurmen, R. J. R. W. Peters, R. J. M. Nolte, R. Brock and J. C. M. van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213–7216 CrossRef PubMed.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- L. M. Dominak, D. M. Omiatek, E. L. Gundermann, M. L. Heien and C. D. Keating, Langmuir, 2010, 26, 13195–13200 CrossRef CAS PubMed.
- K. Funakoshi, H. Suzuki and S. Takeuchi, J. Am. Chem. Soc., 2007, 129, 12608–12609 CrossRef CAS PubMed.
- J. C. Stachowiak, D. L. Richmond, T. H. Li, F. Brochard-Wyart and D. A. Fletcher, Lab Chip, 2009, 9, 2003–2009 RSC.
- T. P. de Souza, A. Fahr, P. L. Luisi and P. Stano, J. Mol. Evol., 2014, 79, 179–192 CrossRef PubMed.
- P. L. Luisi, P. Stano and T. de Souza, Origins Life Evol. Biospheres, 2014, 44, 313–317 CrossRef CAS PubMed.
- E. D'Aguanno, E. Altamura, F. Mavelli, A. Fahr, P. Stano and P. L. Luisi, Life, 2015, 5, 969–996 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sm00551b |
| ‡ Present address: Department of Chemistry, King's College London, Britannia House, 7 Trinity Street, London, SE1 1DB. E-mail: nestor.lopez_mora@kcl.ac.uk |
| This journal is © The Royal Society of Chemistry 2017 |