
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On tuning microgel character and softness of cross-linked polystyrene particles†
Jochen
Schneider
a,
Malte
Wiemann
a,
Anna
Rabe
a and
Eckhard
Bartsch
 *ab
*ab
aInstitut für Physikalische Chemie, Albert-Ludwigs-Universität, D-79104 Freiburg, Germany. E-mail: eckhard.bartsch@physchem.uni-freiburg.de
bInstitut für Makromolekulare Chemie, Albert-Ludwigs-Universität, D-79104 Freiburg, Germany
First published on 18th November 2016
Abstract
Polystyrene (PS) microgel colloids have often been used successfully to model hard sphere behaviour even though the term “gel” invokes inevitably the notion of a more or less soft, deformable object. Here we systematically study the effect of reducing the cross-link density from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (1 cross-link per 10 monomers) to 1:100 on particle interactions and “softness”. We report on the synthesis and purification of 1:10, 1:25, 1:50, 1:75 and 1:100 cross-linked PS particles and their characterization in terms of single particle properties, as well as the behaviour of concentrated dispersions. We are able to tune particle softness in the range between soft PNiPAM-microgels and hard PMMA particles while still allowing the mapping of the microgels onto hard sphere behavior with respect to phase diagram and static structure factors. This is mainly due to a rather homogeneous radial distribution of cross-links in contrast to PNiPAM microgels where the cross-link density decreases radially. We find that up to a cross-link density of 1:50 particle form factors are perfectly described by a homogeneous sphere model whereas 1:75 and 1:100 cross-linked spheres are slightly better described as fuzzy spheres. However the fuzziness is rather small compared to typical PNiPAM microgels so that a hard sphere mapping still holds even for these low cross-link densities. Finally, by varying the reaction conditions – changing from batch to semibatch emulsion polymerization and varying the feed rate or by adjusting the monomer to initiator ratio we can tune the fuzziness or significantly alter the inner structure to a more open, star-like architecture.
:10 (1 cross-link per 10 monomers) to 1:100 on particle interactions and “softness”. We report on the synthesis and purification of 1:10, 1:25, 1:50, 1:75 and 1:100 cross-linked PS particles and their characterization in terms of single particle properties, as well as the behaviour of concentrated dispersions. We are able to tune particle softness in the range between soft PNiPAM-microgels and hard PMMA particles while still allowing the mapping of the microgels onto hard sphere behavior with respect to phase diagram and static structure factors. This is mainly due to a rather homogeneous radial distribution of cross-links in contrast to PNiPAM microgels where the cross-link density decreases radially. We find that up to a cross-link density of 1:50 particle form factors are perfectly described by a homogeneous sphere model whereas 1:75 and 1:100 cross-linked spheres are slightly better described as fuzzy spheres. However the fuzziness is rather small compared to typical PNiPAM microgels so that a hard sphere mapping still holds even for these low cross-link densities. Finally, by varying the reaction conditions – changing from batch to semibatch emulsion polymerization and varying the feed rate or by adjusting the monomer to initiator ratio we can tune the fuzziness or significantly alter the inner structure to a more open, star-like architecture.
1 Introduction
Colloidal suspensions are frequently used to investigate the physical principles of condensed matter systems. Among the phenomena being researched are for example the glass transition1–4 and particle crystallisation.5–9 Of special interest are so-called hard sphere like or nearly hard sphere like particles which provide an easy theoretical treatment and easy computer simulations compared to systems with more complex interactions.10 The most common systems under study are suspensions of sterically stabilised poly-(methylmethacrylate) (PMMA) particles, as a suspension medium, usually a mixture of organic solvents, is used to match the refractive index and the density of the particles to allow measurements using light scattering11–13 and microscopy14–17 while avoiding sedimentation. The major drawback of these systems lies within the usage of solvent mixtures. On the one hand it can be hard to keep the composition of the dispersion media constant over time, e.g. due to non-uniform evaporation of the individual solvents. On the other hand the common solvents added to provide density matching of PMMA particles also induce a degree of charging, which is difficult to screen out in organic solvents.18In a different approach spherical particles of cross-linked polymer chains are used. These so called microgels19 swell in a good solvent, which leads to an easier matching of both refractive index and density. The most common microgels are prepared from N-isopropylacrylamide cross-linked with N,N′-methylenebisacrylamide (PNiPAM) – well known for its temperature dependent swelling in water.20,21 Besides the currently very popular PNIPAM microgels, PMMA22,23 or polystyrene (PS)7,24–28-based microgels are also frequently used as colloidal model systems. While the swelling of the microgel particles provides advantages in terms of refractive index and density matching it may also introduce a certain amount of softness to the system. This typically leads to deviations from the hard sphere behaviour. For example if the structure of a PNiPAM microgel suspension is mapped onto hard spheres, the mapping fails at volume fractions above 0.35.29 In addition, the phase diagram of PNiPAM-microgels as well as of PMMA-microgels shows a fluid-crystal coexistence region, which is smaller than the one expected for hard spheres.21,22 A notable exception are PS core–PNIPAM shell particles which expose hard sphere behaviour in rheological experiments after appropriate mapping.30,31 Nevertheless in the case of PS it could been shown that microgels can be synthesised, where their structural properties25,32 and dynamic properties32,33 are in good agreement with hard sphere like systems.
If one aims at controlling the particle softness, the key parameters are the swelling ratio of the particles and the homogeneity of cross-linking. In the case of PNiPAM-microgels the reactivity of the cross-linking agent is much higher than the reactivity of the NiPAM monomer. This leads to the formation of a highly cross-linked, dense core and a corona of gradually decreasing density (a so called fuzzy sphere).34 Thus the outer areas of these microgels behave more like a soft polymer coil than a solid sphere. If the reactivities of the cross-linking agent and the monomer are similar, a more homogeneous cross-linking is achieved. In the case of PS microgels homogeneously cross-linked microgels of low polydispersity can be prepared if 1,3-diisopropenylbenzene (DIPB) is used to form the polymer network,35 leading to less soft particles than e.g. PNiPAM-microgels.
For this reason PS microgels have been widely used as effective hard spheres to study e.g. glass transition,24–27,36 crystallisation kinetics7,28,37–39 or collective33 and single40 particle diffusion. While the assumption of hard sphere behaviour of the PS microgels is supported by many studies7,36,39,40 systematic differences between particles of different cross-linking ratios can be observed in mixtures of PS microgels and free non-absorbing polymers.41 Here it has been observed that the addition of a free PS polymer to 1:50 cross-linked PS microgel particles shifts the glass transition to much higher values26 than that observed for a comparable PMMA colloid–PS free polymer system.42 In contrast, increasing the cross-link density to 1:10 yields a shift which is comparable to that of the PMMA system.41 In this context it is not yet clear, whether these deviations may be attributed to an intrinsic softness of the microgel particles or to interactions between the polymer and the microgel network. In microgel systems particle softness often plays an important role in the particle interactions and the physical behaviour of the dispersions.21,43 Thus, a systematic investigation of particle interactions in the absence of a polymer is necessary to clarify the role of softness effects in mixtures of PS microgels and a free non-absorbing polymer.
Therefore we studied systematically the effect of varying the cross-link density on the static properties of PS microgels and compared their behaviour with the known one of hard sphere systems. For this purpose we prepared a series of PS microgels with different amounts of cross-linking agents. These particles were prepared via semibatch emulsion polymerisation according to ref. 44. All systems under study showed a low enough polydispersity to undergo a first order freezing transition. Rheological measurements were used to probe particle softness. The static structure factor of samples located below the freezing point was determined from static light scattering (SLS) data to check whether softness effects influence the structure of the dispersions. To evaluate the structural properties of our PS microgels in comparison to hard sphere like PMMA particles and soft sphere like PNiPAM microgels we characterised the polydispersity and particle shape of the unswollen particles by means of transmission electron microscopy (TEM) and SLS. Furthermore the internal structure of the swollen particles was analysed by means of SLS and the obtained particle form factors were comparatively fitted by the homogeneous sphere and the fuzzy sphere model.
Since styrene and DIPB usually form homogeneously cross-linked particles, we also varied the reaction conditions to evaluate whether this allows a specific control of the inner structure of the particles. Therefore we prepared particles via batch emulsion polymerisation and semibatch emulsion polymerisation using different feeding rates to study the influence of the polymerisation technique on the incorporation of DIPB, i.e. the fuzziness of the particles.
2 Experimental section
2.1 Materials
Styrene was obtained from Merck KGaA, dried over CaH2, distilled under reduced pressure and stored at −18 °C under an argon atmosphere. CaH2 was obtained from Fluka in 95% purity and used without further purification. Argon was purchased from Air Liquide GmbH in 99.9990% purity. Sodium dodecyl sulfate (SDS) was obtained from VWR International in 98% purity and further purified twice by recrystallisation from ethanol. Ethanol was purchased from VWR International (England), dried over potassium and distilled under reduced pressure in an argon atmosphere. Sodium bicarbonate (NaHCO3 99% purity), potassium peroxodisulfate (K2S2O8 99% purity), and 1,3-diisopropenylbenzene (DIPB 97% purity) were obtained from Sigma-Aldrich. DIPB was distilled under reduced pressure and stored at −18 °C under an argon atmosphere. For all polymerizations deionised water was used after filtration through an ultra filtration unit of 0.02 μm pore size (3×) and degassing.For the purification of the synthesised particles the following solvents were used. Methanol was purchased in technical grade from Riedel-de Haën, and tetrahydrofurane (THF) was purchased in analytical grade from Acros. Both were distilled under reduced pressure. Benzene was obtained in analytical grade from Carl Roth GmbH and used as received, as was cyclohexane (CH), which was purchased from Merck KGaA, also in analytical grade.
For the characterisation of the synthesised particles 2-ethylnaphthalene (2-EN) (99% purity) was purchased from Sigma-Aldrich and distilled under reduced pressure. Toluene (>99.5% purity) was purchased from Sigma-Aldrich and used as received. Before application these two solvents were filtered thrice using PTFE filters from Carl Roth GmbH with a pore size of 0.02 μm.
The emulsion polymerisation was carried out under an argon atmosphere in a three-necked round-bottomed flask fitted with a teflon-paddle stirrer, a gas inlet and a bubble counter. SDS and NaHCO3 were weighed in and dissolved in water (350 g). The solution was then heated to the reaction temperature of 72 °C while stirring at 350 rpm. Meanwhile a mixture of styrene and DIPB of desired composition was prepared. In the case of semibatch emulsion polymerisation 1 ml of this mixture was added to the flask. The reaction mixture was stirred for 10 min, and then K2S2O8, dissolved in preheated water (30 g, 72 °C), was added. After about 5–10 min, a blue colour appeared, indicating the start of the polymerisation. Now the remaining mixture of the monomers was added within 3 h (or 5.5 h respectively). In the case of batch emulsion polymerisation the styrene–DIPB mixture was added at once before adding K2S2O8 after 10 min.
After an additional 20 min the reaction mixture turned turbid, independent of the reaction conditions. The mixture was allowed to react for 24 h at 72 °C, while a slow stream of argon was maintained. After cooling down to room temperature, the dispersion was filtered through a nylon filter (pore size 38 ± 3 μm) to remove any coagulum formed during the polymerization. 20 ml of the dispersion were retained for later analysis, while the remaining material was purified. Typically yields of 95% were obtained.
First of all the remaining SDS and ionic components were removed and the particles were then separated from the aqueous phase. For this purpose the dispersion was poured into four 1 l one-necked flasks and frozen at −196 °C to break the dispersion while slowly rotating the flask. The contents were allowed to warm up to room temperature. Then they were centrifuged for 1.5 h at 5500 rpm (Multifuge, 3 S-R, Heraeus). The clear supernatants were discarded and the solids were redispersed in methanol (∼400 ml each). The dispersions were centrifuged (1.5 h, 5500 rpm) again and the slightly turbid supernatants were removed. This step was repeated once. The solid was then dried at 50 °C under reduced pressure (750 mbar). This solid was redispersed in THF (10 ml g−1 powder) by stirring at room temperature for at least one week. The dispersion was precipitated in methanol (10 × the amount of THF), filtered through filter paper and redispersed in THF (∼100 ml). The precipitation procedure was repeated once. After filtration the solid was dried at 50 °C for 12 h at 60 mbar. After weighing the solid it was dispersed in benzene (10 ml g−1 powder) by stirring. The dispersion was once per week ultrasonicated to remove aggregates (4 × 15 min, Bandelin, Sonorex Super RK255H). After three weeks the dispersion was filtered and freeze-dried.
To remove polymer chains that were not incorporated into the microgel-network the dry powder was dispersed in CH‡ (∼150 ml) by stirring at 60 °C for several days and then centrifuged for 1.5 h at 40 °C. The supernatant was removed and the steps were repeated twice.
As the particles show a tendency to aggregate during the centrifugation step in CH the solid residue was redispersed in THF (∼90 ml) and once more precipitated in methanol (1.2 l). After filtration, drying and weighing, the powder was redispersed in benzene and treated as described before. After the final freeze-drying the particles were obtained as a white powder.
To determine the radii and form factors of the unswollen particles in water using SLS and to follow the reaction kinetics via dynamic light scattering (DLS) a small drop of the raw dispersion in water was diluted until only a slight turbidity was visible. To characterise the radii and form factors in the swollen state 1 mg of the dry powder was dispersed in 3 ml of toluene.
All samples in organic solvents were placed in an ultrasonic bath (Bandelin, Sonorex Super RK255H) for 4 × 15 min to dissolve any aggregates formed during the purification steps. They were placed on a rotating wheel for at least 1 week, leaving ample time for the particles to swell to their maximum size. To avoid particle adhesion at the cuvette walls due to solvent dewetting after sample tumbling, the dispersions were quickly spun down by centrifugation (5 min at 1800 rpm). In the case of larger particles (R > 200 nm) the sonication procedure described above did not prove to be efficient enough to dissolve all aggregates. Therefore stock suspensions of those systems were prepared as described above (Φ = 0.56–0.66) and additionally treated for 2 × 15 min in an ultrasonic processor (UP50H – Compact Lab Homogenizer, 50 cycles at 100% amplitude). All further samples of those systems were prepared from the respective stock suspensions by dilution. To ensure that the harsh ultrasonic treatment did not disintegrate the microgel network we prepared dilute suspensions from the stock suspensions prior and after the sonication treatment and performed form factor measurements of both samples. In all cases the form factor prior and after sonication did only differ in the low q-limit where aggregates influence the scattering pattern. The position of the form factor minimum was identical in both cases and therefore an additional swelling due to a potential breaking of chains caused by the ultrasonication could be excluded.
2.2 Instrumentation
To perform static light scattering (SLS) measurements of unswollen and swollen microgel particles, two computer driven SOFICA goniometers (Société Française d'Instruments de Contrôle et d'Analyses, modified by SLS-Systemtechnik) with different light sources were used (JDS Uniphase, He:Ne gas laser, λ = 632.8 nm, 5 mW; Laser Components, diode laser, λ = 405 nm, 1.5 mW). To calculate the normalised scattering intensity the laser intensity was measured with a reference photomultiplier and a toluene reference sample was used to convert the current detected by the photomultiplier to the intensity in reciprocal centimetres. This setup allows measurements over the angular range of 25°–145° in steps of 1°. To remove background scattering the intensity was corrected by subtracting from it the intensity of a measurement of pure solvent.
To perform DLS a 3D cross-correlation setup from LS Instruments was used. The light of a diode laser (Coherent, λ = 532 nm, 100 mW) was scattered into two APDs (Perkin-Elmer, SPCM-AQR-13-FC) mounted on a goniometer able to measure at positions between 15° and 150°. We determined the hydrodynamic radii Rh from second order cumulant fits of the intensity autocorrelation function measured at angles below the first form factor minimum, i.e. in the range 1.5 < qReff < 3.5 (cf. Table S2 of the ESI†).
The measurements using transmission electron microscopy (TEM) were performed on a TEM Leo 912 Omega-microscope by Zeiss. A drop of an aqueous dispersion was spread on a copper TEM grid coated with a carbon film and then dried in a high vacuum. The average radius RTEM and the relative standard deviation 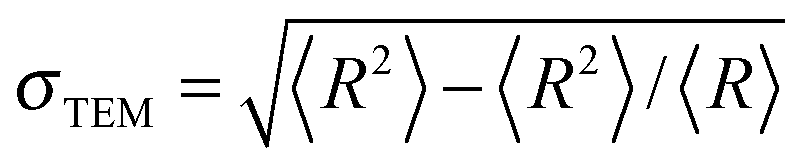 of the particles were determined from the analysis of 100 particles using iTEM software (Fa. Olympus-SIS). Polydispersity was defined as the relative standard deviation.
of the particles were determined from the analysis of 100 particles using iTEM software (Fa. Olympus-SIS). Polydispersity was defined as the relative standard deviation.
All rheological measurements were performed on a stress controlled rheometer (Haake MARS II) in frequency sweep mode in the linear regime covering the frequency range of ω = 0.02–100 rad s−1 with maximum strain at γ = 2%. The samples were tempered to 20.0 ± 0.1 °C for 10 min and measured in a plate–plate geometry (radius 60 mm).
To increase the accessible q-range, P(q) of the swollen microgel particles was measured at 405 nm and 632.8 nm. After subtracting the background scattering the intensities of the two measurements were normalised on top of each other and fitted as I(q) = I(0)·P(q). To reduce multiple scattering effects on the SLS data the samples were further diluted after each measurement and the measurement was repeated. This procedure was continued until the form factors normalized to unit concentration and the derived radii became independent of the sample concentrations. This condition was typically achieved for a volume fraction of Phi = 0.005. The form factor of the swollen particles was either described by the well-known Rayleigh–Debye–Gans result for homogeneous spheres:
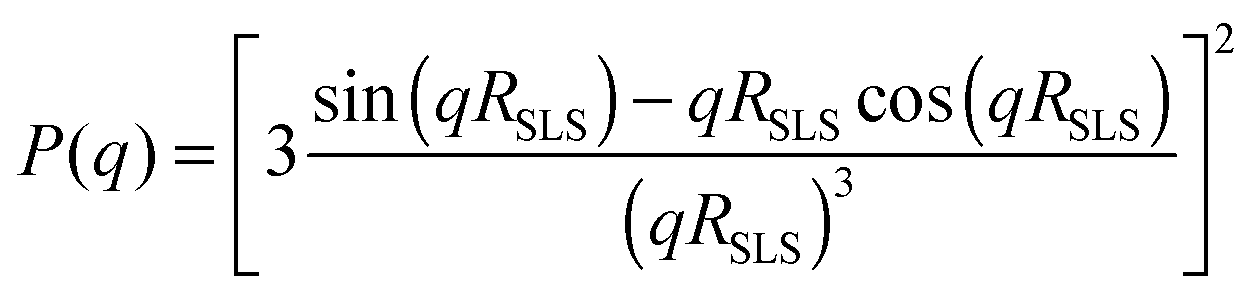 | (1) |
 | (2) |
While the first model assumes a constant radial segment density within the particles, the latter describes a dense core of constant density surrounded by a corona of decreasing density. The radius of a fuzzy sphere is defined as Rfuzzy = RSLS + 2σsurf and corresponds to the distance from the centre of the particle to a position where the density has decayed to zero. All form factors could be fitted with an accuracy of RSLS = ±3 nm, σsurf = ±5 nm, Rfuzzy = ±10 nm, and σR = ±0.02.
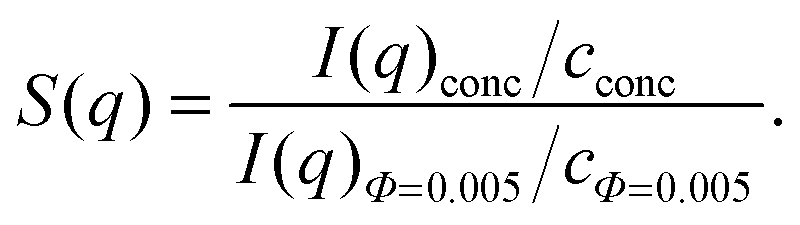 | (3) |
The position of qmReff was calculated using the Verlet–Weis-corrected analytical Percus Yevick integral equation.48,49 Polydispersity was taken into account in accordance with ref. 50 and 51.
2.3 Phase behaviour
The phase behaviour of the particles in 2-EN was characterised as described previously21,25,52 and has been used to determine the volume swelling ratio QHS = (Rswollen/Runswollen)3.Knowledge of QHS is crucial for the calculation of the dispersion volume fraction Φ according to
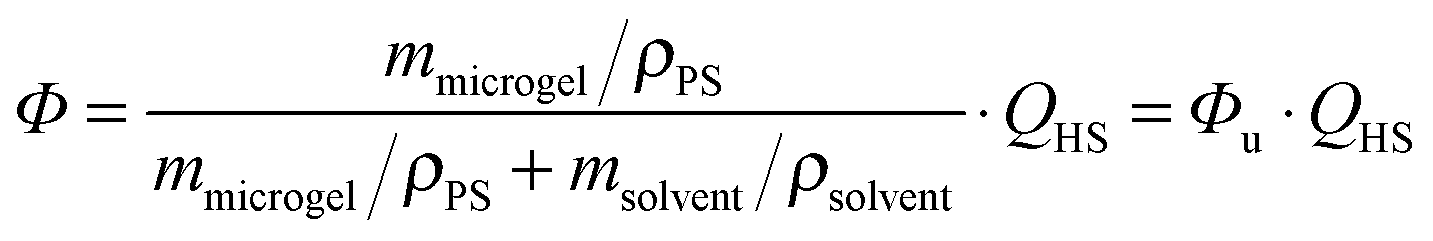 | (4) |
2.4 Determination of the interaction exponent
The softness of particles in a suspension can be related to their elastic plateau modulus (see ref. 43, 53 and references cited there):| GP = G′(ω)|tanδ(ω)→min∝Φm. | (5) |
δ = G′′/G′ with G′ and G′′ being storage and loss modulus, respectively.
If hydrodynamic interactions and structural changes of the particles in the suspension with Φ are neglected and the interaction potential U(r) of the particles is modelled as U(r) ∝ r−n, the interaction exponent n can be calculated as n = 3(m − 1). To obtain the plateau moduli, samples of the particles in 2-EN with volume fractions ranging from Φ = 0.60–0.66 were prepared. An alternative way to determine n uses the observation that the width of the fluid-crystal coexistence region shrinks monotonously with increasing softness, i.e. decreasing n.21,22
3 Results and discussion
3.1 Influence of cross-link density on particle interactions
To investigate the influence of cross-link density on phase behaviour, rheological and structural properties we synthesised particles of approximately 150 nm, 180 nm and 270 nm radius in the swollen state. The systems are referred to as P150-x, P180-x, and P270-x where x indicates the cross-linking ratio 1:x (i.e. one cross-link per x styrene units if 100% cross-link efficiency is assumed). In analogy to previous work25,27,44 the particles investigated in this section were synthesised via semibatch emulsion polymerisation and a feeding duration of 3 h was applied. The influence of the reaction conditions on particle properties will be discussed in Section 3.2. Radii of 150 nm and 180 nm have been commonly used in this group25,27,44 and can be easily accessed by emulsion polymerisation. We also prepared larger particles to get access to a larger range in the qR of the particle form factor P(q). This allowed a more detailed study of the inner structure of the particles. Here 270 nm currently marks the upper limit which can be accessed for all cross-link densities under study. In addition, this range of particle sizes allows us to check for a systematic variation of the swelling ratio QHS and other system characteristics with particle size – an issue raised in previous work.54 To evaluate the influence of cross-link density we varied x from 10 to 100 including 25, 50 and 75.
As the focus of this section lies on the thermodynamic properties, the volume fraction Φ is the key parameter for this work. Thus, we used the first order freezing point to define our Φ-scale. This is illustrated in Fig. 1 where the phase diagram of the system P270-50 is shown as an example.
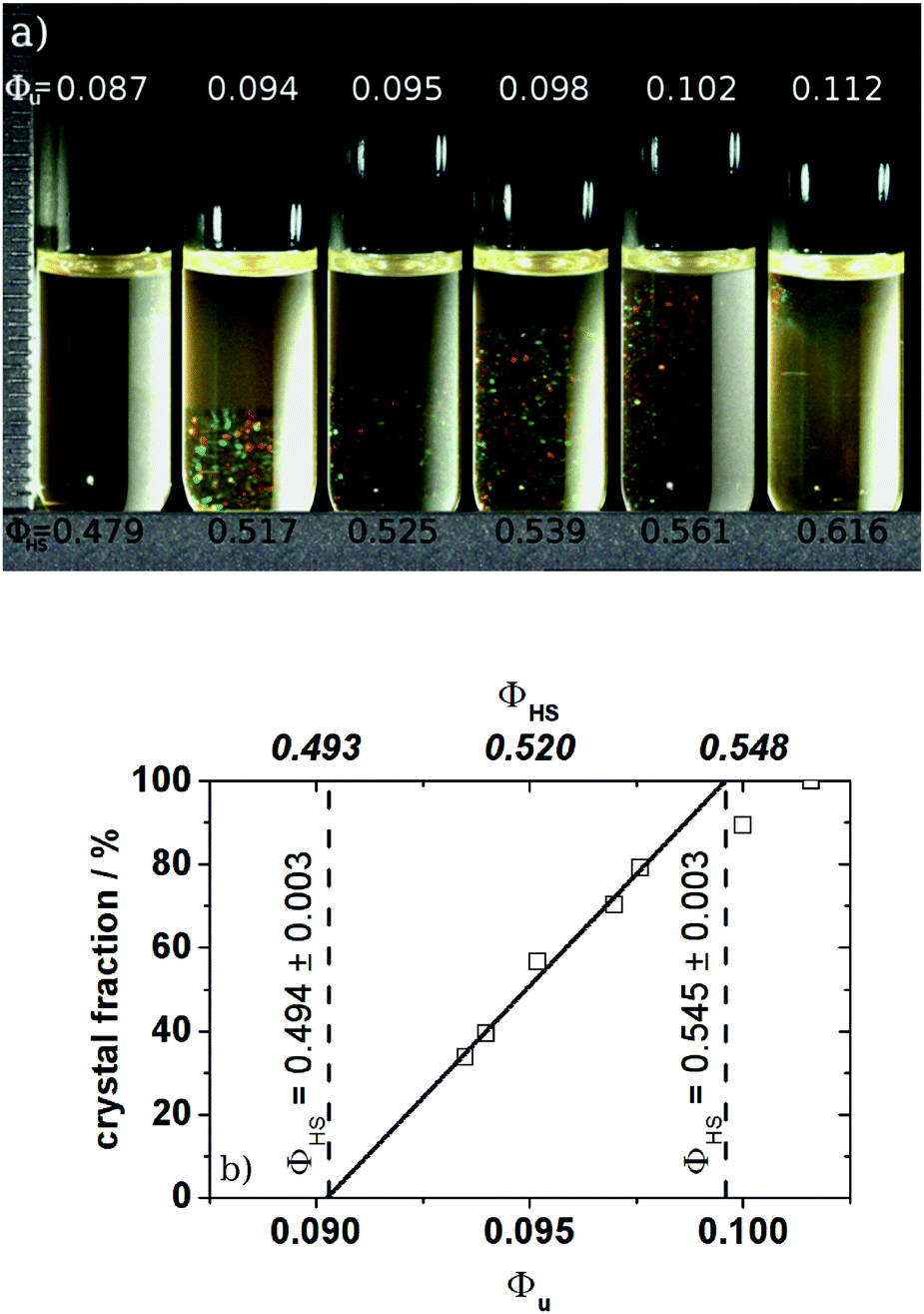 | ||
| Fig. 1 Phase diagram of the 1:50 cross-linked particles P270-50 in 2-EN. (a) Pictures of the different phases; (b) crystal fraction determined from the pictures. The solid line corresponds to the linear extrapolation, defining the coexistence region. The bottom abscissa gives the volume fraction of the unswollen particles, the top one results from mapping Φu at freezing to 0.494 using QHS = 5.5. The dashed lines give the rescaled volume fractions of the freezing and melting points. | ||
All systems exhibit a fluid phase, a coexistence region, a crystalline phase and a glass phase as known from other hard sphere systems. Within the coexistence region the amount of crystals increases with the volume fraction, while the size of the crystallites decreases if the volume fraction is increased beyond Φm ≈ 0.55 towards the glass transition point. From a linear extrapolation of the amount of crystals within the coexistence region as a function of volume fraction the freezing and melting points Φf,u and Φm,u could be obtained. Here Φu denotes the volume fraction of the unswollen particles. The particle swelling ratio QHS can now be determined by mapping Φf,u to the well established value of 0.494 for monodisperse hard spheres.
As shown in Fig. 1 the width of the phase diagram of P270-50 is in good agreement with the one expected for monodisperse hard spheres and the hard sphere value of Φm = 0.545 is obtained. Usually in the case of PNiPAM21 or PMMA microgels22 narrower coexistence regions are found. To our knowledge a similar good agreement between hard spheres and microgels was only found by Eckert et al.55 for PNiPAM microgels exhibiting a swelling ratio of QHS ≈ 17.
They did not quote their cross-link density, but they pointed out that their particles are strongly cross-linked compared to the ones prepared by Senff et al.,21i.e. they used more than 1.4 mol% of the cross-linking agent resulting in a cross-link density of 1:≤144. Unfortunately no further details about the cross-link density were given in ref. 55.
Having a closer look at the other systems studied here, some of them show a slightly lower Φm than 0.545 if Φf is mapped to 0.494. The exact values can be found in Table S3 of the ESI.† Usually this smaller coexistence region is interpreted by means of a softness effect and the values of the interaction parameter n can be determined independently from rheology data by using available correlation plots of the relative width of the coexistence region, i.e. Δρ = (Φm − Φf)/Φf, versus n. For example Paulin et al.22 and later Senff et al.21 could map their narrow coexistence region onto the one of soft spheres. They also showed that the interaction parameters n determined from the phase diagram agrees with the ones measured by rheology. Based on their observations we also tried to determine n from the phase diagram data. However, we did not find a systematic variation of this n with cross-link density or swelling ratio. This is reflected in the variation of Φm with particle size which is reported in Table S3 of the ESI.† For the P150-xx series one finds as expected a systematic increase of the swelling ratio QHS with increasing cross-link density which is accompanied by a concomitant systematic decrease of Φm. This indicates an increasing softness of particle interactions as this is known from computer simulations to lead to a decrease of the width of the fluid-crystal coexistence region.56 However, this correlation is not observed for the P180-xx and the P270-xx series. The reason for this may be the unsystematic variation of size polydispersity within the corresponding particle series. Polydispersity tends to increase the width of the coexistence region57 and, thus, may counteract the narrowing due to particle softness. In contrast, for the P150-xx series the polydispersity seems to increase more systematically with decreasing cross-link density (cf. σR,TEM in Table S2 of the ESI†). Thus, here the polydispersity effect counteracts the effect of softness but the latter dominates – leading to only a moderate decrease of Φm with decreasing cross-link density. Thus, in the case of the here studied microgel systems reliable values of n could only be obtained from rheology data as discussed below.
With respect to the dependence of the swelling ratio on particle size, one observes a somewhat systematic decrease of QHS only for the highest cross-link density: from 2.26 (P150-10) to 2.36 (P180-10) to 3.05 (P270-10) as given in Table S3 of the ESI.† As this variation is almost of the same magnitude as the experimental error in QHS and there is only an unsystematic variation of QHS with particle size for smaller cross-link densities, the particle size seems not to be a significant control parameter for particle softness. As already suggested in the introduction (and demonstrated in the course of this section) we assume that our PS microgels are less soft than the ones studied by Senff et al. or Paulin et al. Thus softness has only a small effect on the phase diagram. In addition, the effect of polydispersity seems to be of similar magnitude to the influence of particle softness and interferes with the determination of the interaction parameter from the phase diagram. As to our knowledge no predictions of the phase behaviour of polydisperse soft spheres exist, we therefore decided to use the simple hard sphere mapping of Φf and probe particle softness via rheological measurements of concentrated microgel suspensions.
If a power law potential U(r) ∝ r−n is assumed, the interaction parameter n can be determined from the plateau-modulus GP as described in Section 2.4 and ref. 43 and 53. In Fig. 2an is plotted as a function of cross-link density 1:x.
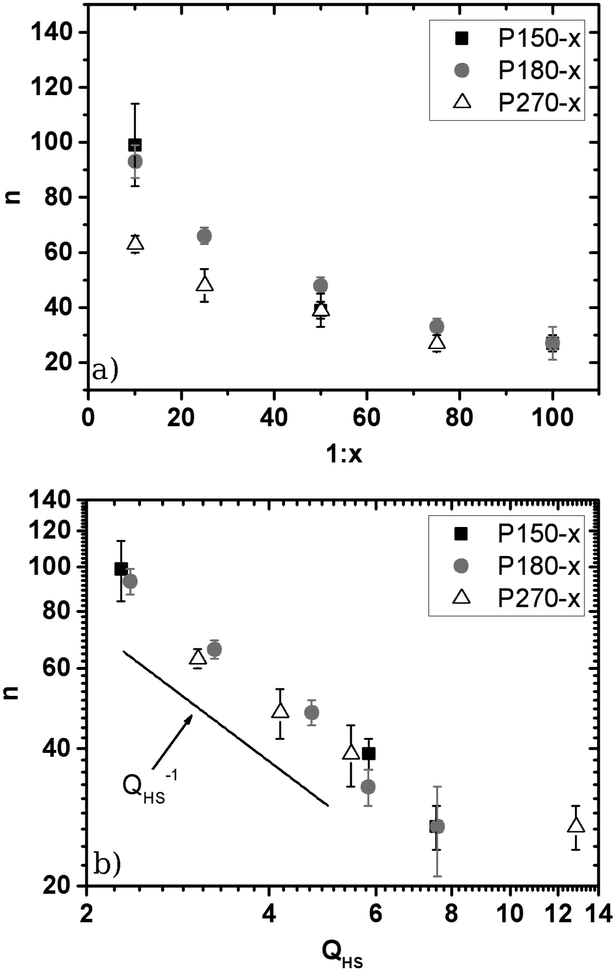 | ||
| Fig. 2 Interaction parameter n derived from rheological measurements under the assumption of an interaction potential U(r) ∝ r−n. (a) Plotted against the cross-link density 1:x; (b) log–log plot against the swelling ratio QHS determined from the phase diagram. The straight line depicts a scaling behaviour of n ∝ QHS−1. | ||
In this representation a decay of n as a function of cross-link density can be observed. The accessible n values vary between n ≈ 100 (1:10), which is not too far from the typical values of hard sphere like PMMA particles,53 and n ≈ 27 (1:100), which is above the upper limit reported for PNiPAM microgels.29
Thus, in general PS microgels exhibit larger n values than PNiPAM microgels and bridge the gap between soft and hard spheres. Stieger et al.29 for example reported n = 22 and 9.5 in the case of 1:34- and 1:141-cross-linked particles (i.e. 5.5 mol% and 1.4 mol% cross-linking agents respectively). The authors attributed the small interaction parameters to the fuzzy corona of their particles. In the case of PNiPAM microgels the amount of cross-linking agent mainly influences the size of the highly cross-linked core and has only a minor influence on the corona. Therefore the cross-link density of PNiPAM-particles provides only a minor contribution to particle softness. However, in our PS microgels a more homogeneous network formation can be assumed leading to an easier control of n via cross-link density. Nevertheless a more detailed look at the size dependence of n and a comparison between different cross-link densities reveals that the cross-link density cannot be the only parameter controlling n as we find some spread of values n for a given cross-link density (cf. Table S3 of the ESI†). For the higher cross-link densities (1:10 and 1:25) there seems to be a systematic decrease of n (increase of softness) upon increasing the particle size at constant cross-link density. Due to the limited database we cannot decide whether there is a clear trend for increased softness upon increasing the particle size, e.g. due to a less efficient cross-linker incorporation, or whether the observed variations are caused by small statistic variations in cross-linking agent incorporation during the semibatch emulsion polymerisation. An interesting result is, however, observed upon plotting the interaction parameter n versus the swelling ratio in Fig. 2b. In the log–log representation one finds a linear dependence of n on QHS−1, indicating a power law behaviour. This result implies that the true control parameter for particle softness is not the nominal cross-link density or the particle size but instead the swelling ratio QHS obtained by mapping the observed fluid-crystal coexistence onto the hard sphere phase diagram. This finding has an important practical advantage. QHS is the key parameter for setting the volume fraction scale for mapping the PS microgels onto the hard sphere system. So far QHS has to be determined from analysis of the fluid-crystal coexistence. Due to the necessity to follow the sedimenting fluid-crystal phase boundary until the long-time linear dependence is clearly expressed and the slow sedimentation of the nearly buoyancy matched microgels, this procedure typically needs up to 3–4 months. The clear n ∝ QHS−1 relation now allows us to determine QHSvia n by the much faster rheology experiments. This relation is valid for all systems except the one with QHS ≈ 13 corresponding to P270-75. As will be shown later the deviation of P270-75 from the simple QHS−1 scaling is most likely caused by differences in the inner structure of this system as compared to the others, resulting in a different swelling to softness relation.
Looking at the absolute values of n it has to be pointed out that all systems studied in this work fulfil the criterion n ≥ 18. For inverse power potentials U(r) ∝ r−n Lange et al.58 demonstrated that a hard sphere mapping can be performed by rescaling the particle number density at freezing to the theoretical values for hard spheres as long as n ≥ 18. By computer simulation they could show that after this mapping, pressure, diffusion coefficients, viscosity and short-range order agree with the known hard sphere behaviour. To cross-check the hard sphere mapping and to examine this prediction experimentally, we probed the structure of our particles in the fluid phase by means of SLS and structure factor analysis and compared it with predictions for hard spheres.50,51 We determined S(q) in the range of 0.20 ≤ Φ ≤ 0.47 and compared the experimental S(q) with calculated values for polydisperse hard spheres. To calculate S(q) we set the volume fraction to Φ = QHSΦu and varied σR in the range determined by TEM and SLS of the unswollen particles in water to properly adjust the height of the structure factor maximum. The absolute values of σR determined by TEM and SLS can be found in the ESI† (Table S2). The effective hard sphere radius Reff was chosen to get a good description of the measured S(q) at all volume fractions. As shown in Fig. 3a the measured structure factor agrees well with the calculated one up to the first order freezing point. The system P150-50 depicted in Fig. 3a showed the best agreement between the measured and calculated S(q).
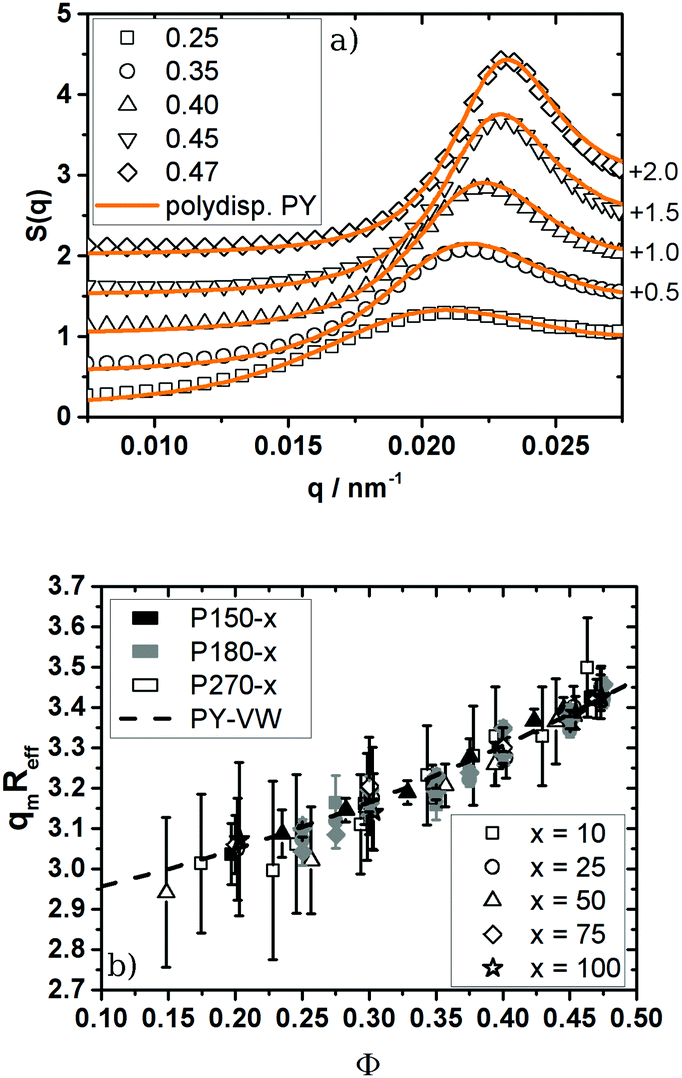 | ||
| Fig. 3 (a) Structure factor of the 1:50 cross-linked particles P150-50. The rescaled volume fractions Φ are given in the key of the figure. Open symbols correspond to experimental S(q), solid lines are calculated S(q) using the Verlet–Weis-corrected Percus Yevick integral equation of polydisperse spheres (σR = 0.08, Reff = 147 nm). For easier comparison S(q) was shifted along the ordinate by adding the value quoted on the right side of the figure. For clarity reasons only every fourth experimental data point is shown. (b) Mapping of the position of the S(q)-maximum in units of qmReff onto the values of monodisperse hard spheres (dashed line). The mapping on polydisperse hard spheres can be found in the ESI† (online). The colour depicts the particle size while the symbols represent the cross-link density 1:x. The experimental errors of qm determination are caused by multiple scattering effects which can result in an artificial maximum in S(q) overlapping with the maximum S(qm). | ||
All other systems showed multiple scattering effects, influencing the shape of S(q) and the absolute peak height. Nevertheless we were still able to determine the maximum of S(q), as in this region single scattering is dominating the scattering pattern.
As shown in Fig. 3b the position of the structure factor maximum qm can be superimposed onto the one of the monodisperse hard spheres. It is also possible to take polydispersity into account but the influence of polydispersity on qm is smaller than the experimental uncertainties of the qm determination. Plots like Fig. 3b for polydisperse hard spheres are shown in the ESI† (Fig. 2).
Our S(q) calculations were performed under the assumption that P(q) is independent of Φ, i.e. in contrast to PNiPAM microgels, no deswelling, overlapping or deformation of the particles could be observed when the volume fraction was increased, even when they are only weekly cross-linked. In addition, the Φ-independence of P(q) in PS microgel systems was recently demonstrated by Burger et al.59 Their SANS-studies of 1:50-cross-linked particles verified that P(q) is constant, even in the glassy state.
In the case of the softer PNiPAM-microgels a similar mapping could only be achieved up to Φ ≈ 0.35,29 while for larger volume fractions Φ and Reff had to be replaced by concentration dependent values to allow the hard sphere mapping. Again, as recently shown by Nägele et al.,60 only the particles used by Eckert et al.55 allowed a similar mapping of S(q) up to Φf in the case of PNiPAM-microgels. Nevertheless there is still one difference between the findings of Eckert et al. and our results. As their particles showed a hydrodynamic radius larger than Reff in our case Rh ≈ Reff. Furthermore Reff agrees well with the swollen radii calculated by multiplying the unswollen radii in water (i.e. the average of RTEM and RMie) by QHS1/3 (i.e. the swelling ratio in one dimension). The detailed comparison of all radii measured for our systems lies beyond the scope of this paper but the interested reader may find further information in Section 2 of the ESI.†
Thus, in general PS microgels show a much smaller degree of softness and a more hard sphere like structure than most microgel systems known from the literature. This is most likely caused by differences in the inner structure between our particles and e.g. PNiPAM microgels. As mentioned before, typical microgels show a distinct density gradient with a densely cross-linked inner core and a fuzzy corona of decaying polymer density. This difference in the inner structure results from differences between the reactivity of the monomer and the cross-linking agent, leading to a higher incorporation of the highly reactive cross-linking agent in the core of the particles. As demonstrated by Antonietti et al.35 styrene and the cross-linking agent DIPB used in this work are of similar reactivity leading to a more homogeneous cross-link density. To assess the validity of this statement for our particles the form factors P(q) of 1:50 and 1:75 PS microgels in the good solvent toluene are reported and analysed in Fig. 4 as limiting cases. The full set of P(q) measurements and corresponding fits can be found in the ESI† (Fig. 2).
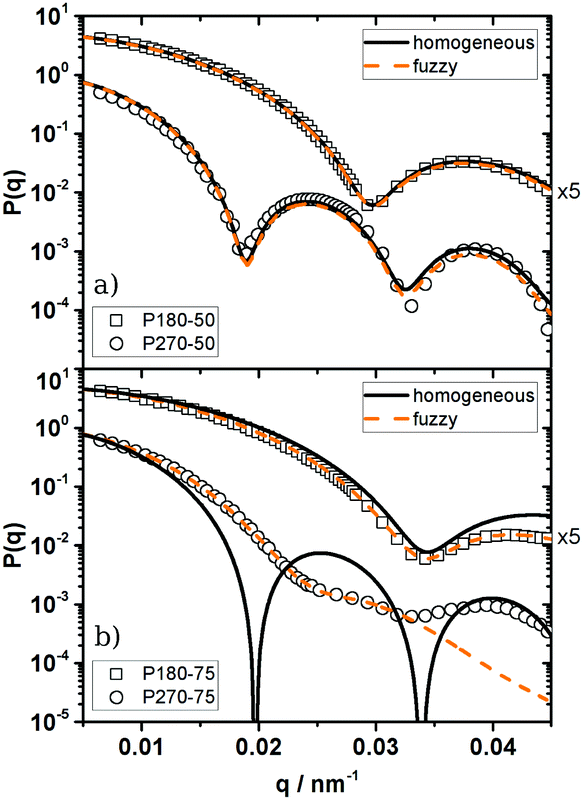 | ||
| Fig. 4 Form factors of particles of different cross-link densities in the good solvent toluene. Symbols: experimental P(q), solid lines: homogeneous sphere model, dashed lines: fuzzy sphere model. For easier comparison P(q) is multiplied by the factors quoted on the right side of the figure. (a) 1:50 cross-linked particles (homogeneous sphere: P180-50: RSLS = 150 nm, σR = 0.06; P270-50: RSLS = 237 nm, σR = 0.04; fuzzy sphere: P180-50: R = 151 nm, σsurf = 7 nm, Rfuzzy = Reff = 165 nm, σR = 0.06; P270-50: R = 237 nm, σsurf = 14 nm, Rfuzzy = Reff = 265 nm, σR = 0.04). (b) 1:75-cross-linked particles (homogeneous sphere: P180-75: RSLS = 130 nm, σR = 0.08; P270-75: RSLS = 228 nm, monodisperse; fuzzy sphere: P180-75: R = 131 nm, σsurf = 20 nm, Rfuzzy = 171 nm, σR = 0.07; P270-75: R = 172 nm, σsurf = 40 nm, Rfuzzy = 252 nm, σR = 0.16). | ||
The 1:50-cross-linked particles can be well described as polydisperse homogeneous spheres (HSs). As depicted in Fig. 4a only small deviations around the form factor minimum can be observed, which are caused by experimental uncertainties of the background correction. It has to be noted that all systems of higher cross-link density are also well described as HSs but the radii determined from the HS fits are systematically smaller than the effective radii determined from the structure factor measurements. Here the discrepancy between Reff and R increases with decreasing cross-link density. For the PNiPAM-particles studied by Eckert et al.55 a fit with the fuzzy sphere model (eqn (2)) was required to achieve a good description of P(q) and a good agreement between Rfuzzy and the effective hard sphere radius Reff was found. Thus the effective hard sphere radius is defined by the outer corona of their fuzzy spheres. Assuming a similar behaviour for our PS microgels it is possible to calculate a fuzzy sphere fit to our data while keeping Rfuzzy = Reff = const. Such a fit is shown in Fig. 4a.
The differences between the homogeneous sphere form factor and the corresponding fuzzy sphere descriptions with respect to the experimental data differ only slightly in the high q-regime. Within the experimental error this difference is indistinguishable. This indicates that even if our particles may not be perfect homogeneous spheres the intrinsic fuzziness will be rather small.
Only if the cross-link density is further decreased, deviations from the HS model can be observed at larger q-values as shown in Fig. 4b. The experimental form factor decays faster than the one predicted by the HS model. This decay is usually attributed to a radial decrease in density. In fact the fuzzy sphere model provides a good description of this decay, at least in the case of the smaller particles.
Due to the similar reactivity of styrene and DIPB the fuzziness of the particles is much smaller than that usually observed for similarly cross-linked PNiPAM-microgels. There the particle density usually decreases over a distance of approximately 2/3 of the particle size.29,34,55 As the cross-linking of the outer corona of the particle stiffens the network a more hard sphere like behaviour can be expected for the PS microgels. For this reason softness plays a minor role in PS microgels leading to larger n values. This allows the mapping of their structure and phase diagram onto the one of the hard spheres, as demonstrated above.
In the case of the larger 1:75-cross-linked particles (P270-75) no form factor minimum can be observed and P(q) exhibits a shoulder at q ≈ 0.027 nm−1 instead of a distinct maximum. Furthermore a maximum at q ≈ 0.04 nm−1 can be noticed. The fuzzy sphere model is only able to describe the experimental P(q) up to q ≈ 0.027 nm−1 and misses the maximum at q ≈ 0.04 nm−1. Moreover the fit provides an unreasonable high polydispersity of σR = 0.16.
Such high polydispersities should prohibit crystallisation but these particles actually crystallise and therefore have to exhibit a much lower polydispersity. Based on the poor description of the experimental P(q) and the model calculation it is obvious that the assumption of a dense core and a fuzzy corona does not hold for P270-75 and the underlying inner architecture must be more complex.
As pointed out by Clara-Rahola et al.,61 highly swollen PNiPAM microgels are better described as star polymers or dendrimers than as fuzzy spheres. Furthermore Willner et al.62 could show that the form factor of large stars show features of the spherical superstructure of the stars. In their particular system of star-like polymers they found the signature of the theoretical second maximum of P(q) of the surrounding homogeneous sphere in the form factor of a 128 arm star. This structural interpretation can be transferred to P270-75. As shown in Fig. 4b the position of the shoulder at q ≈ 0.027 nm−1 as well as the maximum at q ≈ 0.04 nm−1 are in agreement with the first and second maximum of a homogeneous sphere of RSLS = 228 nm. Thus we conclude that these particles are better described by an open, eventually star-like or dendrimer-like, structure than by a fuzzy sphere. This assumption is supported by the large swelling ratio determined from the phase diagram of these particles. Going back to Fig. 2b this also explains why P270-75 does not satisfy the QHS−1 scaling. The change in the inner structure leads to a change in the relation between softness and swelling behaviour.
3.2 Influence of reaction conditions on the inner structure
To understand the origin of the structural difference between P270-75 and the other systems one has to have a closer look at the reaction conditions chosen to prepare our particles. We have to point out that in some cases (especially in the case of larger particles) particle size was adjusted by increasing the amount of styrene/DIPB added, while in other cases the SDS content was used to control particle size. This was necessary, as the variation of the amount of SDS used in the syntheses alone did not provide particles of low polydispersity for all targeted sizes. In this context one has to keep in mind that the increased amount of the styrene/DIPB mixture changes the initiator to monomer ratio which may also influence polymerisation kinetics. To evaluate whether the open structure of P270-75 is solely obtained by the targeted cross-link density of 1:75 or whether the applied reaction conditions play a role, we prepared a series of particles similar to P270-75. As reaction conditions we used batch emulsion polymerisation, our standard semibatch emulsion polymerisation based on a feeding duration of 3 h and a slower semibatch emulsion polymerisation where the monomer–cross-linking agent mixture was added within 5.5 h. To be in better agreement with the original recipe24 18 g of styrene were used in these syntheses. Each synthesis was carried out twice. Once to follow the reaction kinetics and once more to study the inner structure of the particles in the swollen and the unswollen state. In this way we ensured that the sample extraction to follow particle growth did not interfere with the formation of the inner structure studied later on. In Table 1 the details of the synthesis and the characterisation of the inner structure are summarised. For easier comparison the data of P270-75 (feed 3 h, 20 g styrene) were also added to Table 1.
| Reaction conditions | Batch | Feed 3 h | Feed 5.5 h | Feed 3 h |
|---|---|---|---|---|
| Cross-linking density | 1:75 |
1:75 |
1:75 |
1:75 |
| Styrene/g (mmol) | 18.00 | 18.00 | 18.00 | 20.00 |
| (172.9) | (172.9) | (172.9) | (192.1) | |
| DIPB/g (mmol) | 0.1824 | 0.1824 | 0.1826 | 0.2058 |
| (1.153) | (1.153) | (1.154) | (1.300) | |
| K2S2O8/g (mmol) | 0.1304 | 0.1302 | 0.1316 | 0.1306 |
| (0.4824) | (0.4817) | (0.4869) | (0.4832) | |
| SDS/g (mmol) | 0.1997 | 0.2011 | 0.2000 | 0.2116 |
| (0.6924) | (0.6973) | (0.6935) | (0.7337) | |
| NaHCO3/g (mmol) | 0.0952 | 0.0907 | 0.0901 | 0.0899 |
| (1.13) | (1.08) | (1.07) | (1.07) | |
| Water/g | 380 | 380 | 380 | 380 |
| R TEM/nm | 106 | 111 | 115 | 104 |
| R Mie/nm | 107 | 111 | 109 | 104 |
| σ R,TEM | 0.04 | 0.06 | 0.06 | 0.05 |
| σ R,Mie | 0.05 | 0.06 | 0.06 | 0.08 |
| R fuzzy(σsurf)/nm | 196 (6) | 196 (14) | 203 (24) | 252 (40) |
As shown in Fig. 5 the feeding of the monomer–cross-linker mixture results in the expected slower particle growth as compared to a batch emulsion polymerization. While in batch emulsion polymerisation the finite size of the particles is reached after ∼200 min, the feeding of the mixture over a period of 3 h increases this time up to ∼270 min.
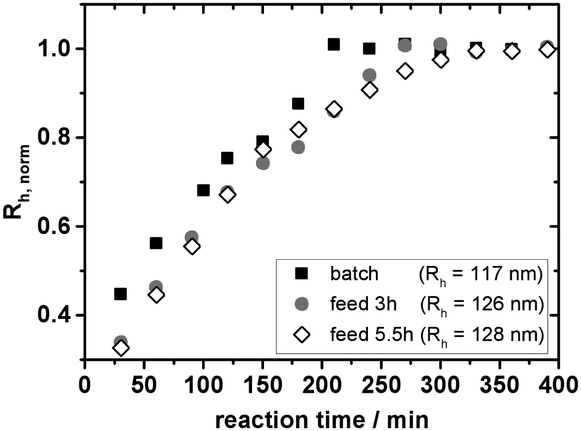 | ||
| Fig. 5 Hydrodynamic radii of three different syntheses of 1:75 cross-linked PS microgels either via batch emulsion polymerisation or semibatch emulsion polymerisation. In the latter case the monomer–cross-linking agent mixture was added during the first 3 h or 5.5 h of the synthesis. The radii are normalised by the value derived from a measurement of a sample taken 24 h after quenching the reaction. Rh quoted in the key of the figure corresponds to this value. | ||
If the feeding rate is decreased even further, the particles require even more time to reach their final size (∼330 min). Fig. 5 shows similar initial slopes of the two semibatch emulsion polymerisations up to approximately 200–250 min. Only after that period a slowing down of the reaction in the case of the slower feeding rate can be observed.
Looking at the absolute values of Rh a small increase in size with decreasing feeding rate can be noticed but these variations are within the accuracy of a repeat synthesis. As our feeding rates are rather high we do not observe the typical decrease in particle size known from semibatch polymerisations under starved feed conditions.63
The form factors of swollen particles prepared in the manner described above are given in Fig. 6, while their radii and polydispersity in the unswollen state are found in Table 1. The radii determined by TEM- and Mie-analysis are in good agreement. Again the variation of the radii between the different batches lies within the variations of a repeated synthesis. The polydispersities determined by TEM and SLS differ up to 3% providing a reasonable match within the experimental uncertainties of these two methods.
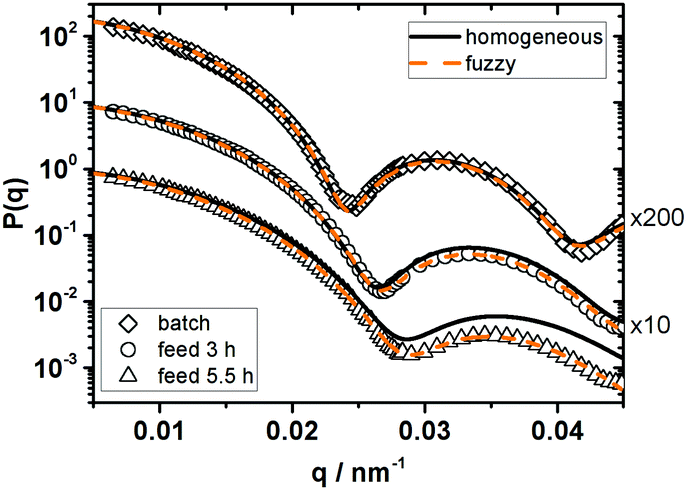 | ||
| Fig. 6 Comparison of the form factors of the three different batches of 1:75 cross-linked particles in the good solvent toluene. For easier comparison P(q) was multiplied by the factors quoted on the right side of the figure. The reaction conditions and feeding durations are given in the key of the figure. Solid lines correspond to fits according to the homogeneous sphere model (diamonds: RSLS = 185 nm, σR = 0.06; circles: RSLS = 169 nm, σR = 0.07; triangles: RSLS = 156 nm, σR = 0.09). Dashed lines correspond to fits according to the fuzzy sphere model (diamonds: R = 184 nm, σsurf = 6 nm, Rfuzzy = 196 nm, σR = 0.05; circles: R = 168 nm, σsurf = 14 nm, Rfuzzy = 196 nm, σR = 0.07; triangles: R = 155 nm, σsurf = 24 nm, Rfuzzy = 203 nm, σR = 0.09). | ||
While the characteristics of the four particle systems are similar in the unswollen state, the situation changes when they are purified and dispersed in the good solvent toluene. As shown in Fig. 6 the form factors of the three batches show systematic differences depending on the reaction conditions but none of the systems show a similar form factor like P270-75.
Changing from batch synthesis to a feed time of 3 h and further on to a feed time of 5.5 h the position of the minimum in P(q) shifts to higher q-values and fitting the HS model to the experimental P(q) leads to increasing deviations between experiment and model description. If the fuzzy sphere model is applied to fit the experimental data, the determined overall size of these three systems is identical within the experimental uncertainties (Rfuzzy = 200 nm), while the fuzziness σsurf increases with decreasing feeding rate. The three systems are slightly smaller than P270-75 (Rfuzzy ≈ 252 nm) indicating a less pronounced swelling.
In the case of the particles prepared via batch emulsion polymerisation it is difficult to distinguish between the homogeneous sphere and the fuzzy sphere fit but the size determined from both models already differs by 11 nm. As discussed in Section 3.1 this indicates that our light scattering experiments are not able to reliably resolve such a small degree of fuzziness.
The increase in fuzziness may either be attributed to a reduced incorporation of the cross-linking agent in the particles in the outer corona or the cross-linking reaction of the already incorporated DIPB is reduced, if the monomer–cross-linker mixture is fed to the reaction. At this point we can only speculate about the detailed reasons for this behaviour.
The increasing fuzziness due to a gradient in the DIPB incorporation is actually counterintuitive as in emulsion copolymerisations a change from batch to semibatch conditions in general increases the homogeneous incorporation of both comonomers.63,64 For example, in the case of PNiPAM-microgels, changing from batch to semibatch emulsion polymerisation reduces the fuzziness of the particles as the depletion of the more reactive cross-linker is lowered by continuously feeding the monomer–cross-linker-mixture to the reaction.65
The second effect, i.e. an increase in fuzziness due to a lowered cross-linking efficiency of the already incroporated DIPB, may also be observed, if the second double bond of DIPB does not react immediately but requires an additional initiation and propagation step. In this case newly formed radicals have to diffuse into the particles to initiate the cross-linking step and even a kind of post-cross-linking after the particles reached their final size may take place. If particle growth is slowed down due to the feeding of the monomer–cross-linker mixture the amount of initiator available to start the post-cross-linking will be reduced. This would also lead to a reduced cross-linking efficiency.
If the lack of post-cross-linking is the key to the fuzziness of the semibatch particles one has to have a more detailed look at the initiator which initiates the polymerisation.
The amount of initiator available at a given particle size differs if the growth rate is changed. Based on the decomposition rate of the initiator suggested by Chern et al.66 (1.1 × 10−4 s−1) it can be estimated that in the case of the batch emulsion polymerisation ∼73% of the initiator are consumed when the particles reach their final size. In the case of the semibatch emulsion polymerisation this value increases to ∼81% and ∼89%, respectively. Thus there is less initiator available when the particles reach their final size in the case of the semibatch emulsion polymerisation compared to the faster batch emulsion polymerisation, concomitantly leading to less radicals for post-cross-linking.
Comparing the form factor of P270-75 with the ones of the three systems shown here also indicates that the initiator in fact plays a role in cross-linking efficiency. The open inner structure and large swelling ratio of P270-75 indicates a lower cross-link density than the three systems prepared from 18 g of styrene, while its feeding rate was similar to the system where the monomer–cross-linker mixture was added within 3 h. Thus the feeding rate alone cannot cause the structural differences. On the other hand the number ratio between the initiator and the comonomers (i.e. styrene plus DIPB) is 0.0025 in the case of P270-75 in comparison to 0.0028 in the case of the three 18 g batches. An additional hint to the importance of a reduced initial initiator to monomer ratio for the final inner structure can be taken from the data of the 1:10 cross-linked systems. P270-10 (initiator to comonomer ratio of 0.0020) shows a larger swelling ratio (QHS = 3.05) than the two other 1:10-cross-linked systems (0.0028, QHS = 2.28 and QHS = 2.36).
Of course so far this hypothesis of an influence of the initiator to comonomer ratio is only indicated by indirect evidence based on the relation between the inner structure of our particles and the reaction conditions. To prove our assumptions direct measurements of the cross-link efficiency e.g. by NMR-spectroscopy of PS microgels with a labelled cross-linking agent would provide additional insights, e.g. information about the number of unreacted double-bonds depending on reaction conditions and the amounts of monomer and initiator used during the reaction. In addition, resolving the inner structure in more detail using small angle neutron scattering and analyzing the obtained form factors using more involved model functions which include blob scattering67 or contributions from network fluctuations68 would be helpful as well. As this work mainly focusses on the influence of cross-linking density and its influence on the thermodynamic behaviour of PS microgels such investigations will be addressed in future work.
4 Conclusions
The thermodynamic properties and inner structure of PS microgels of different size and cross-link density were studied. Their suitability as hard sphere model systems was investigated by probing phase diagrams, rheological properties, structure factors and form factors. Furthermore we demonstrated the influence of the reaction conditions on the inner structure of the particles.Analysing the volume fraction dependence of the plateau modulus in the solid state in terms of a power law we find interaction exponents n ranging from 99 for 1:10 cross-linked particles down to 27 for a cross-link density of 1:100. Thus, the particle softness of PS microgels is located between soft PNiPAM-microgels and hard PMMA particles. We observe a simple linear dependence of n on the inverse of the volume swelling ratio QHS, the latter being determined by mapping the experimental phase diagram of the particles onto that of hard spheres. This implies that QHS is the main control parameter for particle softness, irrespective of nominal cross-link density or particle size. Furthermore, the measured structure factors S(q) (up to the first order freezing point) could be mapped onto those of polydisperse hard spheres. Therefore we adjusted the volume fraction scale using QHS. This is consistent with computer simulation results by Lange et al., which indicate that such a mapping works as long as n ≥ 18. Form factor analysis revealed that the similar reactivities of styrene and the employed cross-linking agent 1,3-diisopropenylbenzene result in homogeneously cross-linked particles with a low degree of fuzziness. In general particles with a higher cross-link density than 1:75 could be described as homogeneous spheres. We could also demonstrate that it is possible to tune the fuzziness of the particles by varying the reaction conditions. The most homogeneously cross-linked particles were provided by batch emulsion polymerization, while in a semi-batch process, i.e. feeding the monomer–cross-linker into the reaction vessel, a small fuzzy corona can be introduced. In the latter case the thickness of the corona increases upon decreasing the feed rate. Nevertheless, even for the smallest cross-linking density the fuzziness is much smaller than that usually observed for PNiPAM-microgels. We identified this behaviour as the reason for the nearly hard sphere like behaviour of our systems. Thus, PS microgels are a versatile tool to model hard sphere systems. A reduction of the cross-link density down to 1:100 while maintaining the hard sphere character will allow access to large sphere sizes useful for crystallization and optical microscopy studies. Here the simultaneous buoyancy and refractive index match can be achieved without application of special solvent mixtures which often introduce unwanted side effects.
The small deviations from true hard spheres also indicate that the differences between microgels of varying cross-link densities in colloid–polymer mixtures are not related to the intrinsic softness of our particles. Thus these deviations are most likely caused by interactions between the free polymer and the microgel network.
Acknowledgements
It is a pleasure to thank H. Moschallski, C. Stilke, O. Thorwarth and C. Wagner for particle synthesis and characterization measurements. Additionally we would like to thank Ch. Friedrich, K. Hasis and C. Weis for their support of the rheological measurements as well as R. Thomann who performed the TEM-measurements. We gratefully acknowledge financial support from the German Research Foundation (DFG) through the IRTG ‘Soft matter science’ (GRK 1642) and through DFG project Ba1619/2-2.Notes and references
- P. N. Pusey and W. van Megen, Phys. Rev. Lett., 1987, 59, 2083–2086 Search PubMed.
- L. Berthier and G. Biroli, Rev. Mod. Phys., 2011, 83, 587–645 Search PubMed.
- G. L. Hunter and E. R. Weeks, Rep. Prog. Phys., 2012, 75, 066501 Search PubMed.
- C. P. Royall and S. R. Williams, Phys. Rep., 2015, 560, 1–75 Search PubMed.
- P. N. Pusey and W. van Megen, Nature, 1986, 320, 340–342 Search PubMed.
- W. Hornfeck, D. Menke, M. Forthaus, S. Subatzus, M. Franke, H.-J. Schöpe, T. Palberg, J. Perlich and D. Herlach, J. Chem. Phys., 2014, 141, 214906 Search PubMed.
- R. Beyer, M. Franke, H. J. Schöpe, E. Bartsch and T. Palberg, J. Chem. Phys., 2015, 143, 64903 Search PubMed.
- W. van Megen, S. M. Underwood, J. Müller, T. C. Mortensen, S. I. Henderson, J. L. Harland and P. Francis, Prog. Theor. Phys. Suppl., 1997, 126, 171–180 Search PubMed.
- T. Palberg, J. Phys.: Condens. Matter, 2014, 26, 333101 Search PubMed.
- C. P. Royall, W. C. K. Poon and E. R. Weeks, Soft Matter, 2013, 9, 17–27 Search PubMed.
- P. N. Pusey, in Colloidal Suspensions, ed. J.-P. Hansen, D. Levesque and J. Zinn-Justin, Liquids, Freezing and the Glass Transition, Elsevier, Amsterdam, 1991, pp. 765–941 Search PubMed.
- P. Zakharov and F. Scheffold, Light Scatt. Rev., 2010, 4, 433–467 Search PubMed.
- R. Xu, Particuology, 2015, 18, 11–21 Search PubMed.
- V. Prasad, D. Semwogerere and E. R. Weeks, J. Phys.: Condens. Matter, 2007, 19, 113102 Search PubMed.
- M. Jenkins and S. Egelhaaf, Adv. Colloid Interface Sci., 2008, 136, 65–92 Search PubMed.
- R. Besseling, L. Isa, E. R. Weeks and W. C. Poon, Adv. Colloid Interface Sci., 2009, 146, 1–17 Search PubMed.
- P. J. Lu and D. A. Weitz, Annu. Rev. Condens. Matter Phys., 2013, 4, 217–233 Search PubMed.
- A. Yethiraj and A. van Blaaderen, Nature, 2003, 421, 513–517 Search PubMed.
- Microgel Suspensions – Fundamentals and Applications, ed. A. Fernandes-Nieves, H. Wyss, J. Mattson and D. A. Weitz, Wiley-VCH Verlag, Weinheim, 2011 Search PubMed.
- H. Schild, Prog. Polym. Sci., 1992, 17, 163–249 Search PubMed.
- H. Senff and W. Richtering, J. Chem. Phys., 1999, 111, 1705–1711 Search PubMed.
- S. E. Paulin, B. J. Ackerson and M. S. Wolfe, J. Colloid Interface Sci., 1996, 178, 251 Search PubMed.
- M. Schneider, R. Michels, V. Pipich, G. Goerigk, V. Sauer, H.-P. Heim and K. Huber, Macromolecules, 2013, 46, 9091–9103 Search PubMed.
- T. Eckert and E. Bartsch, Phys. Rev. Lett., 2002, 89, 125701 Search PubMed.
- T. Eckert and E. Bartsch, Faraday Discuss., 2003, 123, 51–64 Search PubMed.
- T. Eckert and E. Bartsch, J. Phys.: Condens. Matter, 2004, 16, S4937–S4950 Search PubMed.
- N. Willenbacher, J. S. Vesaratchanon, O. Thorwarth and E. Bartsch, Soft Matter, 2011, 7, 5777 Search PubMed.
- A. Kozina, D. Sagawe, P. Díaz-Leyva, E. Bartsch and T. Palberg, Soft Matter, 2012, 8, 627–630 RSC.
- M. Stieger, J. S. Pedersen, P. Lindner and W. Richtering, Langmuir, 2004, 20, 7283–7292 Search PubMed.
- J. J. Crassous, M. Siebenbürger, M. Ballauff, M. Drechsler, O. Henrich and M. Fuchs, J. Chem. Phys., 2006, 125, 204906 Search PubMed.
- M. Siebenbürger, M. Fuchs and M. Ballauff, Soft Matter, 2012, 8, 4014 Search PubMed.
- M. Franke, PhD thesis, Johannes Gutenberg-Universität Mainz, 2014.
- M. Franke, S. Golde and H. J. Schöpe, AIP Conf. Proc., 2013, 1518, 214 Search PubMed.
- M. Stieger, W. Richtering, J. S. Pedersen and P. Lindner, J. Chem. Phys., 2004, 120, 6197–6206 Search PubMed.
- M. Antonietti, W. Bremser and M. Schmidt, Macromolecules, 1990, 23, 3796–3805 Search PubMed.
- S. Golde, T. Palberg and H. J. Schöpe, Nat. Phys., 2016, 12, 712–717 Search PubMed.
- T. Palberg, A. Stipp and E. Bartsch, Phys. Rev. Lett., 2009, 102, 038302 Search PubMed.
- S. Iacopini, T. Palberg and H. J. Schöpe, J. Chem. Phys., 2009, 130, 084502 Search PubMed.
- M. Franke, S. Golde and H. J. Schöpe, Soft Matter, 2014, 10, 5380 Search PubMed.
- A. Kasper, E. Bartsch and H. Sillescu, Langmuir, 1998, 14, 5004–5010 Search PubMed.
- M. Wiemann, N. Willenbacher and E. Bartsch, Colloids Surf., A, 2012, 413, 78–83 Search PubMed.
- K. N. Pham, S. U. Egelhaaf, P. N. Pusey and W. C. K. Poon, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 69, 011503 Search PubMed.
- Z. Zhou, J. V. Hollingsworth, S. Hong, G. Wei, Y. Shi, X. Lu, H. Cheng and C. C. Han, Soft Matter, 2014, 10, 6286 Search PubMed.
- E. Bartsch, T. Eckert, C. Pies and H. Sillescu, J. Non-Cryst. Solids, 2002, 307–310, 802–811 Search PubMed.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, Wiley-VCH Verlag, Weinheim, 1983 Search PubMed.
- H. Kaiser, bhmie (Python), http://code.google.com/p/scatterlib/ (July 2016).
- A. P. Philipse, C. Smits and A. Vrij, J. Colloid Interface Sci., 1989, 129, 335 Search PubMed ; details can be found in M. Wiemann, PhD thesis, Albert-Ludwigs-Universität Freiburg, 2013.
- J. K. Percus and G. J. Yevick, Phys. Rev., 1958, 110, 1–13 Search PubMed.
- L. Verlet and J.-J. Weis, Phys. Rev. A: At., Mol., Opt. Phys., 1972, 5, 939–952 Search PubMed.
- P. van Beurten and A. Vrij, J. Chem. Phys., 1981, 74, 2744 Search PubMed.
- D. Frenkel, R. J. Vos, C. G. de Kruif and A. Vrij, J. Chem. Phys., 1986, 84, 4625 Search PubMed.
- S. E. Paulin and B. J. Ackerson, Phys. Rev. Lett., 1990, 64, 2663–2666 Search PubMed.
- N. Koumakis, A. Pamvouxoglou, a. S. Poulos and G. Petekidis, Soft Matter, 2012, 8, 4271 Search PubMed.
- C. C. Stilke, PhD thesis, Albert-Ludwigs-Universität Freiburg, 2011.
- T. Eckert and W. Richtering, J. Chem. Phys., 2008, 129, 124902 Search PubMed.
- R. Agrawal and D. A. Kofke, Mol. Phys., 1995, 85, 23–42 Search PubMed.
- M. Fasolo and P. Sollich, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 14–20 Search PubMed.
- E. Lange, J. B. Caballero, A. M. Puertas and M. Fuchs, J. Chem. Phys., 2009, 130, 174903 Search PubMed.
- S. Burger, E. Bartsch, P. Lindner and M. Werner, Institut Laue-Langevin (ILL), 2015, DOI: 10.5291/ILL-DATA.9-12-416.
- J. Riest, T. Eckert, W. Richtering and G. Nägele, Soft Matter, 2015, 11, 2821–2843 Search PubMed.
- J. Clara-Rahola, A. Fernandez-Nieves, B. Sierra-Martin, a. B. South, L. A. Lyon, J. Kohlbrecher and A. Fernandez Barbero, J. Chem. Phys., 2012, 136, 214903 Search PubMed.
- L. Willner, O. Jucknischke, D. Richter, J. Roovers, L.-L. Zhou, P. M. Toporowski, L. J. Fetters, J. S. Huang, M. Y. Lin and N. Hadjichristidis, Macromolecules, 1994, 27, 3821–3829 CrossRef CAS.
- B. Li and B. W. Brooks, Polym. Int., 1992, 29, 41–46 Search PubMed.
- C. Chern, Prog. Polym. Sci., 2006, 31, 443–486 Search PubMed.
- S. Meyer and W. Richtering, Macromolecules, 2005, 2, 1517–1519 Search PubMed.
- C.-S. Chern and C.-H. Lin, Polymer, 2000, 41, 4473–4481 Search PubMed.
- S. Gupta, M. Camargo and J. Stellbrink, et al. , Nanoscale, 2015, 7, 13924 Search PubMed.
- D. W. Holley, M. Ruppel, J. W. Mays, V. S. Urban and D. Baskaran, Polymer, 2014, 55, 58 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm02007k |
| ‡ Depending on temperature, CH can act as a good solvent or a bad solvent for PS. As the theta temperature of PS microgels is significantly larger than the one of linear PS chains (>60 °C as compared to 35 °C) the particles can be swollen at higher temperatures to allow the non-connected chains to leave the microgels and deswollen at lower temperature to ease sedimentation of the microgels while keeping the non-connected chains in solution. |
| § To perform the Mie-calculation the python port by H. Kaiser of the source code published in ref. 45 was used to determine the radius RMie. The code is part of the scatterlibproject46 (published under GNU GPL v3). |
| This journal is © The Royal Society of Chemistry 2017 |