
Rational in situ tuning of a supramolecular photocatalyst for hydrogen evolution†
S.
Kaufhold
 a,
D.
Imanbaew
b,
C.
Riehn
bc and
S.
Rau
*a
a,
D.
Imanbaew
b,
C.
Riehn
bc and
S.
Rau
*a
aUlm University, Inorganic Chemistry I, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: sven.rau@uni-ulm.de
bTU Kaiserslautern, Department of Chemistry, Erwin-Schrödinger-Str. 52–54, 67663 Kaiserslautern, Germany
cLandesforschungszentrum OPTIMAS, Erwin-Schrödinger-Str. 46, 67663, Kaiserslautern, Germany
First published on 11th September 2017
Abstract
A new heterodinuclear ruthenium platinum complex containing an N-heterocyclic carbene bridging ligand was synthesised and used for photocatalytic hydrogen evolution. The molecular integrity of the unit was proven by mercury poisoning and the lability of terminal halido ligands verified with ESI-MS. In situ ligand exchange enhances the catalytic performance leading to an almost 500-fold increase in the turnover frequency and a significantly improved overall turnover number.
The use of molecular photocatalysts for the water splitting reaction is a hot topic in solar energy storage chemistry.1–3 This is further fuelled by the idea of generating dye-sensitised photoelectrochemical cells that combine the merits of separated half-reactions in single cells with the benefits of molecular catalysts.4,5 This type of catalyst should ultimately be more efficient than heterogeneous systems that, due to their bulk nature, require a high amount of catalyst material, making their application costly if rare noble metals are employed. Additionally, intramolecular catalysts should ultimately be more efficient than homogenous intermolecular systems that are diffusion limited.6–8 An intramolecular photocatalyst combines the three essential components of a photocatalytic system into one unit – the photosensitiser (PS), an electron relay or, in this case, a bridging ligand (BL) and the catalytic centre (CC).9,10 Another major benefit of such a catalyst concept is the structure–property correlation that can be drawn.7 The single subunits of the catalyst can be altered individually by synthetic means and the changes in the photophysical and photochemical properties as well as the catalytic performance can be benchmarked. Finally, a comparison of the results will enable the rational design of optimised catalysts. Herein we present a proof of principle study on a dinuclear Ru–Pt complex used for light-driven generation of molecular hydrogen from aqueous solution. Ruthenium polypyridine complexes are often used as PSs in photocatalytic reactions as they exhibit beneficial photophysical properties for this kind of application, such as strong absorbance/high quantum yields in the visible spectral region and long excited state lifetimes.11,12 Pd and Pt moieties are among the most frequently used CCs. However, the instability of Pd CCs has been an issue in several cases.13,14 In order to improve the stability of the link between PS and CC, binding motifs other than the often used NN-donor spheres seem more feasible. Especially N-heterocyclic carbene (NHC) ligands have received tremendous attention during the last few decades in organometallic chemistry and catalysis alike, and recently also for the development of photocatalytic assemblies, due to their outstanding stabilisation properties.15 Therefore we designed [(tbbpy)2Ru(μ-bbip)PtCl2]Cl2 (tbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine and bbip = 1,3-dibenzyl-1H-imidazo[4,5-f][1,10]phenanthrolin-2-ylidene) RubbipPtCl2, a system incorporating the photophysical properties of ruthenium polypyridine complexes and the exceptional stability of a NHC binding motif for the catalytically active Pt centre (Fig. 1).
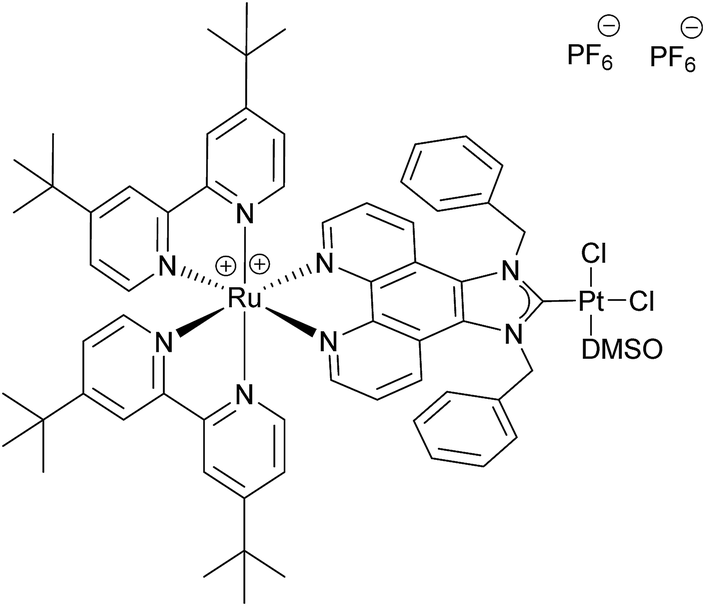 | ||
| Fig. 1 Schematic structure of RubbipPtCl2. | ||
For the synthesis of RubbipPtCl2 we avoided transmetallation from a silver carbene complex as this approach has been proven to result in product contamination by silver impurities that can have a significant influence on the catalytic performance.16 This, in turn, hinders a clear structure–property–correlation and therefore a silver free synthetic strategy was adopted for the synthesis of RubbipPtCl2. NaOMe was applied as a base to deprotonate the imidazolium moiety of Rubbip in the presence of PtCl2(DMSO)2 in order introduce the CC in a one-pot reaction (see ESI chapter S2† for details on synthesis and characterisation). The air and moisture stable product can be purified by Sephadex column chromatography and/or recrystallisation. Characterization was performed by means of nuclear magnetic resonance (NMR), electrospray ionisation high-resolution mass spectrometry (ESI-HRMS) and absorption and emission spectroscopy (Fig. 2). These findings compare well to those of previously investigated RubbipM complexes.17 Notably the emission maximum of RubbipPtCl2 (637 nm) is redshifted in comparison to [Ru(bpy)3]2+ (607 nm). This leads to the assumption that the emissive state is located on the bbip moiety, giving strong evidence of a direct electron transfer to the CC, in analogy to related complexes.17
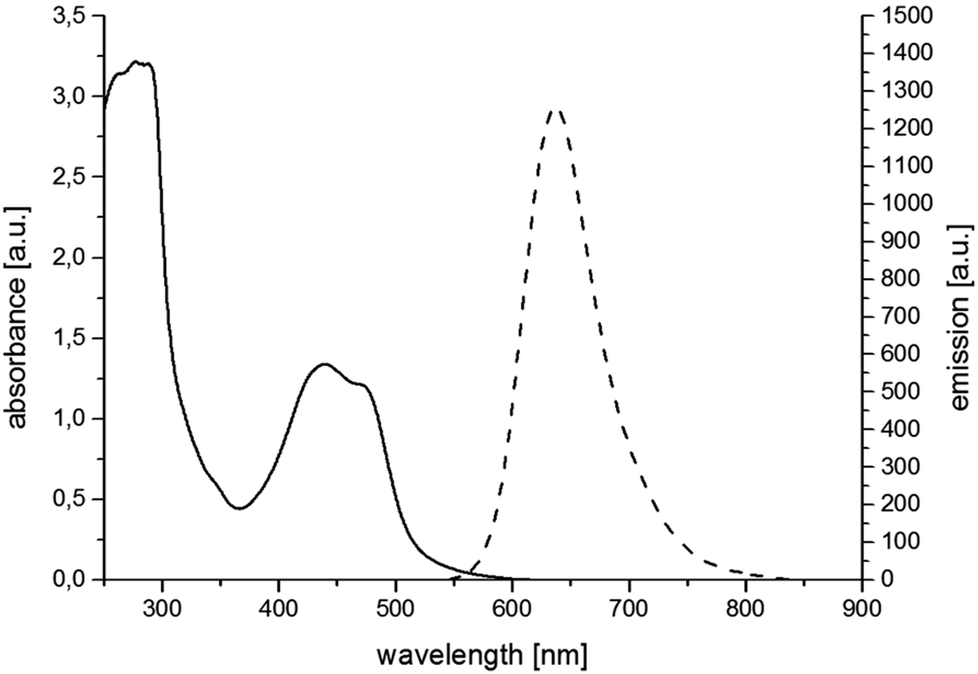 | ||
| Fig. 2 Absorption and emission spectra of RubbipPtCl2 in MeOH. | ||
Following spectroscopic characterisation, the catalytic performance of RubbipPtCl2 in light-driven hydrogen evolution was assessed. As seen in Fig. 3 the catalytic activity is quite low and after 30 h a turnover number (TON = n(H2)/n(cat)) of about 2.5 is attained, with a corresponding turnover frequency (TOF = TON per time) of about 2 d−1 under the given conditions. The molecular integrity of the catalyst was investigated by applying a Hg-poisoning test.18 Addition of Hg did not lead to substantial changes within the first 30 h of catalysis (Fig. 3, red graph). Also, the lack of an induction period and relatively linear kinetics point towards an intramolecular catalytic mechanism. In earlier studies the structurally similar compound [(tbbpy)2Ru(tpphz)PtCl2]2+ (tpphz = tetrapyrido-[3,2-a:2′3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine) RutpphzPtCl2 was synthesized and investigated in our group and gave a TON of 7 in 10 h under similar catalysis conditions.14 In the case of RutpphzPt the manipulation of the CC by exchange of the terminal chlorido to iodido ligands by synthetic means led to a drastic increase in the maximum TON (7 for RutpphzPtCl2vs. 276 for RutpphzPtI2).19
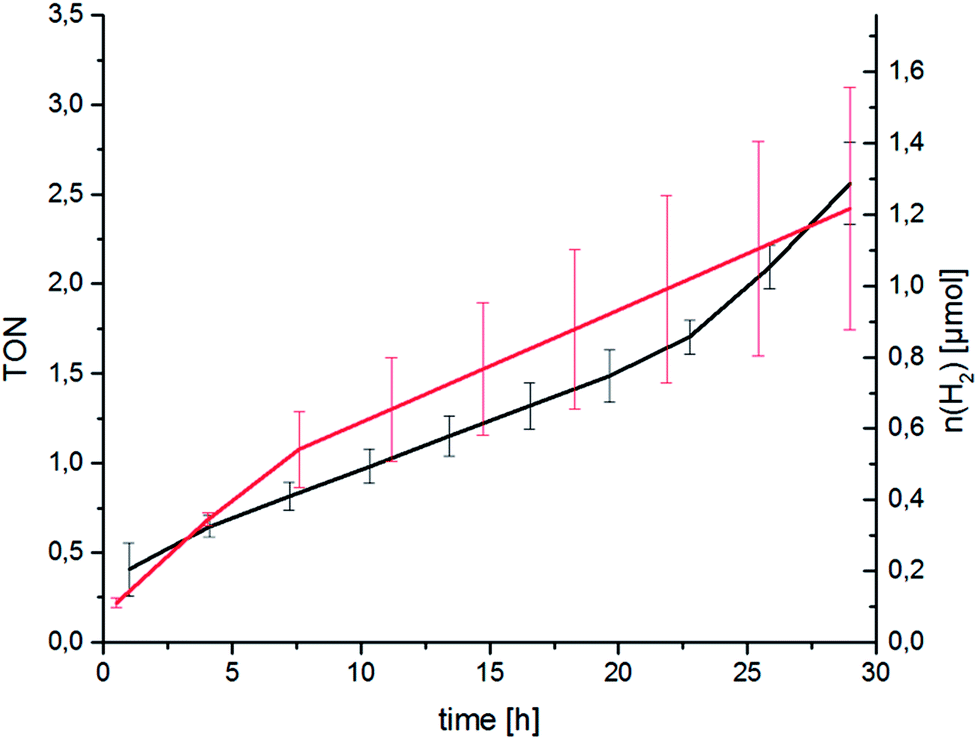 | ||
Fig. 3 Catalytic performance of RubbipPtCl2 without (black graph) and with the addition of Hg (100 μL, red graph). Conditions c(cat) = 67 μM, in MeOH/NEt3/H2O = 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1, irradiation with two LED arrays emitting at λ = 470 ± 20 nm. Averaged over 2 runs, error bars show the standard deviation. :3:1, irradiation with two LED arrays emitting at λ = 470 ± 20 nm. Averaged over 2 runs, error bars show the standard deviation. | ||
Sakai and co-workers argued that the dz2 orbital of Pt catalysts plays a crucial role in the mechanism of hydrogen evolution and that destabilisation of this orbital by increased electron density on the Pt centre can improve catalytic activity.7,20 Thus the stronger π-donating properties of iodido ligands in comparison to chlorido ligands seem to be a feasible explanation for the observations made for the RutpphzPt system.
We were eager to expand this concept on the RubbipPt system as well. Unfortunately the synthesis and purification of RubbipPtI2 proved to be arduous and the compound could not be generated in sufficient purity using the above mentioned approach. However, while characterising RubbipPtCl2 we noticed that ligand dissociation takes place in methanolic solutions of RubbipPtCl2 (Fig. 4) leading allegedly to the exchange of chloride vs. methoxide as observed by ESI-MS (Fig. S3-1†). This is in contrast to the behaviour of RutpphzPt for which stable coordination of halido ligands was found.14,19 Based on this observation we assumed that an in situ introduction of iodido ligands seems feasible if a sufficient amount of iodide is present in the solution. If applicable to RubbipPt, this should result in improved catalytic performance. Indeed, upon addition of 200 equivalents of tetrabutylammonium iodide (TBAI) to the catalytic solution a very strong increase in TOF (41 h−1 for the first 160 min) and TON (120 after 4 h) is observed (Fig. 5). To the best of our knowledge this TOF is among the highest reported for Ru–Pt intramolecular photocatalysts only outperformed by the system reported by Schulz and Pryce and co-workers.21 Again the mercury poisoning test was performed to assess the molecular integrity of the catalyst. Addition of Hg only led to a minor detrimental effect on catalytic activity, decreasing the maximum TON to 90 after 4 h. The TOF levels off earlier but is almost identical within the first hour to the TOF obtained from the non-poisoned catalysis run. Influences on the catalytic performance by addition of mercury have been witnessed and discussed before and do not necessarily imply the presence of a colloidal species.14,22 Possible explanations for the observed changes are unwanted side reactions such as direct photoreactions of the catalyst with Hg or the formation of Hg2+ during catalysis, which in turn may act as a quencher of excited states and thus have a negative influence on TONs.14,22 In addition to quenching, oxidised Hg-species may possibly remove iodide from the catalysis solution by formation of HgI2 and thereby impede catalytic performance.
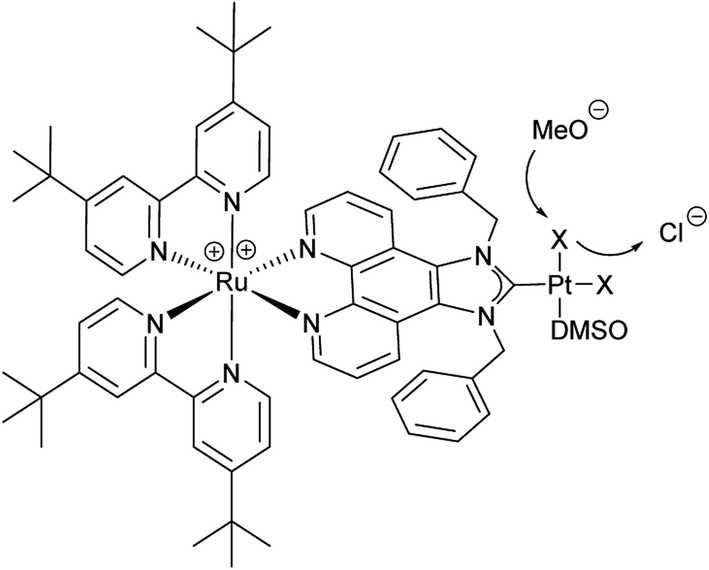 | ||
| Fig. 4 Scheme of the observed exchange of terminal chlorido vs. methoxido ligand. | ||
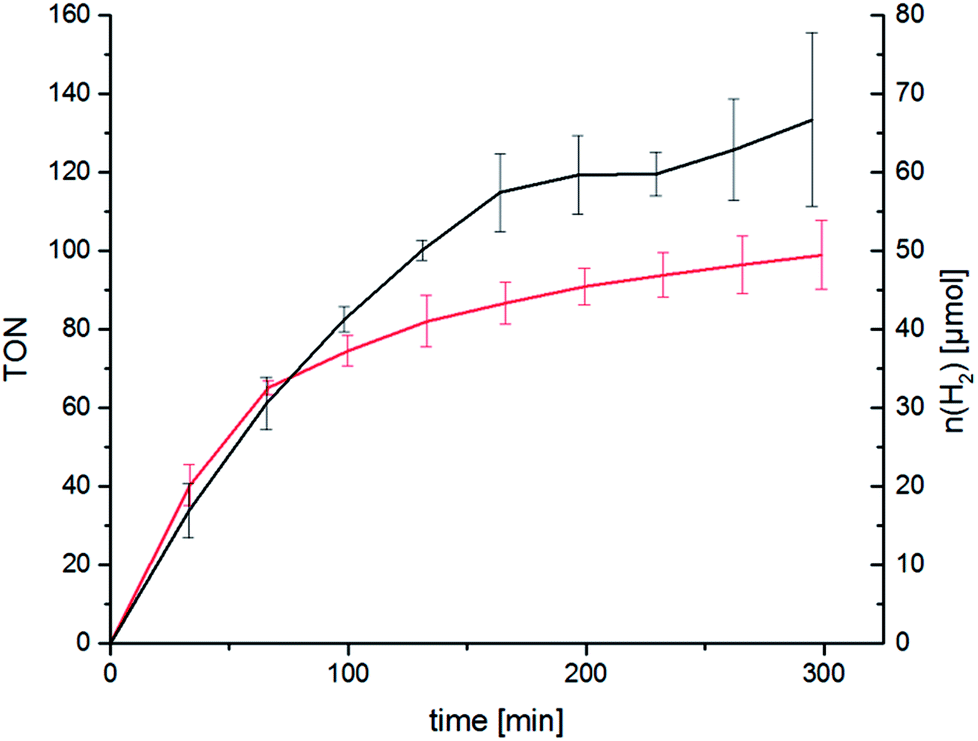 | ||
| Fig. 5 Catalytic performance of RubbipPtCl2 in the presence of 200 equivalents TBAI without (black) and with the addition of Hg (200 μL, red). Conditions: c(cat) = 67 μM, in MeOH/NEt3/H2O = 6:3:1, irradiation with two LED arrays emitting at λ = 470 ± 20 nm. Averaged over 2 runs, error bars show the standard deviation. | ||
Further catalysis experiments were carried out to elucidate the influence of halide ions and halide ion concentration in catalysis solutions. Addition of another 200 eq. TBAI after catalysis ceased, did not lead to reactivation of the high catalytic activity (Fig. S3-9†). The use of 50, 20, 10 and 2 eq. of TBAI led to a stepwise decline in catalytic performance compared to 200 eq. (Fig. S3-6†). This indicates that a certain concentration of iodide is needed for efficient ligand exchange. With 2000 eq. TBAI added, the maximum TON ≈ 110 is also somewhat lower, which might be correlated with alteration of interactions between the catalyst and substrate due to high ion concentration. The use of other halides did not lead to such a pronounced enhancement as that for TBAI. Addition of 200 eq. TBABr yielded 2.5 turnovers after about 6 h, while TBACl and TBAF did not lead to a noteworthy increase compared to catalysis runs without additives (TON ≈ 0.5 after 6 h, Fig. S3-7†). Seemingly only the strong π-donor iodide has a significant influence on the catalytic activity of the Pt CC, while bromide only leads to a minor improvement.
In order to underlay the theory of an exchange of the terminal halido ligands 1H-NMR and ESI-MS were carried out. The addition of TBAI to an NMR-sample of RubbipPtCl2 in deuterated methanol led to a pronounced splitting of the signal of the benzylic CH2-protons and concomitantly a high-field shift of the CH3-protons of the DMSO ligand, while only little changes in the aromatic region were observed (Fig. S3-2 and S3-3†). The change in 1H-NMR signals is a strong indication for an altered chemical environment at the catalytic centre as both CH2 and CH3-groups of DMSO are in close proximity to the CC and thus should react sensitively on any changes. Due to the low solubility of RubbiPtCl2 in methanol other straightforward or convenient methods to investigate the ligand exchange like 195Pt-NMR or infrared spectroscopy could not be applied. However, additional evidence for a change in the ligand sphere of the Pt CC was obtained from ESI-MS investigations on RubbipPtCl2 (Fig. 6). In pure methanolic solution the main signal at m/z = 689.1 refers to the species RubbipPt(Cl)(OMe) with one chlorido ligand exchanged for a methoxido ligand [M – Cl + OMe]2+. If 10 equivalents of TBAI are added to a methanolic solution of RubbipPtCl2, immediately an additional signal at m/z = 737.1 is observed that can be assigned to a RubbipPt(Cl)(I) species [M – Cl + I]2+. Over the course of several hours a third signal at m/z = 782.5 rises in intensity, whereas the other two slowly decrease Fig. 6 and, S3-4†. This signal can be assigned to RubbipPtI2 with two iodido ligands coordinated [M – 2Cl + 2I]2+. Notably, the signal at m/z = 737.1 (0 h) shifts to a lower m/z in the mass spectra of the aged solution (24 h), resulting in a signal at m/z = 735.1, which we assign to a RubbipPt(I)(OMe) species [M – 2Cl + OMe + I]2+. It is thus conceivable that the progression from RubbipPtCl2 to RubbipPtI2 in methanol proceeds via consecutive exchange of the chlorido ligands by methoxido ligands, which are rapidly replaced by iodido ligands. Very similar results were obtained when 100 equivalents of TBAI were added, but with a significantly increased exchange rate (Fig. S3-5†).
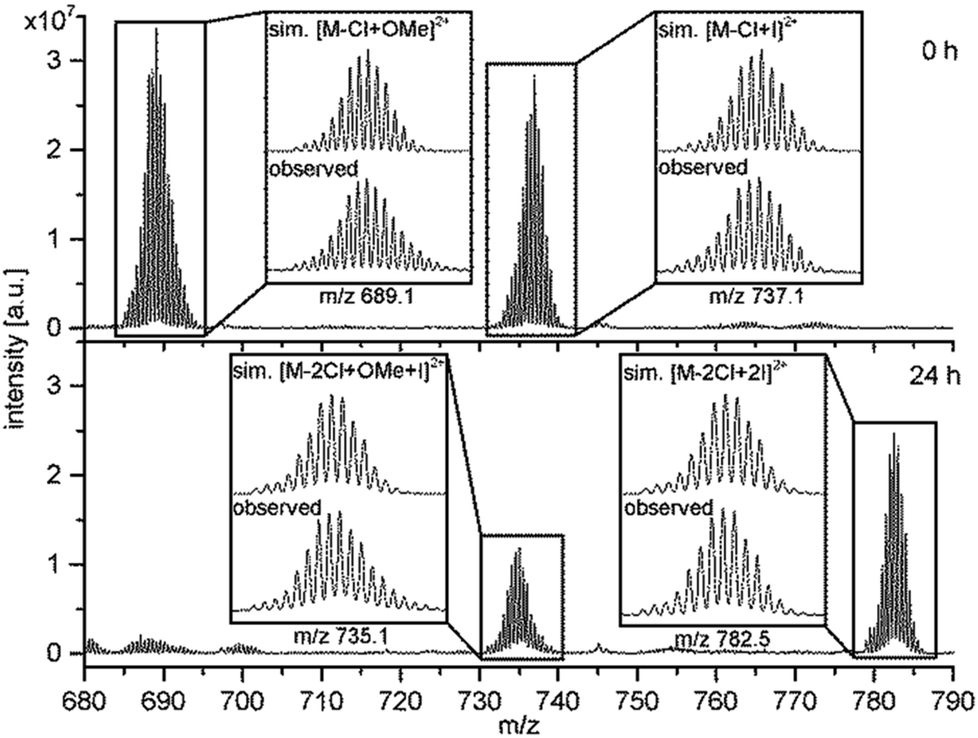 | ||
| Fig. 6 ESI-MS spectra of RubbipPtCl2 in methanol with added TBAI (10 eq.) indicate ligand exchange. Recorded for a freshly prepared solution and after 24 h. The mass isolated signals are shown along simulations of the respective isotope patterns and are in good accordance with each other. | ||
Spurred by our mass spectrometric investigations, natural population analysis (NPA) was performed for RubbipPtCl2 and its conceivable in situ ligand exchange products RubbipPtX2 (X = OMe, F, Br, I). The positive natural charge density at the Pt CC decreases for the series of F > OMe > Cl > Br > I (Table S3-1†). Analogously, electron occupancy in the 5dz2 orbital increases in the same direction, i.e. electron density was calculated to be highest for RubbipPtI2. These results support the idea of facilitated hydrogen reduction by higher electron density at the CC and are in good qualitative agreement with our experimental findings.
In the case of 200 eq. TBAI being added to the solution, a dependency of the catalytic performance on the concentration of RubbipPtCl2 was found (Fig. S3-8†). Lower concentrations also lead to a decrease in TONs (c = 34 μM, TON ≈ 50; c = 17 μM, TON ≈ 20), pointing towards a di-(or even oligo)merization-dependent catalysis mechanism as proposed by Sakai et al.7 The TON also drops at higher concentrations (c = 268 μM TON ≈ 80) which might be due to the strong absorbance of RubbipPt of the used irradiation wavelength and thus less than ideal light penetration through the entire catalysis solution.23,24 Additionally the influence of photooxidised iodide species like I3− on the catalytic performance is conceivable under our catalytic conditions. Here, however, no evidence for the formation of I3− could be found utilizing a photometric test (see Fig. S3-10 and S3-11†).25
As described above we think that ligand exchange from chloride ligands to iodido ligands during catalysis is feasible and that the improvement of the catalytic performance can be attributed to an in situ ligand exchange. However, the time span in which the catalytic system is active when TBAI is present is quite short as the hydrogen evolution levels off after about 2 h. Long-time catalysis runs of RubbipPtCl2 without added TBAI, however, showed that the catalytic system in principle can be active for a very long time. Stable turnover was achieved over 450 h leading to a maximum TON of 37 (Fig. 7). Addition of Hg led to a slight decrease in the maximum TON to about 25, as the catalytic activity levels off earlier (after about 300 h), which can be explained by the interruptive mechanisms discussed earlier.
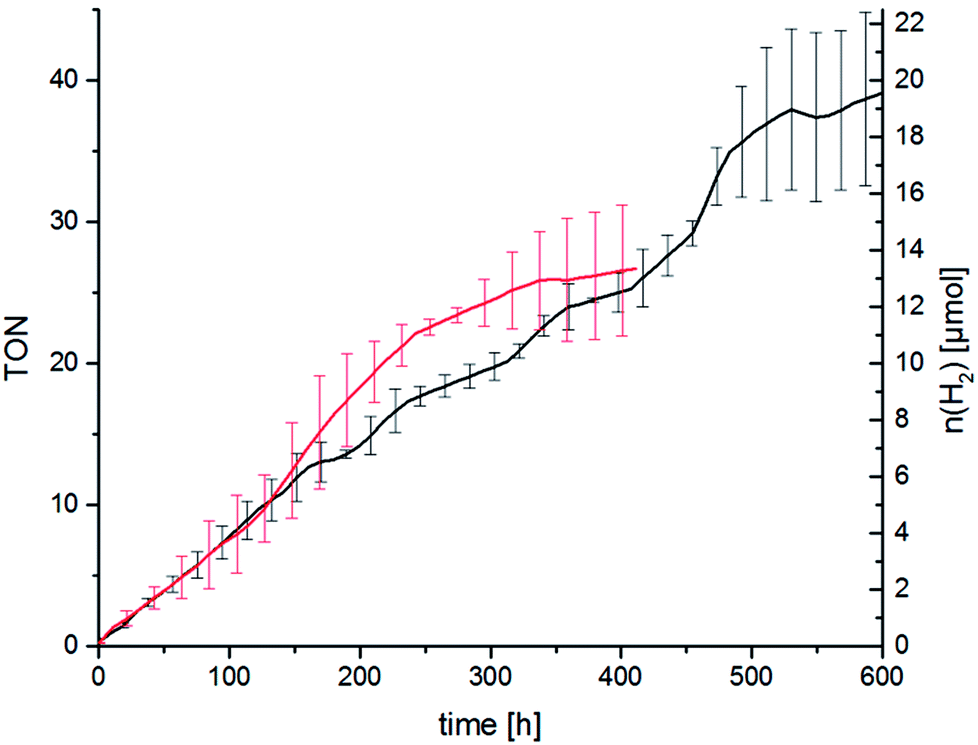 | ||
| Fig. 7 Long-time catalytic performance of RubbipPtCl2 without and with addition of Hg (100 μL), averaged over 2 runs. Conditions c(cat) = 67 μM, in MeOH/NEt3/H2O = 6:3:1 irradiation with two LED arrays emitting at λ = 470 ± 20 nm. | ||
Conclusion
With RubbipPtCl2 we were able to design a NHC-bridged supramolecular photocatalyst for the generation of molecular hydrogen, which exhibits improved activity under catalytic conditions in comparison to the model system RutpphzPtCl2. More importantly, the catalytic activity of the RubbipPt system could be tuned in situ by addition of an iodide salt to the catalysis solution. We correlate this to the lability of the terminal chlorido ligands in methanolic solution which enables an efficient chlorido vs. iodido ligand exchange. This could be exploited to gain a 3× increase in the maximum TON and an almost 500× increase in TOF in the period of the highest activity. To the best of our knowledge this is the first report on an in situ improvement of a homogeneous photocatalyst. However, only either long term stability or high efficiency could be achieved for RubbipPt. The search for a ligand environment that provides both, activation and stabilisation of the catalytic centre is crucial. Until now it is not yet clear how to achieve this effectively. It seems that a certain increase in electron density in the Pt dz2 orbitals is needed for improved catalytic performance. Additionally it might be beneficial if the donor ligand would bind more strongly to the CC in order to keep the activation active for a longer period of time.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
D. I. and C. R. thank the DFG (SFB/TRR 88 “3MET”; project C4), the Stiftung Rheinland-Pfalz für Innovation (project 965) and the Landesgraduiertenförderung Rheinland-Pfalz for funding.References
- M. D. Kärkäs, E. V Johnston, O. Verho and B. Akermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- R. J. Detz, K. Sakai, L. Spiccia, G. W. Brudvig, L. Sun and J. N. H. Reek, ChemPlusChem, 2016, 81, 1024–1027 CrossRef CAS.
- N. Coutard, N. Kaeffer and V. Artero, Chem. Commun., 2016, 52, 13728–13748 RSC.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 CAS.
- H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227–2242 RSC.
- M. Schulz, M. Karnahl, M. Schwalbe and J. G. Vos, Coord. Chem. Rev., 2012, 256, 1682–1705 CrossRef CAS.
- A. Inagaki and M. Akita, Coord. Chem. Rev., 2010, 254, 1220–1239 CrossRef CAS.
- Y. Halpin, M. T. Pryce, S. Rau, D. Dini and J. G. Vos, Dalton Trans., 2013, 42, 16243 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, Springer, Berlin, Heidelberg, 2007, pp. 117–214 Search PubMed.
- P. Lei, M. Hedlund, R. Lomoth, H. Rensmo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2008, 130, 26–27 CrossRef CAS PubMed.
- M. G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 5044–5048 CrossRef CAS PubMed.
- S. Kaufhold, L. Petermann, R. Staehle and S. Rau, Coord. Chem. Rev., 2015, 304–305, 73–87 CrossRef CAS.
- S. Kaufhold, L. Petermann, D. Sorsche and S. Rau, Chem.–Eur. J., 2017, 23, 2271–2274 CrossRef CAS PubMed.
- K. Peuntinger, T. D. Pilz, R. Staehle, M. Schaub, S. Kaufhold, L. Petermann, M. Wunderlin, H. Görls, F. W. Heinemann, J. Li, T. Drewello, J. G. Vos, D. M. Guldi and S. Rau, Dalton Trans., 2014, 43, 13683–13695 RSC.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- M. G. Pfeffer, T. Kowacs, M. Wächtler, J. Guthmuller, B. Dietzek, J. G. Vos and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 6627–6631 CrossRef CAS PubMed.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS.
- N. Das, G. S. Bindra, A. Paul, J. G. Vos, M. Schulz and M. T. Pryce, Chem.–Eur. J., 2017, 23, 5330–5337 CrossRef CAS PubMed.
- R. Okazaki, S. Masaoka and K. Sakai, Dalton Trans., 2009, 6127–6133 RSC.
- T. Sreethawong, T. Puangpetch, S. Chavadej and S. Yoshikawa, J. Power Sources, 2007, 165, 861–869 CrossRef CAS.
- H. Yang, J. Yan, Z. Lu, X. Cheng and Y. Tang, J. Alloys Compd., 2009, 476, 715–719 CrossRef CAS.
- L. Petermann, R. Staehle, T. D. Pilz, D. Sorsche, H. Görls and S. Rau, Eur. J. Inorg. Chem., 2015, 2015, 750–762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on instrumentation, synthetic procedure, 1H-NMR and ESI-(HR)MS measurements, and DFT calculations and additional catalysis results. See DOI: 10.1039/c7se00401j |
| This journal is © The Royal Society of Chemistry 2017 |