
Tailoring oxygen redox reactions in ionic liquid based Li/O2 batteries by means of the Li+ dopant concentration
Laura
Cecchetto
a,
Alvaro Y.
Tesio
 ab,
Mara
Olivares-Marín
ae,
Marc Guardiola
Espinasa
c,
Fausto
Croce
d and
Dino
Tonti
*a
ab,
Mara
Olivares-Marín
ae,
Marc Guardiola
Espinasa
c,
Fausto
Croce
d and
Dino
Tonti
*a
aInstitut de Ciència de Materials de Barcelona, Consejo Superior de Investigaciones Científicas (ICMAB-CSIC), Campus UAB, ES 08193 Bellaterra, Barcelona, Spain. E-mail: dino.t@csic.es; Tel: +34 935 801 853 ext. 362
bINQUIMAE, Departamento de Química Inorgánica, Analítica y Química Física, Facultad de Ciencias Exactas y Naturales, Ciudad Universitaria, Pabellón 2, AR-1428 Buenos Aires, Argentina
cMATGAS, Campus UAB, ES 08193 Bellaterra, Barcelona, Spain
dDipartimento di Farmacia, Università “d'Annunzio” Chieti-Pescara, Via dei Vestini 31, 66100 Chieti, Italy
eDepartamento de Ingeniería Mecánica, Energética y de los Materiales, Universidad de Extremadura, Centro Universitario de Mérida, 06800, Mérida, Spain
First published on 25th October 2017
Abstract
The reusability, non-volatility and non-corrosiveness of ionic liquids (ILs), as well as their ease of isolation and a large electrochemical stability window, make them an interesting choice as environmentally friendly electrolytes for metal/air batteries. ILs have been described as designer solvents as their properties and behaviour can be adjusted to suit an individual reaction need. In the framework of this study we applied a conceptually similar design approach and showed that a simple parameter such as the concentration of a Li+ dopant dramatically affects the reaction yields of Li/O2 based energy storage devices. We studied the effect of Li+ concentration from 0.1 to 1 M in a LiTFSI:PYR14TFSI ionic liquid electrolyte on the kinetics of the oxygen reduction reaction (ORR) and on the formation rate of different Li–O species at two different temperatures, finding that the discharge capacity, rates and product distribution change in a non-linear way. At 60 °C, the highest rates and up to one order of magnitude larger capacities were observed at intermediate LiTFSI concentrations, implying a complete mechanism switch from surface to volume phase mediation for Li2O2 precipitation. At room temperature the same evolution was observed, even if in this case the surface mediation remained predominant at all concentrations. These results suggest the possibility to optimise the ionic liquid based Li/O2 battery performances in terms of discharge capacity and lithium use, by tuning the temperature and alkali cation concentration.
1. Introduction
Lithium–oxygen (Li/O2) rechargeable batteries utilizing aprotic electrolytes surpass all other practically interesting electrochemical battery storage devices in terms of theoretical specific energy. Consequently, these batteries have recently received considerable attention,1–8 being an important candidate for energy storage devices for electric vehicles, and other large scale high-energy storage applications. In spite of these promising prospects, the practical exploitation of Li/O2 systems in organic electrolytes is still far away, due to the numerous scientific challenges that need to be addressed. The main critical points are a low efficiency due to a significant overpotential and a poor reversibility upon cycling. These are mainly related to the formation of the insoluble and insulating Li2O2 product,9–12 whose morphology and structure can greatly affect the cell overpotential and reversibility, and to the presence of parasitic products mostly from solvent decomposition, arising from the reaction of the intermediates of oxygen reduction with the organic electrolytes.13–16 Moreover, as volatile organic solvents are highly polluting and difficult to recycle, their replacement in large energy storage devices also represents an advantage to the environment. The reusability, non-volatility and non-corrosiveness of ionic liquids, as well as their conductivity and a large electrochemical stability window, make them an interesting choice as environmentally friendly electrolytes for rechargeable Li-ion batteries17,18 as well as for metal–air batteries.19 Besides their importance as “green” solvents,20 different studies have shown that ionic liquid electrolytes are generally more stable, compared to other organic electrolytes, to superoxide radical anion attack,21 and that their use could lower the overpotential22,23 while increasing the rechargeability of Li/O2 cells.24 In particular, Elia et al. recently demonstrated exceptional energy efficiency in the order of 82%.23 However, their high viscosity generally introduces mass transport limitations that may result in a film-like coating and poor discharge capacity.25–27 In effect these limitations can be reduced by stirring the electrolyte and operating above room temperature.25 Nonetheless, stirred cells require larger electrolyte volumes, which tend to increase costs and lower the energy density. Quaternary ammonium ionic liquid electrolytes, in particular 1-methyl-1-butylpyrrolidinium bis(tri-fluoromethane sulfonyl)imide (PYR14TFSI), are the most studied in combination with Li/O2 cells. Several investigations have already been reported on the O2 reaction in cells containing PYR14TFSI pure21,28–30 or with different concentrations of LiTFSI,25,28,29,31–33 where glassy carbon29 or porous carbons25,28,31,32 were used as cathodes. The presence of Li+ has been found to remarkably affect the O2 electrochemistry;25,28 however the effect of its concentration has not been discussed in detail. The aim of this work was to tune a PYR14TFSI electrolyte and optimize it for Li/O2 batteries, by adjusting the parameters that affect interactions with O2 and its reduction products. We especially investigated the effect of Li+ concentration at two different temperatures on the formation rate of different Li–O species. We performed a galvanostatic and cyclic voltammetry study and SEM morphological characterisation at room temperature (RT) and at 60 °C with a PYR14TFSI solution containing different concentrations of LiTFSI. We show that the peroxide/superoxide ratio and even the discharge mechanism in a PYR14TFSI based electrolyte can be modulated by controlling the Li salt concentration and the temperature, showing the possibility to largely control Li/O2 batteries by these two parameters.2. Experimental methods
The electrochemical cell was based on ISO-KF standard high vacuum components supplied by ITL Vacuum Components. A detailed description of the cell is reported in a previous study.31 The electrodes consisted of a layer of commercial carbon black (Super P, M.M.M. Carbon) supported on a stainless steel mesh (AISI 316, 180 mesh per inch, ADVENT Research Materials Ltd). Super P (90 wt%) was mixed with 10 wt% of polyvinylidene fluoride (PVDF) as a binder in N-methylpyrrolidone (NMP). The slurry obtained was used to impregnate the mesh, which was finally dried at 100 °C for 12 h. The carbon load used was close to 1 mg cm−2. The electrolyte used was 1-methyl-1-butylpyrrolidinium bis(tri-fluoromethane sulfonyl)-imide (PYR14TFSI, 99.5%, Solvionic) containing LiTFSI (99.95%, SigmaAldrich). PYR14TFSI was stored in a dry box and used as received, while LiTFSI was dried at 120 °C under vacuum for 48 h before use. The separator was a fiberglass filter (FilterLab MFV1, 260 μm thick) soaked with ∼100 ml of electrolyte and the anode was a Li metal foil (Sigma Aldrich, 0.4 mm thick, cut to approx. 0.8 cm2). A nickel foil was used as a current collector. All lithium–oxygen cells were assembled in an argon-filled glovebox. Subsequently, oxygen was forced to pass continuously through the cell for at least 20 min before starting the electrochemical measurements, and a constant flow (5 ml min−1) was maintained during the measurements. Tests at room temperature were done in a lab with the temperature set at 25 °C. For tests at 60 °C, the cells were placed in a heating cabinet designed specifically for this purpose.3. Results and discussion
To compare the oxygen reduction reaction (ORR) activity using LiTFSI concentrations between 0.1 M and 1 M, we performed cyclic voltammetry at scan rates of 100 mV s−1 and 20 mV s−1 at room temperature (RT) and 60 °C under an oxygen flow. The results are reported in Fig. 1. | ||
| Fig. 1 Cyclic voltammetry curves obtained at different LiTFSI molar concentrations in PYR14TFSI at RT and at 60 °C with an applied scan rate of 100 mV s−1. | ||
The typical peak position observed and the cyclic voltammetry shapes are very similar to what was previously observed by Allen et al. in a lithium oxygen cell containing 0.025 M LiTFSI in PYR14TFSI on Au.33 The agreement is remarkable, if we take into account that in our case we are using not an ideal substrate, but a common porous carbon electrode. We therefore propose the same reaction scheme to explain the different peaks observed in our study and use the same labelling, as summarized in Table 1. The cathodic peak C1 may correspond to more than one reduction reaction of O2, occurring almost at the same potential, leading to LiO2, Li2O2 or in some cases to Li2O. Lithium peroxide can be obtained from superoxide via a second electrochemical reduction and/or via chemical disproportionation. The different species generated in the C1 region of the cathodic scan are oxidized at different potentials, giving rise to different anodic peaks. During the anodic scan, lithium peroxide, considered the most stable in aprotic electrolytes, is oxidized to O2 and Li+ in a two-electron reaction corresponding to peak A3, while peak A2 has been previously assigned to the oxidation of lithium superoxide to O2 and Li+24,28 as also confirmed by Raman measurements.34,35 In the 1 M solutions, where viscosity is expected to be particularly high, the anodic peaks are hardly visible, and instead a sort of diffusion plateau is observed; in this case, labels A2 and A3 barely mark their expected positions. According to the peak attribution illustrated in Table 1, the ratio between A2 and A3 can provide qualitative information on the relative amounts and stability of LiO2 and Li2O2 formed in C1. This ratio between the formed products may affect the capacity and reversibility of the Li/O2 cell in the different electrolytes.24
| Li+ + O2 + e → LiO2 | EC C1 | LiO2 → Li+ + O2 + e | EC A2 |
| Li+ + LiO2 + e → Li2O2 | EC C1′ | Li2O2 → 2 Li+ + O2 + 2e | EC A3 |
| 2LiO2 → Li2O2 + O2 | Ch |
At both temperatures the onset potentials, as well as the anodic and cathodic current densities, are affected by the Li+ content. In Fig. 2 are reported the absolute values of the cathodic current recorded at 2 V at RT and 60 °C in the 100 mV s−1 scans. At RT the lowest cathodic currents are observed in the 1 M LiTFSI:PYR14TFSI solution. Moreover, upon increasing the LiTFSI content from 0.1 M up to 0.8 M the cathodic current progressively increases. This is associated with an increase of the anodic peak contribution at a higher potential (peak A3), confirming the rapid LiO2 conversion to Li2O2, in accord with what was already observed by Allen et al.24 At 60 °C the cathodic current increases steeply, as a function of the LiTFSI concentration in the PYR14TFSI solution, reaching a maximum value in the 0.6 M solution and then decreasing at higher LiTFSI concentrations. In this case, when the cathodic peak is increased, a greater A2 peak contribution is observed, while peak A3 becomes negligible compared to what is observed at RT. Thus, in spite of the similar trend of larger cathodic currents at intermediate concentrations, the rise of temperature makes the LiTFSI concentration in the PYR14TFSI solution a more critical parameter for the ORR at the electrode interface.
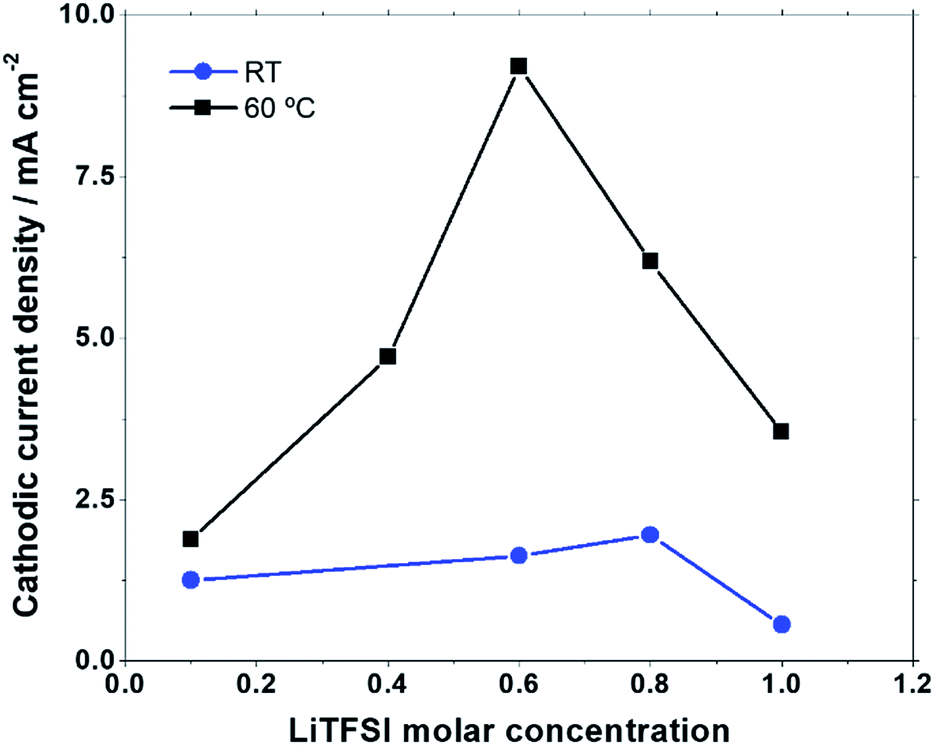 | ||
| Fig. 2 Absolute value of cathodic current at 2 V in cyclic voltammograms at a scan rate of 100 mV s−1 as a function of LiTFSI molar concentration at RT and at 60 °C. | ||
Recent studies have explained that the formation of solid Li2O2 resulting from O2 reduction proceeds by an electrode surface or solution pathway depending on the influence of the solvent donor number on the solubility of the LiO2 intermediate.36 With LiO2 partitioned between LiO2 dissolved in the electrolyte and LiO2 adsorbed on the electrode, the ORR can take place on the electrode surface or in the electrolyte bulk, depending on the electrolyte stabilisation properties. In our case, the increase of the A2 anodic peak with temperature and with LiTFSI concentration could be explained in terms of an increase of soluble LiO2, most likely due to a faster desorption, at the expense of its disproportionation at the immediate surface. LiO2 solubilisation strongly increases its further production and a larger number of molecules would remain within their diffusion length from the electrode surface. The weak temperature-dependence of the A2 overvoltage compared to A3 further supports the attribution of peak A2 to mainly solubilised LiO2. The increase of peak A2 at the expense of A3, observed at 60 °C, would thus suggest that the LiO2 dissolution in the electrolyte is promoted at higher temperatures so that a larger extent of the solution phase mechanism for Li2O2 precipitation takes place. It is worth noticing that the A2/A3 ratio takes smaller values as the scanning speed decreases (Fig. 1f), consistent with peak A2 being related to a LiO2 soluble species, whose diffusion far from the electrode would be favoured at lower scan rates.
Galvanostatic tests at different Li+ concentrations reflect cycling voltammetry results. Fig. 3 depicts the behaviour at RT and 60 °C of the discharge capacity at different LiTFSI concentrations, applying a current of 0.1 mA cm−2. At both temperatures, the graphics show a “volcano” shape evidencing optimal performances for a concentration of 0.6 M. The low capacity at RT compared to the work of Elia et al.23 could be mainly attributed to our cathode geometry, which in our case was flooded and not supported on a gas-diffusion layer. Nevertheless, as is possible to see from Fig. 4, we also observed a remarkably low charge voltage (especially at 60 °C) compared to the literature using other organic electrolytes.6,7,23 While a smaller particle size may play a role, we attribute this lower charge voltage mainly to the smaller quantity of secondary products wrapping the deposits discharged in room temperature ionic liquid-based electrolytes, as recently shown by us using soft X-ray transmission microscopy.37
 | ||
| Fig. 3 Discharge capacity values as a function of LiTFSI molar concentration in PYR14LiTFSI at RT and at 60 °C. | ||
 | ||
| Fig. 4 Voltage (vs. Li+/Li0) as a function of specific capacity (A h g−1) for 0.1 M, 0.6 M and 1 M LiTFSI concentrations in PYR14LiTFSI at RT and 60 °C. | ||
Remarkably, the volcano shape obtained for the discharge capacity is similar to the behaviour already observed for the cathodic current as a function of the LiTFSI content (Fig. 2). We can therefore conclude that the non-linear evolution of the Li+ availability for the ORR at the interface equally affects the cathodic current and discharge capacity when increasing the Li+ concentration. This evolution is analogous to that reported by Liu et al. as a function of LiTFSI concentration in TEGDME38 although the maximum is observed here with a lower Li+ concentration, which can be attributed to the stronger effect of Li+ on viscosity as discussed above. Nevertheless, the most intriguing aspect is the large capacity increase obtained at intermediate concentrations. These values – ranging from 1500 mA h g−1 to 3000 mA h g−1 at 60 °C – correspond to those previously observed with the same electrodes in the presence of a solution-mediated mechanism.26 The theoretical capacity for a film-like discharge on Super P can be estimated to be 1200 mA h g−1 considering a specific surface area of 62 m2 g−1, the Li2O2 film thickness and the capacity density;6,27 however this is considered a limit value that is usually not approached, as it is difficult to homogeneously cover the entire carbon surface.
Therefore, the observed higher capacities can only be explained by a solution phase mechanism. In effect, the SEM images of the electrode surfaces after discharge at 60 °C in 0.1 M, 0.6 M and 1 M LiTFSI solutions (Fig. 5) are consistent with a mechanism change for intermediate concentrations.
 | ||
| Fig. 5 Secondary electron images of the electrode surface after discharge at 0.1 M, 0.6 M and 1 M LiTFSI concentrations in PYR14TFSI at 60 °C. | ||
A heterogeneous precipitate characterised by large particles is evident with the 0.6 M solution, while in the other two cases just the texture of the supporting carbon can be appreciated, as expected with the formation of a nm thick conformal Li2O2 film. The role of soluble LiO2 in obtaining a solution-mediated mechanism and thus a large discharge capacity is highlighted by the evident A2 peak at intermediate concentrations at 60 °C. We report in Fig. 6 the discharge capacity as a function of the A2/A3 ratio (considering the respective absolute peak currents) at RT and 60 °C. In effect, a remarkable linear trend is observed at 60 °C, while at RT no clear correlation can be noticed.
 | ||
| Fig. 6 Discharge specific capacity obtained as a function of the A2/A3 ratio, at RT and 60 °C and linear trend of points obtained at 60 °C. | ||
In the latter case, the relatively low capacities and the high peak A3 contribution suggest a main surface precipitation process that is expected to partly mask the effect of the Li+ concentration on LiO2 solubility, although the highest discharge capacity was still observed for a Li+ concentration of 0.6 M, where the anodic peak A2 reaches its limiting value.
We can therefore conclude that a complete mechanism switch from surface to volume phase mediation takes place at intermediate concentrations at 60 °C, while at RT the same tendency is likely, even if the surface mediation remains present at all concentrations.
Several causes can be considered for this mechanism change, and the present knowledge of the system makes a precise description difficult. Both concentration and temperature affect viscosity, the electrolyte association, and the cathode wettability by the electrolyte.39,40 Viscosity determines the electrolyte conductivity and mass transport of reactants and products. The electrolyte association interacts with the solvation of possible soluble intermediates. Wettability in turn affects the available area for charge transfer, and may influence the nucleation of Li2O2 deposits on the surface of the electrode. The increase of temperature is generally expected to enhance the electrochemistry via these effects, while the effect of concentration is not monotonic. However, the switch from a surface- to a solution-mediated mechanism necessarily involves enhanced LiO2 solubilisation.
Mass transport is likely responsible of the generally improved kinetics at higher temperatures, when viscosity decreases41 and the Li+ mobility substantially increases.42 It could also be involved in the mechanism switch at 60 °C. In effect, previous studies in this IL system showed that electrolyte stirring can largely affect the discharge capacity25 by inducing a similar change of the peroxide deposition mechanism.26 This indicates that depletion of either oxygen or Li+ hinders LiO2 formation and desorption. The strong sensitivity to the LITFSI concentration suggests that even if it is present in a larger concentration than O2 (which is in the order of 2–3 mM (ref. 28)), Li+ could be at least as limiting. This can be explained by the Li+ diffusivity being more than one order of magnitude smaller and more dependent on LiTFSI concentration than oxygen.42,43 Our results, suggesting faster ORR kinetics at the intermediate concentrations used, could be ascribed to a higher Li+ availability at the electrode interface. However, when the LiTFSI content further increases, the coordination of Li+ with TFSI− neighbouring anions starts forming bridged structures limiting mass transport.41,44,45 In this case, the slower diffusion prevails over the increased Li+ content, and the ORR rate declines.
In addition to its effect on mass transport, we think that the Li+ content could also affect electrolyte association, which also plays a role in LiO2 solubility, as has been shown recently in the literature.46,47 Sharon et al.47 especially highlighted how the level of lithium salt association, which strongly depends on its concentration in the electrolyte, could affect LiO2 stability: the more associated Li salt would improve the Li+ soft acid character and thus its affinity for the O2− soft base. This favours LiO2 desorption, and again its solution phase disproportionation, as long as the increased LiTFSI bridging affects viscosity. Further studies are needed to demonstrate what phenomena govern the reaction path in this system.
4. Conclusions
A cyclic voltammogram study performed with a Super P electrode with LiTFSI:PYR14TFSI ionic liquid solutions at different concentrations yielded features similar to those previously reported in the literature on Au or glassy carbon. According to the Li+ content and temperature, it is possible to significantly affect the reaction product ratio as well as the reduction and oxidation voltage and kinetics. The delivered discharge capacity is also affected. At a given temperature, the Li+ concentration affects the delivered current as well as the reaction product ratio. We could confirm that higher temperatures improve the kinetics and also favour superoxide stability for given Li+ concentrations. The highest capacities are observed at an intermediate concentration of 0.6 M at RT and 60 °C. This dependence could involve both mass transport and an electrolyte association effect acting on the way LiO2 is partitioned between the electrode surface and the electrolyte bulk, affecting where Li2O2 precipitation takes place, with dramatic effects on capacity. These results show that by tuning the Li+ concentration and temperature it is possible to switch from a surface-mediated to a solution-phase Li2O2 growth mechanism for optimal Li/O2 cell performances. Further investigations focusing on a more precise understanding of the mechanisms and on the effect of temperature and other electrolyte additives may still largely improve the discharge capacity, reversibility and efficiency; nevertheless the present study confirms that ionic liquids are a promising choice for metal–oxygen batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Commission in the Seventh Framework Programme FP7-2010-GC-ELECTROCHEMICAL STORAGE, under contract no. 265971 “Lithium–Air Batteries with split Oxygen Harvesting and Redox processes” (LABOHR), with a contribution from the Spanish Ministry of Economy, Industry and Competitiveness, through the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2015-0496). M. O. acknowledges CSIC for a JAE-DOC research contract cofunded by the European Social Fund. A. Y. T. acknowledges a research postdoctoral grant from CONICET in particular to the program “Estadías Breves en el Exterior para Becarios Postdoctorales”. We are thankful for fruitful discussions on ionic liquids with Dr Gianni Appetecchi, and on ionic liquid-based Li/O2 batteries with Prof. John Owen and all LABOHR partners.References
- K. M. Abraham and Z. Jiang, A polymer electrolyte-based rechargeable lithium/oxygen battery, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS.
- J. Read, Characterization of the lithium/oxygen organic electrolyte battery, J. Electrochem. Soc., 2002, 149, A1190–A1195 CrossRef CAS.
- T. Kuboki, T. Okuyama, T. Ohsaki and N. Takami, Lithium-air batteries using hydrophobic room temperature ionic liquid electrolyte, J. Power Sources, 2005, 146, 766–769 CrossRef CAS.
- T. Ogasawara, A. Debart, M. Holzapfel, P. Novak and P. G. Bruce, Rechargeable Li2O2 electrode for lithium batteries, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, Lithium-air battery: promise and challenges, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, A Critical Review of Li/Air Batteries, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS.
- R. Black, B. Adams and L. F. Nazar, Non-aqueous and hybrid Li-O2 batteries, Adv. Energy Mater., 2012, 2, 801–815 CrossRef CAS.
- N. Garcia-Araez and P. Novák, Critical aspects in the development of lithium-air batteries, J. Solid State Electrochem., 2013, 1–15 Search PubMed.
- J. Read, K. Mutolo, M. Ervin, W. Behl, J. Wolfenstine, A. Driedger and D. Foster, Oxygen Transport Properties of Organic Electrolytes and Performance of Lithium/Oxygen Battery, J. Electrochem. Soc., 2003, 150, A1351–A1356 CrossRef CAS.
- I. Kowalczk, J. Read and M. Salomon, Li-air batteries: a classic example of limitations owing to solubilities, Pure Appl. Chem., 2007, 79, 851–860 CrossRef CAS.
- S. S. Zhang, D. Foster and J. Read, Discharge characteristic of a non-aqueous electrolyte Li/O2 battery, J. Power Sources, 2010, 195, 1235–1240 CrossRef CAS.
- C. Tran, X. Q. Yang and D. Qu, Investigation of the gas-diffusion-electrode used as lithium/air cathode in non-aqueous electrolyte and the importance of carbon material porosity, J. Power Sources, 2010, 195, 2057–2063 CrossRef CAS.
- B. M. Gallant, R. R. Mitchell, D. G. Kwabi, J. Zhou, L. Zuin, C. V. Thompson and Y. Shao-Horn, Chemical and morphological changes of Li-O2 battery electrodes upon cycling, J. Phys. Chem. C, 2012, 116, 20800–20805 CAS.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, The lithium-oxygen battery with ether-based electrolytes, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, Twin problems of interfacial carbonate formation in nonaqueous Li-O2 batteries, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, The carbon electrode in nonaqueous Li-O2 cells, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed.
- A. Fernicola, F. Croce, B. Scrosati, T. Watanabe and H. Ohno, LiTFSI-BEPyTFSI as an improved ionic liquid electrolyte for rechargeable lithium batteries, J. Power Sources, 2007, 174, 342–348 CrossRef CAS.
- J. H. Shin and E. J. Cairns, N-Methyl-(n-butyl)pyrrolidinium bis(trifluoromethanesulfonyl)imide-LiTFSI–poly(ethylene glycol) dimethyl ether mixture as a Li/S cell electrolyte, J. Power Sources, 2008, 177, 537–545 CrossRef CAS.
- M. Kar, T. J. Simons, M. Forsyth and D. R. Macfarlane, Ionic liquid electrolytes as a platform for rechargeable metal-air batteries: a perspective, Phys. Chem. Chem. Phys., 2014, 16, 18658–18674 RSC.
- M. J. Earle and K. R. Seddon, Ionic liquids. Green solvents for the future, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- J. Herranz, A. Garsuch and H. A. Gasteiger, Using rotating ring disc electrode voltammetry to quantify the superoxide radical stability of aprotic Li-air battery electrolytes, J. Phys. Chem. C, 2012, 116, 19084–19094 CAS.
- L. Cecchetto, M. Salomon, B. Scrosati and F. Croce, Study of a Li-air battery having an electrolyte solution formed by a mixture of an ether-based aprotic solvent and an ionic liquid, J. Power Sources, 2012, 213, 233–238 CrossRef CAS.
- G. A. Elia, J. Hassoun, W. J. Kwak, Y. K. Sun, B. Scrosati, F. Mueller, D. Bresser, S. Passerini, P. Oberhumer, N. Tsiouvaras and J. Reiter, An advanced lithium-air battery exploiting an ionic liquid-based electrolyte, Nano Lett., 2014, 14, 6572–6577 CrossRef CAS PubMed.
- C. J. Allen, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, Oxygen Electrode Rechargeability in an Ionic Liquid for the Li-Air Battery, J. Phys. Chem. Lett., 2011, 2, 2420–2424 CrossRef CAS.
- S. Monaco, F. Soavi and M. Mastragostino, Role of oxygen mass transport in rechargeable Li/O2 batteries operating with ionic liquids, J. Phys. Chem. Lett., 2013, 4, 1379–1382 CrossRef CAS PubMed.
- M. Aklalouch, M. Olivares-Marín, R.-C. Lee, P. Palomino, E. Enciso and D. Tonti, Mass-transport Control on the Discharge Mechanism in Li–O2 Batteries Using Carbon Cathodes with Varied Porosity, ChemSusChem, 2015, 8, 3465–3471 CrossRef CAS PubMed.
- M. Olivares-Marín, P. Palomino, E. Enciso and D. Tonti, Simple Method to Relate Experimental Pore Size Distribution and Discharge Capacity in Cathodes for Li/O2 Batteries, J. Phys. Chem. C, 2014, 118, 20772–20783 Search PubMed.
- S. Monaco, A. M. Arangio, F. Soavi, M. Mastragostino, E. Paillard and S. Passerini, An electrochemical study of oxygen reduction in pyrrolidinium-based ionic liquids for lithium/oxygen batteries, Electrochim. Acta, 2012, 83, 94–104 CrossRef CAS.
- F. De Giorgio, F. Soavi and M. Mastragostino, Effect of lithium ions on oxygen reduction in ionic liquid-based electrolytes, Electrochem. Commun., 2011, 13, 1090–1093 CrossRef CAS.
- N. Mozhzhukhina, A. Y. Tesio, L. P. M. De Leo and E. J. Calvo, In Situ Infrared Spectroscopy Study of PYR14TFSI Ionic Liquid Stability for Li–O2 Battery, J. Electrochem. Soc., 2017, 164, A518–A523 CrossRef CAS.
- M. Olivares-Marín, P. Palomino, J. M. Amarilla, E. Enciso and D. Tonti, Effects of architecture on the electrochemistry of binder-free inverse opal carbons as Li-air cathodes in an ionic liquid-based electrolyte, J. Mater. Chem. A, 2013, 1, 14270–14279 Search PubMed.
- F. Soavi, S. Monaco and M. Mastragostino, Catalyst-free porous carbon cathode and ionic liquid for high efficiency, rechargeable Li/O2 battery, J. Power Sources, 2013, 224, 115–119 CrossRef CAS.
- C. J. Allen, J. Hwang, R. Kautz, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, Oxygen Reduction Reactions in Ionic Liquids and the Formulation of a General ORR Mechanism for Li–Air Batteries, J. Phys. Chem. C, 2012, 116, 20755–20764 CAS.
- D. Zhai, H. H. Wang, J. Yang, K. C. Lau, K. Li, K. Amine and L. A. Curtiss, Disproportionation in Li-O2 batteries based on a large surface area carbon cathode, J. Am. Chem. Soc., 2013, 135, 15364–15372 CrossRef CAS PubMed.
- J. Yang, D. Zhai, H. H. Wang, K. C. Lau, J. A. Schlueter, P. Du, D. J. Myers, Y. K. Sun, L. A. Curtiss and K. Amine, Evidence for lithium superoxide-like species in the discharge product of a Li-O2 battery, Phys. Chem. Chem. Phys., 2013, 15, 3764–3771 RSC.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J. M. Tarascon and P. G. Bruce, The role of LiO2 solubility in O2 reduction in aprotic solvents and its consequences for Li-O2 batteries, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- M. Olivares-Marín, A. Sorrentino, E. Pereiro and D. Tonti, Discharge products of ionic liquid-based Li-O2 batteries observed by energy dependent soft X-ray transmission microscopy, J. Power Sources, 2017, 359, 234–241 CrossRef.
- Y. Liu, L. Suo, H. Lin, W. Yang, Y. Fang, X. Liu, D. Wang, Y. S. Hu, W. Han and L. Chen, Novel approach for a high-energy-density Li-air battery: tri-dimensional growth of Li2O2 crystals tailored by electrolyte Li + ion concentrations, J. Mater. Chem. A, 2014, 2, 9020–9024 CAS.
- H. Liu and L. Jiang, Wettability by Ionic Liquids, Small, 2016, 12, 9–15 CrossRef CAS PubMed.
- A. M. Cione, O. A. Mazyar, B. D. Booth, C. McCabe and G. K. Jennings, Deposition and Wettability of [bmim][triflate] on Self-Assembled Monolayers, J. Phys. Chem. C, 2009, 113, 2384–2392 CAS.
- Q. Zhou, P. D. Boyle, L. Malpezzi, A. Mele, J. H. Shin, S. Passerini and W. A. Henderson, Phase Behavior of Ionic Liquid-LiX Mixtures: Pyrrolidinium Cations and TFSI- Anions – Linking Structure to Transport Properties, Chem. Mater., 2011, 23, 4331–4337 CrossRef CAS.
- F. Castiglione, E. Ragg, A. Mele, G. B. Appetecchi, M. Montanino and S. Passerini, Molecular Environment and Enhanced Diffusivity of Li+ Ions in Lithium-Salt-Doped Ionic Liquid Electrolytes, J. Phys. Chem. Lett., 2011, 2, 153–157 CrossRef CAS.
- Y. Li, Z. Zhang, D. Duan, Y. Sun, G. Wei, X. Hao, S. Liu, Y. Han and W. Meng, The correlation of the properties of pyrrolidinium-based ionic liquid electrolytes with the discharge–charge performances of rechargeable Li–O2 batteries, J. Power Sources, 2016, 329, 207–215 CrossRef CAS.
- O. Borodin, G. D. Smith and W. Henderson, Li+ cation environment, transport, and mechanical properties of the LiTFSI doped N-methyl-N-alkylpyrrolidinium +TFSI – Ionic liquids, J. Phys. Chem. B, 2006, 110, 16879–16886 CrossRef CAS PubMed.
- Y. Saito, T. Umecky, J. Niwa, T. Sakai and S. Maeda, Existing condition and migration property of ions in lithium electrolytes with ionic liquid solvent, J. Phys. Chem. B, 2007, 111, 11794–11802 CrossRef CAS PubMed.
- I. Gunasekara, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, A study of the influence of lithium salt anions on oxygen reduction reactions in Li-Air batteries, J. Electrochem. Soc., 2015, 162, A1055–A1066 CrossRef CAS.
- D. Sharon, D. Hirsberg, M. Salama, M. Afri, A. A. Frimer, M. Noked, W. Kwak, Y. K. Sun and D. Aurbach, Mechanistic Role of Li+ Dissociation Level in Aprotic Li-O2 Battery, ACS Appl. Mater. Interfaces, 2016, 8, 5300–5307 CAS.
| This journal is © The Royal Society of Chemistry 2018 |