
Carbide-derived carbon beads with tunable nanopores from continuously produced polysilsesquioxanes for supercapacitor electrodes†
Benjamin
Krüner‡
 ab,
Christina
Odenwald‡
c,
Aura
Tolosa
ab,
Anna
Schreiber
a,
Mesut
Aslan
a,
Guido
Kickelbick
*c and
Volker
Presser
*ab
ab,
Christina
Odenwald‡
c,
Aura
Tolosa
ab,
Anna
Schreiber
a,
Mesut
Aslan
a,
Guido
Kickelbick
*c and
Volker
Presser
*ab
aINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, 66123 Saarbrücken, Germany
cInorganic Solid State Chemistry, Saarland University, 66123 Saarbrücken, Germany. E-mail: guido.kickelbick@uni-saarland.de
First published on 17th July 2017
Abstract
The MicroJet reactor technique is an excellent continuous method to produce spherical and homogeneous organically modified silica (ORMOSIL) particles in a large scale (10–15 g min−1). We applied this method to manufacture polyorganosilsesquioxanes with different ratios of phenyl and vinyl functional groups, which were later pyrolyzed to obtain silicon oxycarbides. Such polymer-derived ceramic (PDC) materials are highly suited as precursor for carbide-derived carbon (CDC) synthesis. Chlorine etching of PDC at high temperatures removed silicon and oxygen, yielding the formation of nanoporous carbon. Pure poly(phenyl-silsesquioxane) spheres lost their shape during the thermal process by undergoing further condensation reactions. Yet, the spherical shape was conserved during thermal processing after adding vinyl functionalities. The ratio of vinyl and phenyl functionalities controlled the pore structure and the total CDC yield, enabling an increase from 2 mass% to 22 mass%. The total pore volume varied between 1.3-2.1 cm3 g−1 and the specific surface area between 2014–2114 m2 g−1. The high surface area and large pore volume makes these materials attractive for high power supercapacitor electrodes. The specific capacitance of the best sample at low rates in 1 M tetraethylammonium tetrafluoroborate in acetonitrile was 116 F g−1 (at 5 mA g−1) and still 80 F g−1 at very high rates (at 100 A g−1).
1. Introduction
Highly porous carbon materials are attractive for many applications such as gas storage,1,2 catalysis,3 or electrochemical energy storage.4 Electrical double-layer capacitors, also known as supercapacitors, are particularly efficient energy storage devices, capitalizing on rapid and reversible ion electrosorption.5 Due to fast polarization, it is possible to achieve a high specific power, but only a moderate specific energy.4 Beside high power ratings, supercapacitors exhibit a very long lifetime and high Coulombic efficiency.6Carbide-derived carbons (CDCs) are a broad family of carbon materials,7 which can be obtained from metal carbides,8 carbonitrides,9 or oxycarbides.10,11 Depending on the precursor and the synthesis temperatures it is possible to produce CDC materials with different pore architectures11 and carbon structures.12 Oxycarbides, like SiOC, are promising precursors to synthesize highly porous CDCs with a high BET specific surface area (SSA) of 3089 m2 g−1 and a large pore volume of 1.8 cm3 g−1.13 SiOC-CDC micrometer-sized particles have been obtained from commercial polymethyl- and polymethylphenylsilsesquioxanes by pyrolysis of monoliths, followed by chlorine etching at 1200 °C of the grinded monoliths.10 Thereby, it was possible to obtain BET SSA up to 2600 m2 g−1 and a total pore volume up to 1.7 cm3 g−1. Polymethylsilsesquioxane can also be used in an emulsion process to obtain spherical particles with a diameter of 50–600 nm, 1–10 μm, and 10–100 μm, which were successfully converted to polymer-derived ceramics (PDCs).14 Sub-micrometer CDCs with a diameter of 20–200 nm and a SSA of ∼2300 m3 g−1 were successfully obtained by emulsion soft templating using a commercial allylhydriopolycarbosilane precursor.15 The specific capacitance of this material in 1 M tetraethylammonium tetrafluoroborate (TEA-BF4) in acetonitrile (ACN) was 130 F g−1 and 110 F g−1 in aqueous 1 M Na2SO4.15 The small size of the individual particles is beneficial to achieve a high power within supercapacitor applications. Yet, a disadvantage of the mentioned methods to produce PDC precursors for CDC synthesis is the need to add a hazardous metal crosslinking agent like hydrogen hexachloroplatinate(IV),15 zirconium acteylacetonate,16 or zinc acetylacetonate.13 The crosslinking agent is necessary because the oligomers can melt before converted to PDCs.13
Wet chemical preparation methods for the synthesis of defined nano- and microparticles are commonly used as batch processes which exhibit several disadvantages like a variation in the product quality from batch-to-batch and a limited and complex scale-up. Continuous preparation methods offer in general a better controllability and reproducibility and are attractive for industrial scale production.17,18 There are few continuous pathways to obtain spherical particles from oxycarbides. For example, electrospraying yields spherical particles and can be carried out with a commercial polymethylphenylsilsesquioxane.13 The diameter of these particles was 1–6 μm, which is significant larger than soft-templated emulsion beads. The BET SSA of these micrometer spheres was up to ∼2230 m2 g−1 and the specific capacitance was 112 F g−1 for 1 M TEA-BF4 in ACN.
An advanced continuous method is the MicroJet reactor technique.19 Thereby, two reagent solutions are forced with high pressure through micro-nozzles into the reactor which is exposed to a constant gas flow. A fast and homogenous mixing is obtained due to the high shear forces that are generated by the impinging jets. The gas flow pushes the products out of the reactor into a reservoir, which also effectively avoids clogging. The MicroJet reactor technique was already used to synthesize inorganic nanoparticles, such as TiO2,20 BaSO4,21 ZnO, Fe3O4, or CaHPO4.22
In this work, we synthesized for the first time polyorganosilsesquioxane spheres with different ratios of phenyl and vinyl groups from organotrialkoxysilanes by using a MicroJet reactor. Poly(benzyl)silsesquioxane or poly(phenyl)silsesquioxane beads obtained by a sol–gel process have a softening point at ∼50 °C and ∼140 °C.23,24 This property makes them unsuitable to produce PDCs that conserve the initial shape during pyrolysis. By adding vinyl groups to the system, it was possible to obtain a higher condensation degree and to conserve the spherical shape of the particles after the pyrolysis to obtain SiOC-PDCs. Afterwards the samples were treated in chlorine gas to obtain highly porous SiOC-CDCs. The variation of the organic groups of the silanes has a strong influence for the yield and porosity of the CDCs, which relates directly to the rate performance in the supercapacitor.
2. Experimental description
2.1. Materials
Phenyltrimethoxysilane (PTMS; 97%) and vinyltrimethoxysilane (VTMS; 99%) were obtained from ABCR GmbH. Ammonia (25%) was purchased from VWR International GmbH. All chemicals were used as received.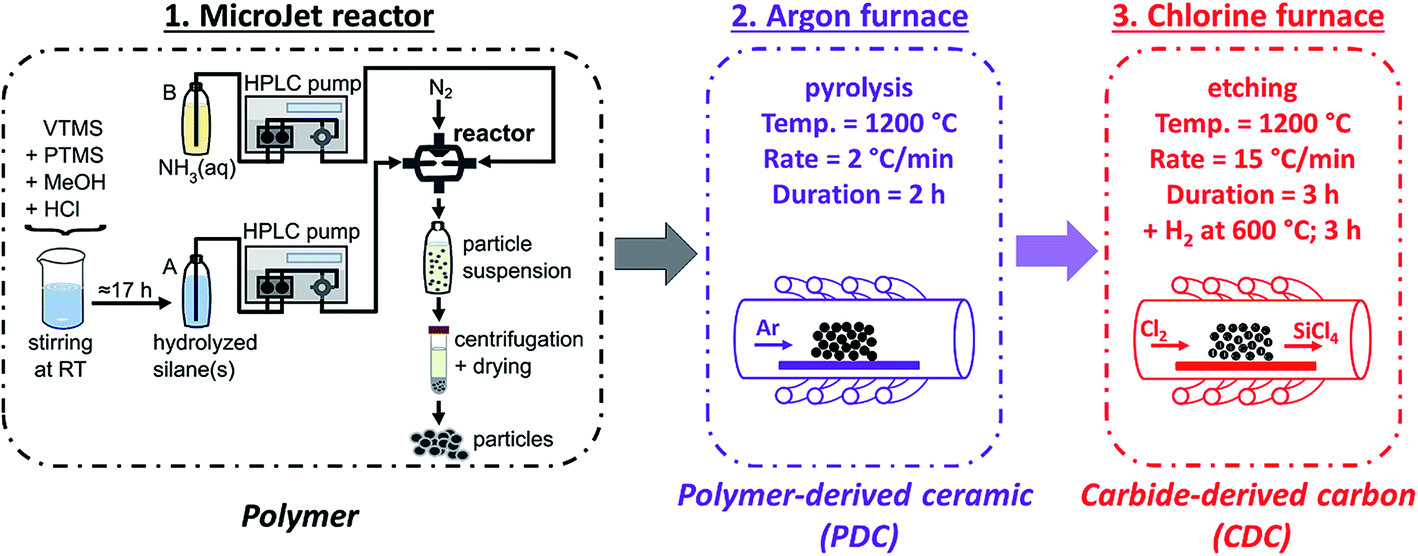 | ||
| Fig. 1 Schematic overview of the synthesis method: (1) the production of the polymer beads with the MicroJet reactor, (2) the pyrolysis, and (3) the chlorine treatment. VTMS: vinyltrimethoxysilane; PTMS: phenyltrimethoxysilane; MeOH: methyl alcohol; RT: room temperature; (A and B): solution A or solution B; HPLC: high performance liquid chromatography. | ||
| Solution A | Solution B | |||||
|---|---|---|---|---|---|---|
| n (VTMS) (mmol) | n (PTMS) (mmol) | n (MeOH) (mol) | n (H2O) (mol) | n (HCl) (mmol) | C(NH3) (mol L−1) | |
| Vi-SiO1.5 | 138.8 | — | 3.2 | 2.8 | 0.03 | 2.2 |
| Ph0.25Vi0.75-SiO1.5 | 104.1 | 34.7 | 2.8 | 2.8 | 0.03 | 2.2 |
| Ph0.5Vi0.5-SiO1.5 | 69.4 | 69.4 | 3.0 | 2.8 | 0.03 | 2.2 |
| Ph0.75Vi0.25-SiO1.5 | 34.7 | 104.1 | 3.2 | 2.8 | 0.03 | 2.2 |
The particles were synthesized at room temperature in a MicroJet reactor of stainless steel (Synthesechemie Dr. Penth GmbH). The precursor solutions A and B were transported through the system with two HPLC-pumps (LaPrep P110 preparative HPLC pumps, VWR International GmbH) with a flow rate of 250 mL min−1. The solutions were forced through opposing nozzles (nozzle diameter 300 μm) into a reaction chamber where they collide as impinging jets and a fast mixing occurs effected by the high jet velocities. A nitrogen gas flow helped to transport the product out of the reactor and avoided clogging. The outlet tube was 1.5 mm in diameter and ca. 85 cm in length. The product suspension was collected in a polyethylene flask. The particles were isolated by centrifugation (8000 rpm, 7012 G, 10 min) and dried in vacuum at 30 °C.
2.2. Material characterization
Solid-state CP-MAS NMR spectra were recorded at 25 °C using a Bruker AV400WB spectrometer (13C 100.62 MHz, 29Si 79.50 MHz). A contact time of 2.0 ms and a variable power contact time (ramp 10050) were used. The spin rate was 13 kHz and the delay time 3–6 s. For 13C NMR, adamantane, and for 29Si NMR, octakis(trimethylsiloxy)silsesquioxane was used as external standards. The intensity of all NMR spectra was normalized.Fourier transform infrared (FT-IR) spectroscopy was applied on the dried polysilsesquioxane samples in attenuated total reflectance (ATR) mode on a Vertex 70 spectrometer (Bruker Optics). The measurements were accomplished in the wave number range of 500–4500 cm−1 and under ambient air. For each spectrum, 32 scans were averaged with a spectral resolution of 4 cm−1.
The XRD patterns were measured with a D8 Advance diffractometer (Bruker AXS) using Cu-Kα radiation (40 kV and 40 mA) and a Goebel mirror with point focus and a two-dimensional X-ray detector (Vantec-500). The sample holder was a sapphire single crystal.
Raman spectroscopy was carried out with a Renishaw inVia Raman microscope with a wavelength of 532 nm and a grating of 2400 lines mm−1. The spectral resolution was 1.2 cm−1, the numeric aperture 0.9, and the incident power on the sample ∼0.02 mW. A spectrum was recorded for 30 s and averaged over 10 accumulations. The D- and G-mode with the amorphous contribution of carbon in the Raman spectra were fitted using five Voigt functions for the SiOCs and four Voigt functions for the CDCs.
The thermogravimetric measurements combined with a mass spectrometer (TGA-MS) were carried out with a STA449F3 Jupiter and QMS 403C Aëolos from Netzsch. The heating rate for the measurement was 10 °C min−1 to 1200 °C in Argon (purity: 5.0).
Scanning electron microscopy (SEM) was performed in a field emission scanning electron microscope JSM-7500F from JEOL. The samples were sputtered with gold to increase the surface conductivity. The size of 150 individual spheres was measured to obtain an average diameter with the image analysis software ImageJ.25 A X-MAX silicon detector from Oxford Instruments was used to perform the energy dispersive X-ray spectroscopy (EDX) attached to the SEM chamber. The operating voltage for the SEM and EDX was 5 kV.
High-resolution transmission electron microscopy (TEM) was executed with a JEM-2100F from JEOL at 200 kV. The polymer samples were dispersed and sonicated in water to deposit them on a lacey carbon film on a copper grid (Gatan). Ethanol was used instead of water to perform this step for the CDCs.
Nitrogen gas sorption analysis (N2 GSA) was performed with an Autosorb iQ system from Quantachrome. The samples were degassed at 100 mbar at 200 °C for 1 h and at 300 °C for 20 h. The temperature during the measurement was −196 °C and the relative pressure was varied between 5 × 10−7 to 1.0 in 76 steps. A quenched-solid density functional theory (QSDFT) kernel assuming slit-like pores (QSDFT) was applied to obtain the pore size distribution from the adsorption isotherms.26 The Brunauer–Emmett–Teller (BET) surface area was calculated with the BET equation in the linear region of the isotherm between 0.02 and 0.3 partial pressure.27 The value of the total pore volume was obtained by the adsorbed volume at a relative pressure of 0.95 and the average pore size (d50) represents the pore size with a half of the total pore volume.11 The data analysis of the nitrogen sorption isotherms was performed with ASiQwin 3.0 from Quantachrome.
Contact angle measurements of the CDC electrodes were carried out with an OC 25 system from Data Physics with demineralized water (volume per drop: 3 μL) in air.
2.3. Electrochemical measurements
Free-standing carbon electrodes were prepared by dispersing the CDC powder in ethanol and adding 5 mass% of polytetrafluoroethylene binder (PTFE, 60 mass% in water, Sigma-Aldrich). The slurry was crushed in a mortar until a doughy carbon paste was formed, which was rolled with a rolling machine from MTI Corporation to a thickness of ∼110 μm. The electrodes were dried for 48 h at 120 °C in a vacuum oven at 2 × 103 Pa. For electrochemical measurements, electrode discs with a diameter of 8 mm were punched out (geometrical information about the electrodes is given in ESI, Table S1†). Custom-built symmetrical two-electrode cells were assembled with carbon-coated aluminum current collectors (type Zflo 2653, Coveris Advanced Coatings) for the organic electrolyte (1 M TEA-BF4 in ACN; BASF, battery grade).28 Graphite foil current collectors were used for the aqueous electrolyte (1 M Na2SO4 prepared with Milli-Q water; pH 5.2). A glass fiber separator (GF/A from Whatman) with a diameter of 13 mm was used between the electrodes. The cell for the organic electrolyte was placed in a vacuum oven (2 × 103 Pa) at 120 °C for 16 h.All electrochemical measurements were carried out in a climate chamber (Binder) at 25 ± 1 °C with a VMP300 potentiostat/galvanostat from BioLogic. Electrochemical impedance spectra were recorded in a symmetrical full cell for 100 kHz to 100 mHz at 0 V with ten points per decade and averaged over five measurements. The electrical serial resistance (ESR) and the electrical distribution resistance (EDR) were normalized to the geometrical area of the electrode. The capacitance from cyclic voltammograms (CV) was calculated with eqn (1) and from galvanostatic cycling with potential limitation (GCPL) with eqn (2). The specific capacitance was normalized to the active mass (95%) of one electrode. The IR-drop of the GCPL measurements was obtained after a resting time of 10 s. The electrochemical performance is benchmarked against PTFE bonded film electrodes from YP-80F (Kuraray Co.), which is referred to as AC (activated carbon).
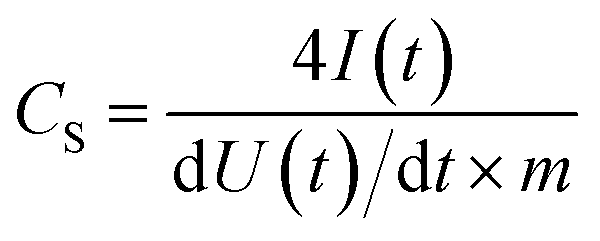 | (1) |
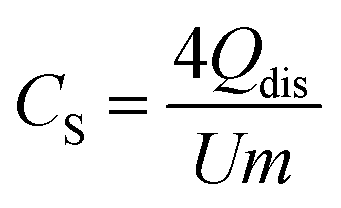 | (2) |
Stability testing via voltage floating for 100 h was also conducted in the climate chamber at 25 °C at 1.4 V for the aqueous and 2.7 V for the organic electrolyte. Every 10 h, galvanostatic cycling was performed at a cell voltage of 1.2 V (aqueous) or 2.5 V (organic). After voltage floating, the electrodes with the aqueous electrolyte were washed with Milli-Q water for further post mortem analysis.
3. Results and discussion
3.1. Particle characterization
FT-IR measurements of the polymer particles confirm the presence of the desired organic groups (Fig. S1†). All samples exhibit peaks from the inorganic backbone in the range of 935–1230 cm−1 from –Si–O–Si– vibrations. In addition, the samples show vibration bands from the organic groups. At 2800–3095 cm−1, in each spectrum C–H stretching vibrations are present and at 1600 cm−1 peaks from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations. Samples with phenyl groups show characteristic signals from Si–C-vibrations at 1430 cm−1 and 1130 cm−1. Samples that contain vinyl groups also show well-defined modes at 1275 cm−1 from Si–C vibrations and peaks at 1410 cm−1 that can be assigned to CH2 bending vibrations.29–31 The increasing vinyl content of the samples Ph0.75Vi0.25-SiO1.5, Ph0.5Vi0.5-SiO1.5, Ph0.25Vi0.75-SiO1.50, and Vinyl-SiO1.5 is reflected by the increasing intensity of the Si-vinyl vibration (1275 cm−1) compared to the Si-phenyl vibration (1430 cm−1).
C vibrations. Samples with phenyl groups show characteristic signals from Si–C-vibrations at 1430 cm−1 and 1130 cm−1. Samples that contain vinyl groups also show well-defined modes at 1275 cm−1 from Si–C vibrations and peaks at 1410 cm−1 that can be assigned to CH2 bending vibrations.29–31 The increasing vinyl content of the samples Ph0.75Vi0.25-SiO1.5, Ph0.5Vi0.5-SiO1.5, Ph0.25Vi0.75-SiO1.50, and Vinyl-SiO1.5 is reflected by the increasing intensity of the Si-vinyl vibration (1275 cm−1) compared to the Si-phenyl vibration (1430 cm−1).
13C NMR spectra (Fig. 2A) of the prepared polysilsesquioxane particles show characteristic signals for the vinyl and phenyl groups. For the samples with both organic groups overlapping peaks were obtained. Samples exhibit no or only weak signals from residual alkoxy groups (at ca. 60 ppm), which suggests that almost all alkoxy groups were hydrolyzed during polymerization and that in T2 units OH-groups are present in addition to the vinyl and phenyl groups. 29Si NMR spectra (Fig. 2B) give important information about the network structure. The peaks at ≈−73 ppm and ≈−82 ppm of the organically modified silica (ORMOSIL) particles can be assigned to T2 and T3 species, meaning silicon atoms that are connected to the network with two or three bonds. The spectra also show that the T2 to T3 ratio decrease with an increasing vinyl content. This behavior confirms the incorporation of vinyl groups into the network, which leads to an enhanced cross-linking that is desired to obtain a better thermal stability.
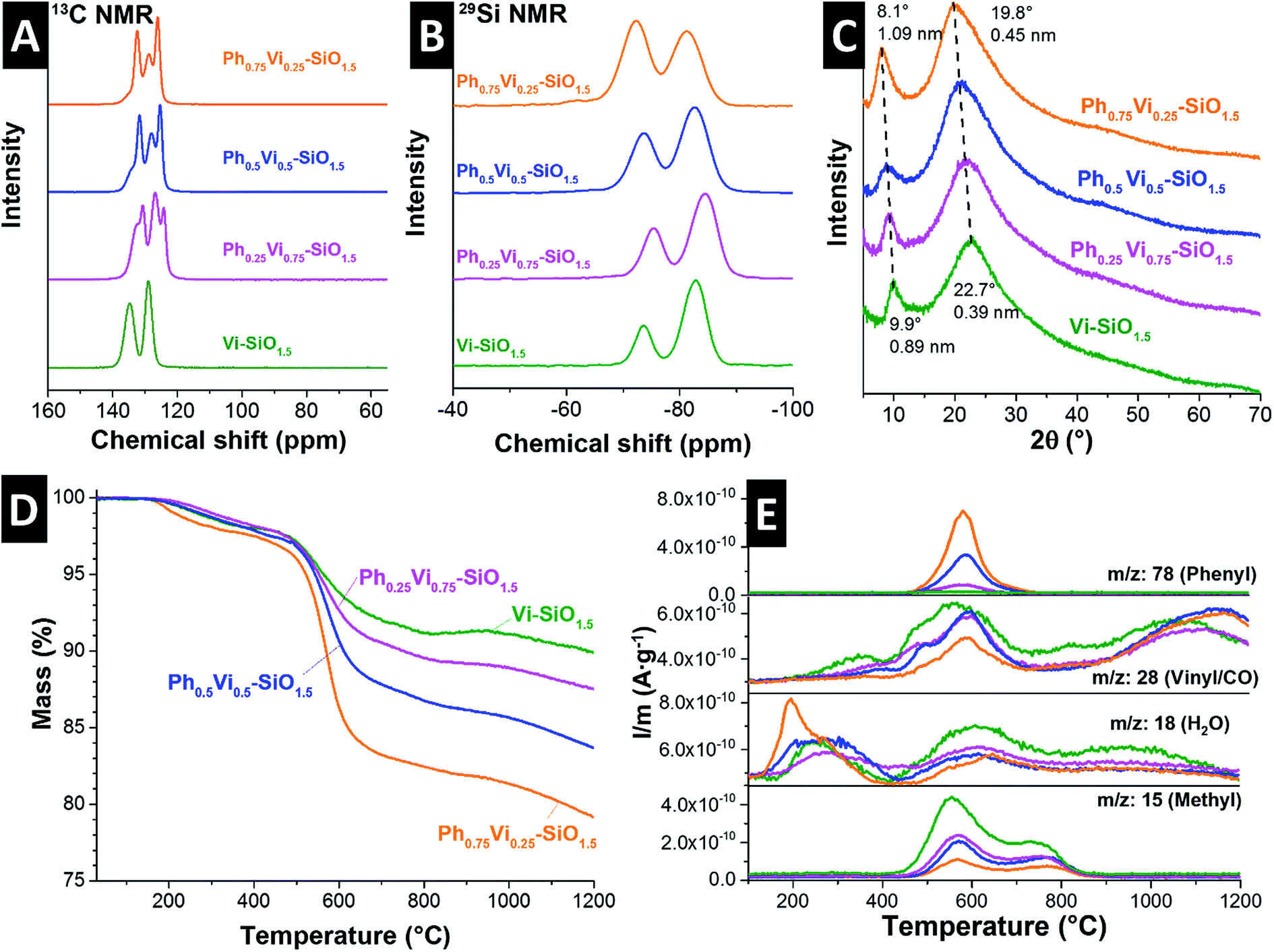 | ||
| Fig. 2 Solid-state 13C NMR (A), 29Si NMR (B), and XRD pattern (C) of the four polymer beads. TGA curve of the pyrolysis process (D) and the corresponding mass spectra of selected leaving groups (E). | ||
The XRD pattern of the mixed polymers in Fig. 2C show a shift of very broad peaks to higher 2θ values with an increasing amount of vinyl groups. Polyphenylsiloxanes can have a ladder like structure and two peaks in the XRD pattern can be identified with the plane-to-plane peak at ∼7.3° 2θ and the chain-to-chain peak of the Si–O–Si frame at ∼18.8° 2θ.32 Polyvinylsiloxanes with a ladder structure exhibit a plain-to-plain peak at ∼10° 2θ.33 The corresponding chain-to-chain peak is at around 23° 2θ due to the lower required space of the vinyl group compared to the phenyl group.33 The measured peak positions of the Vi-SiO1.5 is similar to the reported values in the literature.33 The three samples with mixtures synthesized with different ratios of phenyl- and vinyltrimethoxysilane show peaks in between the pure polyphenyl- and polyvinylsiloxanes, which indicates a homogeneous mixture. From the peak width, it is possible to estimate domain sizes by use of the Scherer equation which were below 5 nm.34 These small domain sizes point out the mainly amorphous character of the synthesized polymers. The absence of long-range order can also be seen in the transmission electron micrographs in the ESI, Fig. S2.†
The pyrolysis of the polysiloxanes was monitored with TGA-MS (Fig. 2D and E, ESI Fig. S1C and D†). All four polymers show a degradation of the ladder structure around 560 °C (Fig 2D), which would be an untypical degradation behavior for a cage structure.35 The mass loss of the vinyl-polymer is the lowest and the mass loss increases constantly with the amount of added phenyl-groups. The mass spectra of the most important evolving groups are plotted in Fig. 2E, while all relevant leaving groups of Ph0.5Vi0.5-SiO1.5 are displayed in the ESI (Fig. S1C and D†). The mass spectra in Fig. 2E identify evolving groups during heating. Water (m/z: 18) is removed from the samples in two main temperature regimes: first, between 135 °C and 430 °C, and second, at temperatures from 450 °C to 800 °C, where also the other organic groups are being removed. Samples with more phenyl groups (m/z: 78 at ∼570 °C) or vinyl groups (m/z: 28 at ∼550 °C) show a larger loss of these functional groups. The atomic mass of 28 can also be related to carbon monoxide, which is formed at higher temperatures (>900 °C) with residual oxygen. Another major leaving group is the methyl group formed by the decomposition of organic chains from the vinyl groups, which shows two peaks at 550 °C and 750 °C.
3.2. Properties of the polymer-derived ceramics and carbide-derived carbons
The chemical composition of the SiOCs is denoted in Table 2. Of our samples, the carbon content of the Vi-SiOC is the lowest with 32.2 ± 4.8 mass% and increases with an increasing amount of phenyl groups to 50.8 ± 1.8 mass% for Ph0.5Vi0.5-SiOC. Yet, the carbon content is not increasing when the amount of phenyl groups is increased from 50% to 75%, which can be explained with the higher mass loss of Ph75Vi25-SiOC.| C (mass%) | O (mass%) | Si (mass%) | |
|---|---|---|---|
| Vi-SiOC | 32.2 ± 4.8 | 31.5 ± 4.0 | 36.3 ± 8.2 |
| Ph0.25Vi0.75-SiOC | 41.9 ± 2.4 | 26.2 ± 1.6 | 31.9 ± 4.0 |
| Ph0.5Vi0.5-SiOC | 50.8 ± 1.8 | 25.2 ± 0.8 | 24.0 ± 2.5 |
| Ph0.75Vi0.25-SiOC | 50.2 ± 1.5 | 25.9 ± 1.0 | 23.9 ± 2.4 |
| Vi-SiOC-CDC | 97.9 ± 0.3 | 1.9 ± 0.3 | 0.3 ± 0.1 |
| Ph0.25Vi0.75-SiOC-CDC | 98.4 ± 0.7 | 1.3 ± 0.4 | 0.4 ± 0.3 |
| Ph0.5Vi0.5-SiOC-CDC | 98.4 ± 0.2 | 1.4 ± 0.1 | 0.2 ± 0.1 |
| Ph0.75Vi0.25-SiOC-CDC | 97.1 ± 0.4 | 2.9 ± 0.4 | n.d. |
As can be seen from our EDX results (Table 2), the removal of silicon and oxygen by chlorine treatment at high temperatures was successful and we only found a low amount of residual silicon and oxygen. The mass losses after pyrolysis and chlorine treatment of samples with different ratios of phenyl and vinyl functional groups are quite different (Table 3). The sample Vi-SiOC had the lowest mass loss after pyrolysis of only 11 mass% and shows the highest mass loss after the etching of 97.6 mass%. The total yield of Vi-SiOC-CDCs synthesis amounts to only 2.2 mass%. The addition of phenyl groups increases the total yield to 21.9 mass%, which can be explained by the higher carbon and aromatic content introduced by the phenyl groups.
| Mass loss after pyrolysis (mass%) | Mass loss after chlorine gas treatment (mass%) | Total mass loss (mass%) | |
|---|---|---|---|
| Vi-SiOC-CDC | 11.0 | 97.6 | 97.8 |
| Ph0.25Vi0.75-SiOC-CDC | 13.0 | 85.1 | 87.1 |
| Ph0.5Vi0.5-SiOC-CDC | 15.0 | 76.1 | 79.6 |
| Ph0.75Vi0.25-SiOC-CDC | 20.7 | 72.4 | 78.1 |
The Raman spectra and XRD pattern of the SiOCs and CDCs with different ratios of vinyl- and phenyltrimethoxysilanes are very similar; therefore, Fig. 3 only depicts data for the Ph0.5Vi0.5-SiOC samples (and the remaining Raman spectra and XRD patterns are found in ESI, Fig. S3†). The SiOC Raman spectra (Fig. 3A) show the presence of incompletely graphitic carbon, due to the position of the D- and G-mode at 1331 cm−1 and 1603 cm−1. The Vi-SiOC shows a higher amount of amorphous carbon than the other SiOC samples, which can be recognized by having a look to the broader full-width at half-maximum (FWHM) of 181 cm−1 and 111 cm−1, compared to 159 cm−1 and 62 cm−1 of Ph0.5Vi0.5-SiOC (Table 4). The ID/IG ratios of the SiOCs ranged between 4.27 and 4.67.
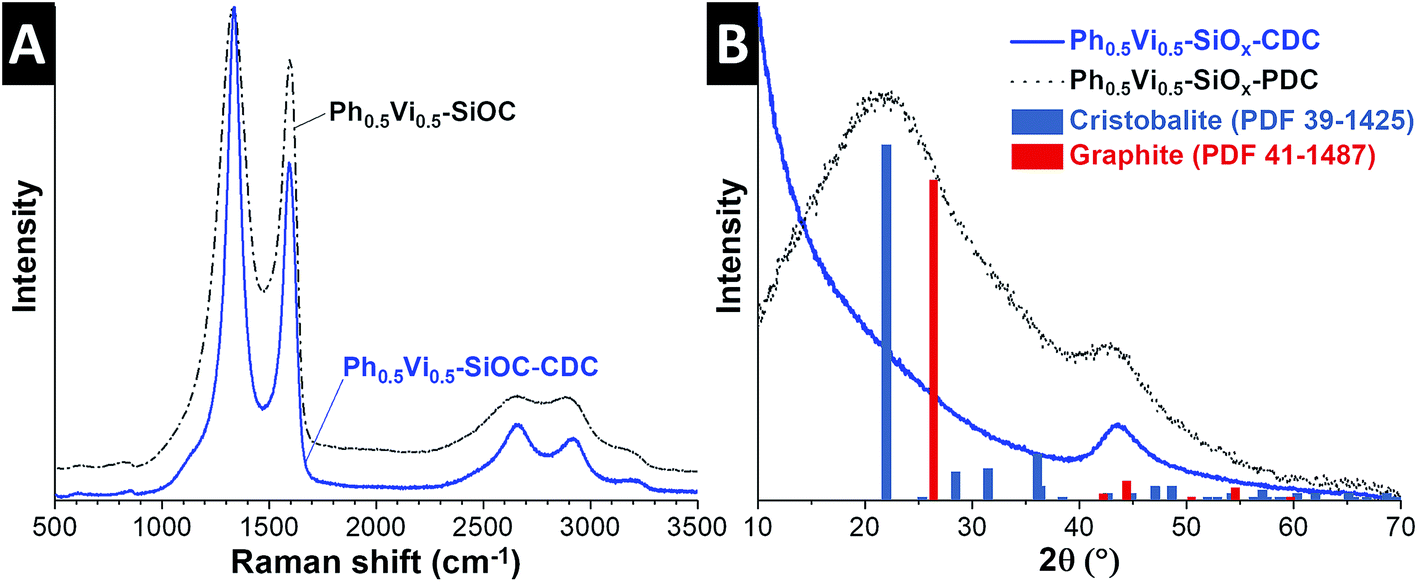 | ||
| Fig. 3 Raman spectra (A) and XRD pattern (B) of the SiOC and CDC from Ph0.5Vi0.5-SiOC. | ||
| Mode | Position (cm−1) | FWHM (cm−1) | I D/IG | |
|---|---|---|---|---|
| Vi-SiOC | D | 1334 | 181 | 4.27 |
| G | 1587 | 111 | ||
| Ph0.25Vi0.75-SiOC | D | 1331 | 162 | 4.67 |
| G | 1601 | 67 | ||
| Ph0.5Vi0.5-SiOC | D | 1333 | 159 | 4.58 |
| G | 1603 | 62 | ||
| Ph0.75Vi0.25-SiOC | D | 1324 | 167 | 4.40 |
| G | 1599 | 66 | ||
| Vi-SiOC-CDC | D | 1333 | 85 | 2.80 |
| G | 1593 | 65 | ||
| Ph0.25Vi0.75-SiOC-CDC | D | 1337 | 89 | 2.40 |
| G | 1597 | 66 | ||
| Ph0.5Vi0.5-SiOC-CDC | D | 1337 | 84 | 2.55 |
| G | 1596 | 68 | ||
| Ph0.75Vi0.25-SiOC-CDC | D | 1339 | 76 | 2.56 |
| G | 1601 | 67 |
The Raman spectra of the CDCs (Fig. 3A) show nanocrystalline graphitic carbon nature. This can also be recognized by the position of the D- and G-mode at 1337 cm−1 and 1596 cm−1.36 The FWHM decreased significantly to 84 cm−1 for the D-mode and 68 cm−1 for the G-mode. The ID/IG ratios of the CDCs are lower (2.40–2.80) compared to the SiOCs. It appears that the amount of amorphous carbon is reduced by comparing the SiOC with the CDC. Yet, it is reasonable to assume that amorphous carbon formed by the pyrolysis did not entirely disappear after the chlorine gas treatment. Instead, additional nanocrystalline carbon is formed by the chlorine treatment at 1200 °C by converting the SiOC to CDC; this leads to a relative lower amount of the amorphous phase compared to the carbon with more structural ordering.37,38 Due to the high temperature during the CDC formation, the carbon of all CDCs has a similar (narrow) FWHM and positions of the D- and G-mode (Table 4).
The XRD pattern of the SiOC in Fig. 3B show mainly two broad signals at 22° 2θ and 44° 2θ. The broad peak at 22° 2θ is related to the short-range order of SiO4 tetrahedra and the 44° 2θ is related to (101)-graphite. The results from EDX, Raman spectroscopy, and XRD show that all PDCs contain amorphous silicon oxide and carbon, which is consistent with the literature.39,40 The XRD pattern of the CDCs in Fig. 3B show no signal related to amorphous silicon oxide; only the peak at 44° 2θ from (101)-graphite is visible. The results of Raman spectroscopy and the X-ray diffraction indicate only carbons are present in the CDC.
Fig. 4 illustrates the morphology of the CDCs by SEM and TEM images. The average diameters of the particles obtained by the SEM images are given in Table 5. The Vi-SiOC-CDCs are the smallest with the narrowest distribution wide (0.68 ± 0.09 μm). Particles with vinyl and phenyl groups are larger and have a broader size distribution of 2.20 ± 0.48 μm (Ph0.25Vi0.75-SiOC-CDC), 2.54 ± 0.58 μm (Ph0.5Vi0.5-SiOC-CDC), and 1.81 ± 0.28 μm (Ph0.75Vi0.25-SiOC-CDC). The broad size distribution of the particles favors effective packing in the electrode, which may benefit the electrical conductivity. On the surface of the particles, small fractures are noticeable where the particles were agglomerated. The TEM images of all CDC samples show disordered carbon, which is consistent with the XRD pattern (Fig. 3B).
 | ||
| Fig. 4 Scanning and transmission electron micrographs of Vi-SiOC-CDC (A), Ph0.25Vi0.75-SiOC-CDC (B), Ph0.5Vi0.5-SiOC-CDC (C), and Ph0.75Vi0.25-SiOC-CDC (D). | ||
| Average particle size (μm) | SSADFT (m2 g−1) | SSABET (m2 g−1) | Total pore volume (cm3 g−1) | Average pore size (nm) | |
|---|---|---|---|---|---|
| Vi-SiOC-CDC | 0.68 ± 0.09 | 2044 | 2473 | 2.06 | 2.9 |
| Ph0.25Vi0.75-SiOC-CDC | 2.20 ± 0.48 | 2198 | 2905 | 1.67 | 1.7 |
| Ph0.5Vi0.5-SiOC-CDC | 2.54 ± 0.58 | 2114 | 2729 | 1.40 | 1.5 |
| Ph0.75Vi0.25-SiOC-CDC | 1.81 ± 0.28 | 2014 | 2554 | 1.27 | 1.4 |
Ph0.75Vi0.25-SiOC-CDC, Ph0.5Vi0.5-SiOC-CDC, and Ph0.25Vi0.75-SiOC-CDC have a type I isotherm resulting from a high amount of micropores measured with N2 GSA in Fig. 5A. Only Vi-SiOC-CDC has a type IV isotherm because it has also a high amount of mesopores, which increase the pore volume. Therefore, Vi-SiOC-CDC shows the highest total pore volume with 2.06 cm3 g−1. The total pore volume is steadily reduced by adding a higher amount of phenyl groups to the siloxane to 1.67 cm3 g−1 for Ph0.25Vi0.75-SiOC-CDC, 1.40 cm3 g−1 for Ph0.5Vi0.5-SiOC-CDC, and 1.27 cm3 g−1 for Ph0.75Vi0.25-SiOC-CDC. Also, the average pore size was reduced from 2.9 nm to 1.4 nm. Yet, the DFT SSA remains rather constant with values between 2014 m2 g−1 and 2198 m2 g−1. In Table 5, we provide values obtained from the N2 GSA. We clearly see that the CDC porosity can be modified in a controllable way by adjusting the functional groups of the silanes which were used as precursor.
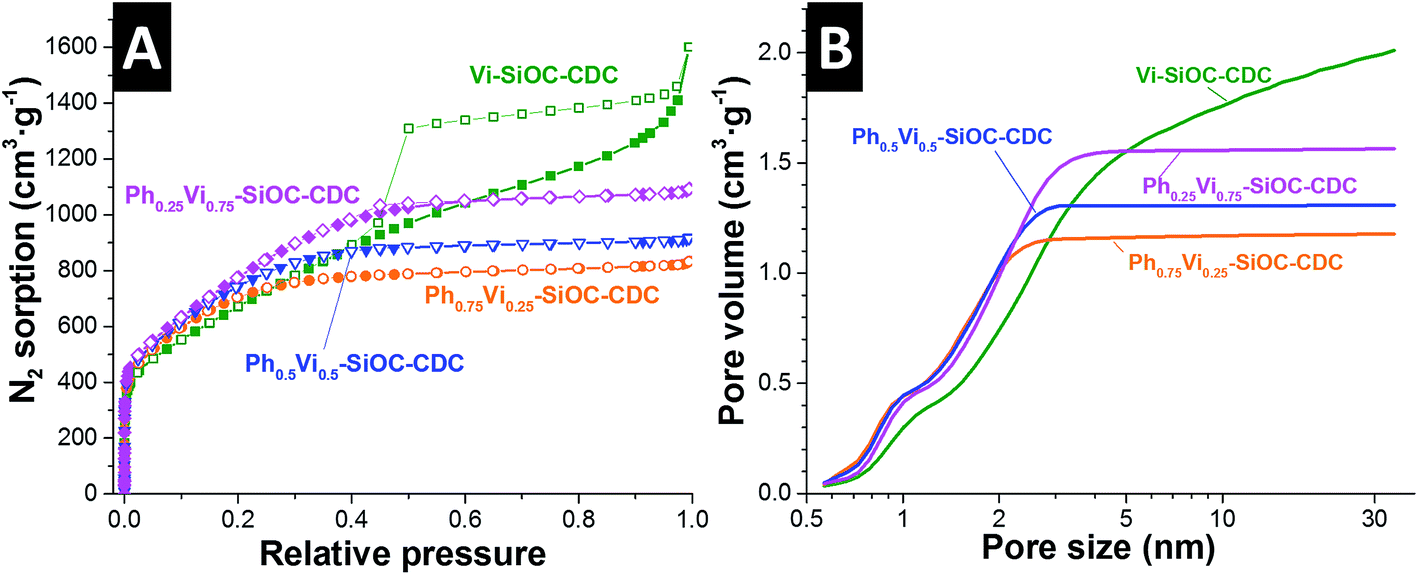 | ||
| Fig. 5 Nitrogen gas sorption isotherms recorded at −196 °C of the CDC samples (A) and the corresponding pore size distributions applying a QSDFT model assuming slit-like pores (B). | ||
3.3. Supercapacitor performance
The electrochemical performance of the SiOC-CDC electrodes was tested in a symmetrical supercapacitor cell using the most commonly used organic electrolyte (1 M in TEA-BF4 in ACN). The CVs with the organic electrolyte in Fig. 6A shows a typical rectangular shape, typical for nanoporous carbon.28 A significant difference of the mass-normalized CVs of the different samples is not distinguishable. The specific capacitances measured in GCPL mode are denoted in Table 6. Ph0.25Vi0.75-SiOC-CDC has a slightly higher specific capacitance in TEA-BF4 in ACN with 116 F g−1 than the other materials (Vi-SiOC-CDC: 111 F g−1; Ph0.5Vi0.5-SiOC-CDC: 106 F g−1; Ph0.75Vi0.25-SiOC-CDC: 112 F g−1). Commercial microporous activated carbon (AC; see ref. 41 for AC properties) has a similar specific capacitance of 110 F g−1 under the same measurement conditions. The variable porosity of the SiOC-CDCs influences the rate handling behavior much stronger than the total capacitance plotted in Fig. 6B. While the AC has only a residual capacitance of 15% at an ultrahigh specific current of 100 A g−1, the sample Ph0.75Vi0.25-SiOC-CDC retains twice as much capacitance under the same conditions (31%). With an increasing mesoporous fraction and an increasing pore volume, the capacitance retention at 100 A g−1 increases to 56% for Ph0.5Vi0.5-SiOC-CDC and Ph0.25Vi0.75-SiOC-CDC and to up to 72% for Vi-SiOC-CDC. We can explain the superior power handling ability of Vi-SiOC-CDC also by the smaller particle size.42 In Table 6, we see a comparison of the performance of the MicroJet SiOC-CDCs with literature values. There is a clear advantage of the MicroJet SiOC-CDCs of retaining a high specific capacitance at high specific currents (10 A g−1 or 100 A g−1). Even materials optimized for high power handling, such as electrospun CDC fiber mats,13 show a higher capacitance loss at 10 A g−1 or 100 A g−1 than electrodes made from Vi-SiOC-CDC when using a similar thickness and measurement conditions. The Coulombic efficiencies are plotted in the ESI (Fig. S4A†) and we see all values approaching 99% at around 1 A g−1.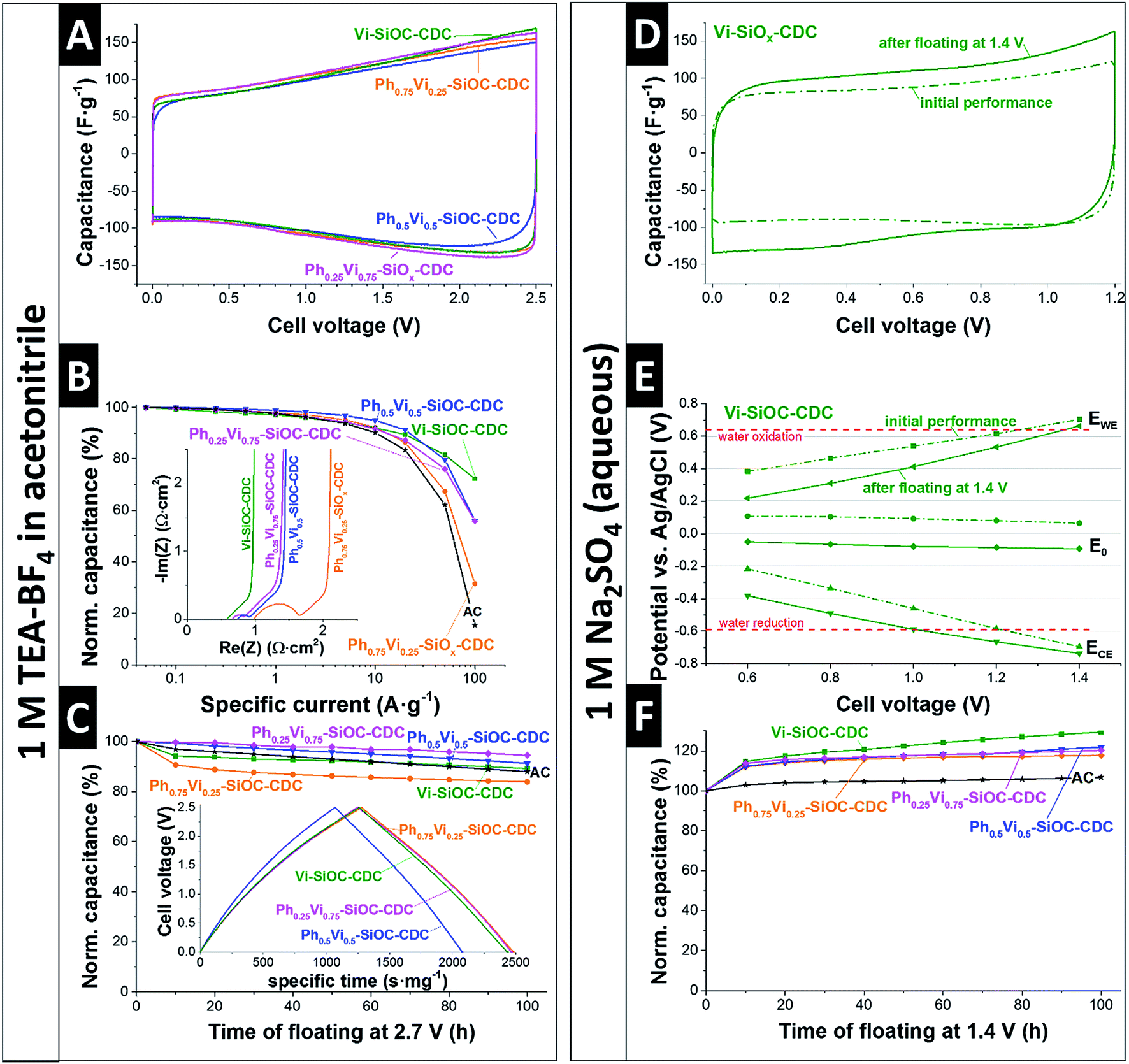 | ||
| Fig. 6 Cyclic voltammograms (A), rate handling behavior with Nyquist plot as inset (B), and the stability test operating with voltage floating at 2.7 V including the GCPL curve to 2.5 V at 0.05 A g−1 as inset (C) of the CDCs in TEA-BF4 in ACN. Cyclic voltammograms (D) of Vi-SiOC-CDC before and after the voltage floating test at 1.4 V (F), and monitored potentials of the positive and negative electrode including the zero-charge potential (E) in aqueous Na2SO4. | ||
| 1 M TEA-BF4 in ACN | Aqueous 1 M Na2SO4 | SSADFT (m2 g−1) | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Low-rate capacitance (F g−1) | Capacitance loss at 10 A g−1 (%) | Capacitance loss at 100 A g−1 (%) | Initial specific capacitance (F g−1) | After floating, specific capacitance (F g−1) | |||
| Vi-SiOC-CDC | 111 (at 0.05 A g−1) | 8 | 28 | 98 | 126 | 2044 | This work |
| Ph0.25Vi0.75-SiOC-CDC | 116 (at 0.05 A g−1) | 8 | 44 | 113 | 135 | 2198 | This work |
| Ph0.5Vi0.5-SiOC-CDC | 106 (at 0.05 A g−1) | 5 | 44 | 116 | 135 | 2114 | This work |
| Ph0.75Vi0.25-SiOC-CDC | 112 (at 0.05 A g−1) | 8 | 69 | 104 | 123 | 2014 | This work |
| AC | 110 (at 0.05 A g−1) | 10 | 85 | 143 | 152 | 1756 | This work |
| Emulsion CDC-NS-70-30 | 130 (at 0.05 A g−1) | 8 | n.r. | 103 | n.r. | 2298 | Ref. 15 |
| Electro-sprayed SiOC-CDC beads | 117 (at 0.1 A g−1) | 20 | 95 | n.r. | n.r. | 2227 | Ref. 13 |
| Electrospun SiOC-CDC fiber mat | 130 (at 0.1 A g−1) | 14 | 37 | n.r. | n.r. | 2394 | Ref. 13 |
| Activated carbon black (BP2000) | 86 (at 0.1 A g−1) | 9 | n.r. | n.r. | n.r. | 1389 | Ref. 55 |
| Carbon onions | 24 (at 0.1 A g−1) | 10 | n.r. | n.r. | n.r. | 404 | Ref. 59 |
| N-doped activated Lignin-derived carbon | 147 (at 0.1 A g−1) | 19 | n.r. | n.r. | n.r. | 2353 | Ref. 60 |
The rate handling ability is influenced by the resistance of the electrodes. Using electrochemical impedance spectroscopy (EIS), we quantified the electrical serial resistance (ESR) and electrical distribution resistance (EDR), as seen in Table 7. Vi-SiOC-CDC, which shows the best rate behavior, has the lowest ESR with 0.59 Ω cm2. Ph0.25Vi0.75-SiOC-CDC and Ph0.5Vi0.5-SiOC-CDC have a slightly higher ESR of 0.66 Ω cm2 and 0.74 Ω cm2, respectively. Ph0.75Vi0.25-SiOC-CDC, which was also the CDC with the lowest performance at high rates has the highest ESR of 0.98 Ω cm2. An increasing ESR value correlates with a reduced performance at high specific currents. The EDR value of Vi-SiOC-CDC is also the lowest with 0.35 Ω cm2. The other EDR values are very close and vary in the range of 0.44–0.51 Ω cm2 without a systematic trend.
| ESR (Ω cm2) | EDR (Ω cm2) | |
|---|---|---|
| Vi-SiOC-CDC | 0.59 | 0.35 |
| Ph0.25Vi0.75-SiOC-CDC | 0.66 | 0.51 |
| Ph0.5Vi0.5-SiOC-CDC | 0.74 | 0.49 |
| Ph0.75Vi0.25-SiOC-CDC | 0.98 | 0.44 |
Also, the performance stability of the system is very high, as can be seen in Fig. 6C. After voltage floating at 2.7 V for 100 h in 1 M TEA-BF4 in ACN, all materials show a residual specific capacitance between 84% and 95%. The commercial AC has a similar residual capacitance of 89%.41 Yet, a comparison with literature values is difficult because the electrochemical stability is influenced by the carbon structure,41 presence and type of functional groups,41 the electrochemical operation window,43 measurement conditions,44 and other cell components like the current collector.45
In addition to a common organic electrolyte, we also benchmarked the supercapacitor performance in an aqueous medium. We see important differences of the supercapacitor performance by using an aqueous electrolyte as compared to the organic electrolyte. The initial CV of Vi-SiOC-CDC in Fig. 6D shows a typical rectangular shape of a supercapacitor with a relatively low specific capacitance of only 98 F g−1. All other CVs are very similar (ESI, Fig. S4†). The capacitance was not reduced after voltage floating at 1.4 V for 100 h; yet, the specific capacitance of Vi-SiOC-CDC increased to 126 F g−1 instead of an assumed loss of capacitance (Fig. 6F). It was already shown that aqueous supercapacitors with Na2SO4 can have a very high stability at high potentials which exceed the thermodynamic stability window of water of 1.2 V.46 The improved performance after floating can be related to (i) progressing wetting,47–50 and (ii) reversible faradaic redox-reactions of oxygen functional groups,51 and reversible hydrogen reaction.52–54
A three-electrode cell with an Ag/AgCl reference electrode provides information about the potential development in the symmetrical setup (Fig. 6E). The initial electrodes have a zero-charge potential of +64 mV versus Ag/AgCl at a cell voltage of 1.4 V. The potential at the positive electrode is 0.70 V versus Ag/AgCl, exceeding the thermodynamic water oxidation of 0.64 V versus Ag/AgCl.46 Also, the potential at the negative electrode is at −0.70 V versus Ag/AgCl below the limit of water reduction at −0.59 V versus Ag/AgCl.46 The high potential at the positive electrode leads to the assumption that an irreversible oxidation of the carbon took place during the voltage holding at 1.4 V. The zero-charge potential was reduced from +64 mV to −93 mV versus Ag/AgCl after the longtime test which can be explained with an increase of oxygen containing functional groups on the carbon surface introduced by the oxidation at high cell voltages.
The aqueous Vi-SiOC-CDC cell was disassembled after the stability test to perform a post mortem analysis via gas sorption analysis, EDX, and contact angle measurements. By comparing the nitrogen sorption isotherms of the powder (Fig. 5A) with the isotherms of the electrodes (Fig. 7A) it is striking that the porosity is reduced. The addition of PTFE-binder leads to a smaller porosity due to an additional mass and pore blocking.55 By comparing the isotherms of the initial electrode with the electrodes after the voltage floating in Fig. 7A, a significant pore volume loss is evident. Table S2† (ESI) supplies the detailed values obtained from the isotherms. The DFT surface area was reduced from 1756 m2 g−1 to 980 m2 g−1 at the negative and 897 m2 g−1 at the positive electrode. The total pore volume also decreased from 1.83 m2 g−1 to 1.23 m2 g−1 at the negative, and 1.19 m2 g−1 at the positive side, which might also be influenced by residual salt from the electrolyte. We see from the normalized pore size distribution in Fig. 7B (i.e., normalized to 100%) that the pore volume is reduced mainly in the micropore range. The reduction of the micropores explains the loss of surface area of 49% compared to a relative low reduction of the total pore volume of 35% of the positive electrode. This is an indication for surface functionalities which block small micropores for N2 during the GSA measurement. The increase of oxygen content further supports this assumption (Fig. 7D).
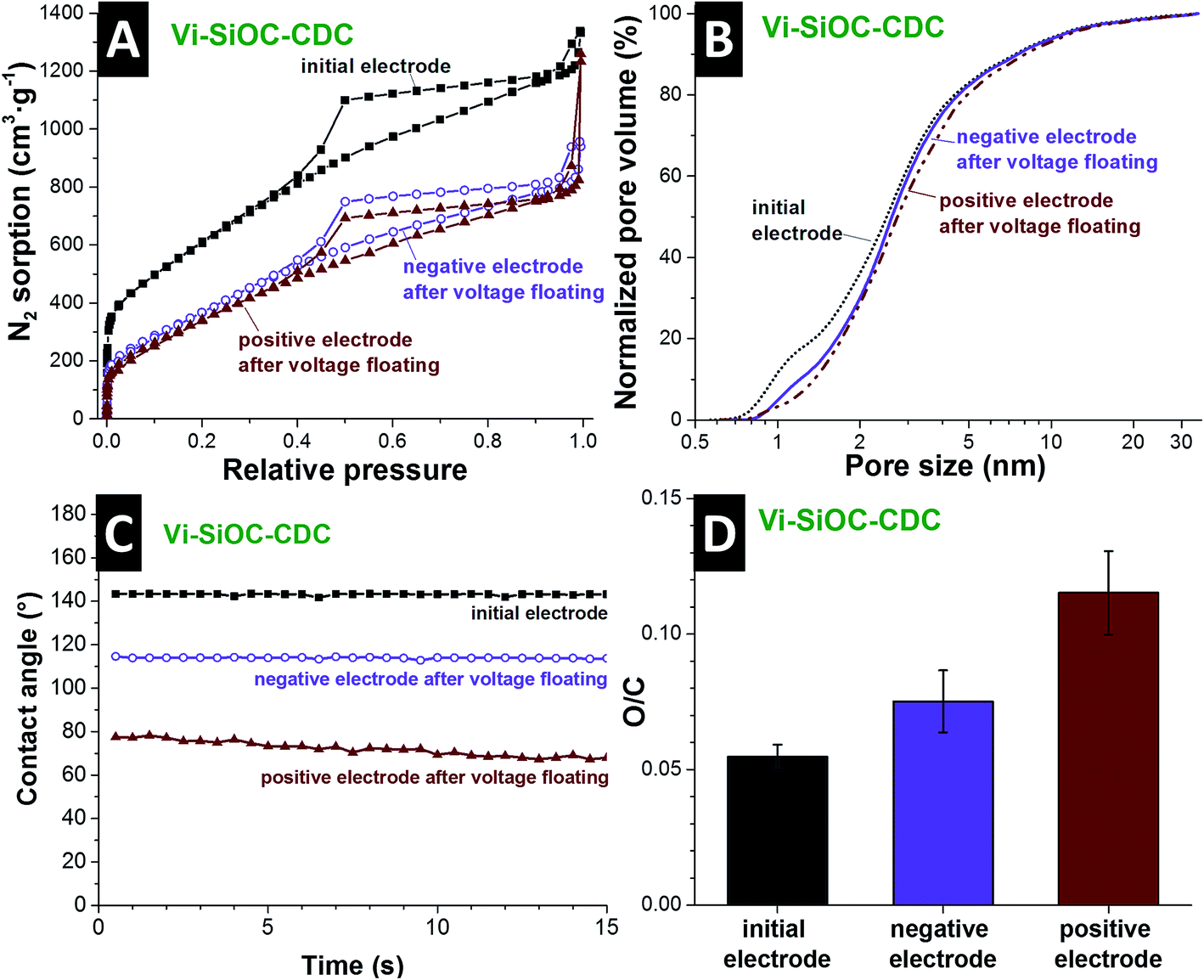 | ||
| Fig. 7 Post mortem analysis of the Vi-SiOC-CDC electrodes after the voltage floating in aqueous 1 M Na2SO4 at 1.4 V cell voltage compared to the initial electrodes. N2 gas sorption isotherms (A), normalized pore size distribution (B), contact angle (C), and the oxygen/carbon ratio measured via EDX (D). | ||
Post mortem EDX analysis was conducted, showing an increase in the oxygen-to-carbon ratio from 0.055 of the initial electrode to 0.075 on the negative, and 0.115 on the positive electrode after the voltage floating (ESI, Table S3†). The increase of oxygen-containing functional groups also influences the wetting behavior of the electrodes (Fig. 7C). Initially, the Vi-SiOC-CDC electrode exhibits a contact angle with water of 143° after 1 s. This is relatively high compared to other highly porous carbon materials, like the AC with an initial contact angle of 63° (ref. 56) or CO2-activated novolac-derived carbons with 121° (ref. 57). The contact angle of the negative electrode after the voltage floating is reduced to 114°. At the positive electrode, oxidation mainly takes place during electrochemical operation, leading to an even lower contact angle of 77° after 1 s and faster water absorption. Besides functional surface groups of the carbon, the contact angle is also influenced by the hydrophobic character of the PTFE-binder,58 which allows only a relative comparison of the samples. The oxidation of the carbon surface and the permanent operation during the testing led to an increase of the wettability of the carbon. Thereby, we enabled enhanced access for the aqueous electrolyte to the surface of the CDCs, which led to an increase of the capacitance after the voltage floating of 16–29%.
4. Conclusions
In this study, phenyl- and vinyltrimethoxysilane mixtures with four different ratios were used to synthesize polysiloxane polymer beads. The MicroJet reactor technique allows continuous manufacturing of polysiloxanes beads with a constant quality at rates of 10–15 g min−1. These beads were found as highly suited for pyrolysis, to obtain SiOC, and chlorine gas treatment, to obtain CDC by removal of non-carbon elements and still conserving the spherical morphology. An increase of phenyl groups increased the total yield after pyrolysis and etching from 2.2 mass% to 21.9 mass%, which is relevant for the economic efficiency to produce highly porous carbon materials. A possible way to increase the yield might be the use of organic functional groups with a higher amount of carbon, like naphthyl or anthracenyl groups. Also, the combination of the pyrolysis and chlorine treatment should be considered to improve the process.By varying the ratio of phenyl and vinyl groups it was possible to produce highly porous CDCs with DFT SSA ranging between 2014-2198 m2 g−1 with a total pore volume in the range of 1.27–2.06 cm3 g−1 without any additional activation step. A higher amount of vinyl groups leads to a higher total pore volume. The increased mesoporosity influences mainly the rate handling behavior of the supercapacitor, while the specific capacitance at low ranges (5 mA g−1) is in 1 M TEA-BF4 in ACN very similar for all synthesized CDCs (106–116 F g−1). The highest residual capacitance of 72% at high current rates (100 A g−1) was also obtained from the sample with the highest amount of mesopores.
The Ragone chart (Fig. 8) illustrates the specific energy of the CDC materials of 25 W h kg−1 at low specific powers and the excellent performance at high specific power. In the best case (Vi-SiOC-CDC), the specific energy only decreases slightly to 12 W h kg−1 at high specific powers of 41 kW kg−1. For comparison: AC shows a very low rate handling ability with only 1 W h kg−1 at 25 kW kg−1 (which is lower than any of the samples studied in this work). The low wettability of the relatively graphitic carbons is unfavorable for aqueous electrolytes, but a long-time testing shows an increase of the specific capacitance of almost 30% when floated at 1.4 V for 100 h. Functional groups are formed by the operation, which improves the wettability as well as the specific capacitance.
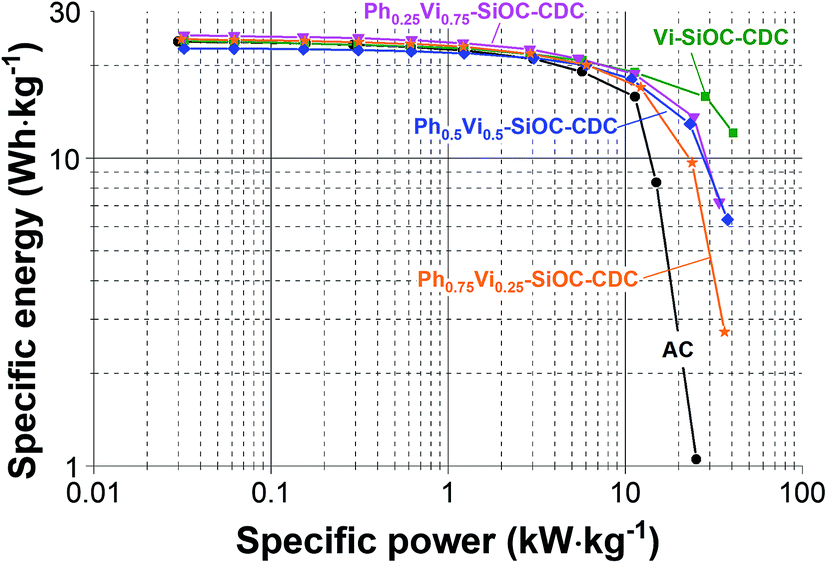 | ||
| Fig. 8 Ragone chart of the four spherical CDC materials and the AC as comparison, measured in 1 M TEA-BF4 in ACN (electrode thickness: 110–130 μm). | ||
In conclusion, the high porosity of the SiOC-CDCs is attractive for electrochemical energy storage with supercapacitors. The excellent rate handling behavior and the high stability in organic electrolyte documents suitability for common supercapacitors.
Acknowledgements
The INM authors kindly acknowledge the continuing support of Prof. Eduard Arzt (INM). We also thank Robert Drumm (INM) for the TGA-MS, Dr Ingrid Grobelsek (INM) for the help with the SEM, Dr Michael Zimmer (UdS) for CP-MAS NMR, and Juhan Lee, Simon Fleischmann, and Nicolas Jäckel (all INM) for fruitful discussions and their support. The INM acknowledges funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework.References
- L. Schlapbach and A. Züttel, Hydrogen-Storage Materials for Mobile Applications, Nature, 2001, 414(6861), 353–358 CrossRef CAS PubMed.
- R. E. Morris and P. S. Wheatley, Gasspeicherung in Nanoporösen Materialien, Angew. Chem., 2008, 120(27), 5044–5059 CrossRef.
- A. Taguchi and F. Schüth, Ordered Mesoporous Materials in Catalysis, Microporous Mesoporous Mater., 2005, 77(1), 1–45 CrossRef CAS.
- P. Simon and Y. Gogotsi, Materials for Electrochemical Capacitors, Nat. Mater., 2008, 7(11), 845–854 CrossRef CAS PubMed.
- M. Lu, F. Beguin, E. Frackowiak, Supercapacitors: Materials, Systems and Applications, John Wiley & Sons, 2013 Search PubMed.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. P. Grey, B. Dunn and P. Simon, Efficient Storage Mechanisms for Building Better Supercapacitors, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- V. Presser, M. Heon and Y. Gogotsi, Carbide-Derived Carbons–From Porous Networks to Nanotubes and Graphene, Adv. Funct. Mater., 2011, 21(5), 810–833 CrossRef CAS.
- R. Dash, G. Yushin and Y. Gogotsi, Synthesis, Structure and Porosity Analysis of Microporous and Mesoporous Carbon Derived from Zirconium Carbide, Microporous Mesoporous Mater., 2005, 86(1), 50–57 CrossRef CAS.
- S.-H. Yeon, P. Reddington, Y. Gogotsi, J. E. Fischer, C. Vakifahmetoglu and P. Colombo, Carbide-Derived-Carbons with Hierarchical Porosity from a Preceramic Polymer, Carbon, 2010, 48(1), 201–210 CrossRef CAS.
- C. Vakifahmetoglu, V. Presser, S.-H. Yeon, P. Colombo and Y. Gogotsi, Enhanced Hydrogen and Methane Gas Storage of Silicon Oxycarbide Derived Carbon, Microporous Mesoporous Mater., 2011, 144(1), 105–112 CrossRef CAS.
- V. Presser, J. McDonough, S.-H. Yeon and Y. Gogotsi, Effect of Pore Size on Carbon Dioxide Sorption by Carbide Derived Carbon, Energy Environ. Sci., 2011, 4(8), 3059–3066 CAS.
- S. Welz, M. J. McNallan and Y. Gogotsi, Carbon Structures in Silicon Carbide Derived Carbon, J. Mater. Process. Technol., 2006, 179(1), 11–22 CrossRef CAS.
- A. Tolosa, B. Krüner, N. Jäckel, M. Aslan, C. Vakifahmetoglu and V. Presser, Electrospinning and Electrospraying of Silicon Oxycarbide-Derived Nanoporous Carbon for Supercapacitor Electrodes, J. Power Sources, 2016, 313, 178–188 CrossRef CAS.
- V. Bakumov, M. Schwarz and E. Kroke, Emulsion Processing and Size Control of Polymer-Derived Spherical Si/C/O Ceramic Particles, Soft Mater., 2007, 4(2–4), 287–299 CrossRef.
- M. Oschatz, M. Zeiger, N. Jäckel, P. Strubel, L. Borchardt, R. Reinhold, W. Nickel, J. Eckert, V. Presser and S. Kaskel, Emulsion Soft Templating of Carbide-Derived Carbon Nanospheres with Controllable Porosity for Capacitive Electrochemical Energy Storage, J. Mater. Chem. A, 2015, 3(35), 17983–17990 CAS.
- A. Guo, M. Roso, M. Modesti, J. Liu and P. Colombo, Preceramic Polymer-Derived SiOC Fibers by Electrospinning, J. Appl. Polym. Sci., 2014, 131(3), 39836 CrossRef.
- S. A. Khan, A. Günther, M. A. Schmidt and K. F. Jensen, Microfluidic Synthesis of Colloidal Silica, Langmuir, 2004, 20(20), 8604–8611 CrossRef CAS PubMed.
- N. Jongen, M. Donnet, P. Bowen, J. Lemaître, H. Hofmann, R. Schenk, C. Hofmann, M. Aoun-Habbache, S. Guillemet-Fritsch and J. Sarrias, Development of a Continuous Segmented Flow Tubular Reactor and the “Scale-out” Concept–In Search of Perfect Powders, Chem. Eng. Technol., 2003, 26(3), 303–305 CrossRef CAS.
- B. Penth, Kontinuierliche Fällung von Nanoskaligen Produkten in Mikroreaktoren, German Patent DE102006004350 A1, 2007.
- B. Dittert, A. Gavrilović, S. Schwarz, P. Angerer, H. Steiner and R. Schöftner, Phase Content Controlled TiO2 Nanoparticles using the MicroJetReactor Technology, J. Eur. Ceram. Soc., 2011, 31(14), 2475–2480 CrossRef CAS.
- A. Rüfer, K. Räuchle, F. Krahl and W. Reschetilowski, Kontinuierliche Darstellung von Bariumsulfat-Nanopartikeln im MicroJet-Reaktor, Chem. Ing. Tech., 2009, 81(12), 1949–1954 CrossRef.
- A. Betke and G. Kickelbick, Bottom-Up, Wet Chemical Technique for the Continuous Synthesis of Inorganic Nanoparticles, Inorganics, 2014, 2(1), 1–15 CrossRef CAS.
- A. Matsuda, T. Sasaki, K. Hasegawa, M. Tatsumisago and T. Minami, Thermal Softening Behavior of Poly (Phenylsilsesquioxane) and Poly (Benzylsilsesquioxane) Particles, J. Ceram. Soc. Jpn., 2000, 108(1261), 830–835 CrossRef CAS.
- K. Katagiri, K. Hasegawa, A. Matsuda, M. Tatsumisago and T. Minami, Preparation of Transparent Thick Films by Electrophoretic Sol-Gel Deposition Using Phenyltriethoxysilane-Derived Particles, J. Am. Ceram. Soc., 1998, 81(9), 2501–2503 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 Years of Image Analysis, Nat. Methods, 2012, 9(7), 671 CrossRef CAS PubMed.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Quenched Solid Density Functional Theory Method for Characterization of Mesoporous Carbons by Nitrogen Adsorption, Carbon, 2012, 50(4), 1583–1590 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60(2), 309–319 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Graphitization as a Universal Tool to Tailor the Potential-Dependent Capacitance of Carbon Supercapacitors, Adv. Energy Mater., 2014, 4(13), 1400316 CrossRef.
- R. K. Sharma, S. Das and A. Maitra, Surface Modified Ormosil Nanoparticles, J. Colloid Interface Sci., 2004, 277(2), 342–346 CrossRef CAS PubMed.
- A. Arkhireeva and J. N. Hay, Synthesis of Sub-200 nm Silsesquioxane Particles using a Modified Stöber Sol–Gel Route, J. Mater. Chem., 2003, 13(12), 3122–3127 RSC.
- J. Macan, K. Tadanaga and M. Tatsumisago, Influence of Copolymerization with Alkyltrialkoxysilanes on Condensation and Thermal Behaviour of Poly (Phenylsilsesquioxane) Particles, J. Sol-Gel Sci. Technol., 2010, 53(1), 31–37 CrossRef CAS.
- J. F. Brown Jr, L. H. Vogt Jr, A. Katchman, J. W. Eustance, K. M. Kiser and K. W. Krantz, Double Chain Polymers of Phenylsilsesquioxane, J. Am. Chem. Soc., 1960, 82(23), 6194–6195 CrossRef.
- Z. Li, X. Cao, H. Xu, P. Xie, M. Cao and R. Zhang, Synthesis and Characterization of Reactive Ladderlike Polyallylsilsesquioxane and Polyvinylsilsesquioxane, React. Funct. Polym., 1999, 39(1), 1–7 CrossRef CAS.
- P. Scherrer, Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen, Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, 1918, 2, 3 Search PubMed.
- S.-S. Choi, A. S. Lee, S. S. Hwang and K.-Y. Baek, Structural Control of Fully Condensed Polysilsesquioxanes: Ladderlike vs Cage Structured Polyphenylsilsesquioxanes, Macromolecules, 2015, 48(17), 6063–6070 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Interpretation of Raman Spectra of Disordered and Amorphous Carbon, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61(20), 14095 CrossRef CAS.
- V. Presser, L. Zhang, J. J. Niu, J. McDonough, C. Perez, H. Fong and Y. Gogotsi, Flexible Nano-felts of Carbide-Derived Carbon with Ultra-high Power Handling Capability, Adv. Energy Mater., 2011, 1(3), 423–430 CrossRef CAS.
- Y. Gao, V. Presser, L. Zhang, J. J. Niu, J. K. McDonough, C. R. Pérez, H. Lin, H. Fong and Y. Gogotsi, High Power Supercapacitor Electrodes Based on Flexible TiC-CDC Nano-felts, J. Power Sources, 2012, 201, 368–375 CrossRef CAS.
- A. Scarmi, G. D. Sorarù and R. Raj, The Role of Carbon in Unexpected Visco (an) Elastic Behavior of Amorphous Silicon Oxycarbide Above 1273K, J. Non-Cryst. Solids, 2005, 351(27), 2238–2243 CrossRef CAS.
- H.-J. Kleebe and Y. D. Blum, SiOC Ceramic with High Excess Free Carbon, J. Eur. Ceram. Soc., 2008, 28(5), 1037–1042 CrossRef CAS.
- N. Jäckel, D. Weingarth, A. Schreiber, B. Krüner, M. Zeiger, A. Tolosa, M. Aslan and V. Presser, Performance Evaluation of Conductive Additives for Activated Carbon Supercapacitors in Organic Electrolyte, Electrochim. Acta, 2016, 191, 284–298 CrossRef.
- C. R. Pérez, S. H. Yeon, J. Ségalini, V. Presser, P. L. Taberna, P. Simon and Y. Gogotsi, Structure and Electrochemical Performance of Carbide-Derived Carbon Nanopowders, Adv. Funct. Mater., 2013, 23(8), 1081–1089 CrossRef.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, A Reliable Determination Method of Stability Limits for Electrochemical Double Layer Capacitors, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS.
- D. Weingarth, A. Foelske-Schmitz and R. Kötz, Cycle versus Voltage Hold–Which is the better Stability Test for Electrochemical Double Layer Capacitors?, J. Power Sources, 2013, 225, 84–88 CrossRef CAS.
- J. Busom, A. Schreiber, A. Tolosa, N. Jäckel, I. Grobelsek, N. J. Peter and V. Presser, Sputtering of Sub-Micrometer Aluminum Layers as Compact, High-Performance, Light-Weight Current Collector for Supercapacitors, J. Power Sources, 2016, 329, 432–440 CrossRef CAS.
- M. Bichat, E. Raymundo-Piñero and F. Béguin, High Voltage Supercapacitor Built with Seaweed Carbons in Neutral Aqueous Electrolyte, Carbon, 2010, 48(15), 4351–4361 CrossRef CAS.
- J. Lee, D. Weingarth, I. Grobelsek and V. Presser, Use of Surfactants for Continuous Operation of Aqueous Electrochemical Flow Capacitors, Energy Technol., 2016, 4(1), 75–84 CrossRef CAS.
- K. Fic, G. Lota and E. Frackowiak, Effect of Surfactants on Capacitance Properties of Carbon Electrodes, Electrochim. Acta, 2012, 60, 206–212 CrossRef CAS.
- M. Aslan, D. Weingarth, P. Herbeck-Engel, I. Grobelsek and V. Presser, Polyvinylpyrrolidone/Polyvinyl Butyral Composite as a Stable Binder for Castable Supercapacitor Electrodes in Aqueous Electrolytes, J. Power Sources, 2015, 279, 323–333 CrossRef CAS.
- J. Lee, S. Kim and J. Yoon, Rocking Chair Desalination Battery Based on Prussian Blue Electrodes, ACS Omega, 2017, 2(4), 1653–1659 CrossRef CAS.
- K. Fic, M. Meller, J. Menzel and E. Frackowiak, Around the Thermodynamic Limitations of Supercapacitors Operating in Aqueous Electrolytes, Electrochim. Acta, 2016, 206, 496–503 CrossRef CAS.
- E. Frackowiak and F. Beguin, Electrochemical Storage of Energy in Carbon Nanotubes and Nanostructured Carbons, Carbon, 2002, 40(10), 1775–1787 CrossRef CAS.
- J. Lee, A. Tolosa, B. Krüner, N. Jäckel, S. Fleischmann, M. Zeiger, D. Kim and V. Presser, Asymmetric Tin–Vanadium Redox Electrolyte for Hybrid Energy Storage with Nanoporous Carbon Electrodes, Sustainable Energy Fuels, 2017, 1, 9 Search PubMed.
- J. Lee, B. Krüner, A. Tolosa, S. Sathyamoorthi, D. Kim, S. Choudhury, K.-H. Seo and V. Presser, Tin/Vanadium Redox Electrolyte for Battery-like Energy Storage Capacity Combined with Supercapacitor-like Power Handling, Energy Environ. Sci., 2016, 9(11), 3392–3398 CAS.
- N. Jäckel, D. Weingarth, M. Zeiger, M. Aslan, I. Grobelsek and V. Presser, Comparison of Carbon Onions and Carbon Blacks as Conductive Additives for Carbon Supercapacitors in Organic Electrolytes, J. Power Sources, 2014, 272, 1122–1133 CrossRef.
- M. Aslan, M. Zeiger, N. Jäckel, I. Grobelsek, D. Weingarth and V. Presser, Improved Capacitive Deionization Performance of Mixed Hydrophobic/Hydrophilic Activated Carbon Electrodes, J. Phys.: Condens. Matter, 2016, 28(11), 114003 CrossRef CAS PubMed.
- B. Krüner, P. Srimuk, S. Fleischmann, M. Zeiger, A. Schreiber, M. Aslan, A. Quade and V. Presser, Hydrogen-treated, Sub-micrometer Carbon Beads for Fast Capacitive Deionization with High Performance Stability, Carbon, 2017, 117, 46–54 CrossRef.
- A. Kolodziej, K. Fic and E. Frackowiak, Towards Sustainable Power Sources: Chitin-Bound Carbon Electrodes for Electrochemical Capacitors, J. Mater. Chemi. A, 2015, 3(45), 22923–22930 RSC.
- M. Zeiger, N. Jäckel, D. Weingarth and V. Presser, Vacuum or Flowing Argon: What is the Best Synthesis Atmosphere for Nanodiamond-Derived Carbon Onions for Supercapacitor Electrodes?, Carbon, 2015, 94, 507–517 CrossRef CAS.
- C. Schneidermann, N. Jäckel, S. Oswald, L. Giebeler, V. Presser and L. Borchardt, Solvent-Free Mechanochemical Synthesis of Nitrogen-Doped Nanoporous Carbon for Electrochemical Energy Storage, ChemSusChem, 2017, 10(11), 2416–2424 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00265c |
| ‡ These authors contributed equally |
| This journal is © The Royal Society of Chemistry 2017 |