
Formation of an external char layer during subcritical water hydrolysis of biomass†
Zijian
Ma
a,
Patricia
Guerra
a,
Maksim
Tyufekchiev
a,
Azadeh
Zaker
a,
Geoffrey A.
Tompsett
a,
P. C. Torres
Mayanga
b,
Tânia
Forster-Carneiro
b,
Peng
Wang
c and
Michael T.
Timko
 *a
*a
aDepartment of Chemical Engineering, Worcester Polytechnic Institute, 100, Institute Road, Worcester, MA 01609, USA. E-mail: mttimko@wpi.edu
bSchool of Food Engineering, University of Campinas (UNICAMP), Rua Monteiro Lobato, no. 80, Campinas 13083-862, SP, Brazil
cBruker Optics, Bruker Corporation, Billerica, MA, USA
First published on 5th September 2017
Abstract
Flow-through subcritical water hydrolysis (FT-SWH) consists of flowing hot liquid water over a fixed bed packed with biomass particles to produce fermentable carbohydrates and other valuable small molecules. In this work, we studied FT-SWH of green coffee powder as a model lignocellulosic feed, investigating temperatures in the range from 150 to 250 °C and 22.5 MPa. Batch SWH was performed as a basis of comparison for the FT-SWH tests. The focus of the study was characterization of the treated solids; specifically, bulk (primarily thermogravimetric analysis, TGA) and surface (both attenuated total reflectance infrared Fourier transform spectroscopy, ATR-FTIR, and Raman microscopy) methods were used to investigate the hemicellulose, cellulose, and lignin content of the solids. Bulk analysis indicated that the solids treated under FT-SWH conditions retained substantial holocellulose content, even when treated under the most aggresisve FT-SWH conditions studied here (250 °C). On the other hand, surface analysis indicated that the treated materials were primarily composed of char, with the surface content of bound carboxylic acids also increasing with increasing hydrolysis temperature. To understand the apparent discrepancy between bulk and surface analysis, the cross sections of the treated samples were analyzed using both FTIR and Raman microscopy. These techniques confirmed that the surface consisted of a char material, whereas the particle interior was primarily holocellulose. Formation of the external char layer seems to prevent hydrolysis of the internal holocellulose, limiting sugar yields. In comparison with FT-SWH, batch SWH at a similar severity factor resulted in reduced sugar yields, decreased the onset temperature for formation of detectable char quantities, and increased char formation. Comparison with batch conditions suggests that flow conditions reduce external char formation but do not prevent it. This study explains the benefits of FT-SWH and inform development and refinement of pretreatment technologies that can reduce char formation rates.
Introduction
Biomass is the most abundant source of renewable carbon on the planet, and replacing petroleum-based transportation fuels with biofuels presents both technical challenges and opportunities for sustainable development in the 21st century.1 The costs of producing biofuels must be reduced to be competitive with petroleum, and economical and efficient decomposition of biomass biopolymers remains one of the most significant bottlenecks preventing production of low-cost biofuels.2 In the past decade, many technologies have been evaluated for biopolymer deconstruction, most of them based on sequential pretreatment and enzyme hydrolysis steps. Pretreatment steps are designed to open the biomass structure, by removing hemicellulose, re-organizing lignin, and de-crystallizing cellulose.3 Enzymes are then able to access the remaining hemicellulose and cellulose, hydrolyzing it to produce fermentable sugars.3Of the various pretreatment and hydrolysis technologies, dilute acid pretreatment (DAP) has received the most attention and is the furthest toward commercialization.4 DAP suffers from several drawbacks.5 First, the liquid acids are generally too dilute to be recovered post reaction.6 As a result, the acid must be neutralized to form the salt, which becomes a waste by-product. In addition, the acid itself poses a corrosion risk. DAP co-produces compounds with cytotoxicity,6 which must be removed prior to fermentation.7 Finally, DAP is not a stand-alone technology and must be coupled with enzyme hydrolysis to convert cellulose and hemicellulose into simple sugars; the enzyme hydrolysis step adds significant cost to the overall process, largely due to the need for excess enzyme.8 For these reasons, work continues to identify superior pretreatment and deconstruction technologies.
Subcritical water hydrolysis (SWH) offers advantages compared to DAP, chiefly the elimination of auxiliary chemicals.9 Specifically, unlike DAP, SWH does not require acids nor does it generate waste salt byproducts. Instead, SWH utilizes temperatures greater than 150 °C to achieve rapid hydrolysis rates.10 Lachos-Perez, et al.11 recently reviewed the field of SWH, and several studies have shown that SWH can achieve sugar yields comparable to DAP.2,12 Despite potential technical benefits and clear environmental advantages, SWH is not yet economically competitive.13 A common problem attributed to SWH is sugar degradation, whereby sugars released following hydrolysis of cellulose and hemicellulose further break down to form hydroxymethylfurfural, furfural, and other compounds.2,3 Cantero et al.14 reduced sugar degradation losses by carefully controlling thermal history using a flow-through slurry reactor; unfortunately, this approach requires pumping a dense biomass slurry, which is a difficult technical challenge on its own.15
Flow-through SWH (FT-SWH) has received increasing attention in the past several years as an approach to convert hemicellulose and a portion of cellulose to fermentable sugars and sugar oligomers.13,16 Unlike the slurry-fed reactor studied by Cantero et al.,14 in which both the biomass and water continuously pass through the heated reactor, FT-SWH continuously passes subcritical water over a fixed bed of biomass particles. Thus, FT-SWH eliminates the need to feed biomass slurry under pressure to the reactor. FT-SWH continuously removes soluble sugars from the reactor as they are produced, partially preventing their degradation,17 and reducing lignin re-deposition.16 While dilution and energy use are potential drawbacks to FT-SWH, Archambault-Léger et al.13 reported that FT-SWH was more economical than dilute acid ($1.01–1.19 per L), steam explosion ($0.86–1.18 per L), or batch-mode hot liquid water ($1.13–1.27 perL) pretreatment. Further, Archambault-Léger et al.13 indicated that neither dilution nor energy use were limiting factors, provided adequate heat integration.
While most prior work has focused on controlling sugar degradation using FT or slurry reactor designs, less attention has been paid specifically to the role of reactor design on char formation. In fact, Lachos-Peres et al.18 used thermogravimetric analysis and Fourier-transform infra-red (FTIR) to study sugarcane straw treated in a FT-SWH reactor. Despite observing optimal sugar yields at 190 °C, the treated solids retained substantial holocellulose content (approximately 50 wt%) and even hemicellulose content even when the biomass was treated at 260 °C.18 Based on these observations, Lachos-Peres et al.18 surmised that hydrolysis was limited in part by char formation, but could not determine a mechanism to explain this link.
In this work, we characterized green coffee powder before and after SWH treatment using various methods, including Raman microscopy. Raman microscopy is a powerful tool for investigating the C–C bonds that dominate char structure,19 providing a potential method to probe the chemistry of char formed on biomass during SWH and differentiating char from re-deposited lignin. Unfortunately, many plant-based materials20 and substances containing char-like structures21 fluoresce when examined using Raman lasers. Unlike many biomass types, green coffee powder and its treated residues do not fluoresce intensely when irradiated by visible lasers,20 permitting use of Raman microscopy to characterize green coffee powder samples. Mayanga-Torres et al.22 previously studied FT-SWH of green coffee powder, reporting modest sugar yields (9 g per 100 g of biomass for total reducing sugars and <1 g/100 for glucose). Therefore, the point of the present work was not to study green coffee powder as a potential energy source, so much as it was to use green coffee powder as a model biomass type to understand char formation and its potential impact on sugar yields.
Here, we used Raman and IR spectroscopy to analyze the surface composition of green coffee powder. TGA provided information on the bulk composition for direct comparison with the surface characterization data. Green coffee powder particles were sectioned and analyzed using both Raman and IR microscopy to obtain chemical composition information on particle exterior and interior to reconcile surface spectroscopy and bulk TGA. The results from this work motivate and inform efforts to design new batch and FT-SWH hydrolysis technologies.
Experimental description
Materials
Brazil Cooxupé (Regional Cooperative of Coffee Growers, Guaxupé Ltda) donated green coffee powder from the species Coffea arabica. Green coffee powder is generated by mechanical abrasion, either by friction during transportation or during sieving processes used during quality control. Prior to testing, green coffee powder was stored at −18 °C. Samples were knife milled and sieved (Marconi, model MA 340, Piracicaba, Brazil) to reduce their size to <1 mm sieve. This process resulted in formation of a fraction of particles too fine to be retained in the reactor; these particles were swept from the reactor prior to heating and are not included in the analysis.De-ionized (DI) water (>17.9 MΩ cm resistivity) was obtained from an in-house purification system. All solvents were obtained from Sigma Aldrich as HPLC grade.
FT-SWH protocol
The protocol and reactor configuration used for FT-SWH is described in the literature and is not described in detail here.22,23 The ESI† provides a schematic representation of the hydrolysis processes (Fig. SI-1†). For all experiments, 5.0 g of green coffee powder were placed inside a stainless steel tube that served as the reactor. All system components were filled with water and pressurized to the desired set point (>22.5 MPa) using a pump. This pressure was selected to ensure that liquid phase conditions existed at all times within the reactor, and included a significant safety factor to account for pressure drop across the biomass bed. The system was heated for approximately 20 min to the desired temperature without flow. Once the set point temperature had been reached, flow was resumed at constant rate (10 mL min−1). Liquid hydrolyzate was collected in glass vials every 2 min for 36 min, then stored prior to analysis. The process was then terminated by releasing pressure and cooling the reactor. Solid samples were collected and dried overnight prior to analysis. Experiments were performed in duplicate. Mass balance closure was >80% in all FT-SWH experiments except those performed at 250 °C. The remaining mass was divided between soluble products which could not be quantified and solids that were lost. At 250 °C, mass balance closure was approximately 60%; at these conditions, the hydrolyzate contained many soluble compounds that could not be quantified, likely explaining the lack of mass balance closure. Because mass balance closure is incomplete at 250 °C, data collected at this temperature should be regarded as semi-quantitative. They are included here to show trends.Batch SWH protocol
For batch SWH, 3.4 g of green coffee powder and 34 g of DI water were loaded into a 4590 micro bench top reactor with an Inconel vessel (approximately 100 cm3 internal volume) obtained from Parr Instrument Company. The reactor was heated with continuous stirring (250 RPM) to the desired temperature (200 °C) and maintained at this temperature for 36 min. Reactor heat-up required approximately 20 min. To quench the reaction, the reactor was rapidly cooled (5–10 min) by submerging it in liquid water. These conditions were selected to match the severity factor and temperature at which char was first observed on the biomass treated under FT-SWH. Including heat-up and cool-down, the severity factor investigated for both FT-SWH and batch SWH were nearly the same (4.0 ± 0.1 for 175 °C and 4.7 ± 0.1 for 200 °C). The hydrolyzate and solid were separated, the solid dried overnight in an oven at 60 °C and the hydrolyzate was filtered and stored at −10 °C prior to subsequent analysis. Batch experiments were performed in duplicate.Hydrolyzate analysis
The hydrolyzate obtained from FT-SWH was analyzed using the high performance liquid chromatography (HPLC) procedure described by Mayanga-Torres et al.22 The hydrolyzate obtained from batch SWH was analyzed using a similar HPLC procedure; the most important aspects of the analysis are provided here. The hydrolyzate obtained from the batch SWH was analyzed using an Agilent 1200 series HPLC system equipped with Diode Array Detector (DAD), Refractive Index Detector (RID), using a Bio-Rad Aminex HPX-87H column. The DAD was used to quantify UV-Vis active compounds such as hydroxymethylfurfural (HMF), while the RID was used to quantify glucose. Hydrolyzate yields are expressed as either grams of recovered product per 100 grams of biomass feed (g/100 g) or micrograms of product per grams of feed (μg g−1).Solids characterization
The National Renewable Energy Laboratory (NREL) methodology was used to determine the moisture content, ash, extractives, protein, holocellulose, and lignin content of green coffee powder.24–28 Mayanga-Torres et al.22 previously published the results, reporting the holocellulose content as 62 ± 6 wt%; total lignin as 36 ± 3 wt%; protein as 11.2 ± 0.3 wt%; and ash as 4.6 ± 0.3 wt%, all on a dry basis.A Netzsch TGA (TG 209 F1 Libra) was used for analysis of green coffee powder and hydrolyzed materials products. An alumina crucible was used for holding samples, and the nitrogen flow was 20 mL min−1. The initial oven temperature was 35 °C and it was increased to 800 °C at a constant rate of 5 °C min−1. Vendor software converted raw data into differential thermograms (DTG).
IR spectra were obtained using a Nicolet spectrometer (Magna 550). Dry powder samples were placed in a Specac “Golden Gate” diamond ATR cell. All spectra were collected with 512 scans and then averaged. The resolution was 4 cm−1 over the wavenumber range from 600 to 4000 cm−1.
Raman spectra of raw and treated samples were obtained using a Horiba XploRa Raman microscope. The instrument utilized a 785 nm laser line at 25–100 mW, with 1800 grating, and a 100× objective lens. Spectra were obtained using a 5 second scan time and averaged over 100 scans. Spectra of 4–5 particles were obtained to observe the homogeneity of the sample. Representative spectra are presented herein.
MagicPlot software was used to fit Raman, FTIR, and DTG data. In all cases, the software was used to obtain a satisfactory initial guess using literature data for band locations as a guide. The software then performed a rigorous non-linear least squares regression to optimize the fit.
A stand-alone FTIR microscope (LUMOS, Bruker) was used to measure individual particles/areas in the prepared cross sections. Using the fully automated ATR (attenuated total reflectance) mode of LUMOS, the area of interest was precisely analyzed with assistance of predefined apertures. Each spectrum was collected for 32 scans with 4 cm−1 spectral resolution from 600 to 4000 cm−1. Multiple spots were examined and representative spectra are presented here.
Histologistics Inc. (151 W. Main Street LOWR, Dudley, MA 01571) cross-sectioned the solid residues. The coffee particles were first embedded in liquid paraffin (60 °C) using metal base moulds and cooled to 0 °C for 30 minutes. The blocks were then cut on a Reichert-Jung microtome to 20 μm thick cross-sections. Sections were floated on a water bath at a temperature of 40 °C for 30 seconds, separated (if necessary), and removed using a positively charged slide. All slides were air dried at room temperature for a minimum of 48 hours before further analysis.
Results
In previous work, Mayanga-Torres et al.22 studied FT-SWH treatment of green coffee powder, reporting that cumulative yields of TRS obtained from FT-SWH of coffee powder increased for approximately 20–30 min, and then reached a maximum. Treatment for greater than 30 min did not lead to increased TRS yields, suggesting that a limit had been reached. Table 1 summarizes the maximum glucose and HMF yields reported previously by Mayanga-Torres et al. at temperatures ranging from 150 to 250 °C.22 In all cases, glucose yields were less than 1 g per 100 g of biomass. Table 1 includes severity factors, calculated based on the temperature history of the reactor.7 The maximum glucose yield was obtained at 150 °C, at a severity factor of 3.2 ± 0.1. In comparison, HMF yield was always <50 μg per gram of biomass and the maximum yield was observed at 175 °C. Increasing temperature did not increase the HMF yield, suggesting its degradation.| Reactor config. | Sev. factora | Rxn. T. (°C) | Gluc. yieldb (g/100 g) | HMF yieldc (μg g−1) | Source |
|---|---|---|---|---|---|
| a Calculated from known temperature history, to within ±0.1 units. b Reported in g per 100 g of biomass feed. c Reported in μg per g of biomass feed. | |||||
| FT | 3.2 | 150 | 0.47 | 9.7 ± 0.9 | Mayanga-Torres et al.22 |
| FT | 4.0 | 175 | 0.335 | 49 ± 2 | Mayanga-Torres et al.22 |
| FT | 4.7 | 200 | 0.202 | 19 ± 1 | Mayanga-Torres et al.22 |
| FT | 6.2 | 250 | 0.155 | 16 ± 3 | Mayanga-Torres et al.22 |
| Batch | 4.0 | 175 | 0.2 | 1900 ± 500 | This work |
| Batch | 4.7 | 200 | 0.027 | 1800 ± 100 | This work |
The data in Table 1 are highly representative of the SWH data reported in the literature, especially with 2 important features often held in common: (1) observation of an optimal temperature less than 200 °C and (2) observation of a maximum cumulative sugar yield obtained after approximately 30 min, with further increases not leading to increased sugar accumulation. The observation of an optimal temperature for sugar yield is especially interesting as this would typically be attributed to sugar degradation. However, under FT conditions, sugar degradation is minimized and sugar yields should be expected to increase monotonically with increasing temperature – or at least increase monotonically with increasing temperature and then reach a stable value. This is not what is observed, suggesting that another process than sugar degradation may limit yields obtained under FT conditions. To understand more fully, the residual solids produced under FT-SWH conditions were analysed using TGA, FTIR, and Raman microscopy.
Treated solids were analyzed using TGA as a bulk technique to determine relative amounts of hemicellulose, cellulose, lignin, and other major components in the treated samples. The ESI† contains measurement details, including the raw TGA as Fig. SI-2.† Fig SI-3† compares the DTG curves obtained from analysis of samples treated under FT-SWH conditions at different temperatures. The DTG thermograms consist of several clear peaks corresponding to the main components of biomass (hemicellulose, cellulose, and lignin).29 In addition, a peak appears at 175–200 °C that is assigned as “semi-volatile” content of the coffee powder (specifically SV1), presumably lipids and other extractable content.30 The “semi-volatile” component disappears after treatment at >150 °C; however, a new band, at 200–220 °C appears in samples treated at 200 °C. The volatility of the new band is consistent with semi-volatile content, but would seemingly arise from a different source than SV1 for two reasons: (1) the peak is not present in the initial sample and (2) the temperature range of the new band is distinct from the original semi-volatile component. Accordingly, the new band is termed “SV2”. Samples treated at 200 and 250 °C contain a second additional peak, appearing at 400–450 °C that is attributed to char, in accordance with observations made by Kong et al.31
Having identified the main components of the feed and SWH-treated biomass, the raw DTG data were then curve fit, as described previously in the literature,5 to quantify the main components of the feed and treated biomass. Fig. SI-4–SI-6† provide representative DTG curve fitting and quality control results while Table SI-1† provides the DTG temperatures used for the fitting, which were maintained at constant values (±20 °C) to ensure compositional relevance while providing good statistical fits to the data.
Fig. 1 provides the composition results deduced from TGA. Fig. 1 is based on analysis of the recovered biomass and has implicit units of grams per gram of recovered biomass. The data in Fig. 1 show many important trends. SV1 decreases with increasing hydrolysis temperature, consistent with extraction of native semi-volatile compounds during treatment. Interestingly, hemicellulose content decreases with increasing treatment temperature, but does not vanish even when the biomass is treated at 250 °C. Cellulose exhibits a similar trend compared to hemicellulose, decreasing with increasing treatment temperature but not vanishing. As mentioned previously, SV2 is an important component (>2 wt%) for samples treated at 200 and 250 °C. Interestingly, the SV2 content is greater in the sample treated at 200 °C than the one treated at 250 °C, possibly consistent with formation of SV2 as a reaction intermediate. Lastly, char is detected in samples treated at 200 and 250 °C, with the amount of char increasing with increasing reaction temperature. Char formation is observed at temperatures at which sugar yields are rapidly decreasing (Table 1).
 | ||
| Fig. 1 TGA data obtained from analysis of coffee powder treated under FT-SWH conditions at different temperatures. | ||
Fig. 1 indicates that samples treated at 200 and 250 °C contain substantial residual cellulose and hemicellulose content. Previous work suggests that pure xylan should hydrolyze at temperatures less than 230 °C,32 making the persistence of hemicellulose in the powder puzzling. In contrast with hemicellulose, FT-SWH treatment at 150 °C removes SV1 from the coffee powder. Moreover, the appearance – and disappearance – of SV2 and the growing importance of char in the treated samples require explanations. Given that previous work had suggested a link between char formation and observations of maximum sugar yields,18 we focused our analysis on the char.
Raman microspectroscopy is a powerful tool for studying the C–C bonds that compose char.19 Unlike many other biomass types, which fluoresces strongly when irradiated by the visible wavelength lasers (523 and 785 nm) used most commonly for Raman microspectroscopy, green coffee powder does not fluoresce when analyzed using Raman microspectroscopy. Fluorescence is a much more intense phenomenon than Raman scattering and samples that strongly fluoresce cannot be analyzed using typical Raman instruments.21 Some recently developed Raman instruments, namely those that use 2-photon methods33 or that substitute UV lasers21 for visible lasers, can overcome sample fluorescence; however, these instruments have their own limitations or are still under development. The fact that green coffee powder (and especially its treated versions) does not fluoresce significantly, therefore, provides an opportunity to use Raman microspectroscopy that many other samples do not.
Fig. 2 provides representative Raman spectra obtained from analysis of the raw green coffee powder and hydrolyzed samples. Table SI-2† provides detailed Raman band assignments for coffee powder and treated samples; the most important spectral features are discussed here. The spectrum of raw green coffee powder has bands that can readily be associated with the backbone C–O and C–C bonds of cellulose34 and the triglyceride bonds of lipids.35 With treatment at increasing hydrolysis temperatures, the intensities of bands associated with cellulose and triglycerides decrease and are replaced by strong bands at approximately 1360 and 1600 cm−1. The new bands can be identified with the symmetric and asymmetric modes of aromatic rings, commonly observed in the Raman spectra of chars.36
 | ||
| Fig. 2 Raw Raman spectra obtained for feed and feed treated at different FT-SWH temperatures (as indicated). Specific bands are highlighted for chemical content. | ||
Raman spectra are quantitative in the sense that the intensity of the band is proportional to the concentration of the underlying bond vibration; however, the bond oscillator strength is, in general, unknown, making actual quantification difficult. Nonetheless, as a semi-quantitative method, Raman intensities can still provide useful insight. For the material treated at 250 °C, Raman microscopy indicates that the surface of the treated coffee powder is nearly exclusively char. This is an apparent contradiction to the TGA results (Fig. 1) which show that char is a minor component of the treated biomass. Taken together, therefore, Fig. 1 and 2 suggest compositional differences between the bulk and the surface, and formation of an external char layer on a carbohydrate-rich internal core can explain the apparent contradiction between TGA and Raman characterization. The remainder of this work seeks to test this explanation.
Raman microscopy is ideally suited for investigation of C–C bonds; however, IR active vibrations provide more information on polar bonds present in important functional groups, especially carbonyls. Since polar functional groups may provide evidence of the char formation mechanism, ATR-IR was used to analyze raw and treated coffee samples. Fig. SI-6† provides representative ATR-IR spectra obtained for the raw material and treated samples. The IR spectra of the raw material contain bands associated with cellulose,37 lignin,38 triglycerides,39 and chlorogenic acid40 – a specific compound present in coffee. Table SI-3† provides detailed IR band assignments. The intensities of chlorogenic acid and triglyceride bands decrease after FT-SWH treatment, again consistent with the decreased SV1 content suggested by TGA. The intensities of cellulose bands decrease with treatment, but unlike when analyzed via Raman microscopy, cellulose bands do not disappear from the IR spectra after treatment at temperatures greater than 200 °C. The difference between Raman and IR analysis may be attributable to the deeper penetration depth typical of IR (>2 μm) compared with Raman (<2 μm) and/or differences in Raman and IR cross sections. As a result, sub-surface cellulose may impact IR spectra more than Raman spectra or cellulose may simply be more IR active than char.
The IR spectra in Figure SI-6† show a band initially present at approximately 1700 cm−1 that appears to shift to approximately 1740 cm−1 after FT-SWH treatment. We attribute both bands to carbonyls. The band initially present at 1700 cm−1 can be attributed to triglyceride carbonyls, whereas the band that appears at 1740 cm−1 is associated with carboxylic acids. Triglycerides apparently are removed from the biomass during treatment and play at most a limited role in charring. Carboxylic acids, on the other hand, may lead to char formation.41,42 Accordingly, we sought to understand the presence of carboxylic acid more precisely.
As with Raman spectroscopy, IR is semi-quantitative. However, IR can be used to compare signals arising from similar functional groups that should have comparable oscillator strengths. Accordingly, we fit the bands associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds attributed to lignin and triglycerides and the CO bands associated with carboxylic acid and triglycerides (Figure SI-7†). Comparison of intensities of bands arising from the CC bonds of lignin and triglycerides is performed primarily to establish the reliability of the method since FT-SWH treatment is expected to remove triglycerides but not affect lignin. Analysis of the CO bands then provides insight on the surface content of carboxylic acids, which may be associated with char formation.
C bonds attributed to lignin and triglycerides and the CO bands associated with carboxylic acid and triglycerides (Figure SI-7†). Comparison of intensities of bands arising from the CC bonds of lignin and triglycerides is performed primarily to establish the reliability of the method since FT-SWH treatment is expected to remove triglycerides but not affect lignin. Analysis of the CO bands then provides insight on the surface content of carboxylic acids, which may be associated with char formation.
Fig. 3a is a plot of the lignin to triglyceride ratio deduced from IR spectra. IR indicates that the raw powder contains roughly equal amounts of lignin and triglycerides. This is not entirely consistent with TGA results as these indicate that SV1, which is presumably the source of triglycerides, is a minority component while lignin composes about 35% of the original biomass. The apparent discrepancy between TGA and IR may be attributable to differences in the spatial distribution of triglycerides and lignin. As expected based on the relative chemical stability and solubility of lignin and triglycerides, IR indicates that FT-SWH treatment increases the amount of lignin relative to triglycerides. Fig. 3a shows that the lignin to triglycerides ratio increases monotonically with increasing treatment temperature, taking a value of approximately 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for samples treated at 250 °C. Because lignin is a thermally stable biopolymer with limited water solubility and triglycerides are present as extractable oils, the monotonic increase in the lignin:triglyceride ratio is consistent with expectations and establishes semi-qualitative interpretation of IR spectra.
:1 for samples treated at 250 °C. Because lignin is a thermally stable biopolymer with limited water solubility and triglycerides are present as extractable oils, the monotonic increase in the lignin:triglyceride ratio is consistent with expectations and establishes semi-qualitative interpretation of IR spectra.
 | ||
| Fig. 3 Semi-quantitative analysis of IR spectra: (a) lignin/triglycerides ratio and (b) triglycerides/carboxylic acid ratio. | ||
Fig. 3b plots the ratio of carboxylic acid to triglycerides deduced from IR spectra. In the raw sample, the triglyceride content is nearly twice that of the acids. With increasing FT- SWH hydrolysis temperature, the triglyceride to acid ratio decreases monotonically, reaching a value of about 0.4:1 for the sample treated at 250 °C. On its own, this analysis cannot establish if the acid content is increasing or if the triglyceride content decreases; in fact, Fig. 3a combined with Fig. SI-6† suggest that both are occurring in parallel. Nonetheless, the carboxylic acid content clearly does not decrease with increasing reaction temperature. Likewise, because the samples were prepared under flow conditions, the carboxylic acids must be covalently bound to the surface – or at least physically trapped near it. Soluble acids would simply be removed from the reactor with the exiting products.
Several plausible phenomena can explain the presence of carboxylic acids on the treated biomass surface. Cellulose itself is known to possess carboxylic acid functional groups, resulting from partial oxidation by air or other environmental oxidants.43 Likewise, many hemicellulose types possess acidic side chains.44 Moreover, simple carbohydrates undergo dehydration and ring-opening reactions in hydrothermal water to produce carboxylic acids.45 Lastly, the presence of surface-bound acids is consistent with the formation of acidic hydrolyzates reported previously under FT-SWH conditions.22,23 The acidic pH (typically <4) may in part be due to de-protonation of bound acids on the biomass surface.
The carboxylic acid band may be associated with the appearance of SV2. Specifically, Figueiredo et al.46 analyzed activated carbon using TGA coupled with mass spectrometry to conclude that surface-bound carboxylic acid groups volatilize at approximately 175–225 °C, in good agreement with the volatilization temperature of SV2. Therefore, the combination of ATR-IR and TGA suggests formation of carboxylic acids on the biomass surface during treatment. These acids may play a role in char formation or char accumulation on the particle surface. For example, acidic conditions catalyze sugar degradation and formation of char-like humins.47 Moreover, free acid groups may provide reactive anchoring points for char deposition on the biomass particles.
Raman microscopy and ATR-IR analysis of the original powders provide useful information; however, the usefulness of spatially resolved chemical composition data is limited when analyzing powders. For this reason, representative samples were cross sectioned to obtain chemical information on outer surfaces and particle interiors. The sectioned samples were then analyzed using both ATR-IR microscopy and Raman microscopy, techniques chosen to provide complementary chemical information with similar sample penetration depths (approximately 1 μm for both methods).48
Fig. 4 is a composite containing both a representative microscope image and several IR spectra obtained in different locations. Particle images show a clearly defined peripheral edge that appears black which surrounds a larger core that appears lighter brown. The peripheral edge is approximately 10 μm thick. The dark color of the periphery is qualitatively similar to that of char, whereas the interior is visibly similar to the biomass feed. IR spectra support the conclusions suggested by visual inspection of the analysis. Specifically, Fig. 4 shows that IR spectra from the particle interior consistently exhibit three main bands in the range from 1000 to 1200 cm−1 and attributable to carbohydrates.49 In contrast, spectra of the particle exterior exhibit only a broad band in this range, attributable to the Si–O band from the glass mounting slide.50 Likewise, spectra of the particle exterior exhibit a broad band at 1750 cm−1 that can be attributed to carboxyl groups, whereas this band is much sharper in the particle interior. All of these observations support the existence of a compositionally distinct external surface on the FT-SWH treated biomass.
 | ||
| Fig. 4 Micrograph and ATR-IR spectra obtained for a cross sectioned biomass particle treated under FT-SWH conditions at 200 °C. | ||
Fig. 5 contains a representative image obtained via Raman microscopy along with spectra obtained from the interior and exterior of the particle. Unlike the IR image, the Raman microscopy image does not have sufficient contrast to define visually a particle interior and exterior. Nonetheless, spectra obtained from the exterior and interior are clearly differentiated. A dotted line has been added to Fig. 5 to highlight the transition from particle exterior to interior. The Raman spectra provide further support. The spectra from the particle exterior is dominated by bands at 1350 and 1600 cm−1, previously attributed to the characteristic bands of char materials.36 For comparison, Fig. 5 contains a spectrum obtained from analysis of a glucose hydrochar, which contains prominent features that closely resemble those of the char surface.
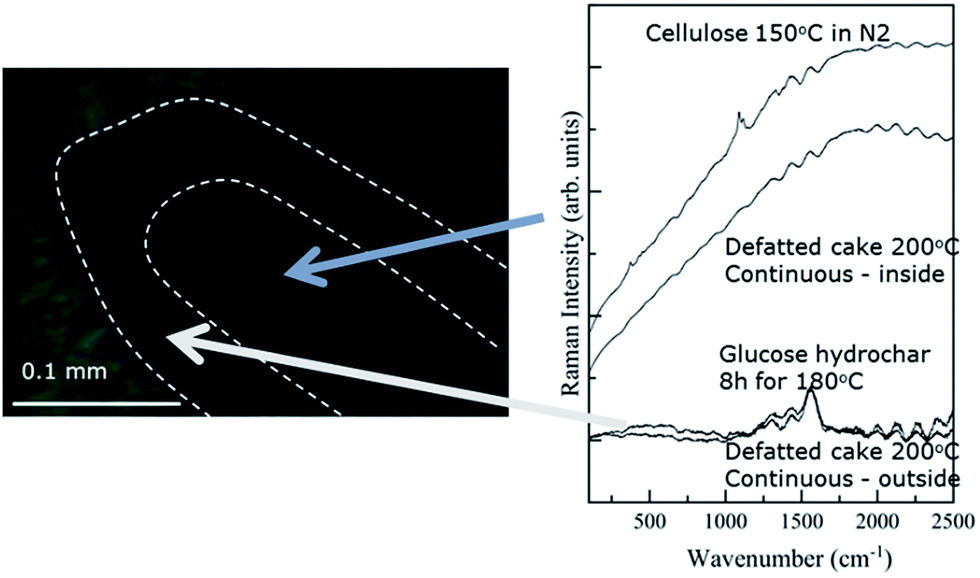 | ||
| Fig. 5 Micrograph and Raman spectra obtained for a cross sectioned biomass particle treated under FT-SWH conditions at 200 °C. | ||
Fig. 5 shows that the spectrum obtained from the particle interior is dominated by a broad, nearly featureless signal that is attributable to fluorescence. As mentioned previously, the fluorescence signal obscures that from Raman scattering. That stated, the structure of the fluorescence signal obtained from the particle interior is qualitatively very similar to the signal arising from cellulose treated under N2 at 200 °C, which is shown for comparison. The resemblance between the spectrum obtained from the particle interior and that arising from heat-treated cellulose suggests that the interior continues to undergo thermal degradation after surface char formation. Thermal degradation reactions appear to produce fluorophoric compounds which then dominate the Raman signal. In fact, thermal degradation reactions, rather than hydrolysis, likely take place in the particle interior as the char layer prevents diffusion of water and/or acids into the holocellulose core, effectively ending hydrolysis.
The results presented here clearly establish the formation of a char layer on the external surfaces of biomass particles treated under FT-SWH conditions. As a final experiment, we compared some of the important results obtained from samples treated under FT-SWH conditions to those obtained for a sample treated under batch SWH conditions. To capture the effects of thermal history, the severity factors used for the FT-SWH and batch experiments were similar (4.0 ± 0.1 at 175 °C and 4.7 ± 0.1 at 200 °C). Thus, the main difference between the experiments should be the presence (FT-SWH) and absence (batch) of flow.
Table 1 provides carbohydrate and degradation compound yields obtained for batch SWH of coffee powder. As expected, glucose yields are much greater for the FT-SWH treatment than the batch treatment, whereas yields of the HMF decomposition product are greater under batch conditions. Moreover, glucose yields are much more temperature dependent under batch conditions than FT, as increasing the severity from 4.0 to 4.7 decreases glucose yields obtained under batch conditions by nearly an order of magnitude, yet glucose yield decreases by less than a factor of 2 under FT-SWH conditions.
Fig. 1 compares the TGA results obtained for batch-treated samples with those treated under FT-SWH conditions, showing differences between the treatments in both SV and char composition. In terms of SV, Fig. 1 indicates that the batch-treated sample at 175 °C retains SV1 content, whereas the FT-treated sample does not. Subsequent analysis using ATR-FTIR (Fig. SI-8†) confirmed that the batch-treated sample retained greater chlorogenic acid and triglyceride content than the FT-treated sample, consistent with the TGA data which indicated more effective extraction of SV contents under flow conditions than batch. After treatment at 200 °C under batch conditions, TGA no longer indicates presence of SV1, consistent with its extraction and/or degradation. Therefore, FT conditions permit extraction of SV content at lower temperatures than batch.
In addition to differences in SV content, batch treatment increases the char yield relative to that obtained under FT-SWH conditions. First, the onset temperature associated with formation of detectable char is reduced by at least 25 °C under batch conditions compared to FT, as TGA indicates the presence of char in the batch-treated sample at 175 °C but only in FT-treated samples treated at ≥200 °C. For samples treated at 200 °C, batch-treated samples contain nearly double the char as obtained at the same conditions under flow. Fig. SI-9† provides the DTG data obtained from batch treatment compared to data obtained from FT treatment, showing the increased char content of the batch-treated samples. Due to heavy charring at 200 °C, batch runs at 250 °C were not performed; however, data at 175 and 200 °C strongly suggest that the biomass will be highly charred at 250 °C.
Comparison between batch and flow conditions suggests that char formation occurs at least in part via re-condensation of reactive products on the biomass surface. Under batch conditions, the reactive products (e.g., aldehydes, furans, acids, etc.) accumulate in the hydrolyzate, and their corresponding re- condensation rates increase over time. Under flow conditions, the reactive products are continuously removed from the reactor with other soluble products, limiting their ability to condense onto the biomass surface.
The surface char explanation suggests an un-expected benefit of flow-through conditions for biomass hydrolysis. Previous work explained the benefits of flow primarily as reducing losses due to sugar degradation.17 The results presented here show that limiting surface char formation may also be important. Fig. 6 is a conceptual schematic of the process. When initially exposed to SWH conditions, holocellulose compounds undergo hydrolysis reactions. As hydrolysis proceeds, surface-bound carboxylic acid groups are formed or exposed to the reaction media while concurrently sugar degradation produces char precursor compounds, either in the hydrolyzate or on the surface itself. With continued treatment, a char layer forms on the particle surface, preventing further hydrolysis or at least greatly reducing hydrolysis rates.
 | ||
| Fig. 6 Conceptual schematic of char layer formation on the exterior surfaces of biomass particles during SWH treatment. | ||
The formation of an external char layer is similar to the explanation forwarded by Mosteiro-Romero et al.51 to model hydrolysis rates of spruce wood milled to different particle sizes. In particular, Mosteiro-Romero et al.51 found that simple reaction-diffusion models could not explain observed hydrolysis rates and invoked the existence of an oily film composed of non-polar products that are water insoluble, as suggested previously by Vogel52 from his studies of biomass gasification in supercritical water. In fact, at the conditions of SWH, most presumptive hydrolysis and degradation products are water soluble.32,53 Instead, our model explains that the external layer is physically adhered or perhaps even chemically bonded to the particle surface.
The char formation explanation is distinct from lignin re-deposition previously reported for samples treated under batch and flow-through pretreatment.16,54 Lignin deposits are typically present as droplets on the biomass surface when observed using SEM.54 In fact, Mayanga-Torres et al.22 did not report surface droplets in their SEM images of coffee treated under FT-SWH conditions. Instead, a uniform layer, approximately 10–100 μm thick, covers the biomass particle and can be observed using visible microscopy. Moreover, the Raman spectrum of the surface is not consistent with lignin,55 again suggesting an entirely different composition and origin.
The external char layer model forwarded to explain results observed at SWH conditions has some power to explain the success of kindred pretreatment approaches. First, the benefit of flow is likely to remove soluble precursor molecules before they can condense on the surface. Just as clearly, reducing the initial particle size can increase sugar yields, though this strategy has limits as found by Mosteiro-Romero et al.51 Likewise, slurry flow hydrolysis reactors, in which biomass particles continuously enter and exit the reactor, limit the residence time of the solid inside the reactor, preventing it from accumulating char on their surfaces.14 Moreover, slurry flow reactors can tune residence times and heat-up times more carefully than batch or even flow-through reactors and may provide greater access to conditions which favor hydrolysis over carbonization.14 Careful use of acids and bases, for example as used in dilute acid pretreatment,2 may improve sugar yields by adjusting the relative rates of hydrolysis,56 char precursor formation,57,58 and char formation reactions.59 Solvents, for example tetrahydrofuran used in co-solvent enhanced lignin fractionation (CELF), may help solubilize char as it forms60 or they may reduce the surface density of char precipitation sites on the biomass surface. Lastly, solid acids, which purportedly target oligosaccharides over monosaccharides, may reduce the rates of precursor formation relative to hydrolysis.61
Conclusions
Green coffee powder was used as a model biomass feed for FT-SWH. Bulk analysis of the treated particles by TGA contradicted surface analysis by IR and Raman spectroscopy; the former indicated that the treated biomass contained substantial amounts of residual cellulose and hemicellulose whereas the latter indicated that the material was nearly entirely carbonized. The particles were cross sectioned to understand the discrepancy, with the finding that a char coating covered the external surfaces with a thickness of about 10 to 100 μm and the interior was predominantly cellulose. Flow through conditions succeed in decreasing formation of the char layer relative to batch conditions, likely by reducing the concentrations of char precursor molecules that accumulate at the surface under batch conditions. The formation of an external char layer likely explains the success of flow-through, slurry feed, acid promoted, and solvent enhanced hydrolysis and pretreatment technologies, as each of these methods likely reduces char formation rates or prevents accumulation of char on the surface. Future work on biomass hydrolysis should be guided in part on avoiding the accumulation of char on particle surfaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded in part by the U.S. National Science Foundation (#1342320 and #1554283) and the São Paulo Research Foundation – FAPESP (2011/19817-1). Dr Sergey Shilov (Bruker) assisted with sample preparation. Dr Glenn Gaudette (WPI) provided advice on sample microtoming.References
- M. M. Bugge, T. Hansen and A. Klitkou, Sustainability, 2016, 8, 691 CrossRef.
- T. Zhang, R. Kumar and C. E. Wyman, Carbohydr. Polym., 2013, 92, 334–344 CrossRef CAS PubMed.
- C. K. Nitsos, K. A. Matis and K. S. Triantafyllidis, ChemSusChem, 2013, 6, 110–122 CrossRef CAS PubMed.
- D. C. Elliott, P. Biller, A. B. Ross, A. J. Schmidt and S. B. Jones, Bioresour. Technol., 2015, 178, 147–156 CrossRef CAS PubMed.
- J. Cai, W. Wu, R. Liu and G. W. Huber, Green Chem., 2013, 15, 1331–1340 RSC.
- M. Góral, B. Wiśniewska-Gocłowska and A. Mączyński, J. Phys. Chem. Ref. Data, 2006, 35, 1391–1414 CrossRef.
- N. Abatzoglou, E. Chornet, K. Belkacemi and R. P. Overend, Chem. Eng. Sci., 1992, 47, 1109–1122 CrossRef CAS.
- W. L. Luyben, Energy Fuels, 2008, 22, 4249–4258 CrossRef CAS.
- H. A. Ruiz, R. M. Rodriguez-Jasso, B. D. Fernandes, A. A. Vicente and J. A. Teixeira, Renewable Sustainable Energy Rev., 2013, 21, 35–51 CrossRef CAS.
- I. Cybulska, H. Lei and J. Julson, Energy Fuels, 2009, 24, 718–727 CrossRef.
- D. Lachos-Perez, A. Brown, A. Mudhoo, J. Martinez, M. T. Timko, M. A. Rostagno and T. Forster-Carneiro, Biofuel Res. J., 2017, 14, 611–626 Search PubMed.
- S. G. Allen, D. Schulman, J. Lichwa, M. J. Antal, E. Jennings and R. Elander, Ind. Eng. Chem. Res., 2001, 40, 2352–2361 CrossRef CAS.
- V. Archambault-Léger, Z. Losordo and L. R. Lynd, Biofuels, Bioprod. Biorefin., 2015, 9, 95–108 CrossRef.
- D. A. Cantero, M. D. Bermejo and M. J. Cocero, ChemSusChem, 2015, 8, 1026–1033 CrossRef CAS PubMed.
- E. J. Berglin, C. W. Enderlin and A. J. Schmidt, Review and assessment of commercial vendors/options for feeding and pumping biomass slurries for hydrothermal liquefaction, Pacific Northwest National Laboratory (PNNL), Richland, WA (US), 2012 Search PubMed.
- S. Bhagia, H. Li, X. Gao, R. Kumar and C. E. Wyman, Biotechnol. Biofuels, 2016, 9, 245 CrossRef PubMed.
- V. Archambault-Léger, X. Shao and L. R. Lynd, ChemSusChem, 2014, 7, 2721–2727 CrossRef PubMed.
- D. Lachos-Perez, P. Mayanga-Torres, J. Martinez, M.-J. Cocero, G. A. Tompsett, P. Guerra, M. T. Timko, M. A. Rostagno and T. Forster-Carneiro, Bioresour. Technol., 2017, 243, 1069–1077 Search PubMed.
- A. C. Ferrari, J. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. Novoselov and S. Roth, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- N. Gierlinger and M. Schwanninger, Spectroscopy, 2007, 21, 69–89 CrossRef CAS.
- C. Li and P. C. Stair, Catal. Today, 1997, 33, 353–360 CrossRef CAS.
- P. C. Mayanga-Torres, D. Lachos-Perez, C. A. Rezende, J. M. Prado, Z. Ma, G. T. Tompsett, M. T. Timko and T. Forster-Carneiro, J. Supercrit. Fluids, 2017, 120, 75–85 CrossRef CAS.
- D. Lachos-Perez, F. Martinez-Jimenez, C. A. Rezende, G. Tompsett, M. Timko and T. Forster-Carneiro, J. Supercrit. Fluids, 2016, 108, 69–78 CrossRef CAS.
- B. Hames, C. Scarlata and A. Sluiter, NREL/TP-510-42625: Determination of protein content in biomass, National Renewable Energy Laboratory, Golden, CO, 2008 Search PubMed.
- R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, NREL/TP-510-42619: Determination of extractives in biomass, National Renewable Energy Laboratory, Golden, CO, 2005 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, NREL/TP-510-42618: Determination of structural carbohydrates and lignin in biomass, National Renewable Energy Laboratory, Golden, CO, 2008 Search PubMed.
- A. Sluiter, B. Hames, D. Hyman, C. Payne, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and J. Wolfe, NREL/TP-510-42621: Determination of total solids in biomass and total dissolved solids in liquid process samples, National Renewable Energy Laboratory, Golden, CO, 2008 Search PubMed.
- A. Sluiter, B. Hames, C. S. R. Ruiz, J. Sluiter and D. Templeton, NREL/TP-510-42622: Determination of Ash in Biomass, National Renewable Energy Laboratory, Golden, CO, 2008 Search PubMed.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- R. Hertz-Schünemann, T. Streibel, S. Ehlert and R. Zimmermann, Anal. Bioanal. Chem., 2013, 405, 7083–7096 CrossRef PubMed.
- L. Kong, P. Miao and J. Qin, J. Anal. Appl. Pyrolysis, 2013, 100, 67–74 CrossRef CAS.
- H. Pińkowska, P. Wolak and A. Złocińska, Biomass Bioenergy, 2011, 35, 3902–3912 CrossRef.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322, 1857–1861 CrossRef CAS PubMed.
- J. H. Wiley and R. H. Atalla, Carbohydr. Res., 1987, 160, 113–129 CrossRef CAS.
- A. B. Rubayiza and M. Meurens, J. Agric. Food Chem., 2005, 53, 4654–4659 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Chem.–Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- N. Reis, A. S. Franca and L. S. Oliveira, Talanta, 2013, 115, 563–568 CrossRef CAS PubMed.
- C. Heitner, D. R. Dimmel and J. A. Schmidt, Lignin and lignans: advances in chemistry, CRC press, 2010 Search PubMed.
- D. Pujol, C. Liu, J. Gominho, M. Olivella, N. Fiol, I. Villaescusa and H. Pereira, Ind. Crops Prod., 2013, 50, 423–429 CrossRef CAS.
- S. Mishra, P. Tandon, P. J. Eravuchira, R. M. El-Abassy and A. Materny, Spectrochim. Acta, Part A, 2013, 104, 358–367 CrossRef CAS PubMed.
- G. Yang, E. A. Pidko and E. J. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS.
- C. Falco, N. Baccile and M.-M. Titirici, Green Chem., 2011, 13, 3273–3281 RSC.
- D. Fengel and G. Wegener, Wood: chemistry, ultrastructure, reactions, Walter de Gruyter, 1983 Search PubMed.
- A. Oosterveld, G. Beldman, H. A. Schols and A. G. Voragen, Carbohydr. Res., 2000, 328, 185–197 CrossRef CAS PubMed.
- A. Kruse and A. Gawlik, Ind. Eng. Chem. Res., 2003, 42, 267–279 CrossRef CAS.
- J. Figueiredo, M. Pereira, M. Freitas and J. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- S. K. Patil and C. R. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- A. McQuillan, Adv. Mater., 2001, 13, 1034–1038 CrossRef CAS.
- L. F. Ballesteros, J. A. Teixeira and S. I. Mussatto, Food Bioprocess Technol., 2014, 7, 3493–3503 CrossRef CAS.
- A. Smith, The Coblentz Society Desk Book of Infrared Spectra, 1982, pp. 1–24 Search PubMed.
- M. Mosteiro-Romero, F. Vogel and A. Wokaun, Chem. Eng. Sci., 2014, 109, 220–235 CrossRef CAS.
- F. Vogel, in Handbook of green chemistry, Heterogeneous Catalysis, ed. P. Anatas and R. Crabtree, Wiley-VCH, Weinheim, 2009, vol. 2, ch. 12, pp. 281–324 Search PubMed.
- H. Pińkowska, P. Wolak and A. Złocińska, Chem. Eng. J., 2012, 187, 410–414 CrossRef.
- H. Li, Y. Pu, R. Kumar, A. J. Ragauskas and C. E. Wyman, Biotechnol. Bioeng., 2014, 111, 485–492 CrossRef CAS PubMed.
- N. Gierlinger, T. Keplinger and M. Harrington, Nature Protocols, 2012, 7, 1694–1708 CrossRef CAS PubMed.
- M. Sasaki, Z. Fang, Y. Fukushima, T. Adschiri and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 2883–2890 CrossRef CAS.
- B. Girisuta, L. Janssen and H. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. M. Kabyemela, T. Adschiri, R. M. Malaluan and K. Arai, Ind. Eng. Chem. Res., 1997, 36, 1552–1558 CrossRef CAS.
- M. Liebeck, C. Pfeifer, A. Drochner and G. H. Vogel, Chem. Ing. Tech., 2013, 85, 516–522 CrossRef CAS.
- T. Y. Nguyen, C. M. Cai, R. Kumar and C. E. Wyman, ChemSusChem, 2015, 8, 1716–1725 CrossRef CAS PubMed.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato and S. Hayashi, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed IR, Raman, and TGA data. See DOI: 10.1039/c7se00260b |
| This journal is © The Royal Society of Chemistry 2017 |