
An overview of a novel concept in biomass pyrolysis: microwave irradiation
Xuesong
Zhang
 *a,
Kishore
Rajagopalan
a,
Hanwu
Lei
b,
Roger
Ruan
c and
Brajendra K.
Sharma
*a
*a,
Kishore
Rajagopalan
a,
Hanwu
Lei
b,
Roger
Ruan
c and
Brajendra K.
Sharma
*a
aIllinois Sustainable Technology Center, University of Illinois at Urbana-Champaign, 1 Hazelwood Drive, Champaign, IL 61820, USA. E-mail: xuesong@illinois.edu; bksharma@illinois.edu
bDepartment of Biological Systems Engineering, Washington State University, Richland, WA 99354-1671, USA
cCenter for Biorefining, Department of Bioproducts and Biosystems Engineering, University of Minnesota, St. Paul, MN 55108, USA
First published on 27th June 2017
Abstract
The increasing demand for renewable fuels and chemicals necessitates the exploration of alternative sources to replace petroleum sources. Biomass has been viewed as the most promising source to produce sustainable fuels and chemicals. Underpinning the key advantages of microwave heating (e.g., rapid and controlled heating, energy saving, and no requirement for agitation or fluidization), microwave-assisted pyrolysis (MAP) is one of the most attractive techniques for the valorization of biomass, which are more amenable to produce three high quality products: bio-oil, gas, and bio-char. In this respect, this article reviews the biomass pyrolysis using microwave irradiation from several points of view, starting from fundamentals of microwave irradiation, types of microwave absorbers, and chemistry of non-catalytic MAP and focusing on chemistry of catalytic MAP plus various categories of catalysts. Recent progress in the experimental studies on both non-catalytic MAP and catalytic MAP of biomass is also demonstrated with emphasis on the bio-oil yield and quality. Additionally, reaction kinetics and future prospects in the light of current studies are also given in this review. Consequently, this review illustrates both the highlights of significant achievements from biomass pyrolysis using microwave irradiation and the milestones that are necessary to be obtained in the future.
 Xuesong Zhang | Xuesong Zhang is a Postdoctoral Research Associate at the Illinois Sustainable Technology Center, University of Illinois Urbana-Champaign. He received his B.E. degree in 2012 from Northwest A&F University (China), and his PhD degree from Washington State University in 2016. His research focuses on the thermochemical conversions including microwave pyrolysis and hydrothermal liquefaction, catalysis, bio-oil upgrading for advanced jet fuels plus valuable chemicals, as well as treatment of aqueous phase from biocrude/bio-oil, and reverse osmosis of treated aqueous phase to recover nontoxic water and value-added products. He has co-authored over 20 relevant peer-reviewed publications. |
 Kishore Rajagopalan | Dr Rajagopalan is an Associate Director at the Illinois Sustainable Technology Center and was appointed as the State Pollution Prevention Scientist in 2013. He has over 30 years of experience in plant operations, green process development, separations, and pollution prevention research. His personal research interests are in the integration of separation research to advance green process development with a special focus on membrane processes. He has a BS in Chemical Engineering and a Ph.D. in Food Science along with a postgraduate diploma in management. He has over 50 peer-reviewed publications and holds 5 U.S. patents. |
 Hanwu Lei | Hanwu Lei received his PhD degree in Biosystems and Agricultural Engineering from University of Minnesota, Twin Cities (USA) in 2006. He is an associate professor of Biological Systems Engineering at Washington State University. His research is on discovering and applying novel approaches for renewable energy and value-added process development from agricultural feedstocks and products. Dr Lei's research activities include biomass thermochemical and biochemical conversions to biofuels and bioproducts. His research is focused on microwave pyrolysis, catalysis processes, bio-oil upgrading, and biomass carbon utilization for jet fuels, phenols, aromatics, cycloalkanes, hydrocarbons, activated carbon, and bio-based chemical production. |
 Roger Ruan | Roger Ruan is a Distinguished Guest Professor of Nanchang University and a Professor and Director of Center for Biorefining at the University of Minnesota. He received his Ph.D. degree from the University of Illinois at Urbana-Champaign in 1991. His current interests are in nonthermal plasma technology development and application, algae and aquaponics system for waste utilization and treatment, solid waste conversion for energy fuels, chemicals, and materials production, and food shelf stability and quality enhancement and safety assurance. He has published over 400 peer-reviewed papers, books and patents. |
 Brajendra K. Sharma | B. K. Sharma joined the Prairie Research Institute – Illinois Sustainable Technology Center at the University of Illinois, Urbana-Champaign in 2009 as a Senior Research Engineer. In February 2016, he was elected a fellow of the Society of Tribology & Lubrication Engineers. His research interests include thermochemical conversion of biomass and waste materials, biolubricants, bio-oil derived chemicals and biobased products. His current research is focused on conversion of waste plastics, tires, and waste lipids into liquid fuels using pyrolysis and hydrothermal liquefaction; recovery of value-added products, antioxidants from bio-oils; upgradation of bio-oils through a low-cost catalytic and extraction process; and bio-oil derived bioasphalts. |
1. Introduction
The depletion of fossil fuel resources and resulting climate changes are of preliminary economic, academic and politic concern worldwide. Alterations to the climate because of the greenhouse effect are threatening to humans and other species, originating from anthropogenic activities including the burning of fossil fuels for power generation.1 In this context, energy consumption is projected to continuously increase with an average annual growth of approximately 1.6% in the next two decades; whilst the supplies of fossil fuel resources for energy generation are limited.2 Alternative solutions are sought to switch from conventional to renewable resources, e.g., biomass. The valorization of biomass is currently an area of utmost interest since the utilization of biomass has the potential to substitute significant amounts of petroleum-derived fuels. Moreover, biomass is abundantly available currently, offering over 10% of the global energy supply, and being recognized as the top four energy sources in the light of world final energy consumption in 2011.1,3Biomass generally refers to wood waste, agricultural crops and their residues, municipal solid waste, food waste, animal waste, algae, aquatic plants etc. For instance, lignocellulosic biomass (e.g., wood, grass, and agricultural residues) is an abundant and carbon-neutral resource found on the earth,2 which makes the production of second generation biofuels highly attractive in a broad sense.4 Lignocellulosic biomass is primarily comprised of cellulose, hemicellulose and lignin. Cellulose and hemicellulose consist of complex polysaccharides, while lignin as highly substituted and mononuclear phenolic amorphous polymers is made up of phenylpropane units.5 Microalgae are another very important biomass source containing proteins, carbohydrates, enzymes, vitamins, and minerals.6 In particular, biofuels evolved from microalgal biomass are identified as third generation biofuels.7,8 It is discerned that microalgae have a much higher capacity to manufacture 30–100 times more energy per hectare in comparison with terrestrial corps.9 Despite some limited use of biomass typically for power generation and low quality bio-crude production, biomass still appears to be the most attractive and feasible form to produce advanced biofuels, fine chemicals and byproducts with net zero carbon emission.10
There are several technologies that have been extensively evaluated and implemented in past decades to valorize biomass, which can be mainly divided into two platforms: thermochemical and biological technologies. A biological pathway generally involves the application of various microorganisms and bio-catalysts for the transformation of biomass into high value-added fuels, chemicals, gases and other valuable outputs.11 Nevertheless, biological conversions of biomass are not cost-effective mainly because of the fact that the biochemical techniques can be merely devoted to the utilization of cellulose and hemicellulose fractions.12 In contrast, thermochemical approaches are obviously more energy efficient and flexible pertaining to feed and products, including pyrolysis, gasification, combustion, reforming and hydrothermal conversions.13 Among the primary thermochemical techniques, pyrolysis is a prominent and well-recognized platform that has been extensively investigated and employed to valorize biomass into valuable products.12,14 Typical pyrolysis is to heat biomass in the absence of oxygen at 400–600 °C, yielding bio-oil, biochar, and biogas. Fast pyrolysis of biomass can garner a very high yield (up to 80 wt% of dry feed) of liquid product retaining main energy (up to 70%) in the so-called bio-oils.15,16 Bio-oil has been considered as the cheapest biofuel and has attracted crucial research concerns in the past two decades.17 Moreover, the technique of biomass pyrolysis is on the verge of commercialization.18
Bio-oil derived from the immediate quenching of pyrolytic vapors is a viscous black liquid and it is composed of over 300 oxygenated compounds.19 Typical bio-oil mainly contains three major classifications of compounds, i.e., sugar-evolved compounds, small carbonyl compounds, and lignin-derived compounds.20 The distribution of these compounds in the bio-oil is dominantly dependent on the category of biomass and pyrolysis severity.21 It is evident that the high water content (15–30 wt%) triggers a low heating value of the bio-oil.20,22 A large variety of carboxylic acids renders low pH values of 2–2.5.23 In general, the instability of bio-oil is mostly attributed to the oxygen-rich nature, thereby rendering the main difference between bio-oils and hydrocarbon fuels.20,24 As a result, these detrimental properties (e.g., high acidity, low heating value, high reactivity, and high viscosity) of bio-oils hamper their straightforward application as transportation fuels.15,20
Enhancing bio-oil quality toward its properties identical to those of petroleum-derived fuels is necessitated.25 Efforts of rejecting its high oxygen content should be devoted to via certain upgrading technologies.15,19 These technologies should be developed as efficient processes that transform biomass into biofuels, showing high compatibility and energy density with the current energy infrastructure.26 Advanced biomass conversion technologies have been undertaken to achieve these goals in the last two decades.15,16 Of these, catalytic deoxygenation has been widely investigated to improve the bio-oil quality and primarily includes two platforms: catalytic cracking and hydrodeoxygenation (HDO).27,28 Catalytic cracking is a regular technique that implements solid acid catalysts in the pyrolysis system and it is performed at atmospheric pressure without the introduction of hydrogen.29 Yet, it is noted that catalytic cracking of bio-oils is not promising owing to the low quality products observed with a low carbon yield and significant coke formation, leading to a short catalyst lifetime.30 HDO of bio-oil adopts petroleum oil hydrotreating protocol by using supported metal catalysts in a pressurized hydrogen atmosphere to achieve desired hydrocarbons.17,31 This technology is attractive and has been developed in terms of the substantial increase in high quality products.32 Yet, the HDO process has to be carried out under high-severity conditions (e.g., elevated reaction temperature and high hydrogen pressure). High operating costs due to costly noble catalysts utilization, substantial hydrogen consumption, and significant catalyst deactivation are endured in the HDO process as well.33
Instead, catalytic fast pyrolysis (CFP) is an alternative approach to directly upgrade hot pyrolytic vapors assisted by a suitable catalyst prior to quenching pyrolytic vapors to gain bio-oil.14 The in situ catalytic fast pyrolysis (referred to as in situ CFP) is conducted by directly combining biomass with the catalyst; when the catalyst placed in a downstream reactor only upgrades the pyrolytic vapors, the process is referred to as ex situ catalytic fast pyrolysis (ex situ CFP).34 CFP has been suggested to be more amenable for directly converting biomass into high grade bio-oils with improved stability since it can get rid of polymerization and re-evaporation of bio-oils.21,23,35–40 It is noteworthy that zeolite catalysts (e.g., HZSM-5) have been considered as the most efficient catalysts to generate considerable petrochemicals (aromatics and olefins).41–45
It is found that conventional pyrolysis suffers from some intrinsic drawbacks such as heat transfer resistance, heat losses to the surroundings, non-selective heating, damage to reactor walls due to continuous electrical heating, etc.46,47 Due to the above-mentioned problems, the integration of microwave irradiation and pyrolysis process is a novel conceptual development to effectively valorize biomass/organic waste as sketched in Fig. 1.11,48,49 MAP technology is emerging as one of the most promising approaches of enhancing and accelerating chemical reactions due to effective heat transfer profiles through microwave irradiation.50,51 Furthermore, MAP encloses the potentials of easy control of reaction conditions, fast and selective heating, low reaction temperatures, and low energy requirements.11,52
 | ||
| Fig. 1 The representative concept of microwave-assisted pyrolysis. Adapted from ref. 11. | ||
Identical to CFP, a novel concept of catalytic microwave-assisted pyrolysis (CMAP) has been presented to generate outstanding products by using catalysts such as HZMS-5. As expected, CMAP not only obtains high quality biofuels and fine byproducts, but also possesses perceived advantages of MAP. Very recently, catalytic microwave-assisted co-pyrolysis (CMACP) is attracting substantial attention worldwide, and it is recognized as one of the most advanced pyrolytic technologies, outperforming conventional CFP in terms of highly desirable products.53–55 Given high grades of bio-oil, bio-char, and biogas derived from the CMAP of biomass, this technique paves a novel and economically feasible pathway for the valorization of biomass on a large scale.53 In the light of these premises, the CMAP of biomass is clearly beneficial for environment and energy recapture.
Numerous comprehensive reviews have reported pyrolysis and catalytic pyrolysis of biomass. Some recent reviews have also highlighted MAP of lignocellulosic biomass for biofuel production.4,51,56–61 To the best of our knowledge, the CMAP of biomass (with the concentration on lignocellulosic biomass and microalgae) has never been reviewed. This is an unprecedented review that aims to shed light on the technology with respect to the CMAP of biomass. Herein, it starts with the principles of microwave irradiation, giving special attention to the comparison between conventional heating and microwave heating. The role and categories of microwave absorbers are also discussed. Furthermore, fundamental chemistry and kinetics of the reactions in CMAP are elucidated as well. In order to understand the state-of-the-art situation of CMAP, up-to-date research efforts regarding CMAP of biomass are summarized. With the consideration of microwave irradiation contribution in catalytic pyrolysis, this review will focus principally upon recent advances in the improvements of product quality and yields for the sake of future commercialization.
2. Microwave irradiation
2.1 Fundamentals of microwave irradiation
Microwaves are generally defined as non-ionizing radiations, namely electromagnetic waves that are comprised of two perpendicular components (i.e., electric and magnetic fields).51 Microwave is recognized as an unconventional source of energy since it can be transmitted, reflected or absorbed toward the reagents.62 In the electromagnetic spectrum as shown in Fig. 2, microwaves fall in between the infrared and radio wave regions of the electromagnetic spectrum with the wavelengths ranging from 0.01 to 1 m, and the corresponding frequency varying from 0.3 to 300 GHz.51,63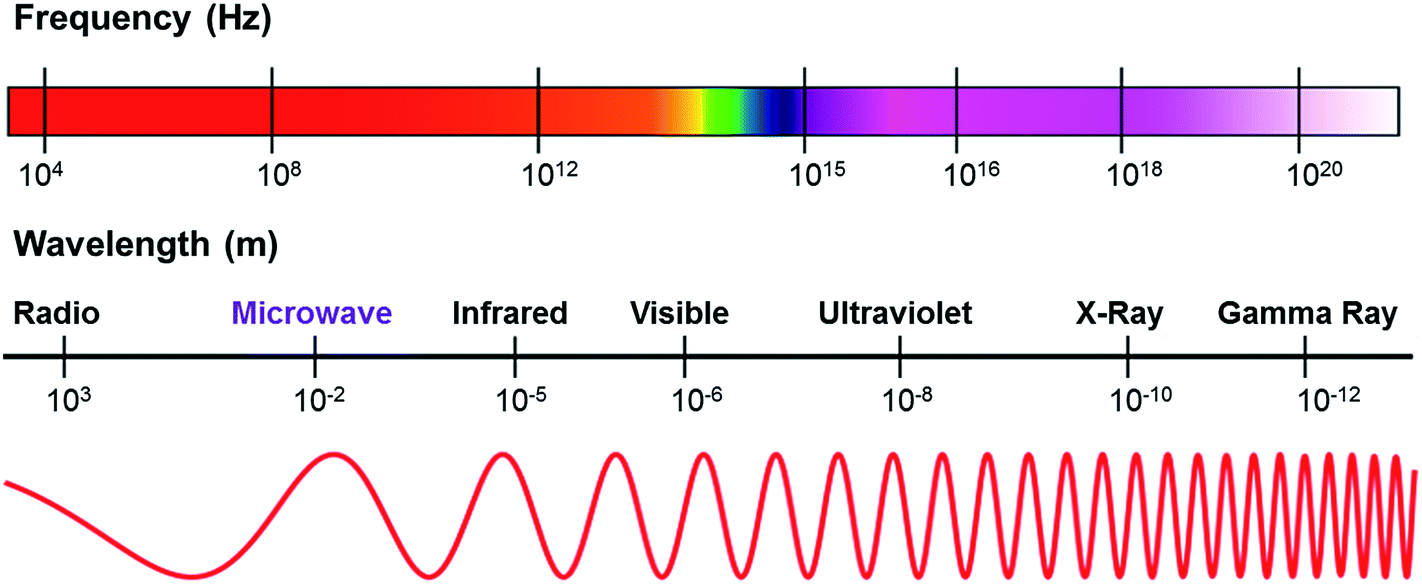 | ||
| Fig. 2 The frequency and wavelength of microwave radiation region. Adapted from ref. 64 and 65. | ||
With this region, the microwave frequencies interfere with the frequencies allocated to cellular phones and telecommunications.57,59,65 To avoid any interference, domestic microwave ovens and most microwave reactors for chemical synthesis are operated at the frequency of 2.45 GHz, corresponding to a wavelength of 12.25 cm.65,66 At 2.45 GHz, designed microwave ovens are prone to heating foods; meanwhile the water inside acts as a good absorber of microwave irradiation.67 The four frequencies of 0.915 GHz, 2.45 GHz, 5.8 GHz, and 24.124 GHz are reserved by the Federal Communications Commission (FCC) for industrial, scientific, and medical (ISM) purposes.51 Most industrial microwave processing for chemical synthesis is conducted at the frequencies of 0.915 GHz and 2.45 GHz, which are assigned for heating applications.59
It is observed that the conversion efficiencies of electrical energy to microwave energy are approximately 85% and 50% for 0.915 GHz and 2.45 GHz, respectively.57 In comparison with 2.45 GHz, the utilization of 0.915 GHz can offer a substantially large penetration depth, which is an essential indicator in the evaluation of the microwave assimilation capacity of materials, design of the microwave cavity size, and the process of scale-up.59 Microwave penetration depth is also affected by material types, microstructure properties, and temperature. For instance, microwave penetration depth in water is just 1.4 cm by using the frequency of 2.45 GHz at 25 °C, whereas its penetration depth is enhanced to 5.7 cm using the same frequency at 90 °C.59 In the case of quartz glass, the microwave penetration depth can reach up to 160 m. As for the microwave penetration depth in biomass, it is mainly dependent on biomass density, composition, water content, etc.59 The microwave penetration depth in representative materials are given in Table 1.
| Microwave transparent | Reflector | Microwave absorber | |||||
|---|---|---|---|---|---|---|---|
| Material | tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ δ |
d p (m) | Bulk metal | d p (μm) | Material | tanδ |
d p (cm) |
| Alumina | 1.0 × 10−3 | 12.65 | Al | 1.7 | Water | 0.15 | 3 |
| Fused quartz | 3.0 × 10−4 | 75.73 | Cu | 1.3 | SiC | 0.37 | 1.93 |
| Borosilicate glass | 1.2 × 10−3 | 15.7 | Au | 1.5 | Carbon black (20 μm) | 0.23 | 5.75 |
| Teflon | 4.8 × 10−4 | 56.4 | Ag | 1.3 | Graphite powder (20–80 μm) | 0.36–0.67 | 1.34–2.09 |
| PVC | 5.6 × 10−3 | 4.03 | Zr | 6.7 | Graphite carbon powder (60–80 mesh) | 0.4–1 | 0.5–0.9 |
| Polystyrene | 3.0 × 10−4 | 76.2 | Activated carbon | 0.31–0.9 | 0.7–3.43 | ||
| Silicon | <1.2 × 10−2 | >3.96 | Charcoal | 0.14–0.38 | 6–11 | ||
2.2 Principles of microwave heating
Microwave heating is a non-contact energy transfer process that transforms electromagnetic energy into thermal energy at certain frequencies, implying fast heating rates, where electromagnetic energy is effectively absorbed by the materials under microwave irradiation.69 Microwave heating consists of two general mechanisms: dipolar polarization and ionic migration as sketched in Fig. 3.59,69–71 It is mainly attributed to the alignment of dipoles (Fig. 3A) or ions (Fig. 3B) in the electric field.68 With regard to polar molecules, the electric field component of microwave results in water and other polar fluid (e.g., ethanol) molecules rotating and aligning in both induced and permanent dipoles with an alternating field, which is named as dipolar polarization.69 The molecule friction and collisions caused by increased molecular rotation and movements thereby lead to microwave heating and heat loss.66 As for ionic migration, dissolved charged particles oscillate back and forth under microwave irradiation, and they also collide with neighboring molecules, thereby generating heat.4 It is noteworthy that the ionic migration mechanism has a more significant influence than the dipolar polarization pertaining to the heat-generating capacity.68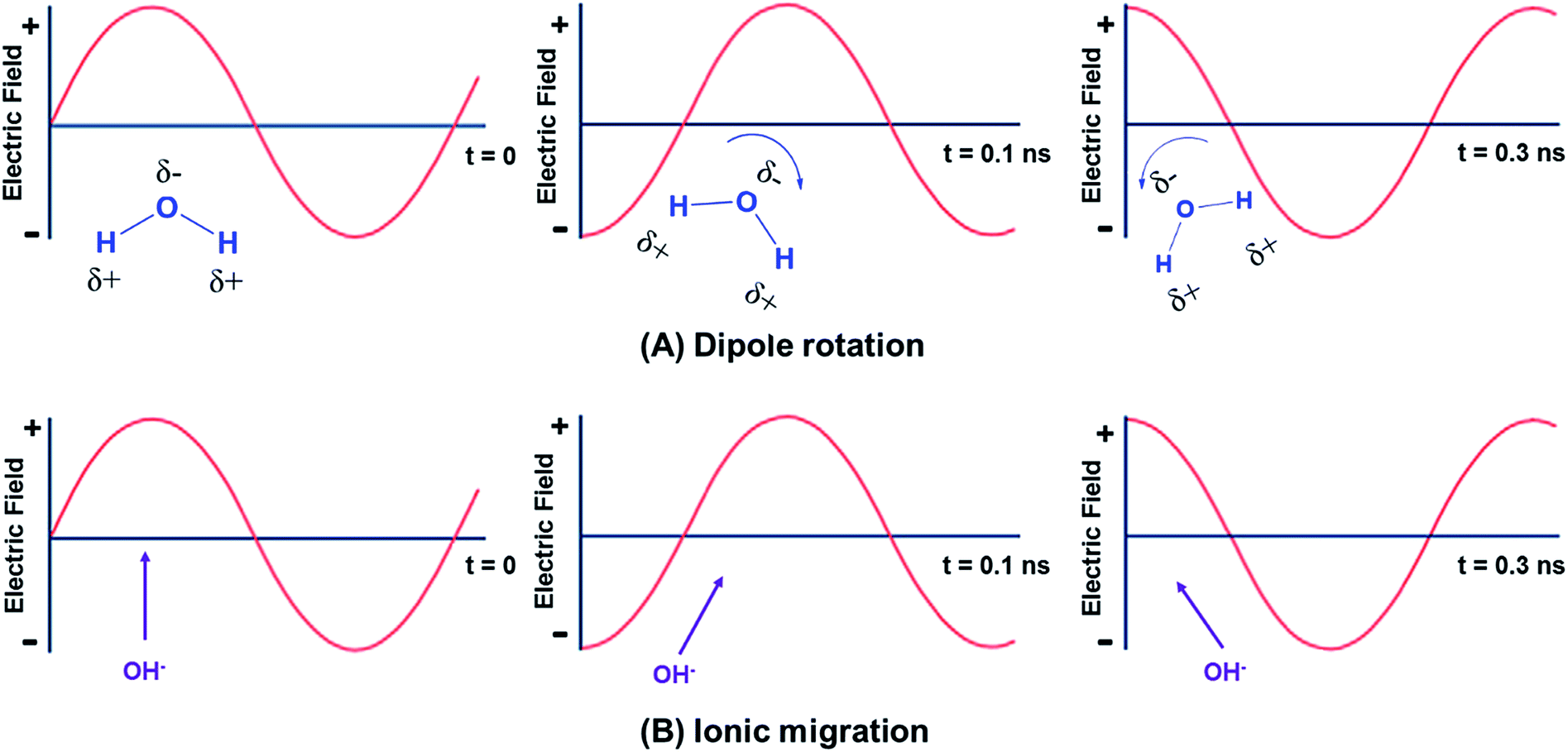 | ||
| Fig. 3 The fundamental mechanism of microwave heating: (A) dipole rotation; (B) ionic migration. Adapted from ref. 68. | ||
As the response of different materials to microwave irradiation is not comparable, only partial materials are amenable to microwave heating. According to the response to microwave irradiation, there are three types into which a material can be broadly categorized: (i) insulators or microwave-transparent materials in which the microwaves pass through without any energy losses (e.g., quartz, Teflon, etc.); (ii) conductors where the microwaves cannot penetrate through and reflect back (e.g., metals); and (iii) absorbers where the microwaves can pass through and be absorbed (e.g., water, oils, etc.).51,72 Since materials that absorb microwave irradiation are usually called as dielectrics or absorbers, microwave heating is also defined as dielectric heating.64
2.3 Comparison between microwave heating and conventional heating for biomass pyrolysis
Conventional thermal heating is widely known as a traditional heating pattern with heat transfer from the surface to the center of a material by conduction, convection, and radiation.4,51 Nevertheless, conventional heating is extremely slow and inefficient, which is relatively dependent on the convection currents and thermal conductivity of the material.51 In contrast, microwave heating, transforming electromagnetic energy into thermal energy, is viewed as one energy conversion rather than heating transfer.51 Since microwaves can penetrate materials and deliver energy, heat can be generated through the volume of materials (referred to as in-core volumetric heating). The formation of hot spots is a vital property of microwave heating, which is derived from the inhomogeneity of microwave field or dielectric characteristics within the feedstock.4,73 Thus, the accurate temperature inside the feedstock is much higher than the measured temperature in the bulk. It is proven that hot spots play a significant role in the yield and quality of microwave processing products. To control or prefer the hot spot phenomenon, several important factors (such as cavity design and operating conditions) can be implemented to modify the phenomenon.73The different mechanisms for microwave and conventional heating patterns are demonstrated in Fig. 4. Unlike conventional heating, microwave heating shows a much higher temperature at the central of the material comparing to the surrounding material. Table 2 lists the specific details regarding the advantages and disadvantages for both microwave heating and conventional heating. There are many perceived merits of microwave heating, including uniform internal heating of large biomass particles, rapid and controlled heating, and no requirement for agitation or fluidization.75 Microwave heating is more efficient than conventional heating because microwave heating can overcome the major issues of conventional heating, including limitations of specific heat and density, heterogenic heating of the surface, and thermal conductivity of materials.76 Moreover, it requires less energy input for heating feedstock in comparison with conventional heating approach.62 It has been estimated that 15% of energy in biomass feed is required to drive a conventional pyrolysis process.25 Approximately 2.7–3.1 kJ g−1 energy input is required for woody biomass pyrolysis (assuming a gross calorific value of 18–20.5 kJ g−1).77 However, the energy consumption for MAP of woody biomass to achieve an acceptable degree of pyrolysis is in the range of 2.2–2.5 kJ g−1;78 increasing the power density to 100 MW m−3 can even allow a decrease in specific energy to as little as 1.07 kJ g−1.79 Underpinning these key advantages, studies of MAP of biomass have been recently reported, and indicate that MAP is a highly scalable technology suitable for distributed conversion of biomass.4,11,51,59
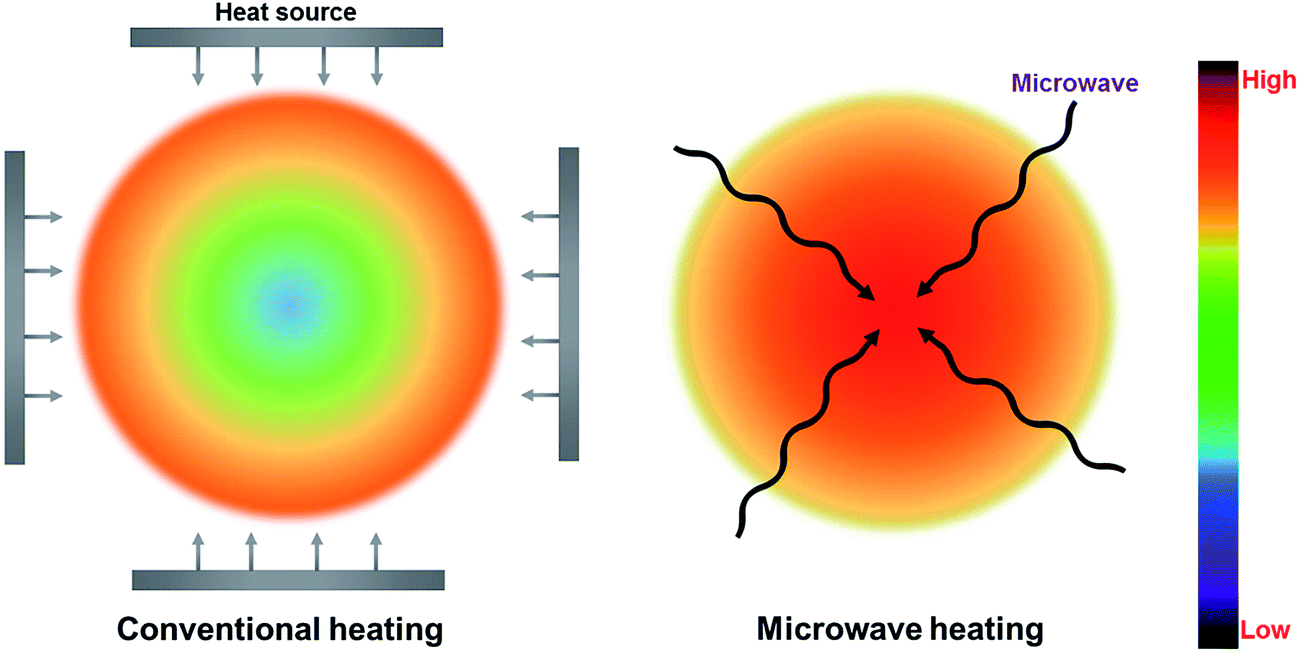 | ||
| Fig. 4 Microwave-induced and conventional heating patterns. Adapted from ref. 74. | ||
| Technique | Advantages | Disadvantages |
|---|---|---|
| Microwave heating | • Broad range of feedstock for valorization and wide range of higher quality products is achieved11,67 | • Inherent issue of temperature measurement and uniformity4,11 |
| • Non-contact and volumetric heating4,51,57,59,66,67 | • Penetration depth inhibits the large reaction vessel for scaling up65,67,80 | |
| • Energy transfer rather than heat transfer4,51,57,67 | • Uncontrolled heating leads to safety concerns65 | |
| • Energy savings11,67,80 | • Low energy conversion efficiency81,82 | |
| • Rapid and efficient heating4,51,57,59,66,67 | • Difficulties of the scaling up of microwave-assisted heating for biomass pyrolysis11 | |
| • Material selective heating57,67 | ||
| • Quick start up and stopping11,57,66,67 | ||
| • Higher level of safety and automation57,67 | ||
| Conventional heating | • High flexibility of feedstock and products11 | • Relatively slow and inefficient convection currents and thermal conductivity4,59,65 |
| • Well developed and easy to scale up11 | • Lower quality products4,11 | |
| • High possibility of continuous processing on a large scale11 | • High energy consumption4,11 |
On the other hand, microwave heating also suffers from some disadvantages as given in Table 2. For example, the current methods used to measure the accurate temperature in a microwave reactor are not effective because of the interference between electromagnetic irradiation and thermocouple sensors.4 The declared efficiency of electric energy as compared to that of microwave energy is approximately over 70% when the modern magnetrons are operated at 2.45 GHz; consequently, the energy conversion efficiency is low, which can amount to 20–60% from the input electric energy to heat by microwave heating.81–84 It has been also assessed that the energy losses in transformers and magnetron during MAP of biomass is ca. 26%, and the power loss caused by the heat loss accounted for approximately 29%.85 In addition, the scaling up of biomass pyrolysis using microwave heating is highly difficult and still in the infancy stage since the limited penetration depths in materials trigger a very restrictive size of the reaction vessel.11 As a consequence, a simple and direct comparison between microwave heating and conventional heating cannot give rise to correct interpretations. To date, the theory with regard to the observed rate enhancement using a microwave heating technique purely due to thermal effects is widely accepted by the literature.86 Even though non-thermal effects may not be thoroughly ruled out, they are much less essential for chemical reaction enhancement.86,87
3. Microwave absorbers
The ability of a specific substance to transform electromagnetic energy into heat plays a vital role in microwave heating; and it is determined by the dielectric properties, i.e., dielectric loss (ε′′) and dielectric constant (ε′).4,59,63 The former measures the efficiency with which electromagnetic energy can be converted into heat, whilst the latter illustrates the capacity of molecule polarization by an electric field.4 The ratio of the dielectric loss (ε′′) to the dielectric constant (ε′) is denoted as the loss tangent factor (tanδ), which is a parameter used to express the overall efficiency of a material to absorb energy from microwave radiation.59 In general, the materials used for microwave heating can be classified as high (tanδ > 0.5), medium (0.1 < tanδ < 0.5), and low microwave absorbing (tanδ < 0.1).63 The dielectric loss tangent of diverse materials is also summarized in Table 1. Materials with low tanδ values cannot be heated to desired temperatures by a rapid heating rate, using sufficient amount of microwave energy. Most biomass show relatively low microwave absorption when they are subjected to microwave-assisted heating.59 Yet raw biomass can be blended with materials with high tanδ values before being fed into a MAP reactor to achieve a rapid heating rate. These materials with high tanδ are usually named as microwave absorbers.
3.1 Solid carbonaceous microwave absorbers
Solid Carbon Based Materials (SCBMs) are regarded as good microwave absorbers. SCBMs can indirectly heat feedstock that are relatively transparent to microwave. A variety of SCBMs have been employed as ideal candidates for microwave absorbers to promote biomass pyrolysis, e.g., charcoal, activated carbon, coke, and graphite.46,94–96 As a matter of fact, generated biochar from MAP of biomass can be reused as cost-effective microwave absorbers. Although biochar formed in the pyrolysis is less active than graphite, biochar is a modest absorber potentially having some impact at some instances. The tanδ of most SCBMs as listed in Table 1 is much higher than that of biomass, which suggests that the addition of SCBMs can garner some perceived benefits: (i) the increase of microwave assimilation capacity of bulk materials; (ii) heat transmitted to the surrounding materials; and (iii) the supply of a rapid heating rate and sufficient temperature at the low microwave powers.59,66
Regarding SCBMs, there are no other contaminations with extra elements; on the contrary, they can even contribute to the enhancement of the calorific value of biochar. However, if additives such as SiC are used as the microwave absorbers, the element of silicon is added into the solid fraction.56 Accordingly, the consequence of this situation has to be carefully considered when using additives as microwave absorbers. Under microwave irradiation, SCBMs heated by microwave energy can generate hot spots termed as micro-plasmas, appearing in the form of small sparks or electric arcs.94 The micro-plasmas is limited to tiny space and usually lasts for fractional second; and, they are of two classification depending on nature and shape: ball lighting and arc discharge effects.97 It is possibly like that the MAP of biomass in the presence of SCBMs may lead to thermal instability and localized heating of biomass. Yet, the choice plus distribution of SCBMs with biomass can be conductive to reducing hot spots.59 Additionally, it is manifested that some SCBMs can also take part in biomass gasification and pyrolysis under microwave irradiation, influencing the yields and quality of products.
3.2 Metal oxide microwave absorbers
Metallic oxides can also act as good microwave absorbers, providing several merits: the effective absorption of microwave energy, the increase of materials' heating rate, and the improvement of devolatilization from biomass.59 Compared with SCBMs, metal oxide microwave absorbers (MOMAs) can substantially affect product yields and quality during the MAP of biomass.59 It is mainly due to the fact that the evolved vapor and gas can simultaneously undergo secondary and catalyzed reactions when contacting MOMAs.The most common metal oxides utilized as microwave absorbers are copper oxide (CuO), magnesium oxide (MgO), calcium oxide (CaO), iron oxide (Fe3O4), nickel oxide (NiO), etc. Moreover, some metals and metal hydroxides can serve as microwave absorbers including elemental iron (Fe), elemental aluminum (Al), sodium hydroxide (NaOH), potassium hydroxide (KOH), iron chloride (FeCl3), zinc chloride (ZnCl2), etc.98 As mentioned above, the concerns of MOMA utilization are the recycling of these absorbers, along with the utilization or disposal of biochar containing the additives.66
4. Microwave-assisted pyrolysis chemistry
4.1 Chemistry of non-catalytic microwave-assisted pyrolysis
MAP is a promising alternative technique to conventional pyrolysis mainly because of dielectric heating. Basically, a heating rate of 10–200 °C s−1 can be achieved by using microwave absorbers, favoring the liquid and gaseous products.25,51 Normally, quartz reactors are employed inside a microwave chamber where biomass is placed; and a product cooling and collection system is introduced, where the condensable bio-oil is collected.99–102 The MAP system is usually purged with N2, He, or CO2 to maintain an oxygen-free environment.103 The inert gases can also assist pyrolysis volatile vapors flowing from the reactor to condensable system to obtain bio-oil. The non-condensable vapors escape as gas at the end of the condensers, while biochar is left in the quartz reactor. As biomass is often a poor absorbing material, microwave absorbers should be introduced and homogeneously blended with biomass prior to the pyrolysis, to gain a rapid heating rate. It is discerned that selecting a proper microwave absorber is key in the improvement of bio-oil quality and fine biochar for soil amendment and remediation applications.104The MAP of biomass reveals a potential for bio-oil production with higher carbon content, higher heating value, and lower oxygen content than conventional pyrolysis of biomass.20,105–107 The MAP of biomass can produce biochar with higher surface area and pore volume than conventional pyrolysis of biomass.11 Furthermore, it is indicated that biochar from MAP is uniform and very clean.108,109 At the same time, part of biochar can be recycled into the microwave reactor, serving as the microwave absorber to recover some sensible heat. Generally, MAP of biomass is a novel technique for producing more porous biochar that is used in sorption applications or as a precursor for activated carbon production. The gaseous product from the MAP of biomass dominantly contains H2, CO, CH4, and CO2, which is thought of as a promising technique for producing H2-enriched fuel gas.107,110 The gaseous product from MAP of biomass can liberate more energy due to the higher heating value than gas from conventional pyrolysis of biomass. It is also proved that increasing reaction temperature can substantially increase H2 and CO concentrations, while CO2 concentration significantly decreases; that is possibly due to the self-gasification of biochar.94
The MAP of biomass can allow a careful control of pyrolysis parameters to maximize bio-oil or gas production; operating pyrolysis parameters can induce and alter partial reaction pathways, garnering different chemical profiles of pyrolytic vapors/bio-oil.11 It has been reported that the discrepancy in product yields is attributable to the difference in biomass characteristics, particle size, reaction temperature, reaction time, reactor design, product vapor residence time, microwave heating manner, microwave power level, etc.57,107,111–115 Among these factors, reaction temperature plays the most critical role in product yields and distribution. In general, the increase of reaction temperature can substantially increase the gas yield; while the bio-oil yield drastically decreases and the alteration of the biochar yield is not obvious. It is suggested that when pyrolysis vapors are released by the devolatilization of biomass in MAP, the heavy intermediates in the vapors can be thermally cracked into permanent gaseous compounds at elevated reaction temperature.57 Prolonged residence time of pyrolysis vapors under microwave irradiation can extend their secondary reactions, resulting in the decrease of bio-oil production but increase of biochar formation.112 Hence, the pyrolysis vapors should be swept out of the microwave chamber instantly to reduce secondary cracking of the hot organic vapors and to deliver water vapor together with water-soluble small polar molecules out of the microwave reactor.4
More importantly, the microwave power level can obviously affect product yields and qualities. The improvement of the microwave power level increases the microwave heating rate, thus promoting the yields of pyrolysis vapors and bio-oil production. Besides, the variation of microwave power level not only affects the biochar yield, but also alters the heating value of biochar. Since the heating values of cellulose and hemicellulose are much lower than that of lignin (the more thermo-resistant biopolymer), the devolatilization of cellulose and hemicellulose in biomass is beneficial for the increase of biochar heating value.4 At relatively low microwave power levels, the degradation of cellulose and hemicellulose is dominant, whereas the thermal degradation of lignin is not remarkable. However, the devolatilization of lignin is considerable at high microwave power levels, resulting in the drop in the heating value of biochar. Some chemical pretreatments, such as using ionic liquids or acids, are also good for the MAP of biomass.4
On the other hand, the regular MAP of biomass is not beneficial for high bio-oil yield and quality, and the thorny challenge is to promote the heating rate during MAP.116 Recently, a novel concept of a fast microwave-assisted pyrolysis (fMAP) utilizing microwave absorbers has been proposed by Ruan's group, as depicted in Fig. 5.116–118 In this new concept with minimal energy input under microwave irradiation, the microwave absorbers particles (e.g., SiC) are kept heated on a fixed bed, and the chamber temperature inside the microwave oven is steady when the biomass is fed. Owing to a simultaneous indirect and direct heating methods: microwave irradiation and thermal conduction from heated absorbers particles, biomass can be loaded in contact with the hot absorbers and it can be almost instantly heated to the desirable temperature. Biomass subjected to the fMAP can be instantaneously converted into pyrolytic volatiles, giving rise to a very high bio-oil yield, and this technique is of great interest for the scale-up.
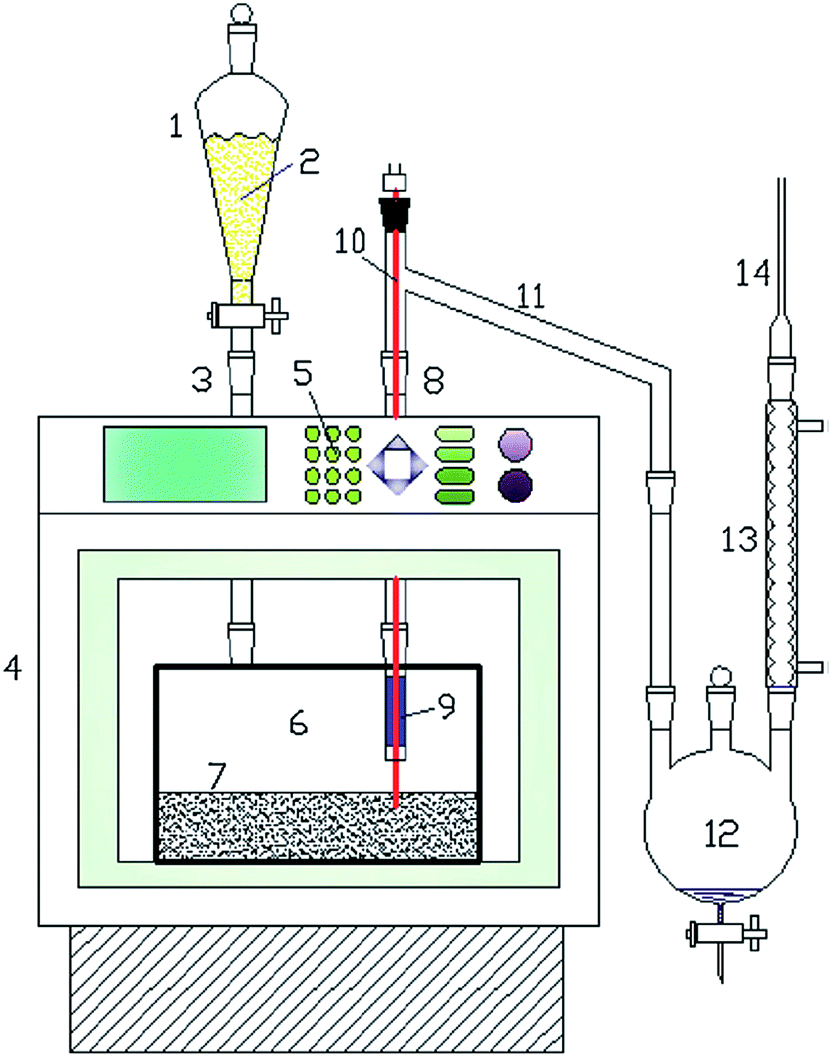 | ||
| Fig. 5 Schematic diagram of fMAP setup. (1) Semi-continuous feeder; (2) biomass; (3) quartz inlet connector; (4) microwave oven; (5) control panel; (6) quartz reactor; (7) microwave absorber bed; (8) quartz outlet connector; (9) thermocouple; (10) quartz connector; (11) connecting tube; (12) bio-oil collector; (13) condenser; (14) gas exporting tube. Reprinted with the permissions from ref. 53. Copyright (2016) Elsevier. | ||
The mechanism corresponding to non-catalytic MAP of biomass has been widely investigated by a number of researchers. In terms of MAP, the rapid and in-core volumetric heating contribute to a very prompt moisture release from the biomass, improving its surface area and the surface or pore structure; meanwhile, a very quick release of organic vapors are favored during MAP.4,11 Moreover, the inverted heat transfer properties of microwave heating, namely higher temperature at the material interior and lower temperatures at the material surface, avoids the direct contact between the hot biochar and released pyrolysis vapors so that char-catalyzed secondary cracking is hampered. In addition, microwave heating is easily controlled and the reaction temperature can be readily maintained at a desired moderate level, which is also beneficial for the fast pyrolysis process. It is noticeable that the non-catalytic MAP of biomass undergoes a sequence of exothermic and endothermic reactions.30 It is found that endothermic reactions are energy-consuming and require prolonged microwave irradiation to achieve complete reactions;85,119 whilst exothermic reactions rapidly elevates the temperature of materials and reduce the microwave energy consumption. It is reported that the maximum temperature of MAP is highly correlated to the heating value of feedstocks and microwave power level.120
More specifically, the overall reaction pathway with regard to non-catalytic MAP of lignocellulosic biomass is presented in Fig. 6. It can be seen that the decomposition of cellulose and hemicellulose is mainly composed of two steps.121 In the first step with the low temperature ranged from 329 to 350 °C, cellulose and hemicellulose undergo a series of depolymerization and dehydration reactions to form furfuran and 2-furancarboxaldehyde. During the second step at the high reaction temperature varied from 400 to 471 °C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O linkage is broken and recombined to generate β-methoxy-(S)-2-furanethanol and tetrahydro-2,5-dimethoxy-furan. Unlike the decomposed pathways of cellulose and hemicellulose, the decomposition of lignin involving depolymerization, dehydration, cracking, and hydrogenation can be relatively complex. Lignin is primarily depolymerized and dehydrated into propenyl-guaiacols at the low temperature from 329 to 350 °C. The propenyl-guaiacols can be further hydrogenated into propyl-guaiacols at 350–400 °C. The cracking of lignin and guaiacols takes place at 350–471 °C, and the positions of C–C bond broken are highly associated with the temperature. In addition, the scission of Cβ–Cγ bond occurs at 350 °C, followed by the scission of Cα–Cβ bond at 400 °C versus C4–Cα bond at 450 °C. Ultimately, the cleavage of C–OCH3 emerges at the temperature of 471 °C.
O linkage is broken and recombined to generate β-methoxy-(S)-2-furanethanol and tetrahydro-2,5-dimethoxy-furan. Unlike the decomposed pathways of cellulose and hemicellulose, the decomposition of lignin involving depolymerization, dehydration, cracking, and hydrogenation can be relatively complex. Lignin is primarily depolymerized and dehydrated into propenyl-guaiacols at the low temperature from 329 to 350 °C. The propenyl-guaiacols can be further hydrogenated into propyl-guaiacols at 350–400 °C. The cracking of lignin and guaiacols takes place at 350–471 °C, and the positions of C–C bond broken are highly associated with the temperature. In addition, the scission of Cβ–Cγ bond occurs at 350 °C, followed by the scission of Cα–Cβ bond at 400 °C versus C4–Cα bond at 450 °C. Ultimately, the cleavage of C–OCH3 emerges at the temperature of 471 °C.
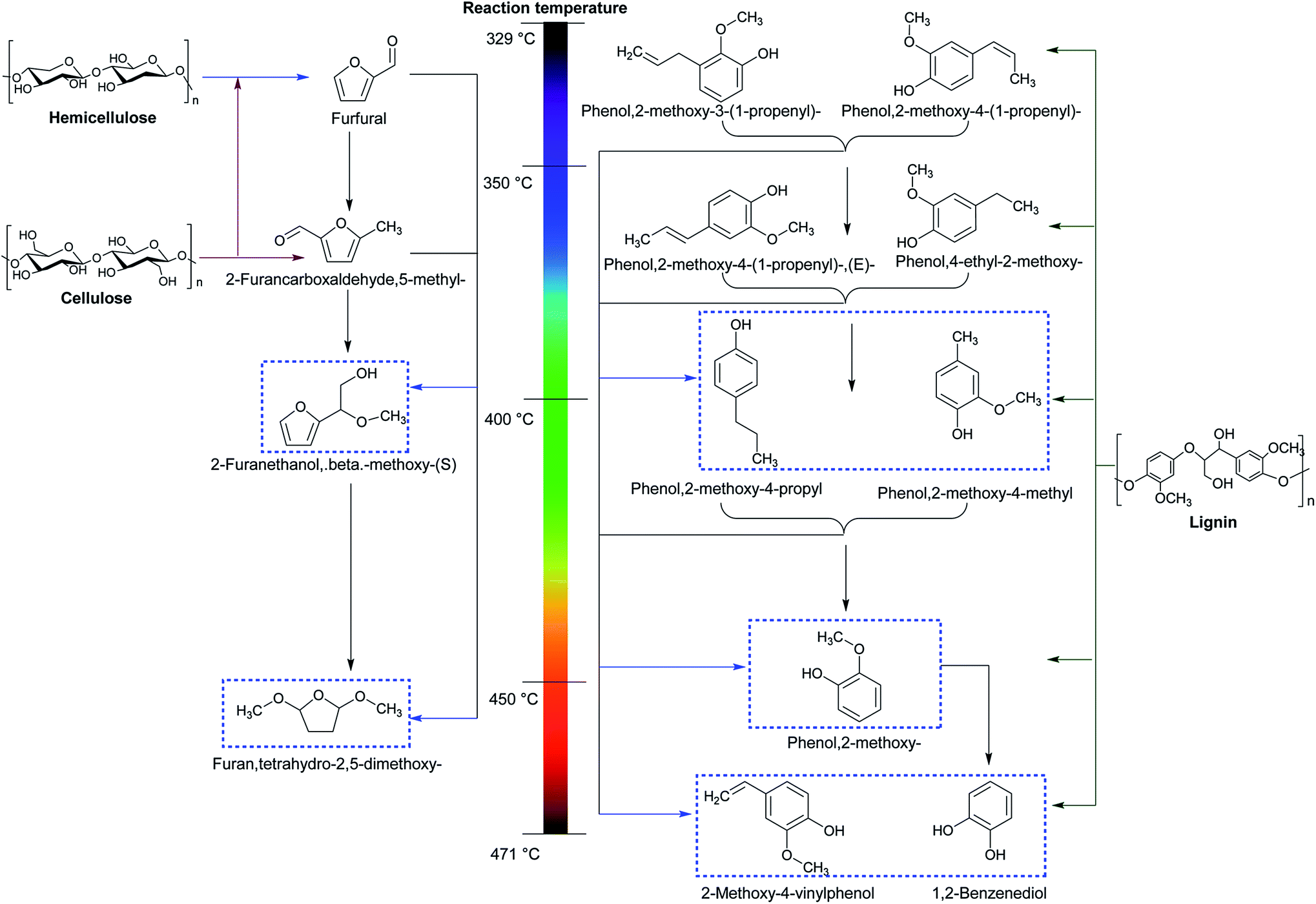 | ||
| Fig. 6 Schematic presentation with respect to non-catalytic microwave-assisted pyrolysis of lignocellulosic biomass. Reproduced by the permissions from ref. 121 Copyright (2012) Elsevier. | ||
To date, it is widely known that simplicity and effectiveness are two key criteria to evaluate a technique for the production of liquid fuel.30 Co-pyrolysis is considered as another ideal process by using diverse materials as feedstock, satisfying the aforementioned criteria.14 There are a number of studies showing that microwave-assisted co-pyrolysis (MACP) of biomass with polymers have successfully improved product yield and liquid quality.122–124 Unlike catalytic cracking and HDO, the MACP technique has a promising application for biomass utilization due to its attractive outcomes.53 Unlike MAP process, MACP is performed by using the mixture of biomass and other materials (e.g., plastics and scum) as certain mass ratios. However, the other operating steps and conditions of MACP process are comparable with those of MAP process.
The synergistic effect between biomass and polymers in MACP process is the main factor for the improvement in target product quantities and qualities. The mechanism corresponding to non-catalytic MACP of biomass and polymers (e.g., plastics) has been investigated.53–55 The mechanism of radical interactions during MACP contributing to synergistic effect is mostly proposed. It is shown that the decomposition mechanisms of biomass and plastics are readily different. The decomposition of biomass under microwave irradiation undergoes a series of exothermic and endothermic reactions; while the thermal degradation of plastics occurs via a sequence of radical mechanisms (initiation, propagation, and termination).53,55 Since the thermal stability of biomass is much lower than that of plastics during MACP process, the free radicals from the decomposition of biomass can enhance the degradation of plastics-derived macromolecules. Meanwhile, the active hydrogen proton derived from the thermal degradation of plastics can react with unstable biomass-derived oxygenates to inhibit the polymerization reactions to form less valuable products.
4.2 Chemistry of catalytic microwave-assisted pyrolysis
CMAP is a relatively emerging technique.125 In terms of chemistry or steps for CMAP, they are comparable with those of CFP. Basically, CMAP is performed by means of introducing a catalyst or additive during the MAP process. The use of catalysts or additives in the CMAP process could enhance the desirable product yield and selectivity.11 Among the widely used catalysts and additives, zeolite-based catalysts (e.g., HZMS-5, γ-Al2O3, and TiO2) and additives (such as activated carbon, Ni2O3, CaO, and Na2CO3) are especially attractive owing to their excellent performances at the improvement of product selectivity and quality in the CMAP process.54,95,126–128Similar to CFP, the CMAP technique is also divided into two categories: in situ CMAP and ex situ CMAP. In situ CMAP of biomass is carried out by means of premixing biomass and catalyst samples into the quartz reactors. Alternatively, a fixed bed reactor filled with microwave absorbers and a catalyst mixture is placed into microwave cavity, which is also named as in situ CMAP. This platform is developed and mainly applied by Ruan and his co-workers.58,116,125,130,131 In this in situ CMAP system, the microwave absorbers are preheated to a set temperature and simultaneously catalysts are maintained at the same temperature by heat conduction; biomass is subsequently fed into the microwave oven and subjected to fast pyrolysis and catalytic reforming. Nonetheless, one main shortcoming of this approach is the uncontrollable catalytic temperature, which cannot optimize the product yields and selectivity. Instead, ex situ CMAP of biomass incorporates an upfront MAP process with a catalytic reforming process.45,116,132–134 Herein, hot pyrolysis vapors derived from the MAP of biomass can be subjected to the packed-bed catalysis reactor which is filled with catalysts for a preferred product selectivity. As expected, the ex situ CMAP of biomass indicates the controllable catalytic temperature, which could optimize the bio-oil yield and selectivity.
Various mechanisms and reaction pathways during the CMAP of different biomass have been investigated. The overall reaction pathways and reaction mechanism regarding the CMAP of cellulose by using HZSM-5 as the catalyst have been identified in the light of these observations from cellulose to high-selectivity aromatic hydrocarbons.129 It is affirmed that cellulose is thermally decomposed and dehydrated to form anhydrosugars, which subsequently undergo dehydration and re-arrangement reactions to generate furanic compounds such as furan and its derivatives with appended side groups as depicted in Fig. 7. Anhydrosugars are not prone to entering the zeolite pores due to the large molecule size,136 thus the dehydration and re-arrangement reactions should occur at the external surface of the modified HZSM-5. The furanic compounds can thereafter diffuse into the modified catalyst pores and go through a series of decarbonylation, decarboxylation, dehydration, and oligomerization reactions to yield both monocyclic aromatics and olefins. Monocyclic aromatics either leave the catalyst pores as final products or further react with oxygenates or other reaction intermediates to form the polycyclic aromatics like naphthalene and its derivatives. In the whole deoxygenation processes, the competing reaction in this phase is the catalytic coke polymerization from thermal decomposition of homogeneous vapour phase and heterogeneous catalytic reactions deactivating strong acid sides and suppressing the diffusion of aromatics. Afterwards, Zhang and his colleagues have investigated the overall reaction mechanism regarding the CMAP of intact biomass as sketched in Fig. 8. Unlike the decomposed pathways of cellulose and hemicellulose in the CMAP system, lignin could be initially decomposed into phenolic compounds (such as phenols and guaiacols); and these phenolic compounds can be catalyzed by HZMS-5 catalyst via dehydration, cracking, and oligomerization reactions to form aromatic hydrocarbons.
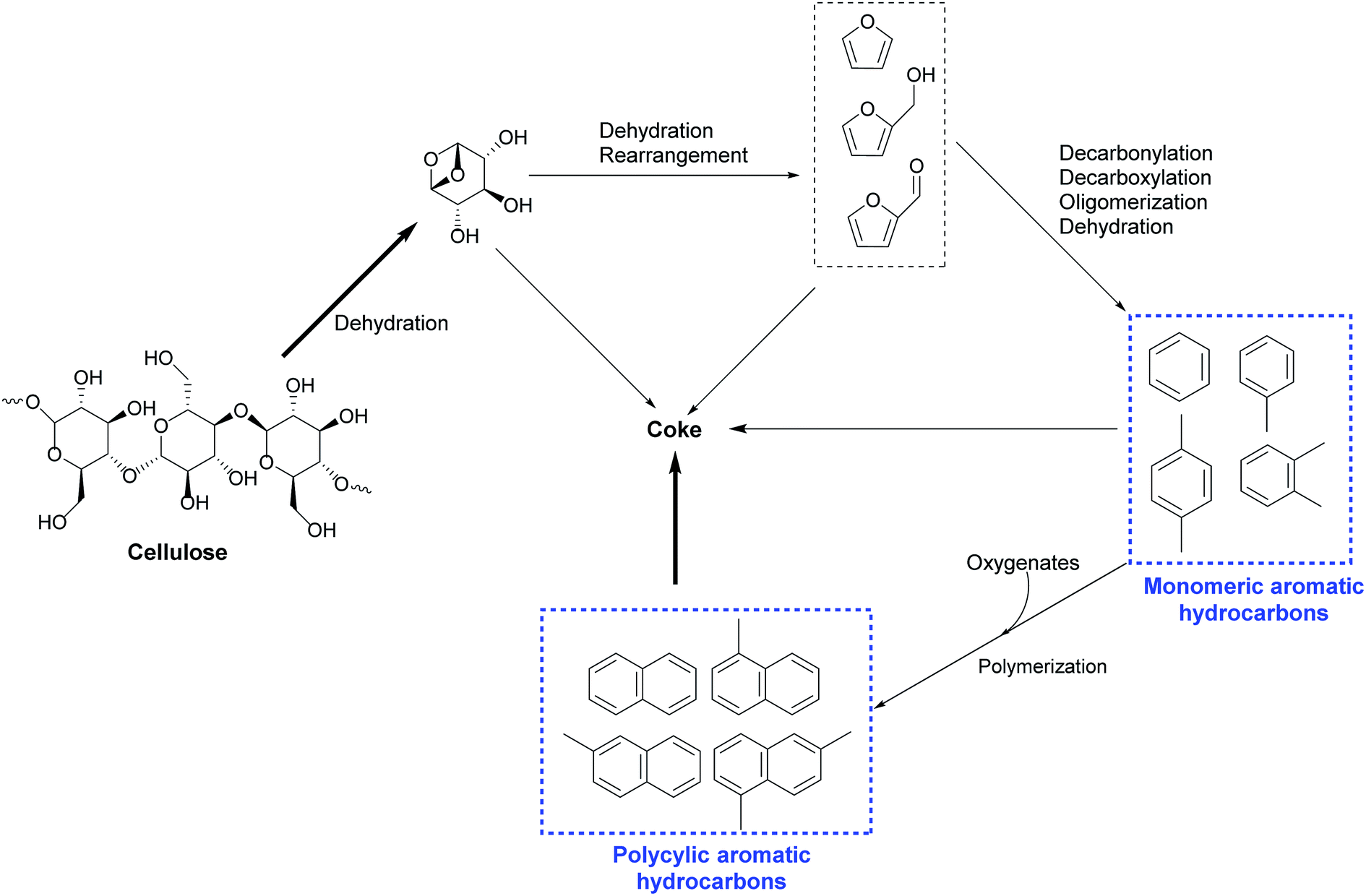 | ||
| Fig. 7 Overall reaction pathway for catalytic microwave pyrolysis of cellulose over modified HZSM-5. Reproduced with the permissions from ref. 129 Copyright (2014) American Society of Agricultural and Biological Engineers. | ||
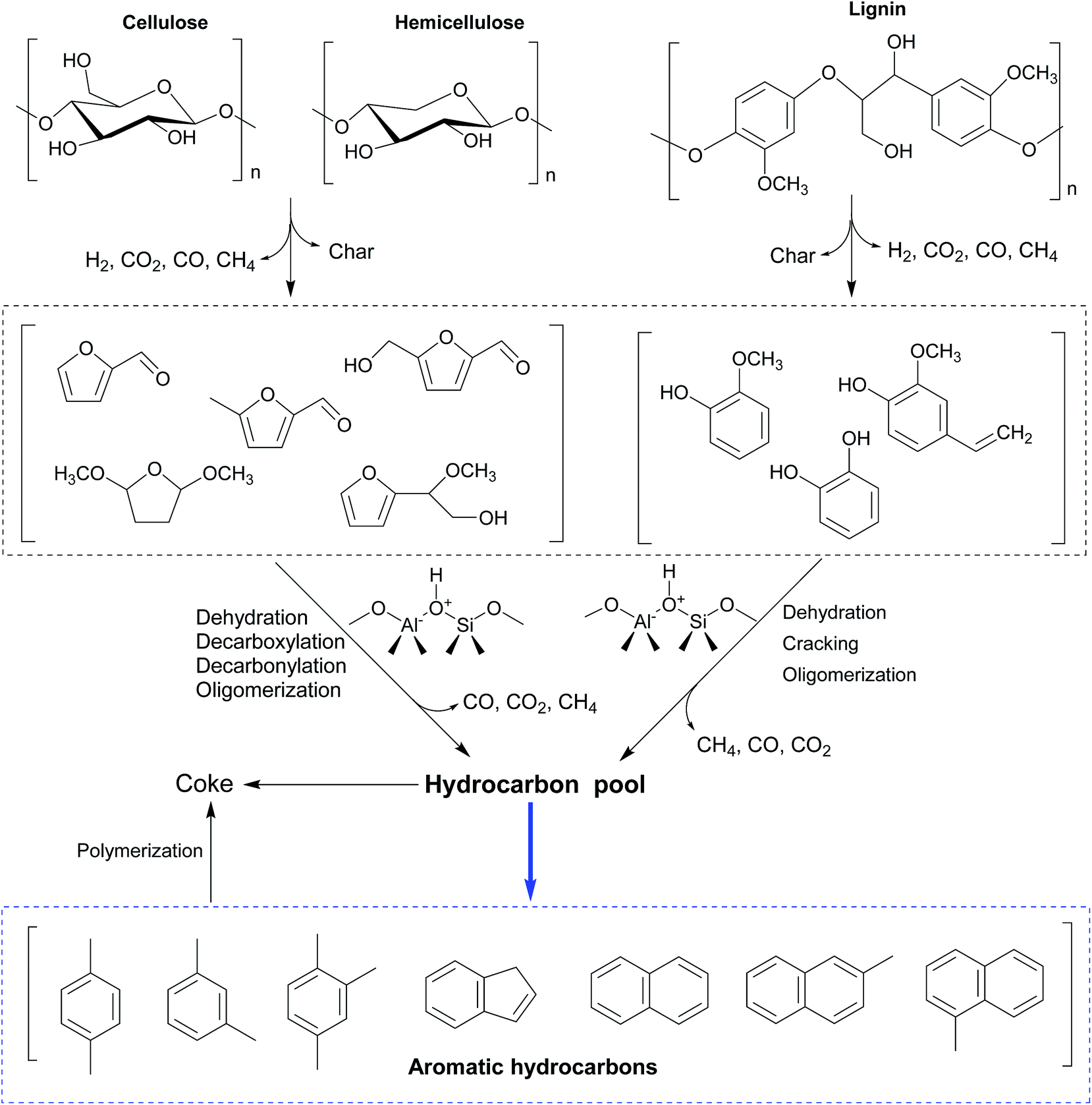 | ||
| Fig. 8 Possible reaction scheme regarding catalytic microwave-assisted pyrolysis of lignocellulosic biomass by using HZSM-5 as the catalyst. Reprinted by the permissions from ref. 135 Copyright (2015) Royal Society of Chemistry. | ||
The reaction mechanism for furfural conversion to xylenes and toluene during CMAP has been investigated as shown in Fig. 9.99 It can be seen that furfural can initially undergo decarbonylation reaction to generate allene (C3H4) and CO. The allene can go through either aromatization to form benzene or oligomerization and crackings giving rise to propylene, hydrogen and cyclopentadiene. The alkylation of benzene can readily take place at high temperatures because of the active feature of benzene. Therefore, propylene serving as the intermediate compound can react with benzene via alkylation to form xylenes, toluene and ethylene. Meanwhile, generated toluene could further react with propylene to produce xylenes through alkylation. From the conversion of furfural into xylenes, the proportion of ethylene as the by-product is regarded as an index of aromatic formation during the CMAP process. More specifically, the furanic compounds go through HZSM-5 framework during the CMAP system that is demonstrated in Fig. 10.
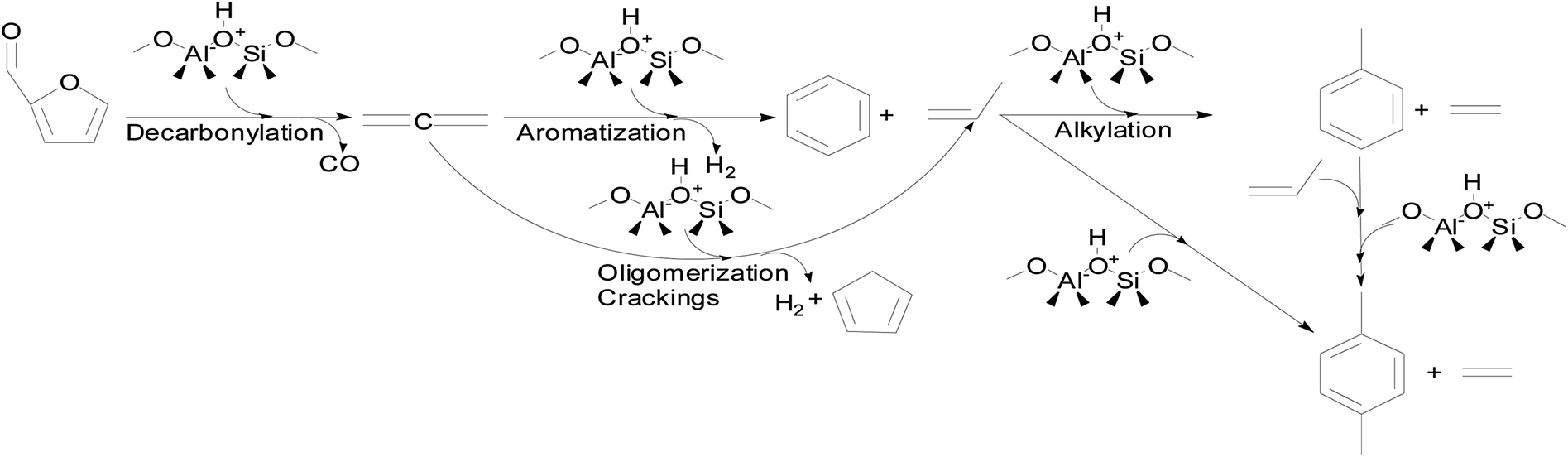 | ||
| Fig. 9 The proposed reaction pathway from furfural to xylenes and toluene in the catalytic microwave-assisted pyrolysis employing modified HZSM-5 as the catalyst. Reprinted by the permissions from ref. 133 Copyright (2015) Royal Society of Chemistry. | ||
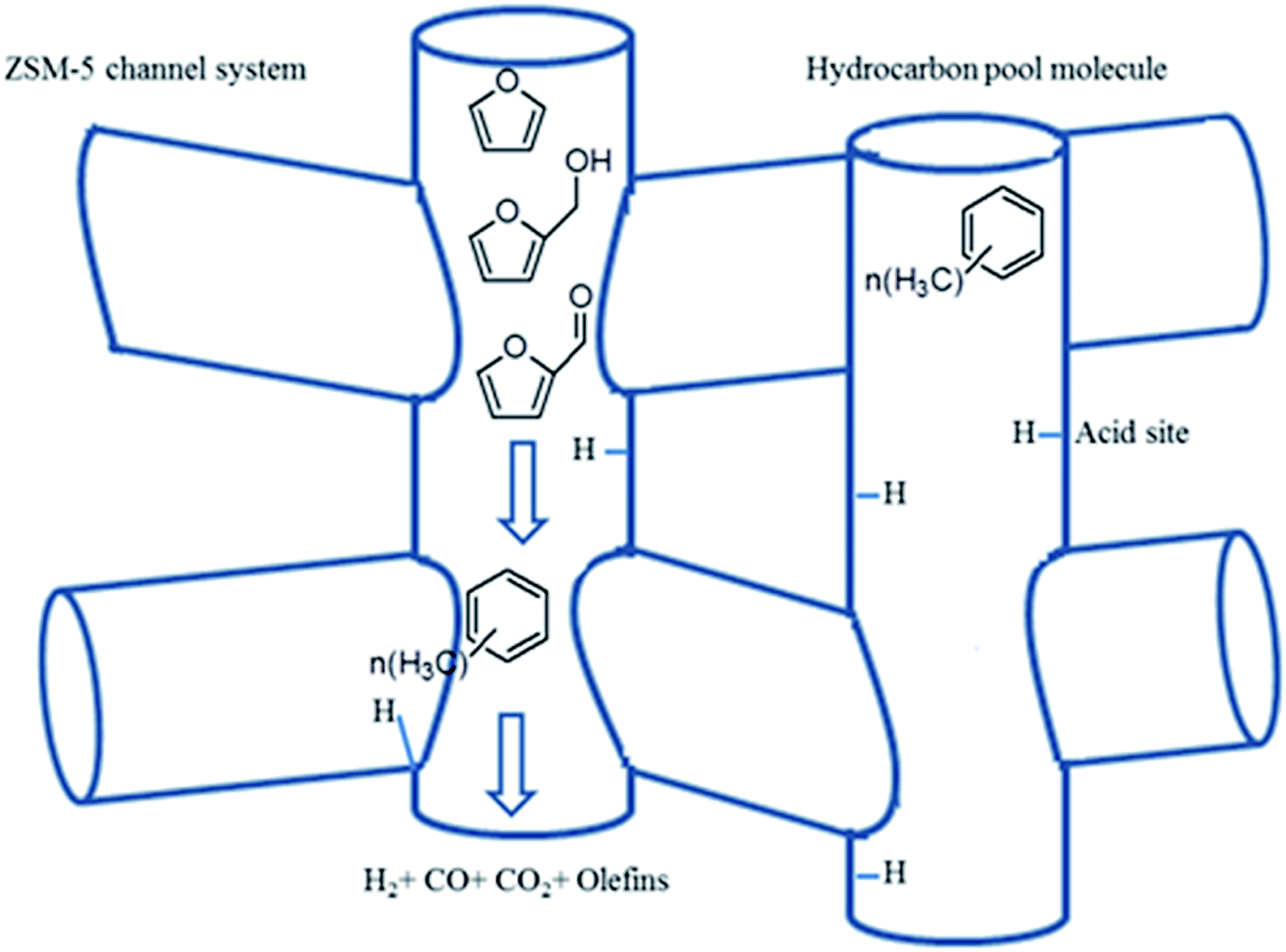 | ||
| Fig. 10 The generation of aromatic hydrocarbons in the HZSM-5 framework during the catalytic microwave-assisted pyrolysis system: the catalyst consists of a local area of zeolite framework with at least one Brønsted acid site and an organic co-catalyst. Reprinted by the permissions from ref. 133 Copyright (2015) Royal Society of Chemistry. | ||
However, CMAP of biomass cannot manufacture a very high yield of liquid organic fraction even in the presence of highly efficient catalysts; nevertheless, high amounts of solid residues, including both biochar and coke, are achieved in the process.45,100,118,125,132,133,137,138 Indeed, a low yield of liquid organic fraction from CMAP is not cost-effective to scale up the process in a biorefinery. It is discerned that the petrochemicals with a low yield of aromatics and high solid formation are mainly related to the oxygen-enriched intrinsic nature and hydrogen deficiency of biomass.2,139,140 In addition, the hydrogen to carbon effective (H/Ceff) ratio plays a vital role in coke formation and converting efficiency of biomass into valuable products.141–143 In order to improve the carbon efficiency of liquid organic fraction and minimize the solid formation, it is reasonable that the addition of high H/Ceff ratio co-reactants with biomass in the CMAP can help mitigate these issues.54,122 Waste plastics represent an abundant and cheap hydrogen sources, which can be implemented to improve the yield of liquid organic fraction and lower the solid formation in the CMAP process.53,55 Accordingly, this technique using two or more feedstock (usually biomass and polymers) in CMAP is referred to as catalytic microwave-assisted co-pyrolysis (CMACP). Unlike non-catalytic MACP, CMACP introduces the catalysts in the process by using either in situ or ex situ manner. The basic experimental steps and reaction conditions of the CMACP process are similar to those of the CMAP process.
The reaction mechanism and overall reaction pathways pertaining to the CMACP can be relatively complicated due to various types of materials employed in the process.142 The CMACP mechanism can be classified into two ways: the mechanism regarding biomass and polymers during thermal decomposition, and the mechanism regarding the pyrolysis vapors at the catalytic sites.140 As for CMACP of biomass and plastics model compounds, it is observed that there is a positive synergy for aromatic production in the co-feeding of cellulose and low-density polyethylene (LDPE) during the process.55 The phenomenon is mainly attributed to the interactions between cellulose-derived furanic compounds and LDPE-derived olefins. It is suggested that furanic compounds (e.g., furan and furfural) derived from cellulose could react with light olefins (e.g., ethylene and propylene) originating from LDPE to generate aromatics through Diels–Alder reactions followed by a dehydration reaction. It is proven that furanic compounds and light olefins act as the diene and dienophile compounds, respectively.141,144,145 The Diels–Alder reactions followed by dehydration reaction can improve the yield of aromatic production and reduce the coke formation through polymerization of furans.41,146 It is also evidenced that LDPE-derived hydrocarbons including as olefins and alkanes can also serve as the hydrogen donor for cellulose-derived oxygenates, leading to the reduction of coke yield in the zeolite-catalyzed conversions.147,148
The CMACP process using real woody biomass with plastics as feedstock has been performed as well.53,54,149 Zhang et al.53 has demonstrated the overall reaction network and mechanism (CMACP of lignocellulosic biomass with hydrocarbon-based plastics in the presence of zeolite-based catalysts) as outlined in Fig. 11. Regarding the dominant route in lignocellulosic biomass, cellulose should first undergo a sequence of dehydration, decarbonylation, and decarboxylation to yield furanic compounds in the thermal degradation.44,133 As such, it is evidenced that hemicellulose is likely to depolymerize into furanic compounds,121 which is identical to the result of cellulose degradation.133 Unlike cellulose and hemicellulose, lignin in lignocellulosic biomass is mainly depolymerized into phenolic compounds. In terms of the degradation of plastics in another route, thermal degradation of plastics usually takes place through two mechanisms (random scission and chain-end scission).150,151 The two abovementioned mechanisms can occur simultaneously, giving rise to free radicals along with the long carbon chains.152 At the same time, the radical fragments can be converted into straight chain hydrocarbons through hydrogen transfer reactions.152 In addition, the hydrogen from the thermal decomposition of plastics can be provided for biomass-derived oxygenates which serve as the strong acceptor, suppressing the char formation.
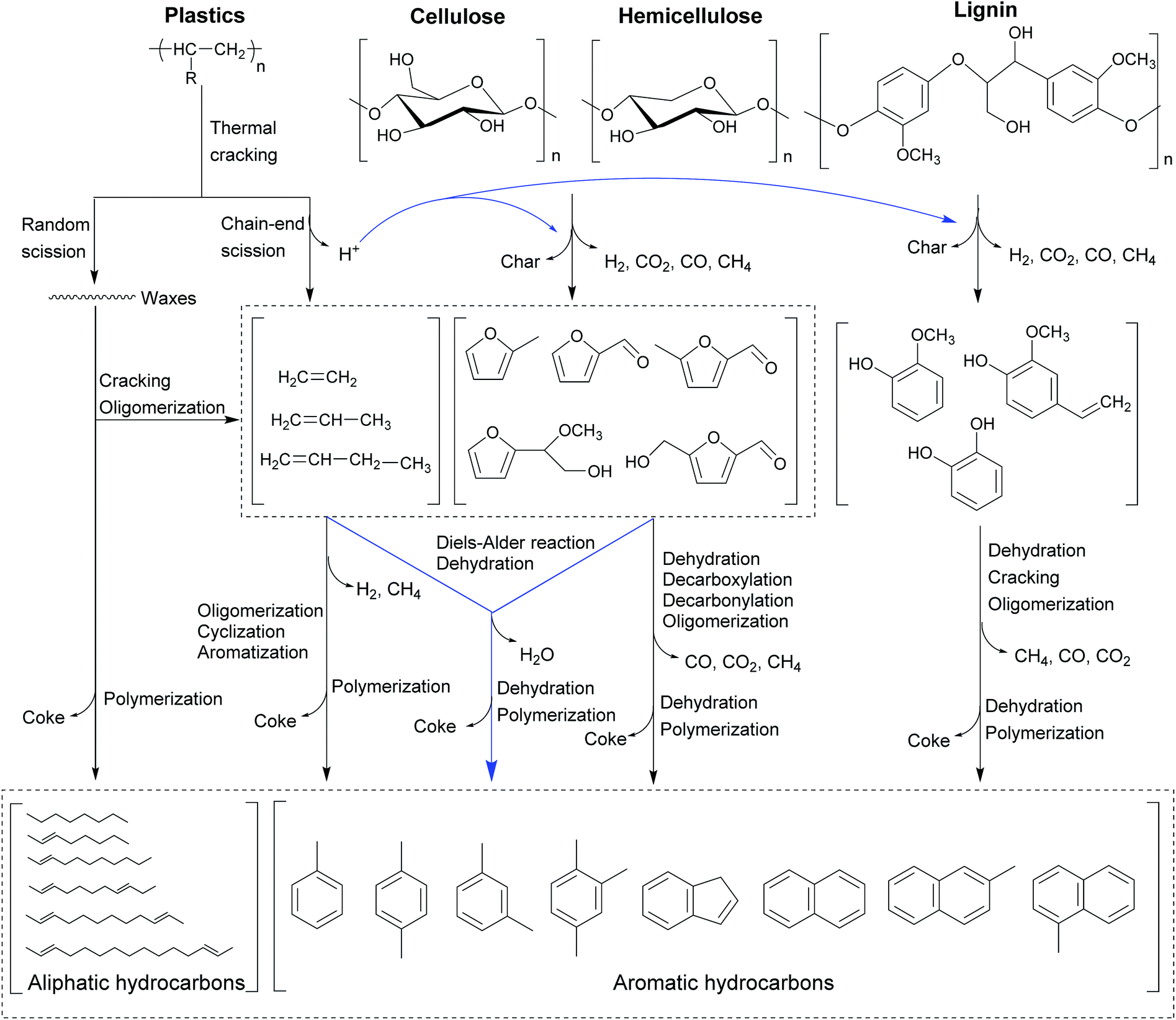 | ||
| Fig. 11 Proposed reaction pathways for catalytic microwave-assisted co-pyrolysis of lignocellulosic biomass and plastics.53 Reprinted with permission from ref. 53 Copyright (2016) Elsevier. | ||
It is worth noting that the waxes with large molecule weight from the thermal decomposition of plastics can predominantly go through catalytic cracking over zeolite catalysts via two carbocationic mechanisms, generating light olefins.150,153 On the other hand, it is found that a small partial amount of waxes could directly undergo the catalytic cracking and oligomerization reactions to produce liquid alkanes and olefins. The light olefins can thereafter react with furanic compounds by the Diels–Alder reaction followed by the dehydration reaction to generate aromatic hydrocarbons. Meanwhile, these plastics-derived light olefins can individually undergo cyclization, aromatization, and oligomerization reactions to form aromatic hydrocarbons.140 As such, these furanic compounds could partially go through decarbonylation, aromatization, and oligomerization reactions on the zeolite catalysts to obtain aromatic hydrocarbons as well. As previously reported, it is confirmed that the interaction via the hydrocarbon pool mechanism could be found in addition to the Diels–Alder reaction.41 Since the carbon yield of liquid organic fraction significantly increases but the coke yield dramatically decreases during the CMACP process, the Diels–Alder reaction is the dominant reaction pathway during the CMACP process compared to the hydrocarbon pool mechanism. Additionally, phenolic compounds can be also transformed into aromatic hydrocarbons over zeolite-based catalysts by the dehydration, cracking, and oligomerization reactions.44,154
During the CMAP of lignocellulosic biomass individually, the hydrogen-deficient oxygenates from the thermal degradation of cellulose and hemicellulose can readily polymerize and react with phenolic compounds to form coke.128,132 Most phenolic compounds from the thermal cracking of lignin are too large to diffuse into zeolite pores.155 Yet, these phenolic compounds are relatively unstable and can easily attach on the catalyst surface, and further polymerize or react with small molecular oxygenates to yield coke.140 On the other hand, the resultant furanic compounds can readily react with plastics-derived light olefins via Diels–Alder reactions rather than polymerization reactions, resulting in the decrease of coke formation during the CMACP process.53 As such, lignin-derived phenolic compounds can abstract hydrogen atoms derived from the degradation of plastics; thus these phenolic compounds are stabilized by the hydrogen donors and the polymerization reaction can be inhibited to generate more coke.53,140 Nonetheless, it should also be kept in mind that the issue of coke formation in the CMACP can only be mitigated, rather than being thoroughly eliminated.53,55 It is reinforced by the overall reaction pathway plus the reaction mechanism during the CMACP process that the plastics-derived olefins and biomass-derived oxygenates can partially go through polymerization reactions to generate coke as well.
4.3 Catalysts for catalytic microwave-assisted pyrolysis
By using K2CO3 and NaOH as the catalyst in the CMAP of biomass, gas is the dominate fraction; that is due to the fact that the catalysts can strongly absorb microwaves, resulting in the extremely high temperature inside biomass. Taking K2CO3 for example, it is a strong polar material, which can absorb more microwave energy. Through employing K2CO3, the activation energy during the CMAP of biomass is much lower than that in the direct MAP of biomass, indicating that the catalytic effect of K2CO3 is the preliminary role in the CMAP of biomass.157 At a high reaction temperature, the increase of gaseous products during CMAP is mainly attributed to the catalysis of K2CO3, which is also used as the catalyst for tar conversion into CO, H2, and CH4. Moreover, the introduction of K2CO3 can promote the gasification reaction between CO2 and C, the dry reforming and cracking reaction of CH4.158 On the other hand, it is suggested that the catalytic performance of K2CO3 is weak at the low temperatures, and the heat and mass transfer resistance is enhanced with the addition of K2CO3 at low temperatures, thereby postponing the pyrolysis reactions. It is also noted that alkaline earth metal chlorides including MgCl2 and ZnCl2 could accelerate the biochar formation at low temperatures but reduce its generation at high temperatures. Sometimes, chlorides salts can favor the liquid production during MAP of biomass.159
K3PO4 mixed with biomass can remain in the biochar after MAP and provide two essential macro-nutrients of K and P for plants. K3PO4 has been also identified as a great microwave absorber, which can remarkably reduce bio-oil acidity. It is found that high load of K3PO4 during MAP enhances the endothermic reactions, resulting in a slow heating rate. Large amounts of gases products are also attributed to the endothermic reaction of CH4 dry reforming caused by high temperatures of K3PO4 particles in comparison with biomass particles, and the accumulation of coke deposited on K3PO4 surface that further enhances the gaseous production. Moreover, it is noted that the element of K is prone to catalyzing gasification reaction and improving the cracking reaction of large molecular weight compounds at high heating rates under microwave irradiation, creating small molecules of gas (e.g., CO and H2). The higher biochar yield from MAP of biomass in the presence of K3PO4 is probably because of the low heating rates and the high presence of K that is responsible for the enhancement of the secondary carbon formation and the inhibition of devolatilization of cellulose and hemicellulose, thereby triggering more biochar production.104
Acidic metal oxides including Al2O3 have been used as the catalyst in CMAP of biomass. The presence of acidic metal oxides can result in the decrease of the liquid product, but the increase of gaseous and solid products. Several studies have suggested that Al2O3 catalyst has high catalytic performance towards the thermal decomposition of biomass or tarry constituents.15,22 Besides AlO4 tetrahedron is included in commercial zeolite catalysts (HZMS-5) that have long been known for biomass conversions and bio-oil cracking.160 By using the γ-Al2O3 as the catalyst in the CMAP of biomass,161 the introduction of γ-Al2O3 promotes the thermal decomposition of biomass to produce organic volatile vapors but has little impact at the variation of gaseous composition at the low temperature comparing to that of no catalyst. On the contrary, the application of γ-Al2O3 results in the increase in the shares of H2 and CH4 relative to the absence of catalyst at the high temperature. These observations imply that γ-Al2O3 exhibits a very high catalytic performance at the cracking of tarry constituents in a high temperature range of >300 °C during CMAP of biomass. Al2O3 is also found to enhance the bio-oil production through either suppressing biochar yield or gas yield or both.159 Even though Al2O3 does not improve the converting efficiency from biomass to pyrolysis vapors (slight variation in the biochar yield), it promotes the formation of liquid products.
Basic metal oxides are well known to be active catalysts for ketonization and aldol condensation of carboxylic acid and carbonyl compounds.2 Alkaline earth metal oxide, e.g., CaO and MgO, are classic base catalysts where oxide ions serve as bases and metal cations behave as Lewis acids.2 It is manifested that the employment of either MgO or CaO as the catalyst in the CMAP of biomass can enhance the gaseous production.54,149,158,161,162 It should be mainly due to several secondary reactions including self-gasification and interactions among hot pyrolytic intermediates occurring during the CMAP of biomass to alter gaseous compositions and the three-phase production distribution. As for the comparisons between CaO and MgO as the catalysts during the comparable CMAP system,162 the addition of CaO in the CMAP of biomass slightly enhance the share of H2, while adding MgO as the catalyst slightly causes the decrease of H2 share. The quantity of CaO has a moderate negative effect on the reaction temperature, suggesting that CaO may affect the dielectric heating of the reaction system; while MgO has a weak negative effect. The high production of H2 and CO implies that the use of CaO as the catalyst makes the equilibrium of CH4 steam reforming move to the right-hand side of the reaction. Therefore, using CaO as the catalyst during the CMAP of biomass is beneficial for the formation of syngas, while MgO exhibits a reverse function in syngas production. It is worth noting that the CaO quantity plays a strongly negative role in solid yield whereas it shows a significantly positive effect on liquid or gaseous yield. The MgO quantity is likely to present considerable influence in gaseous or liquid yield, whilst the impact on solid yield can be negligible.162
Regarding the specific role of CaO in the CMAP process,161 the increasing share of H2 in the gas product is partially due to the thermal cracking of hydrocarbons. It is discerned that CaO can effectively catalyze the reactions of hydroxyl group and therefore facilitate the water–gas shift reaction that results in the decrease of CO during the low-temperature CMAP stage. As an alkaline oxide, CaO can absorb a part of CO2 evolved from the CMAP and water–gas shift reaction to form CaCO3; the decreased share of CO2 and increased share of H2 can be observed.158,161 Accordingly, CaO is a promising catalyst used to produce pure H2 in MAP or gasification. The low yield of CH4 in the gas product is principally due to the gas-phase conversion of CH4 into CH3OH and methyl radical intermediated by CaO. Moreover, with the addition of CaO as the catalyst in the CMAP of biomass, the heat and mass transfer resistance between biomass particles is hampered, which limits the further decomposition of more biomass.158 In addition, CaO as the catalyst also contributes to the deoxygenation of bio-oil; moreover, the production of light phenols in bio-oil is enhanced at the expense of large oxygenated compounds.54 The reaction mechanism is surmised that when pyrolysis vapors pass through the mesoporous CaO catalyst, heavy compounds (e.g., large phenols and anhydrosugars) are cracked into light molecules.
When MgO is used as the catalyst in the CMAP of biomass at the high temperature, the alkylated phenols share in the phenolic compounds decreases whilst phenol share increases with the increases of temperature, implying that the MgO catalyst is less effective at the high temperature.149 It is also noticeable that the phenol is dominant in the phenolic compounds due to high bond dissociation energy of the HO-Ph. However, with the high loading of the MgO catalyst, bio-oil yield gradually decreases. The phenomenon is mainly due to the reason that more phenols derived from MAP undergo alkylation reaction with the increasing addition of MgO, eventually resulting in the drop of bio-oil yield.149 Additionally, the production of chain hydrocarbons is enhanced because the pore size of MgO favors olefins pass.
Apart from acidic and basic metal oxides, transition metal oxides including NiO, Ni2O3, CuO, TiO2, and Fe3O4 have also been researched in the CMAP of biomass.85,95,161–163 It has been widely reported that most transition metal oxides can accelerate the heating rate under microwave irradiation. Ni-based catalysts such as NiO and Ni2O3 show high catalytic activities towards the thermal decomposition of biomass during the CMAP process and improve the yields of both bio-oil and gas, particularly with the case of the Ni2O3 catalyst.161 The introduction of both the Ni-based catalyst results in the higher gas production rate and larger content of combustible gas (H2, CO, and CH4).161 In comparison with the absence of catalysts, the use of NiO and Ni2O3 can contribute to a considerable increase in CO and H2 yields, while causing a significant drop in CH4 and CO2 yields. It is obvious that NiO and Ni2O3 not only facilitate the cracking of organic matter but also enhance the CO2 reforming and stem reforming reactions. It is interesting to note that Ni2O3 presents more excellent catalytic performance toward the CO2 and steam reforming reaction than NiO during the CMAP of biomass.
When NiO and CuO used as the catalysts are compared in the CMAP of biomass,162 the addition of NiO as the catalyst enhances the production of H2 or syngas, while the utilization of CuO slightly reduces the H2 or syngas production. The quantity of NiO has a positive effect on liquid yield while it has a strongly negative effect on gas or solid yield. CuO quantity is also positive correlated to liquid yield while it is medium negative associated with either solid or gas yield. In general, the solid yield is slightly decreased with the increase of NiO or CuO quantity, whereas the liquid yield is substantially promoted with the increasing loading of either NiO or CuO. By using NiO and CuO as the catalyst in the CMAP of biomass, polycyclic aromatic hydrocarbons (PAHs) can be detected by using CuO, while PAHs cannot be observed in the presence of NiO.163 As for TiO2 as the catalyst, it is also good for the enhancement of H2 production during the CMAP of biomass;95 it can promote the degradation of biomass to produce more organic volatiles as well.161
In order to determine the influence of catalyst properties in the reaction chemistry and product distribution during the CMAP of biomass, and to further engineer highly efficient and stable catalysts, the properties of representative zeolite-based catalysts are interpreted in Table 3. In the CMAP of biomass using HZMS-5 as the catalyst, primary pyrolysis vapors pass through the catalyst layer, they can be adsorbed by the catalyst first, and thereafter a series of reactions take place inside the micropores, internal structure, and internal volume, including dehydration, decarbonylation, decarboxylation, isomerization, rejecting the oxygen content in the form of water and light gas (e.g., CO and CO2).166
| Catalyst | S BET (m2 g−1) | V pore (cm3 g−1) | d pore (nm) | A Brønsted (mmol NH3 per g) | A Lewis (mmol NH3 per g) | T acidity (mmol NH3 per g) | Ref. |
|---|---|---|---|---|---|---|---|
| a S BET: BET surface area; Vpore: pore volume; dpore: average pore size; ABrønsted: Brønsted site acidity; ALewis: Lewis site acidity; Tacidity: total acidity. | |||||||
| HZSM-5 | 396.2 | 0.097 | 5.2 | — | — | 0.18 | 53 and 133 |
| HZSM-5 | 308 | — | 2.73 | 0.80 | 0.60 | 1.40 | 125 |
| 1.3-SiO2/HZSM-5 | 302 | — | 2.50 | 0.75 | 0.55 | 1.30 | |
| 2.5-SiO2/HZSM-5 | 290 | — | 2.15 | 0.70 | 0.45 | 1.15 | |
| 3.7-SiO2/HZSM-5 | 283 | — | 1.88 | 0.60 | 0.40 | 1.00 | |
| 5.1-SiO2/HZSM-5 | 278 | — | 1.69 | 0.55 | 0.35 | 0.90 | |
| 5.9-SiO2/HZSM-5 | 268 | — | 1.56 | 0.50 | 0.25 | 0.75 | |
| HZSM-5 | 342 | 0.17 | — | 0.97 | 0.65 | 1.62 | 167 |
| 1EDTA-HZSM-5 | 330 | 0.18 | — | 0.85 | 0.62 | 1.47 | |
| 2EDTA-HZSM-5 | 321 | 0.20 | — | 0.72 | 0.60 | 1.32 | |
| 3EDTA-HZSM-5 | 310 | 0.205 | — | 0.62 | 0.59 | 1.21 | |
| 4EDTA-HZSM-5 | 308 | 0.21 | — | 0.58 | 0.58 | 1.16 | |
| HZSM-5 | 308 | — | 2.75 | — | — | — | 131 |
| PC-HZMS-5 | 288.5 | — | 2.02 | — | — | — | |
| SiO2-HZSM-5 | 289.5 | — | 2.16 | — | — | — | |
| EDTA-HZSM-5 | 289.6 | — | 2.14 | — | — | — | |
To give more insight into the chemical reactions over HZMS-5 catalyst during the CMAP of biomass, the actual mechanism of the CMAP of biomass in the presence of the HZMS-5 catalyst has been proposed according to the hydrocarbon pool model.133 The hydrocarbon pool model has been successfully accepted to demonstrate the reaction mechanisms based on zeolite catalysts.168–170 The co-catalytic hydrocarbon pool generally consisting of entrained organic species within the inorganic zeolite framework provides an alternative pathway towards hydrocarbons with lower energy barriers, forgoing the problematic direct reaction routes.168–170 In this regard, the cooperation of the entrained aromatic component inside the inorganic zeolite framework possibly creates a rather unique reaction route as illustrated in Fig. 10. Herein the inorganic zeolite framework can act as the dehydration, oligomerization, aromatization and methylation center for the generation of aromatic hydrocarbons. Polymethylbenzenes mainly work as the entrained aromatic compounds because naphthenic species usually have a high energy barrier.170 The crucial reaction mechanism of the hydrocarbon pool may be that appropriate intermediates (e.g., furans) can react with entrained hydrocarbon species in the catalyst including a series of complicated steps to form the aromatic hydrocarbons and olefinic products during the catalytic cycle.133
The product distribution and selectivity can be tuned by fabricating the structure or components of zeolite-based catalysts. Varying the Si/Al ratio and doping other metal cations or oxides in zeolite-based catalysts is an effective approach to fabricate the strength and density of active acid sites. A lot of metal modified HZMS-5 catalysts have been tested during the CMAP of biomass. Metal modified HZMS-5 (e.g., Zn/HZMS-5) catalysts are normally prepared by using a wet incipient impregnation method , resulting in the replacement of a portion of protons in HZMS-5 with metal ions.171 Zinc ions and Lewis acid formed by zinc implementation can stimulate H-atom migration through C–H activation, which can catalyze the oligomerization of pyrolysis vapors to form aromatic hydrocarbons.132 It is also deemed that the doping of zinc can accelerate the decarbonylation reaction of intermediates (e.g., furans) to produce aromatic hydrocarbons than the parent HZMS-5 catalyst.171 Hence, Zn/HZSM-5 is viewed as the one of most promising catalysts for aromatic hydrocarbon production.132 Moreover the zinc ions on HZMS-5 can also maintain the function of preventing coke formation over protons.132
It is noted that the formation of aromatic hydrocarbons is mainly attributed to the role of internal acid sites of HZMS-5, and external acid sites are primarily responsible for coke generation because of the shape selectivity of HZMS-5 that triggers it to be difficult to form coke in the internal pores.172 The accumulated coke on the external surface of the catalyst can lead to the blockage of pore opening, causing rapid deactivation. To regard coke formation and improve aromatic hydrocarbon production, the external acid sites of HZMS-5 should be eliminated but meanwhile the internal acid sites should be maintained. One of the promising approaches to lower strong external acid sties but retain the internal weak sites is to selectively remove strong acid sites by dealumination.173 Ethylene diamine tetraacetic acid (EDTA) presents prevalent capacity to eliminate the framework.174
With the EDTA modification, the BET surface area decreases, while the total pore volume rises. The underlying reaction of this phenomenon is given as follows:167 first of all, the aluminium–oxygen tetrahedron goes through hydrolysis reaction to form aluminium hydroxide, and subsequently the aluminium hydroxide can react with the proton acid centers to generate Al(OH)2+ and H2O. Ultimately, the Al(OH)2+ cation is exchanged by Na+ and then undergoes chelation reaction with EDTA to eliminate the external framework aluminums, contributing to the emergence of hydroxyl holes and the increase of pore size, thus resulting in the improvement of total pore volume. Simultaneously, the generation of hydroxyl holes and the increase of pore size cause the loss of original micropores, thereby incurring a drop in the total specific surface area.167 It is also found that the EDTA modified HZMS-5 (referred as EDTA-HZSM-5) can reduce the external strong acid sites, contributing to the enhanced stability of the catalyst and retarding the catalyst deactivation, as well as promoting the catalyst life to accelerate the catalytic reactions during the CMAP process.167
Soon afterwards, the external acid sites of HZMS-5 catalyst was passivated by means of pre-coking treatment, chemical vapor deposition of inert silica (SiO2-CVD), and EDTA chemical modification.131 Pre-coking modified HZSM-5 (referred as PC-HZMS-5) has some coke deposited on its external surface, thereby reducing its external acid sties. It is found that the appropriate pre-coking treatment of HZMS-5 can reduce the coke yield and enhance the target products. HZSM-5 modified by chemical vapor deposition of tetra-ethyl-orthosilicate (TEOS) is designated as SiO2-HZMS-5. As a large molecule compound, TEOS cannot enter the narrow intracrystalline void space of HZMS-5. As a result, the external acid sites of HZSM-5 are replaced by SiO2via thermal decomposition of TEOS; nonetheless, the acid sites on the internal surface are not affected by the treatment.131 EDTA chemical modification has also been implemented to eliminate the outer framework aluminums of HZMS-5. Since EDTA is a bulky compound and it is not capable of entering the internal channels of HZMS-5, the external acid sites of HZMS-5 can be selectively removed by EDTA while the internal acid sites can be held intact.131
It is discerned that all the catalyst treatments of pre-coking, SiO2-CVD and EDTA chemical modifications can lead to a decrease of coke and bio-oil yields, but an increase of water and gas yields compared with parent HZMS-5 in the CMAP of biomass.131 It is illuminated that all these surface modification methods could selectively remove the outer acid sites of HZMS-5. As the removal of external acid sites and the drop in coke yield could prolong the catalyst life and improve catalytic reactions, more pyrolysis vapors can diffuse into the internal pores of HZMS-5 and undergo a sequence of reactions to generate more water and light gas. To conclude, these modified HZMS-5 catalysts, especially SiO2-HZMS-5, show better catalytic activities than parent HZMS-5 in the CMAP of biomass.131
In the CMAP of biomass employing activated carbon as the catalyst, the high concentration of phenols is probably produced from the radical reaction of O–CH3 homolysis where cellulose-derived oxygenated and phenolic compounds derived from lignin function as H-donors and H-acceptors, respectively.52,127,175 As for the reaction mechanism, when superheated water heated under microwave irradiation is subjected to the activated carbon catalyst, carboxylic anhydride can react with water to generate carboxylic acids that become proton (H+) donors and acidic catalyst. It is also shown that the amount of carbonyl group on the activated carbon catalyst increases, which is relatively correlated with guaiacol conversion to phenols.52
The protonation reaction of carbonyl oxygen can be enhanced by acids in bio-oil. Some of the electron density on oxygen which is available for donation to carbonyl carbon is taken up by new O–H bonds.52 Thus, the net effect of protonation can weaken the carbon oxygen π bond, and protonation of the carbonyl renders the carbonyl carbon to be a stronger electrophile. The carbonyl carbon will react much more quickly with whatever nucleophiles are present in bio-oil; the methoxide (O–CH3) that is an electron rich species donates electrons to electron-poor carbon. As a result, O–CH3 homolysis for guaiacol conversions to phenols is catalyzed by activated carbon catalyst. In addition, the protonation mechanism is also reversible; and, the electrons on the newly formed O− atom can collapse back down to reform the carbonyl and kick off a good leaving group (CH3O−).52 The phenols are then produced by the reaction of O–CH3 homolysis in which the guaiacols are stabilized with saturated alkyl- or H-donors.
When using metal modified activated carbon as the catalyst (e.g., ferrum-modified activated carbon),177 the substantial increase of ketones/ethers is obtained, having a close relationship with the drop in guaiacols. It can be explained that furans are subjected to acid-catalyzed ring opening to form aldehydes; and a sequence of condensation reactions such as aldol condensation are facilitated to gain structural stable products. It is also noticed that the amount of phenols in bio-oil increases compared to that from the non-catalytic MAP process in the absence of ferrum-modified activated carbon. This phenomenon is illuminated by catalytic conversions of guaiacols under the impact of hydrogen donors from many reactions, including demethylation, alkylation, demethoxylation, and dehydration.177
5. Practical non-catalytic microwave-assisted pyrolysis of biomass
The non-catalytic MAP of biomass has been extensively investigated in the past decade using various types of microwave absorbers, and most of the studies before 2013 have been reviewed by several studies.4,11,51,56,57,59,66 In this section, the up-to-date non-catalytic MAP of biomass will be primarily interpreted. The product profiles and compositions from the non-catalytic MAP of selected biomass are summarized in Table 4. To determine the quality of bio-oil from MAP, some of the resultant bio-oils are shown in Table 5. These results indicate that the quality of bio-oils from MAP is superior to that of conventional pyrolysis bio-oils. Even though biocrude derived from hydrothermal liquefaction (HTL) of algae could present comparable properties with bio-oil from the fMAP of microalgae, the high energy consumption and high-severity reaction conditions (e.g., high pressure and long reaction time) of HTL are inferior to those of the MAP technique in the industrial application. It is discerned that diverse biomass has been valorized in the MAP process to produce bio-oil, gas, and biochar. With regard to oil palm biomass, two kinds of oil palm biomass (shell and fibers) were selected for the MAP process with the assistance of biochar as the microwave absorber.46 It is found that the microwave heating rate and the yields of bio-oil, gas, and biochar are significantly dependent on the ratio of biomass to microwave absorber. A positive synergistic effect was observed in using microwave absorber and oil palm biomass according to the quality of bio-oil, containing important chemical compounds such as phenols and ketones.| Feedstock | Operating conditions | Results | Ref. | ||
|---|---|---|---|---|---|
| Absorber | Reaction parameters | Bio-oil/gas/solid yield (wt%) | Bio-oil/gas composition | ||
| Rice straw | — | 50–500 W, 105–563 °C, 30 min | 22.56/max. 49.37/28.07 | Mainly alkanes and polars/mainly H2 and CO2 | 178 |
| Rice straw | — | 50–500 W, 105–563 °C | ∼50/10–34/20–50 | Mainly mono-aromatics, aliphatics and PAHs/mainly H2 and CO | 179 |
| Corn stover | — | 700 W, 515–685 °C, 4–22 min | 26.94–36.98/17.71–42.36/— | Dominate phenols, aliphatic hydrocarbons/— | 180 |
| Corn stover | SiC | 750 W, 450–550 °C | Max. 64/max. ∼38/max. 28 | Dominant phenolic compounds/dominant CO and CO2 | 117 |
| Wood sawdust | SiC | 750 W, 450–550 °C | Max. 65/max. ∼25/max. ∼50 | Dominant phenolic compounds/dominant CO and CO2 | |
| Douglas fir pellets | — | 700 W, 350–450 °C, 10–20 min | 31.4–53.9/7.9–15.0/31.2–60.7 | Dominant phenolic compounds and furans/dominant CO and CO2 | 121 |
| Wood | H2O | 600–1200 W, 200 °C | 42.23–47.10/8.39–9.68/43.23–48.39 | Mainly phenolic compounds and furans/mainly CO and CO2 | 181 |
| Oil palm shell | Activated carbon | 180–720 W, 45 min | 16.43–36.75/—/— | Mainly phenolic compounds/— | 182 |
| Oil palm shell | Activated carbon | 800 W, 400–500 °C, 33 min | Max. 28/38–47/28–41 | Dominant phenolic compounds/— | 103 |
| Oil palm shell | Biochar | 450 W, 25 min | Max. 22/max. 29/49 | Mainly phenolic compounds, ketones, and aldehydes/— | 46 |
| Oil palm fibers | Biochar | 450 W, 25 min | Max. 25/max. 30/45 | Mainly ketones, phenols, and carboxylic acids/— | |
| Oil palm EFB pellets | Activated carbon | 300–450 W, 200–560 °C, 25 min | Max. 21/∼50/∼30 | Mainly phenolic compounds/— | 183 |
| Prairie cordgrass | — | 700 W, 530–670 °C, 6–20 min | 20.3–33.1/32.0–53.7/13.8–47.7 | Mainly phenolic compounds and hydrocarbons/— | 184 |
| P. juliflora | Flyash | 280–700 W, 500 °C | 9.60–39.87/35.06–49.58/22.65–28.28 | Mainly phenolic compounds/mainly H2 and C2 hydrocarbon | 185 |
| Aluminum | 560 W, 500 °C | 36.83/37.33/25.84 | Mainly phenolic compounds/mainly H2 and CH4 | ||
| Graphite | 560 W, 500 °C | 31.25/39.79/28.96 | Mainly phenolic compounds/mainly H2 and CH4 | ||
| Char | 560 W, 500 °C | 32.95/42.45/24.60 | Mainly phenolic compounds/mainly H2 and CH4 | ||
| SiC | 560 W, 500 °C | 26.37/47.96/25.67 | Mainly phenolic compounds/mainly H2 and CH4 | ||
| Microalgae (Chlorella sp.) | Biochar | 500–1250 W, 462–627 °C, 20 min | Max. 28.6/∼27/∼25 | Dominant aromatic hydrocarbons and phenols/dominant H2, CH4, and CO | 75 |
| Microalgae (C. vulgaris) | Lignite char | 700 W, 10 min | ∼30/∼50/∼20 | Dominant aromatic hydrocarbons, aliphatic hydrocarbons, and heterocyclic compounds/— | 47 |
| Peanut shells | Lignite char | 700 W, 10 min | ∼10/∼60/∼30 | Dominant phenolic compounds and aromatic hydrocarbons/— | |
| Fe3O4 | 700 W, 10 min | ∼10/∼60/∼30 | Dominant phenolic compounds and aromatic hydrocarbons/— | ||
| Arundo donax | Carbon | 3000 W, 365–493 °C, 18–40 min | 22.1–40.9/4.6–17.2/43.8–62.9 | Mainly aromatics and acetic acid/mainly CO2 and CO | 186 |
| Waste paper | — | 1200 W, <200 °C | 42/15/43 | Mainly carbohydrates and aromatic compounds/— | 105 |
| Soapstock | SiC | 800 W, 400–600 °C, 30 min | 44.18–69.37/9.95–27.28/15.62–39.87 | Mainly aliphatic hydrocarbons/— | 58 |
| Properties | Non-catalytic MAP bio-oil | Conventional pyrolysis bio-oil20 | Biocrude from HTL of algae187,188 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Coffee hulls107 | Corn stover106 | Pine wood sawdust189 | Wheat straw190 | A. donax rhizomes186 | Waste paper105 | Chlorella sp.75 | |||
| a Acid number. b LHV (lower heating value in MJ kg−1). c EHCcalc (MJ mol−1). d TAN (Total Acid Number), mg KOH per g. | |||||||||
| Elemental composition (wt%) | |||||||||
| C | 74 | 60.7 | 48.8 | 58.9 | 26.1 | 49.9 | 65.4 | 54–58 | 77.6–79.2 |
| H | 8.4 | 7.7 | 6.8 | 6.85 | 5.1 | 5.8 | 7.84 | 5.5–7.0 | 10.0–10.6 |
| N | 8.1 | 2.0 | 0.9 | 1.15 | 0.4 | — | 10.28 | 0–0.2 | 4.0–4.7 |
| S | 0.8 | 0.2 | 0 | 0.02 | — | 0.04 | 0 | 0.3–0.5 | |
| O | 8.7 | 29.4 | 43.5 | 33.2 | 68.4 | 44.2 | 16.48 | 35–40 | 5.3–8.0 |
| Water (wt%) | — | 15.2 | 26.2 | <1 | 50.7 | 2.6 | — | 15–30 | 2.8–7.8 |
| Density (kg m−3) | — | 1250 | 1145 | 1200 | 1030 | — | 980 | 1200 | 0.94–0.96 |
| pH value | — | 2.87 | 2.5 | 1.4a | — | — | 9.7 | 2.5 | 59–74d |
| Viscosity (mPa s, 40 °C) | — | 185 | 15.2 | — | 1.90 | — | 60 | 40–100 | 114–355 |
| HHV (MJ kg−1) | 34.4 | 17.5 | 15b | 16–22b | 9.2c | 21.8 | 30.7 | 16–19 | 25.8–33.3 |
A multimode microwave system has been implemented to carry out the MAP of oil palm empty fruit bunch pellets using activated carbon as the microwave absorber. It is found that the ratio of biomass to microwave absorber not only affected the temperature profiles, but also influenced product yields and distributions. The highest bio-oil yield of 21 wt% was gained when using 25% microwave absorber. Phenolic compounds (60–70%) was mainly presented in the bio-oil detected by GC/MS and confirmed by FI-IR analysis. Biochar could be regarded as a potentially alternative solid fuel due to the high HHV of 25 MJ kg−1.183 Ani and his co-workers have also developed a new technique to pyrolyze oil palm shells in a microwave system using an activated carbon as the microwave absorber to solve the problem of bio-oil deposition.103 It is noticed that the temperature profile, product yields and the characteristics of products were relatively dependent upon the stirrer spend and microwave absorber percentage. The maximum bio-oil yield was 28 wt% under the conditions operated at 25% microwave absorber and 50 rpm stirrer speed. Up to 85% selectivity toward phenols in the bio-oil is identified.
Furthermore, the uniformly distributed coconut activated carbon has been applied as a microwave absorber during the MAP of oil palm shell waste biomass to investigate the effects of microwave absorber loading, microwave power, and N2 flow rate on its heating profile, bio-oil yield and its compositions.182 A central composite design (CCD) was implemented to estimate the importance of process parameters on bio-oil yield, suggesting that the N2 flow rate was the most significant effect. It is discerned that the phenol content was dominant in bio-oil, accounting for 32.24–58.09%. It can be summarized that the MAP of oil palm shell with carbon absorber shows the potential to manufacture valuable fuel products. The MAP of oil palm fiber has also been conducted to optimize experimental conditions for maximum H2 and biochar yields according to CCD.191 Both H2 and bio-char yields together were optimized at 450 °C and a N2 flow rate of 200 cm3 min−1 under the microwave power of 400 W. The biochar possessed a large proportion of carbon content (>60 wt%) and higher value of HHV (>20 MJ kg−1); and some pores were found through SEM and BET analysis, which is effective for soil improvement.
The behavior pertaining to MAP of another woody biomass (Larch) has been reported to establish the bio-oil yield and its quality from MAP of biomass.78 It is the first study in biomass pyrolysis to utilize a MAP technique and methodology which was fundamentally scalable, from which the fundamentals of design for a continuous processing system could be predicted to maximize bio-oil yield and its quality. The bio-oil yield from MAP was comparable to that from conventional pyrolysis; however, the corresponding quality was much higher compared with that from conventional pyrolysis due to the rapid heating and quenching during the MAP process. It is also achieved that the amounts of levoglucosan and phenolic compounds were an order of magnitude higher in MAP when compared to those achieved by conventional pyrolysis. Prosopis juliflora (one type of woody biomass) has been also valorized in the MAP process to investigate the effects of microwave power, feedstock particle size, types of microwave absorber, biomass to microwave absorber mass ratio, and biomass loading on bio-oil, gas, and biochar yields, bio-oil compositions, and energy recovery in bio-oil and biochar.185 Five types of microwave absorber, namely, biochar, graphite, aluminum, silicon carbide, and fly ash (an industrial waste), were used in the MAP process. A maximum bio-oil yield of 40 wt% with a HHV of 26 MJ kg−1 was gained using fly ash as the microwave absorber. A mixture of phenolic compounds, aromatic hydrocarbons, carboxylic acids, etc. were included in the bio-oil. Nearly 51% deoxygenation of feedstock were obtained with an atomic O/C ratio of 0.24 in the bio-oil. Besides several organs (rhizomes, stems and leaves) of Arundo donax (one of the several species of the so-called reed) have been subjected to MAP with carbon as microwave absorber based on different reaction conditions.186 A minor yield (4.6%) of gas together with a large yield (up to 62.9%) were observed whilst up to 40.9% of bio-oil yield was gained. The bio-oil contains a large amount of aromatics, acetic acid, furans, etc.
Apart from the above-mentioned lignocellulosic biomass, microalgae (Chlorella sp.) has been subjected to MAP using biochar as the microwave absorber to gain high-quality bio-oil.75 A maximum yield (28.6%) of bio-oil and a higher heating value (HHV) of 30.7 MJ kg−1 were achieved at the microwave power of 750 W. The bio-oil was mainly comprised of aliphatic hydrocarbons, aromatic hydrocarbons, and long chain fatty acids among which aliphatic and aromatic hydrocarbons are admirable compounds for use in petroleum hydrocarbon fuels. Accordingly, it is affirmed that the MAP paves a very promising approach for microalgae conversion into advanced renewable biofuel. Moreover, MAP of algal and lignocellulosic biomass was compared to determine the effect on the product yield and distribution.47 The bio-oil yields from the MAP of peanut shell and C. vulgaris are 11.0 wt% and 27.7 wt%, respectively. It is noted that phenolic compounds are dominant in the bio-oil derived from peanut shells, while C. vulgaris-derived bio-oil primarily contains more nitrogen-containing species. In addition, the concentration of OH, C–H, CO, O–CH3, and C–O functional groups in all biochar samples significantly decreased after the pyrolysis.
Regarding the novel experimental design of CCD for MAP of biomass, other groups (particularly Lei's group) have widely used CCD to predict the optimization of product yields and distributions. In this regard, prairie cordgrass has been applied in the MAP process to determine the influences of pyrolysis temperature and reaction time in the yields of bio-oil, gas, and biochar.184 CCD was performed to estimate the effects of reaction variables on bio-oil and gas yields and establish prediction models. A maximal yield (33.1 wt%) of bio-oil was obtained, and the bio-oil contained a large amount of aliphatic and aromatic hydrocarbons. The MAP of corn stover has been performed by Lei et al. to determine the effects of reaction temperature, reaction time, and feedstock particle size on the yields of bio-oil, gas, and biochar.101 CCD was satisfactorily developed to explain the biofuel conversion yield as a function of these independent variables. It is manifested that most minerals could remain in the biochar according to the mineral analysis. A series of important chemical compounds (e.g., phenols, aromatic and aliphatic hydrocarbons, furans) could be found in the bio-oil. Lei and his co-workers also conducted non-catalytic MAP of Douglas fir sawdust pellets using CCD and response surface analysis to understand the influences of reaction temperature and time on the yields of bio-oil, gas, and biochar. It is obtained that bio-oil and gaseous yields increased with the elevated reaction temperature and prolonged reaction time. The maximum bio-oil yield of 57.8% was gained; moreover, phenolic compounds and furans were main compositions existing in the bio-oil. The gaseous product mostly consisted of CO, CO2, CH4, and short chain hydrocarbons.121
As for the production of H2 from the MAP process, the MAP of rice straw has been conducted by Huang et al.178 It reveals that the microwave power and feedstock particle size are two key parameters affecting product yields and distributions. The bio-oil was primarily composed of alkanes, aromatic hydrocarbon, and phenolic compounds. The gaseous fraction mainly contained H2, CO2, CO, and CH4; and H2 is dominant in gaseous fraction. Thereafter, Huang and co-workers aimed to investigate the productivity of H2-rich fuel gas from non-catalytic MAP of rice straw.110 The primary compositions of gaseous product were also H2, CO2, CO, and CH4; in particular, the proportion of H2 is more than 50 vol%. TA-MS analysis has proven that dielectric heating made the MAP different from conventional pyrolysis. A chemical equation has been nearly balanced to demonstrate gaseous composition produced from MAP of rice straw. From the viewpoint of energy consumption, approximately 60% of the input energy could be transferred and utilized as bioenergy. Besides the MAP of rice straw has been carried out to evaluate the products, mechanisms, and reaction kinetics.179 It is discerned that more gaseous and less biochar products were observed at higher microwave power levels, whilst bio-oil product remains stable with a maximum yield of ∼50 wt%. The atomic H/C and O/C ratios of biochar are much lower than those of feedstock. Although the primary components of gaseous product are also H2, CO, CO2, and CH4, CO became dominant in the gaseous product with the percentage of 57%. The kinetic parameters regarding the MAP of rice straw increased with the augment of microwave power level.
Another study for the production of H2 by feeding rice straw into a MAP system has been reported. The optimal H2 production was observed when rice straw was fed into the system at a microwave power of 1000 W; approximately 40.47 mg of H2 could be produced from each gram of rice straw with the conversion rate of 67.45%.192 Moreover, oil palm fiber was recently used for the production of H2 rich gas through the MAP process on the basis of different reaction parameters.191 It has been suggested that the utilization of a smaller biomass particle size, a higher reaction temperature, a higher microwave power, and a higher N2 flow rate could dramatically produce more H2 rich gas. A maximum H2 yield (10.91 g kg−1) was attained when using the biomass with the smallest particle size (less than 1 mm). TGA and SEM analysis were used to evaluate the properties of biochar, which shows a potential application for soil fertilization due to the existence of a porous hole in its microstructure.
To determine the effect of pyrolysis platforms on the product yields and distribution, woody biomass has been subjected to both microwave pyrolysis and conventional pyrolysis to determine the effects of each process on the product yields and composition with regard to bio-oil, gas, and biochar products.181 Comparable yields (∼46 wt%) of bio-oil production were gained for both cases of microwave pyrolysis and conventional pyrolysis; while a much lower gas yield was observed from microwave pyrolysis due to its fast heating rate. As the heating rate was enhanced, both the peak release of CO and CO2 moved to a higher reaction temperature for both microwave and convention pyrolysis. It is also perceived that the clear release of CH4 was achieved at a higher heating rate of microwave pyrolysis.
A novel concept with regard to the fMAP of corn stover and wood sawdust has been proposed by Ruan's group.117 This process using SiC as the microwave absorber was conducted as a function of reaction temperature, feedstock lading, feedstock particle size, and vacuum degree. A maximum bio-oil yield of 65 wt% from fMAP of wood sawdust was obtained, while the optimal bio-oil yield from the fMAP of corn stover was 64 wt%. It is thus indicated that the fMAP of biomass using SiC as the microwave absorber was a feasible and promising technique to promote the practical values and commercial application outlook of the MAP process. Furthermore, Wang et al. has conducted the fMAP of soapstock with SiC as the microwave absorber to produce hydrocarbon fuels.58 It is worth noting that the utilization of SiC significantly enhanced the hydrocarbon production and the selectivity towards alkanes and aromatics. Both the effects of pyrolysis temperature and feeding rate on hydrocarbon yield and its compositions were investigated; and it is found that the optimal conditions to maximize the hydrocarbon yield of 64.74 wt% were at 550 °C and a feeding rate of 6 g m−1, indicating high selectivity toward alkanes but low selectivity toward alkenes. The properties of the hydrocarbons are comparable with those of 0# diesel in terms of the density and dynamitic viscosity. This research presents that the fMAP of soapstock is a promising technique for commercial production of hydrocarbon fuels.
In addition, a low temperature MAP has been developed for syngas production using macroalgae as feedstock.193 This protocol garnered unprecedented H2 production, with switchable H2/CO ratios from 3 to 1 depending upon pyrolysis conditions. The biochar was exerted as microwave absorber, giving rise to an optimum H2 production with regards to a pure graphite material utilized as comparison. It is also found that arcing effects under microwave irradiation resulted in an interesting pseudo-catalytic effect induced by the metal oxides contained in macroalgae, possibly accounting for the enhanced results. Another low temperature (<200 °C) MAP of waste office paper has been reported to produce bio-oil for the utilization as an adhesive for aluminum–aluminum bonding.105 The yields of organic phase and aqueous phase were 19% and 23%, respectively. Broad categories of compounds indicative of carbohydrates, aromatics, and carbonyl-containing moieties were included in the bio-oil through a sequence of characterization by ICP-MS, ATR-IR, GC-MS and NMR. A liquid–liquid fractionation of the organic phase was carried out to attain an in-depth understanding of the adhesive characteristics of bio-oil. The ‘acidic’ fraction indicates far better adhesion properties than the ‘neutral’ fraction with no bonding achieved for the aqueous fraction. The mixture of the ‘acidic’ and ‘neutral’ fractions reveals better adhesion, thereby implying a possible synergistic effect.
More interestingly, a non-catalytic MACP of sewage sludge with rice straw was investigated elsewhere.124 It is noticed that the introduction of rice straw essentially promoted the performance of microwave heating, achieving a much higher maximum temperature. A maximum temperature (up to 500 °C) was detected from the mixture containing 20 wt% rice straw. It is inferred from the outcome that there was a positive synergistic effect between sewage sludge and rice straw for microwave heating. The high heating temperature can provide another approach for thermal treatment of waste sewage sludge. It is found that the calorific value of the biochar from the MACP of the mixture containing 30–40 wt% rice straw was significantly improved, compared with the biochar from the MAP of sewage sludge individually; and the fixed carbon content (up to 33 wt%) of the biochar was gained from MACP of the blends. It is also discerned that the atomic H/C and O/C of the biochar are very close to those of anthracite coal. Therefore, the biochar from the MACP of sewage sludge and rice straw indicates a high potential to be co-fired with coal or utilized as a replacement.
6. Practical catalytic microwave-assisted pyrolysis of biomass
6.1 Catalytic microwave-assisted pyrolysis of biomass
At the early stage, Chen et al.95 carried out the CMAP of pine wood sawdust using eight inorganic additives (i.e., NaCl, NaOH, Na2CO3, H3PO4, Fe2(SO4)3, Na2SiO3, TiO2, and HZSM-5) in the light of their catalytic effects, as demonstrated in Table 6. It is observed that all catalysts improved the solid production while there was no dramatic change of bio-oil yield. The incondensable gas mainly consisted of H2, CH4, CO and CO2. In particular, alkaline sodium compounds (NaOH, Na2CO3 and Na2SiO3) could significantly favored the formation of H2. The most abundant organic component was acetol in the bio-oils from CMAP in the presence of these additives except H3PO4 and Fe2(SO4)3. On the contrary, H3PO4 and Fe2(SO4)3 mostly favored the formation of furfural and 4-methyl-2-methoxy-phenol in the bio-oils. Thereafter, Ruan and his co-workers have reported the effects of various metal oxides, salts, and acids including K2Cr2O7, KAc, MAl2O3, MgCl2, AlCl3, CoCl2, Na2HPO4, ZnCl2, and H3BO3 on product yields and distribution during in situ CMAP of corn stover and aspen wood.159 The five catalysts of KAc, Al2O3, Na2HPO4, MgCl2, and H3BO3 were found to increase the bio-oil yield by either suppressing biochar or gas yield or both. It is also noticed that the employment of the catalysts considerably simplified the chemical compounds in the bio-oils, improving the liquid product selectivity. Moreover, furfural accounts for approximately 80% based on the GC/MS peak area at 8 g MgCl2 per 100 g biomass level. Thus, it is suggested that these catalysts play a vital role in product yields and selectivity during the in situ CMAP process.| Biomass | Operating conditions | Results | Ref. | |||
|---|---|---|---|---|---|---|
| Absorber | Catalyst/catalyst to biomass ratio | Reaction conditions | Bio-oil yield (wt%) | Bio-oil composition, calorific value (MJ kg−1) and oxygen content (wt%) | ||
|
a WHSV (h−1).
b (WHSV)−1 (h).
c Carbon yield of aromatics.
d Yield of liquid organics.
e The ratio of bentonite:HZSM-5:soapstock.
f Reaction conditions: 500 °C, catalyst to biomass ratio of 1:2.
|
||||||
| Douglas fir pellets | Activated carbon | Activated carbon/1.3:1–4.68–1 |
700 W, 589–757 K, 1.3–14.7 min | 6.8–50.2 | Mainly phenolic compounds | 127 and 175 |
| Douglas fir pellets | Activated carbon | Fe modified activated carbon/1:2 |
700 W, 309–591 °C, 2.3–13.7 min | 23.3–45.2 | Dominant furans, phenols, guaiacols and ketones | 177 |
| Douglas fir pellets | Corn stover biochar | Biochar/1:4–1:1 |
700 W, 480 °C, 10 min | 25.8–37.1 | Dominant phenols and guaiacols | 194 |
| Douglas fir pellets | Biochar | HZSM-5/1.3:1–4.7:1 |
700 W, 332–668 °C, 1.3–14.7 min | 28.43–40.83 | Mainly phenols, guaiacols, and aromatic hydrocarbons | 137 |
| Douglas fir pellets | Biochar | HZSM-5/0.021–0.075b | 700 W, 269–481 °C, 9 min | 32.20–37.75 | Dominant aromatic hydrocarbons | 45 |
| Douglas fir pellets | Biochar | Zn/HZSM-5/0.021–0.075b | 700 W, 269–481 °C, 9 min | 24.75–44.80 | Dominant aromatic hydrocarbons | 132 |
| Cellulose | Activated carbon | HZSM-5/0.045–0.126b | 700 W, 269–481 °C, 10 min | 30.35–36.18 | Dominant mono-cyclic aromatic hydrocarbons | 133 |
| Alkali lignin | Activated carbon | Activated carbon/0.82–2.62a | 700 W, 309–591 °C, 8 min | 15.46–41.48 | Mainly phenols and guaiacols | 52 |
| Hybrid poplar | Activated carbon | HZMS-5/1:4 |
700 W, 500 °C, 10 min | 24.76c | Dominate xylenes and naphthalenes | 128 |
| Loblolly pine | Activated carbon | HZMS-5/1:4 |
700 W, 500 °C, 10 min | 22.91c | Dominate xylenes and naphthalenes | |
| Douglas fir | Activated carbon | HZMS-5/1:4 |
700 W, 500 °C, 10 min | 21.52c | Dominate xylenes and naphthalenes | |
| Mushroom waste | SiC | HZSM-5/1:1 |
750 W, 550 °C | ∼47d | Mainly acids and hydrocarbons | 167 |
| SiC | EDTA-HZSM-5/1:1 |
750 W, 550 °C | Max. ∼45d | Mainly acids and hydrocarbons | ||
| Mushroom substrate | SiC | HZMS-5/1:20 |
750 W, 500 °C, 45 min | ∼18d | Mainly oxygen-containing aliphatic and aromatic compounds | 131 |
| SiC | PC-HZSM-5/1:20 |
750 W, 500 °C, 45 min | 14.8d | Mainly oxygen-containing aliphatic and aromatic compounds | ||
| SiC | SiO2-HZMS-5 | 750 W, 500 °C, 45 min | 16.0d | Mainly oxygen-containing aliphatic and aromatic compounds | ||
| SiC | EDTA-HZSM-5 | 750 W, 500 °C, 45 min | ∼15.5d | Mainly oxygen-containing aliphatic and aromatic compounds | ||
| Corn stover | SiC | HZMS-5/1:100–1:20 |
750 W, 400–700 °C, 45 min | Max. 31.2 | Mainly oxygen-containing aliphatic and aromatic compounds | 116 |
| Corn stover | SiC | HZMS-5/1:20 |
750 W, 500 °C, 45 min | ∼20d | Mainly oxygen-containing aliphatic and aromatic compounds | 125 |
| SiC | SiO2-HZSM-5/1:20 |
750 W, 500 °C, 45 min | 15.6–22.6d | Mainly oxygen-containing aliphatic and aromatic compounds | ||
| Peanut shell | Activated carbon | Activated carbon/1:8–1:4 |
2000 W, 300–600 °C, 50 min | 11.13–16.62 | Dominate phenols | 195 |
| Lignite char | Lignite char/1:8–1:4 |
2000 W, 300–600 °C, 50 min | 13.50–24.26 | Dominate phenols | ||
| Pine sawdust | Activated carbon | Activated carbon/1.5:8–2.5:8 |
2000 W, 300–600 °C, 50 min | 16.00–33.18 | Dominant phenol and aromatic hydrocarbons | |
| Lignite char | Lignite char/1.5:8–2.5:8 |
2000 W, 300–600 °C, 50 min | 20.00–35.38 | Dominant phenol and aromatic hydrocarbons | ||
| Soapstock | SiC | Bentonite/1:1 |
1000 W, 550 °C | ∼48d | Mainly olefins and oxygenates | 196 |
| SiC | HZMS-5/1:1 |
1000 W, 550 °C | ∼43d | Mainly olefins and oxygenates | ||
| SiC | Bentonite and HZSM-5/1:1:2e |
1000 W, 500–600 °C | ∼32–∼43d | Mainly olefins, aromatics, and alkanes | ||
| Chlorella sp. | SiC | HZMS-5/1:2–1:1 |
750 W, 450–500 °C, 30 min | Max. 57 | HHV = 26.80, O = 23.52f | 118 |
| Nannochloropsis | SiC | HZMS-5/1:2–1:1 |
750 W, 450–500 °C, 30 min | Max. 59 | HHV = 27.15, O = 17.16f | |
| Algae | Activated carbon | Activated carbon/1:20–3:20 |
400–800 W, 450 °C | Max. 54.3 | Dominant carboxylic acids, aromatics, and nitrogen-containing compounds | 197 |
Other metal oxides (NiO, CuO, CaO and MgO) have been implemented as catalysts in CMAP of corn stover, lowering the formation of PAHs and thereby rendering the bio-oil to be less toxic.163 The catalytic effects of NiO, CaO, CuO, and MgO on CMAP of sugarcane bagasse were also tested.162 Adding either NiO or CaO slightly promoted the production of H2, whilst either CuO or MgO presented inverse effect. Furthermore, the introduction of either CaO or MgO improved the gaseous production, whereas the addition of either NiO or CuO assisted in the liquid production. Moreover, the additives (SiC, activated carbon, coke, K2CO3, and NaOH) were used as catalysts during the CMAP of sawdust.157 It is observed that the additives indicated obvious effects on product yields and their characteristics. Both K2CO3 and NaOH favored the formation of gaseous products because they could strongly absorb microwaves, resulting in extremely high temperature inside the sawdust. It is also noticed that K2CO3 could simplify the bio-oil compositions, improving the bio-oil quality.
The CMAP of wheat straw using CaO and K2CO3 as catalysts have been operated in a fixed-bed microwave reactor at a constant temperature of 500 °C as well.158 The introduction of CaO gave rise to the lowest concentration of CO2 in the gaseous product, while the employment of K2CO3 facilitated the production of H2 and CO at high temperature. In addition, the synergistic effects were manifested using a mixture of K3PO4 and clinoptilolite or bentonite compared to individual catalyst during the CMAP of switchgrass, implying the increase of the microwave absorption rate, bio-oil and biochar qualities.104 It is worth noting that combining 10 wt% K3PO4 with 10 wt% clinoptilolite remarkably reduced the water content in the bio-oil by 39.5%, whilst the pH of the bio-oil increased by 43% compared with the pH of the bio-oil employing 10 wt% clinoptilolite individually, implying a potential synergistic effect between the catalyst mixtures.
With regard to carbon-based catalysts, Bu et al. has investigated the CMAP of biomass using activated carbon as the catalyst to determine the influence of pyrolytic conditions in the yields of both phenols and phenolics. Very high concentrations of phenols (38.9%) and phenolics (66.9%) were observed in the bio-oil with a catalyst to biomass ratio of 3:1, which were superior to those obtained from the MAP of biomass without activated carbon addition.127,175 Bu and his co-workers thereafter used diverse activated carbon as catalysts during the CMAP of woody biomass to determine the effect on product yields and chemical compositions of bio-oils.198 It is found that wood-based and lignite coal-based activated carbon contributed to high amounts of phenols, containing 74.61% and 74.77% in the bio-oils, respectively. It is also manifested that activated carbon as the catalysts could be reused 3–4 times without any regeneration, producing high concentrations of phenol and phenolic compounds. Besides lignin has also been explored to produce renewable phenols and fuels from CMAP using activated carbon as the catalyst.52 As expected, the main chemical compositions of bio-oils were phenols and guaiacols; up to 45% selectivity toward phenols in the bio-oil was achieved. Unlike the role of activated carbon during the CMAP of woody biomass, ferrum modified activated carbon showed different catalytic performance, primarily producing furans, phenols, guaiacols, and ketones occupying 80–87% of the bio-oils.177
Yu's group has also produced phenolic-rich bio-oil from the CMAP of peanut shells and pine sawdust by using activated carbon and lignite char as catalysts.195 Several parameters (e.g., pyrolysis temperature and biomass to catalyst ratio) have been investigated to garner phenolic-rich bio-oil. It is worth noticing that activated carbon could considerably enhance the production of phenolic compounds in the bio-oil. The maximum phenolic content in the bio-oil was 61.2%, and the formation of nanotubes in peanut shell biomass particles was also found. Another carbon-based material (biochar) has been utilized as the catalyst in the CMAP of biomass to estimate the effect of biochar as a catalyst on CMAP of biomass and bio-oil upgrading.194 It is noted that high amounts of phenols (46%) and hydrocarbons (16%) were gained in the bio-oil and high-quality gas enriched in H2, CO, and CH4 was also achieved. More interestingly, fast pyrolysis oil upgraded by biochar as the catalyst was dominated by phenols (∼37%) and hydrocarbons (43%) at high biochar loadings. Additionally, rice husk biochar and the biochar-supported metallic (Ni, Fe, and Cu) catalysts have been applied in the CMAP of rice husks to maximize the high quality syngas.199 It is shown that the biochar-supported metallic catalysts could promote the microwave absorption and enhance the heating rate. Biochar-supported Ni catalysts revealed the most effective impacts on gas production, with a gas yield of 53.9% and the concentration of target syngas production of ∼70%; while biochar-supported Ni and Fe catalysts played pivotal roles in tar removal.
Typically, zeolite-based catalysts are the most popular catalysts used in the CMAP process. Wang et al. has exerted HZMS-5 as the catalyst for in situ CMAP of Douglas fir pellets to produce high-quality bio-oils.137 It is shown that a series of important and useful chemical compositions (i.e., phenols, guaiacols, and aromatic hydrocarbons) were the most abundant compounds, accounting for 82% in the bio-oil. To produce a high amount of valuable aromatic hydrocarbons, ex situ CMAP of biomass was operated using an upfront microwave pyrolysis process coupled with a fixed-bed catalytic process in the presence of HZMS-5 catalyst.45 Aromatic hydrocarbons were enriched and became the most abundant compounds, reaching 92.6% in the bio-oil. These outcomes affirm that the HZSM-5 catalyst favors the production of aromatic hydrocarbons during the CMAP process. Moreover, Wang and her co-workers leveraged the ex situ CMAP system by adding a more efficient catalyst (Zn/HZMS-5) to further improve the concentration of aromatic hydrocarbons in the bio-oil.132 Both GC/MS and FTIR analysis confirmed that aromatic hydrocarbons were dominant in the bio-oil.
Based on previous work, Zhang et al. has investigated a novel pathway of the CMAP of cellulose integrated with packed-bed catalysis in the presence of a solid phase catalyst (well-modified HZSM-5) to produce gasoline-range aromatics and hydrogen-enriched fuel gas.133 It is found that the chemical compounds of the bio-oils were aromatic hydrocarbons, phenols, and aromatic oxygenates. The maximum selectivity toward aromatic hydrocarbons were 96.6%, in which up to 48.6% of chemical compounds belongs to gasoline-range aromatics. H2 was the primary composition in the gaseous product, occupying approximately 40 vol%. It has been suggested that these findings pave a new route for biorefinery industries to manufacture advanced products (aromatics and hydrogen-rich gases) via microwave-induced technologies. It is noticed that the spent HZSM-5 derived from the catalytic process at 500 °C still presented a high BET surface area, pore volume, and pore surface area (Table 7, entry 2). The results indicate that the elevated temperature promoted the cracking reaction of pyrolytic volatiles on the catalyst towards small molecules. Subsequently, diverse lignocellulosic biomass (hybrid poplar, loblolly pine and Douglas fir) was subjected to CMAP using well-modified HZMS-5 as the catalyst to determine the optimal conditions for the production of C8–C16 aromatics.128 The maximum carbon yield of target aromatics was 24.76%, which is observed from the CMAP of hybrid poplar at 500 °C with a catalyst-to-biomass ratio of 0.25. Similarly, a two-step fMAP of corn stover for bio-oil production in the presence of microwave absorber (SiC) and HZMS-5 catalyst has been recently developed by Zhong's group.116 Two variables (reaction temperature and catalyst to biomass mass ratio) were examined to determine their effects on bio-oil yield and chemical components. It is observed from this study that the pyrolysis temperature of 500 °C was suggested as the optimal conditions for maximum bio-oil yield and highest quality of bio-oil. As a result, the two-step fMAP resulted in higher quality bio-oil with a smaller catalyst to biomass ratio than that of the one-step fMAP process. Likewise, Liu et al. has conducted sequential two-step catalytic fMAP of corn stover to yield high-quality bio-oil using HZMS-5 as the catalyst.134Table 8 reveals the physicochemical properties of bio-oils from the fMAP of corn stover, which suggests that high-quality bio-oil was observed through sequential two-step catalytic fMAP. The fresh, spent and regenerated HZMS-5 catalysts have been characterized and evaluated using the XRD technique to determine the effect of coke on the catalyst structure (Fig. 12). Interestingly, the diffractogram comparison of the catalysts showed obvious similarities, the crystal structure and crystallinity of HZMS-5 catalyst were intact before and after the CMAP and regeneration processes. It is manifested that HZMS-5 show good stability in the CMAP process.
| Entry | Catalyst | S BET (m2 g−1) | V pore (cm3 g−1) | d pore (nm) | A Brønsted (mmol NH3 per g) | A Lewis (mmol NH3 per g) | T acidity (mmol NH3 per g) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a S BET: BET surface area; Vpore: pore volume; dpore: average pore size; ABrønsted: Brønsted site acidity; ALewis: Lewis site acidity; Tacidity: total acidity. b 2H-Z5: 2EDTA-HZMS-5. | ||||||||
| 1 | Parent HZSM-5 | 396.2 | 0.097 | 5.2 | — | — | 0.18 | 53 and 133 |
| 2 | Spent HZMS-5 | 306.3 | 0.063 | 4.6 | — | — | — | |
| 3 | Fresh 2H-Z5b | 321 | 0.20 | — | 0.72 | 0.60 | 1.32 | 167 |
| 4 | 2H-Z5-regeneration 1st | 315 | 0.201 | — | 0.41 | 0.58 | 0.99 | |
| 5 | 2H-Z5-regeneration 2nd | 301 | 0.186 | — | 0.40 | 0.54 | 0.94 | |
| 6 | 2H-Z5-regeneration 3rd | 283 | 0.175 | — | 0.39 | 0.49 | 0.88 | |
| 7 | 2H-Z5-regeneration 4th | 272 | 0.168 | — | 0.33 | 0.46 | 0.79 | |
| 8 | 2H-Z5-regeneration 5th | 259 | 0.159 | — | 0.31 | 0.45 | 0.76 | |
| Properties | Chlorella sp. | Nannochloropsis | Corn stover | |||
|---|---|---|---|---|---|---|
| Non-catalysis | HZMS-5 catalysis | Non-catalysis | HZMS-5 catalysis | Non-catalysis | HZSM-5 catalysisb | |
|
a HHV (MJ kg−1) = (3.55 × C2 − 232 × C − 2230 × H + 51.2 × C × H + 131 × N + 20600) × 10−3.
b Pyrolysis temperature: 550 °C, catalyst bed temperature: 425 °C, catalyst to biomass ratio: 1:5.
c Calculated by difference.
|
||||||
| Elemental composition (wt%) | ||||||
| C | 65.70 | 59.27 | 81.64 | 59.75 | 48.13 | 68.43 |
| H | 9.34 | 7.75 | 8.20 | 6.75 | 8.16 | 6.51 |
| N | 8.45 | 9.46 | 5.24 | 16.34 | 1.66 | 1.62 |
| O | 15.78 | 23.52 | 4.90 | 17.16 | 42.05c | 23.44c |
| Water (wt%) | — | — | — | — | 29.82 | 9.11 |
| Density (kg m−3) | 1000 | 1010 | 1180 | 1180 | — | — |
| pH value | 9.33 | 9.54 | 9.93 | 9.62 | — | — |
| Viscosity (cP, at 40 °C) | 11 | 11 | 1.18 | 1.18 | — | — |
| HHV (MJ kg−1)a | 32.37 | 26.80 | 42.00 | 27.15 | 19.79 | 29.85 |
 | ||
| Fig. 12 The XRD patterns of fresh HZSM-5, spent HZMS-5, and regenerated HZMS-5. Reproduced with permission from ref. 134 Copyright (2017) Elsevier. | ||
Modified zeolite-based catalysts are also preferred to be used during the CMAP process. In this respect, Ruan's group have tested a variety of modified HZMS-5 catalysts during the CMAP of biomass to improve bio-oil yield and quality. For instance, when SiO2 was deposited on HZMS-5, the external acid sites of HZMS-5 dramatically decreased.125 With the increase of the SiO2 deposited amount, the yield of the liquid product from the CMAP of corn stover first increased and then decreased, and the amounts of aliphatic hydrocarbons, aromatic hydrocarbons, and oxygen-containing aromatic compounds first increased until the maximum values and then decreased. In order to minimize coke yield and enhance hydrocarbon production, PC-HZSM-5, SiO2-HZMS-5, and EDTA-HZSM-5 were utilized as catalysts in the CMAP of spent edible mushroom substrates.131 All modifications for HZMS-5 led to a loss of external acid sites of HZMS-5. Of the modified catalysts, SiO2-HZMS-5 was in favor of maximizing bio-oil yield and minimizing coke yield. Furthermore, SiO2-HZMS-5 contributed to a higher relative content of hydrocarbons (especially toluene and xylene) in the oil phase than the other catalysts.
Another study has reported the CMAP of mushroom waste using various EDTA modified HZMS-5 catalysts.167 Among these EDTA modified HZMS-5 catalysts, an EDTA treatment for 2 h performed prominent promise for removing oxygenated chemicals and improving aromatic species as well as suppressing coke formation. The regeneration of the modified HZSM-5 catalyst by EDTA treatment for 2 h was also studied as shown in Table 7 (entry 4–8). Compared to the characteristics of the fresh 2H-Z5, the BET surface area, pore volume, and the total number of acid sites gradually decreased as the regeneration times increased. It is discerned that the highest relative content of hydrocarbons and lowest coke yield were both observed under the third regeneration cycle conditions. Hence, it is concluded that a moderate regeneration time would encourage the yield of target products, and high-quality bio-oil production could be garnered. In addition, two catalysts (bentonite and HZMS-5) combined were used as the catalyst during the CMAP of soapstock.196 The two parameters of catalyst and pyrolysis temperature were tested to understand the effects on product yields and bio-oil chemical compositions. It is found that the combined catalyst can reduce the water content, and bentonite could assist in the increase of bio-oil yield. Up to 84.2% selectivity toward hydrocarbons in the bio-oil was gained, the proportion of oxygenates could decrease to 15.8%, and the N-containing compounds were completely eliminated. It is also noted that the introduction of bentonite could significantly promote the BET surface area of biochar, effectively improving the removal efficiency of Cd2+ by 27.4 wt%.
More importantly, microalgae are also attractive to be valorized through the CMAP system. Borges et al. has studied the fMAP of Chlorella sp. and Nannochloropsis in the presence of a microwave absorber (SiC) and catalyst (HZMS-5).118 The maximum bio-oil yields from the CMAP of Chlorella sp. and Nannochloropsis were 57 wt% and 59 wt%, respectively. The characteristics of bio-oils from fMAP of the two types of microalgae are demonstrated in Table 8. It is discerned that fMAP is a promising technique to valorize microalgae for commercial application and economic outlook. The CMAP of natural algae was also conducted for bio-oil production by using activated carbon as the catalyst.197 The maximum bio-oil yield gained was 54.3%, and the energy yield of bio-products was 236.9%. It is obtained that carboxylic acids were the main composition in the bio-oil, optimizing at 66.6%. This study has indicated that the CMAP of algae provides a route to not only ameliorate the environment but also achieve renewable fuels or chemicals.
Apart from advanced bio-oil production, magnetic biochar has been developed from the CMAP process as well. Magnetic biochar has been synthesized by Mubarak et al. via the CMAP of empty fruit bunch using FeCl3 as the catalyst.200,201 The effects of microwave power, retention time, and impregnation ration of FeCl3 to biomass were investigated. The newly produced magnetic biochar has a very high surface area of 890 m2 g−1, efficiently removing methylene blue with an efficiency of 99.9% from aqueous solution with a maximum adsorption of 265 mg g−1. Likewise, sugarcane bagasse was used to prepare magnetic biochar through the CMAP process using Fe2O3 as the catalyst.202 The optimum conditions to maximize magnetic biochar yield (69%) were at an impregnated Fe2O3 to biomass ratio of 0.45 with a radiation time of 30 min. It is noticed that the magnetic biochar could remove 96.2% of hazardous Cd2+ from aqueous solutions. Moreover, K3PO4, clinoptilolite and/or bentonite have served as catalysts during the CMAP of switchgrass to engineer biochar with decent water holding capacity, cation exchange capacity and fertility of loamy sand soil.203 It is observed that the addition of these catalysts during the CMAP process improved the biochar surface area and plant nutrient contents. The catalysts remaining in the biochar could provide important nutrients for the growth of bioenergy and food crops as well.
6.2 Catalytic microwave-assisted co-pyrolysis of biomass and other polymers
As claimed above, CMACP is one of the state-of-the-art techniques which has recently aroused numerous concerns. Both Ruan's group and Lei's group are paying more attention to this novel technology. Table 9 summarizes the current studies with regard to the CMACP of biomass with other polymers. To be more specific, Zhang and his co-workers conducted the CMACP of cellulose with LDPE to manufacture C8–C16 range aromatic hydrocarbons in the presence of well-promoted HZMS-5 catalysts.55 It is noticeable that the raw organics with enhanced carbon yield (∼44%) were more principally lumped in the jet fuel range at the catalytic temperature of 375 °C with the LDPE to cellulose mass ratio of 3:4. And then Zhang et al. leveraged intact biomass (Douglas fir pellets) co-fed with LDPE through the CMACP system to produce highly valuable aromatic hydrocarbons.53 It is discerned that improved carbon yield (40.54%) of liquid organics primarily lumped in the jet fuel range was also obtained at the catalytic temperature of 375 °C with the LDPE to biomass ratio of 3:4. Accordingly, it is reaffirmed that the CMACP of biomass with plastics indicates a promising way to produce jet fuel range compounds.
| Type of materials | Reaction conditions | Results | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Biomass | Polymers | Catalyst | Polymers to biomass ratio | Temperature (°C) | Bio-oil yield (C%) | Bio-oil composition | |
|
a Aromatic yield (C%).
b The HZMS-5 to MgO ratio of 1:1.
c The HZSM-5 to Cao ratio of 1:1.
d Gas composition.
|
|||||||
| Cellulose | LDPE | HZSM-5 | 0.4:1–1.45:1 |
250–500 | 36.05–46.35a | Dominant xylenes and trimethylbenzenes | 55 |
| Lignin | LDPE | HZMS-5 + MgOb | 1:3–2:1 |
450–600 | 22.8–35.9 | Dominant aromatics and phenolic compounds | 149 |
| Corn stover | Scum | HZMS-5 + CaOc | 1:4–4:1 |
450–650 | 17.4–31.4 | Mainly aromatic and polycyclic aromatic hydrocarbons | 54 |
| Douglas fir pellets | LDPE | HZMS-5 | 0.4:1–1.45:1 |
200–500 | 34.18–42.66a | Mainly xylenes and trimethylbenzenes | 53 |
| Bamboo sawdust | Waste tire | HZSM-5 | 1:4–4:1 |
450–650 | 19.56–37.86 | Mainly aliphatic and aromatic hydrocarbons | 204 |
| Bamboo sticks | HDPE | Ni/Al2O3 | 1:20–1:4 |
600 | 10.3–22.0 | Dominant H2 and COd | 123 |
| Nannochloropsis sp. | Scum | HZMS-5 | 1:4–4:1 |
450–650 | 11.6–27.7 | Mainly aromatic and polycyclic aromatic hydrocarbons | 122 |
Moreover, bifunctional catalysts (HZMS-5 and CaO) have been investigated in the CMACP of corn stover and scum for bio-oil production.54 The effects of reaction temperature, HZSM-5 to CaO ratio, and corn stover to scum ratio on product yields and chemical selectivity were estimated. Maximum bio-oil and aromatic yields were both observed at 550 °C. It is also discerned that scum as a hydrogen donor had a positive synergistic effect with corn stover to improve the bio-oil yield and aromatic hydrocarbons production when the H/Ceff exceeded 1. Besides, the maximum yield (29.3 wt%) of aromatic hydrocarbons was achieved when the corn stover to scum ratio was set at 1:2. Likewise, bifunctional catalysts (HZMS-5 and MgO) were introduced in the CMACP of lignin and LDPE to evaluate the effects of pyrolysis temperature, lignin to LDPE ratio, HZSM-5 to MgO ratio, and feed to catalyst ratio on product yields and distributions.149 500 °C was found to be the optimal conditions in terms of the maximum bio-oil yield. Increasing LDPE proportion could improve the production of aromatics; at the same time methoxyl group substituted phenols were eliminated. Improving the HZMS-5 to MgO ratio could promote the yield of aromatics, but reduce the yield of alkylated phenols. With respect to other catalysts, the CMACP of bamboo with high-density polyethylene (HDPE) was studied using a Ni/Al2O3 catalyst to produce a high yield of H2-enriched syngas.123 It is observed that the bio-oil quality was improved and a large amount of H2 was also produced during the CMACP process. The maximum concentration of H2 could reach 54 vol% in the gaseous fraction when the bamboo wood to HDPE ratio was fixed at 4:1.
As for microalgae biomass, microalgae co-fed with scum in the CMACP using the HZSM-5 catalyst has been reported as well.122 Identical to the above-mentioned studies, effects of pyrolysis temperature, catalyst to feed ratio, and microalgae to scum ratio on bio-oil yield and composition were investigated. It is found that the maximum bio-oil yield and highest share of aromatic hydrocarbons in the bio-oil were gained at 550 °C. The addition of scum in the process not only improved bio-oil yield but also enhanced aromatic production. It has also been suggested that the synergistic effect between microalgae and scum during the CMACP process was remarkable only when the effective hydrogen index of feedstock was larger than ∼0.7. Additionally, waste tires were also co-fed with lignocellulosic biomass (bamboo sawdust) during CMACP using HZMS-5 as the catalyst for bio-oil production.204 Several variables (pyrolysis temperature, catalyst to feed ratio, and biomass to waste tires ratio) were examined to determine their influence in bio-oil production. It is found that the optimum pyrolysis temperature was maintained at 550 °C, indicating the maximum bio-oil yield and lowest share of polycyclic aromatic hydrocarbons. Moreover, waste tires as the hydrogen donor could be beneficial for the promotion of bio-oil production and aromatic hydrocarbons. It should be also noted that the maximum production of aromatic hydrocarbons was garnered at the biomass to waste tires ratio of 1:1.
7. Kinetics of both non-catalytic and catalytic MAP of biomass
The systematic study on behaviors and kinetics of biomass in MAP system is highly important for the effective design of the thermochemical conversion units. Besides, determination of kinetic behaviors of biomass during the MAP process in high temperature regions can still be regarded as one of the most attractive research areas.57 Recently, a few studies have investigated the characteristics and kinetics of biomass in the non-catalytic MAP process.47,121,162,179,184,205 The kinetic scheme to model thermal decomposition of biomass under microwave irradiation can be considered as the following reaction scheme:| Biomass → biochar + volatiles |
In general, the thermal decomposition of biomass at a constant heating rate can be explained as the following formula:
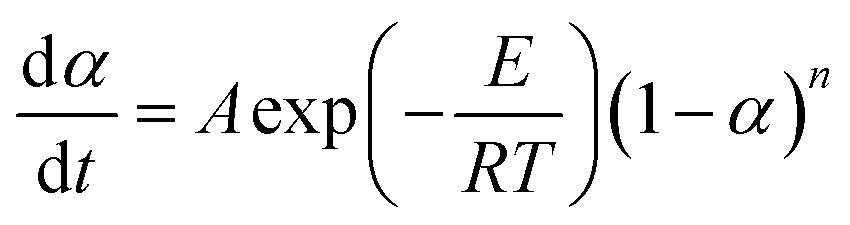
where X0 is the initial mass of biomass; Xt is the mass at time t and Xf is the final mass at the end of MAP. The Coast–Redfern method is typically used to determine the values of kinetic parameters during MAP.47,121,162,179,184,205 For a constant heating rate β during MAP, β = dT/dt, rearranging the above equations and integrating gives:
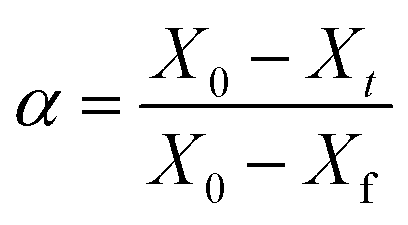
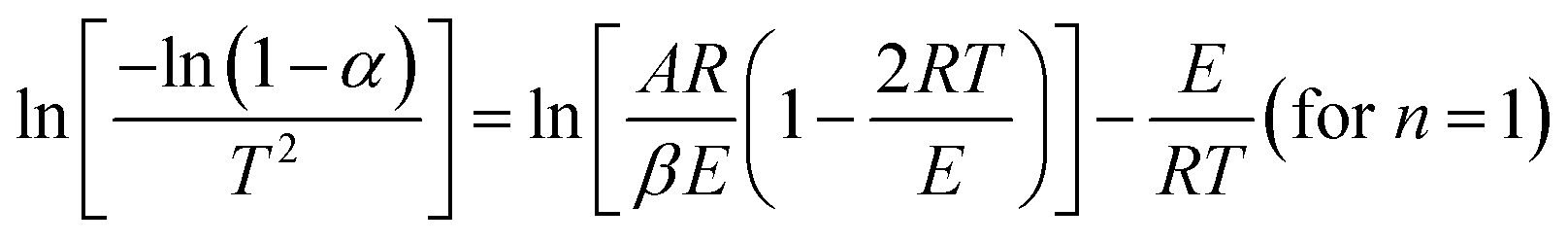
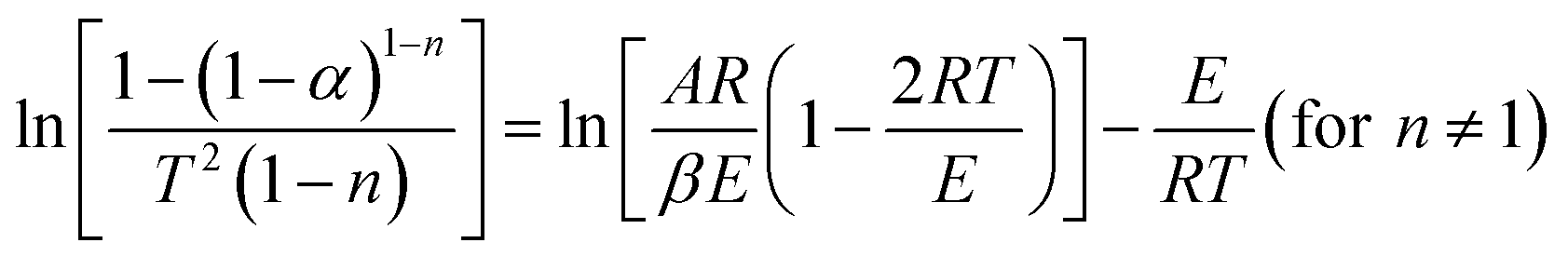
Assuming 2RT/E ≪ 1, simplifying the two above-mentioned equations can be expressed as the following formulas:
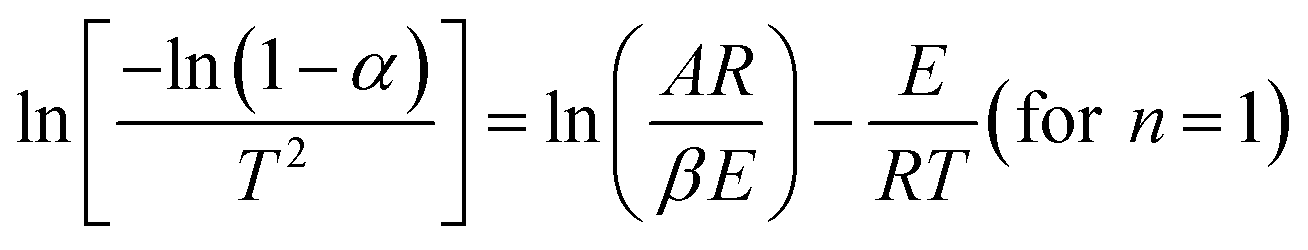
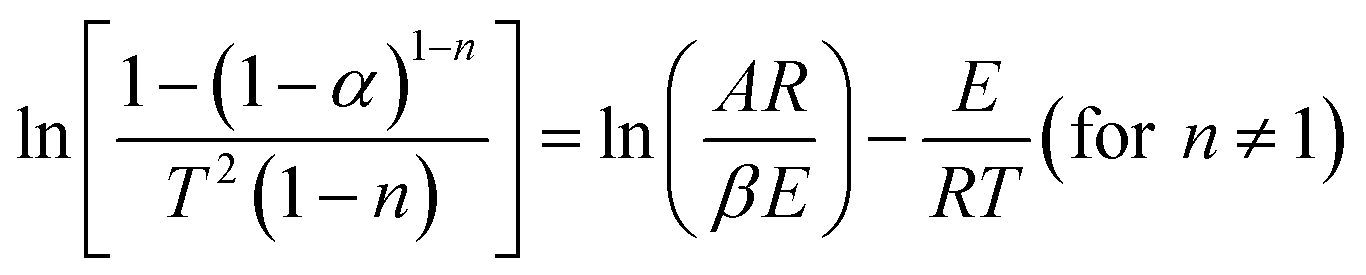
As a result, a plot of ln[(−ln(1 − α)/T2)] against 1/T for n = 1 or ln[(1 − (1 − α)(1−n))/(T2(1 − n))] versus 1/T when n ≠ 1 can give rise to a straight line with a slop of −E/R and an intercept of ln(AR/βE). Therefore, the pre-exponential factor A and the activation energy E can be determined.
Table 10 gives representative studies on kinetic parameters for both non-catalytic MAP and CMAP of biomass. It can be seen that high coefficients of determination (R2) were observed for all studies, implying the significant fit for these reactions. It is also noted that the activation energy (E) and pre-exponential factor of non-catalytic MAP is much lower than those of conventional pyrolysis, indicating the difference in kinetic processes for both heating manners.205 The kinetic parameters of CMAP in the presence of catalysts are also listed in Table 10. As such, the coefficients of determination (R2) of these linear regressions were all larger than 0.99.162 Among the catalysts, CaO reveals the best catalytic performance whilst MgO indicates the worst effect. However, this kinetic analysis is not as satisfactory as it should be because of the difficulty in acquiring a precise relationship between biomass weight and reaction temperature.
| Types of materials | Reaction order n | E (kJ mol−1) | A (s−1) | R 2 | Ref. |
|---|---|---|---|---|---|
| a Assume n = 1. | |||||
| Non-catalytic MAP | |||||
| Douglas fir pellets | 1 | 16.5 | 15.27 | 0.81 | 121 |
| 2 | 21.0 | 1.42 | 0.79 | ||
| 3 | 33.5 | 3.03 | 0.85 | ||
| Rice straw | 1 | 27.1 | 1.06 | 0.97 | 179 |
| Prairie cordgrass | 1 | 3.1 | 2.26 | 0.42 | 184 |
| 2 | 13.3 | 1.82 | 0.91 | ||
| 3 | 12.1 | 1.53 | 0.89 | ||
| Sugarcane bagasse | 1 | 18.9 | 0.18 | 0.99 | 162 |
| Bamboo sawdust | 1.5 | 24.5 | 198 | 0.97 | 205 |
8. Future prospects and outlook
The MAP technology that has obtained plenty of attention has been manifested to be the promising alternative of conventional pyrolysis for the valorization of biomass, and its prospects are really optimistic.60 That is because the input energy and processing time of MAP can be much lower, and the efficiency is much higher than conventional pyrolysis.206,207 Regardless of low energy conversion efficiency from electric energy to thermal energy via microwave irradiation, the energy recovery is very high when the output energy of biofuel is divided by input energy of biomass plus energy for pyrolysis during the MAP process. It is found that the energy recovery during MAP of biomass could achieve 91%, which is much higher than the energy recovery of 35–39% from similar biomass by means of conventional flash pyrolysis techniques.208 In the same regard, the energy recovery by means of conventional pyrolysis of coffee hulls was 84%, while the energy recovery increased up to 99% using a microwave technique.107 According to the report from Lei's group, if biochar generated is supplied for a coal-firing plant with a 35% efficiency to electricity and 90% transmission efficiency to a MAP plant, more than 150% electricity for the MAP plant can be supplied from biochar firing.209 Indeed, the MAP technique is likely to fairly promising for the valorization of biomass on a large scale.To enhance the energy conversion efficiency from electric energy to thermal energy during microwave irradiation, it has been recently found that a higher energy conversion efficiency can be obtained by using single-mode microwave irradiation.210 It is also well known that the energy loss of microwave energy to thermal energy plays a key role in the overall energy conversion efficiency. Hence, the improvement of the energy conversion efficiency in the step will remarkably contribute to enhancing the energy conversion efficiency. In this sense, microwave absorbers used in the MAP system can assist in the reduction of the overall energy consumption. Nevertheless, commonly used microwave absorbers microwave absorbers (e.g., biochar and metal oxides) leading to a sharp increase in dielectric properties at high temperatures allow the thermal runaway effect to take place.211 This effect is hard to control and can induce the occurrence of secondary pyrolysis reactions, such as gasification of biochar matrix and reforming of valuable compounds in the bio-oil.212 As a consequence, commonly used microwave absorbers are presented to lower the temperature threshold of the thermal runaway effect, and these microwave absorbers are thus not viable on an industrial scale for bio-oil production. Instead, the development of the novel microwave absorbers is obviously the other vital direction to alleviate the issues.74 Since the increase of microwave absorption ability of the absorber can facilitate the transformation of microwave energy to thermal energy, thereby enhancing the energy conversion efficiency. It is suggested that the doping of another material or nanoparticles on microwave absorbers, and the fabrication of the layered composites can achieve the promoted dielectric and thermal response. The newly developed microwave absorbers may contribute to the reduction of the thermal runaway effect during the MAP process.
Furthermore, it is widely recognized that there are some other downsides of MAP technology that are required to be overcome and promoted. The most essential attempts are to increase the bio-oil yield and improve the selectivity towards target products.213,214 Current studies have determined the optimized conditions (e.g., temperature range, microwave powers, the type and loadings of microwave absorbers, and catalytic patterns) for maximizing desirable products so far.58,120,215 As every feedstock source is relatively unique, the pyrolysis performance is thus affected.214 To remedy the dilemma of feedstock's influence, there have been some feasible investigations on pretreatment technologies. One of the most promising pretreatment approaches is torrefaction.60 Homogeneity of torrefied biomass allow the MAP process to yield similar and simplified chemical compositions. Nonetheless, future investigations should be carried out to improve the energy efficiency of pretreatment technologies and lower the production cost of the large-scale process.
Given the state-of-the-art outcomes in this field, the motivation is transferred to biomass valorization in large-scale biorefineries. Moreover, when the MAP system is enlarged to the industrial application scale, the ratio of energy for biomass pyrolysis to total energy will be considerably enhanced, i.e. the ratio of heat loss during the process can be dramatically reduced.85 However, the scale-up of the current MAP of biomass is difficult. To uplift the MAP technique to the industrial level, these are some steps that should be considered.216 First of all, the key parameters that affect the process, such as microwave power, reaction temperature, and residence time should be clearly understood. Based on the fundamental understanding and process requirements, a model should be established to determine how the microwave irradiation interacts with biomass in a range of pilot-scale concepts and designs. Ultimately, an ideal amount of microwave absorber should be investigated to rapidly induce the pyrolysis by consuming the least energy.
With careful consideration of current lab-scale findings, a continuous MAP process can be developed and scaled up for future industrial applications. It is discerned that coupling MAP process and continuous flow technology can overcome the main drawbacks of microwaves and give a very promising way to manufacture high value-added chemicals or advanced fuels since the continuous flow has been proved to accelerate process intensification and obtain safe, efficient and sustainable products.80 For example, continuous MAP with an apparatus fitted with magnetrons capable of outputting a maximum microwave power of 5000 W has been developed by Lam et al.217 Waste automotive oil was fed into the continuous MAP system at a feed ratio of 5 kg per hour; and the MAP system presented a positive energy ratio of 8 and a net energy output of over 179 MJ h−1. In terms of microwave reactors for the large scale, it is worth pointing out that large scale microwave reactors are commonly designed and specifically constructed according to applications to maximize energy transfer and process efficiency.86,218 It has been concluded by Moseley et al. that medium to large scale multimode microwave reactors are more energy efficient than conventional heating reactors.218,219 To develop a reliable MAP concept of biomass on a large scale, high power density has been proven to be a key criterion to enable the rapid conversion of biomass without the large amount of heat loss to the surroundings according to batch tests.220,221 It is also suggested that very rapid quenching systems is the most effective alternative to recover bio-oils at the large scale.222 Since the quality of bio-oil production is highly dependent upon the control of residence time of volatiles. The release of pyrolytic volatiles should be conducted rather rapidly (<1 s) to present secondary fragmentation reactions within bio-oils and to maintain an acceptable quality.223,224 As fouling the handling system can result in a decrease in bio-oil yield and may cause damage to the microwave hardware (magnetrons and waveguide), a pressure window will be commonly used to separate the waveguide from microwave cavity where pyrolysis occurs on a large scale.211 Generally, nearby metallic surfaces must be avoided during the scaling up of MAP to prevent the dielectric breakdown induced by an excess of a charge buildup over the dielectric strength of the material.211 In this regard, five various prospective concepts have been proposed by Beneroso et al. pertaining to the scaling up of the MAP of biomass depending on the means by which biomass is transported through the continuous process: (1) rotary kiln concept; (2) conveyor belt concept; (3) rotating ceramic-based disc concept; (4) microwave fluidized bed concept; (5) auger reactor concept.211 These concepts have been widely used in the chemical industries but not within the MAP process. In fact, they have been evaluated for their electromagnetic compatibility and ability to deliver sufficient power density to induce pyrolysis without the utilization of conventional microwave absorbers.
Indeed, the commercialization of the MAP technology is associated with techno-economic feasibility.11 Several studies have evaluated the feasibility of both non-catalytic MAP and CMAP techniques from an economic point of view.225–228 In this regard, Ruan et al. discussed the utilization of small biorefinery systems which are distributed in different local communities.225 The results suggested that a small distributed biomass energy production system was technologically more feasible, economically viable, and suitable than current conventional large-scale biomass energy systems. Extra income can be provided for farmers and truly involved biomass producers. Accordingly, the continuous MAP of biomass and waste feedstock shows crucial potential to integrate biorefineries for the local conversion of biomass residues or waste into fuels and chemicals. Furthermore, the techno-economic analysis regarding ex situ CMAP of biomass for bio-oil production was also evaluated.227 For this case, a distributed mobile microwave pyrolysis system coupled with an ex situ catalysis process is assumed to produce aromatic hydrocarbon enriched bio-oil, gas and biochar. This study revealed that this scenario can gain a high profit annually, and the return of investment (ROI) can reach 45.34% per year. It is also interesting that a slight improvement of bio-oil yield can significantly increase the ROI.
To improve the yield of bio-oil and obtain more advanced fuel or chemicals for the improvement of ROI, the CMACP process integrated with the hydrogenation process paves a more feasible and promising way to valorize both biomass and waste hydrogen-rich polymers (such as waste plastics and waste tires). More specifically, jet fuels are mainly composed of linear- and branched-chain alkanes (C8 – C16).15,229 It is widely proven that cyclic alkanes can be burned very cleanly releasing more energy straight-chain alkanes because cyclic alkanes are defined as compact molecules within a robust ring strain.230,231 To enhance the qualities of jet fuels, jet fuel range cyclic alkanes can sever as additives in civilian jet fuels.15,232 It has been suggested that the promising and simple method for the production of cyclic alkanes is the hydro-cycloaddition of aromatic hydrocarbons.100,128,135,233,234 Based on the studies from the CMACP of biomass with other polymers, aromatic hydrocarbons are mostly lumped in the range of C8–C16.53,55 Moreover, Lei and his colleagues have developed a green catalytic route for the production of renewable cyclic alkanes from woody biomass via the CMAP process and the hydro-cycloaddition process.100,128,135 Taking these outcomes into consideration, hydrogen-rich plastics have been introduced into the CMAP of biomass to enhance the carbon yield of aromatic hydrocarbons.53,55 Therefore, the enhanced carbon yield of aromatic hydrocarbons was hydrogenated into highly valuable cyclic alkanes for potential use as replacements or additives in jet fuels as sketched in Fig. 13.
 | ||
| Fig. 13 Promising routes for the conversion of lignocellulosic biomass into advanced biofuels using the system of catalytic microwave-assisted pyrolysis. Reprinted with permission from ref. 53 Copyright (2016) Elsevier. | ||
It is also observed that the co-feeding of biomass with waste polymers during the CMACP process could lower the coke formation. Yet, the heteroatoms can considerably affect the lifetime of zeolite-based catalysts (e.g., HZMS-5) during the CMACP process. Hence, developing a catalyst that can mitigate the influence is necessitated. To enhance the value of the resultant aromatic hydrocarbons, newly developed catalysts should accelerate tailored reaction routes toward either optimizing the carbon yield of valuable monocyclic aromatics (such as BTX) or jet fuel range precursors. Besides, the ideal catalysts should enable catalysis of a sequence of desired reactions, including dehydration, cracking, decarboxylation, decarbonylation, and C–C coupling.14 These zeolite-based catalysts developed should also own appropriate acid sites and acidity for target aromatic hydrocarbons through certain reaction pathways and to extend their lifetime.
9. Conclusions
To summarize, this study reviews a state-of-the-art technique regarding the MAP of biomass. The sections from Fundamentals of microwave irradiation to Catalysts for catalytic microwave-assisted pyrolysis are specifically demonstrated. Owing to the key features of MAP (e.g. rapid volumetric heating, easy control, and energy saving), MAP has been proven as the more effective pathway to valorize biomass than conventional pyrolysis. It is also noticed that the MAP technology is a well-established one that can be used to enhance pyrolysis for target products. Several types of catalysts including metal oxides and zeolite-based catalysts have been most commonly employed during the CMAP of biomass, significantly affecting the product yields, distributions, and even qualities. Techno-economic analysis of both non-catalytic MAP and CMAP processes suggested that the distributed mobile microwave pyrolysis system whether coupled with the catalysis process or not is profitable.According to the current technique converting aromatic hydrocarbons to cyclic alkanes under very mild conditions, the CMACP integrated with a hydro-cycloaddition process is extremely essential for the production of advanced biofuels or highly valuable chemicals from biomass and waste polymers. Nevertheless, current studies are primarily dependent on the lab scale equipment using biomass with pure plastics to produce aromatics. Apparently, the most achievable and economic method is to directly transform biomass with waste polymers into target aromatic hydrocarbons, rather than pure plastics. In this premise, the CMACP technique remains in its infancy and requires continual research work on the large scale-up of the technique. As a result, the MAP technology paves a promising route for biomass utilization in a biorefinery; however, there are still some challenges for further advances in the MAP technique, including improvement of heating uniformity, scale-up of current technology for commercialization, etc.
Abbreviations
| BTX | Benzene, toluene, and xylenes |
| CCD | Central composite design |
| CFP | Catalytic fast pyrolysis |
| CMACP | Catalytic microwave-assisted co-pyrolysis |
| CMAP | Catalytic microwave-assisted pyrolysis |
| EDTA | Ethylene diamine tetraacetic acid |
| FCC | Federal Communication Commission |
| fMAP | Fast microwave-assisted pyrolysis |
| HDO | Hydrodeoxygenation |
| HDPE | High-density polyethylene |
| HHV | High heating value |
| HTL | Hydrothermal liquefaction |
| ISM | Industrial, scientific, and medical |
| LDPE | Low-density polyethylene |
| MACP | Microwave-assisted co-pyrolysis |
| MAP | Microwave-assisted pyrolysis |
| MOMAs | Metal oxide microwave absorbers |
| PAHs | Polycyclic aromatic hydrocarbons |
| PC | Pre-coking |
| ROI | Return on investment |
| SCBMs | Solid carbon based materials |
| SiO2-CVD | Chemical vapor deposition of inert silica |
| TEOS | Tetra-ethyl-orthosilicate |
Acknowledgements
This study was supported partially by the postdoctoral fellowship of the Illinois Sustainable Technology Center, University of Illinois at Urbana-Champaign, and The Agriculture and Food Research Initiative of National Institute of Food and Agriculture, United States Department of Agriculture (Award Number: 2016-67021-24533; Award Number: 2016-33610-25904).References
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9, 2939–2977 CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- S. Heidenreich and P. U. Foscolo, Prog. Energy Combust. Sci., 2015, 46, 72–95 CrossRef.
- C. Yin, Bioresour. Technol., 2012, 120, 273–284 CrossRef CAS PubMed.
- Q. Bu, H. Lei, A. H. Zacher, L. Wang, S. Ren, J. Liang, Y. Wei, Y. Liu, J. Tang, Q. Zhang and R. Ruan, Bioresour. Technol., 2012, 124, 470–477 CrossRef CAS PubMed.
- A. Raheem, W. A. K. G. Wan Azlina, Y. H. Taufiq Yap, M. K. Danquah and R. Harun, Renewable Sustainable Energy Rev., 2015, 49, 990–999 CrossRef CAS.
- V. Skorupskaite, V. Makareviciene and D. Levisauskas, Algal Res., 2015, 7, 45–50 CrossRef.
- Y. Zhou, L. Schideman, G. Yu and Y. Zhang, Energy Environ. Sci., 2013, 6, 3765 CAS.
- A. Demirbas, Energy Convers. Manage., 2010, 51, 2738–2749 CrossRef CAS.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- R. Luque, J. A. Menéndez, A. Arenillas and J. Cot, Energy Environ. Sci., 2012, 5, 5481–5488 CAS.
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89, S29–S35 CrossRef CAS.
- N. H. Leibbrandt, J. H. Knoetze and J. F. Görgens, Biomass Bioenergy, 2011, 35, 2117–2126 CrossRef CAS.
- X. Zhang, H. Lei, S. Chen and J. Wu, Green Chem., 2016, 18, 4145–4169 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- E. Butler, G. Devlin, D. Meier and K. McDonnell, Renewable Sustainable Energy Rev., 2011, 15, 4171–4186 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- Y.-T. Cheng and G. W. Huber, ACS Catal., 2011, 1, 611–628 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Energy Environ. Sci., 2011, 4, 793–804 CAS.
- D. C. Elliott, T. R. Hart, G. G. Neuenschwander, L. J. Rotness, M. V. Olarte, A. H. Zacher and Y. Solantausta, Energy Fuels, 2012, 26, 3891–3896 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- J. R. Regalbuto, Science, 2009, 325, 822–824 CrossRef PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- Y. Zhang, T. R. Brown, G. Hu and R. C. Brown, Chem. Eng. J., 2013, 225, 895–904 CrossRef CAS.
- K. L. Hew, A. M. Tamidi, S. Yusup, K. T. Lee and M. M. Ahmad, Bioresour. Technol., 2010, 101, 8855–8858 CrossRef CAS PubMed.
- F. Abnisa and W. M. A. Wan Daud, Energy Convers. Manage., 2014, 87, 71–85 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- A. K. Panda, R. K. Singh and D. K. Mishra, Renewable Sustainable Energy Rev., 2010, 14, 233–248 CrossRef CAS.
- T. R. Brown, Y. Zhang, G. Hu and R. C. Brown, Biofuels, Bioprod. Biorefin., 2012, 6, 73–87 CrossRef CAS.
- G. Yildiz, M. Pronk, M. Djokic, K. M. van Geem, F. Ronsse, R. van Duren and W. Prins, J. Anal. Appl. Pyrolysis, 2013, 103, 343–351 CrossRef CAS.
- T. R. Carlson, T. P. Vispute and G. W. Huber, ChemSusChem, 2008, 1, 397–400 CrossRef CAS PubMed.
- T. R. Carlson, G. A. Tompsett, W. C. Conner and G. W. Huber, Top. Catal., 2009, 52, 241–252 CrossRef CAS.
- T. R. Carlson, J. Jae, Y.-C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110–124 CrossRef CAS.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 CAS.
- Y. T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., 2012, 51, 1387–1390 CrossRef CAS PubMed.
- Y. T. Cheng, Z. Wang, C. J. Gilbert, W. Fan and G. W. Huber, Angew. Chem., 2012, 51, 11097–11100 CrossRef CAS PubMed.
- X. Li, H. Zhang, J. Li, L. Su, J. Zuo, S. Komarneni and Y. Wang, Appl. Catal., A, 2013, 455, 114–121 CrossRef CAS.
- C. Dorado, C. A. Mullen and A. A. Boateng, ACS Sustainable Chem. Eng., 2014, 2, 301–311 CrossRef CAS.
- H. Zhang, J. Nie, R. Xiao, B. Jin, C. Dong and G. Xiao, Energy Fuels, 2014, 28, 1940–1947 CrossRef CAS.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727 RSC.
- L. Wang, H. Lei, J. Lee, S. Chen, J. Tang and B. Ahring, RSC Adv., 2013, 3, 14609 RSC.
- A. A. Salema and F. N. Ani, Bioresour. Technol., 2011, 102, 3388–3395 CrossRef CAS PubMed.
- N. Wang, A. Tahmasebi, J. Yu, J. Xu, F. Huang and A. Mamaeva, Bioresour. Technol., 2015, 190, 89–96 CrossRef CAS PubMed.
- V. L. Budarin, P. S. Shuttleworth, J. R. Dodson, A. J. Hunt, B. Lanigan, R. Marriott, K. J. Milkowski, A. J. Wilson, S. W. Breeden, J. Fan, E. H. K. Sin and J. H. Clark, Energy Environ. Sci., 2011, 4, 471–479 CAS.
- V. L. Budarin, Y. Zhao, M. J. Gronnow, P. S. Shuttleworth, S. W. Breeden, D. J. Macquarrie and J. H. Clark, Green Chem., 2011, 13, 2330 RSC.
- P. Lidstrom, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225–9283 CrossRef CAS.
- F. Motasemi and M. T. Afzal, Renewable Sustainable Energy Rev., 2013, 28, 317–330 CrossRef CAS.
- Q. Bu, H. Lei, L. Wang, Y. Wei, L. Zhu, X. Zhang, Y. Liu, G. Yadavalli and J. Tang, Bioresour. Technol., 2014, 162, 142–147 CrossRef CAS PubMed.
- X. Zhang, H. Lei, L. Zhu, M. Qian, X. Zhu, J. Wu and S. Chen, Appl. Energy, 2016, 173, 418–430 CrossRef CAS.
- S. Liu, Q. Xie, B. Zhang, Y. Cheng, Y. Liu, P. Chen and R. Ruan, Bioresour. Technol., 2016, 204, 164–170 CrossRef CAS PubMed.
- X. Zhang, H. Lei, L. Zhu, X. Zhu, M. Qian, G. Yadavalli, D. Yan, J. Wu and S. Chen, Bioresour. Technol., 2016, 214, 45–54 CrossRef CAS PubMed.
- D. J. Macquarrie, J. H. Clark and E. Fitzpatrick, Biofuels, Bioprod. Biorefin., 2012, 6, 549–560 CrossRef CAS.
- Y.-F. Huang, P.-T. Chiueh and S.-L. Lo, Sustainable Environ. Res., 2016, 26, 103–109 CrossRef.
- Y. Wang, L. Dai, R. Wang, L. Fan, Y. Liu, Q. Xie and R. Ruan, J. Anal. Appl. Pyrolysis, 2016, 119, 251–258 CrossRef CAS.
- F. Mushtaq, R. Mat and F. N. Ani, Renewable Sustainable Energy Rev., 2014, 39, 555–574 CrossRef CAS.
- H. M. Morgan, Q. Bu, J. Liang, Y. Liu, H. Mao, A. Shi, H. Lei and R. Ruan, Bioresour. Technol., 2017, 230, 112–121 CrossRef CAS PubMed.
- Y. Zhang, P. Chen, S. Liu, P. Peng, M. Min, Y. Cheng, E. Anderson, N. Zhou, L. Fan, C. Liu, G. Chen, Y. Liu, H. Lei, B. Li and R. Ruan, Bioresour. Technol., 2017, 230, 143–151 CrossRef PubMed.
- H. Zhang, J. Ding and Z. Zhao, Bioresour. Technol., 2012, 123, 72–77 CrossRef CAS PubMed.
- S. Mallakpour and Z. Rafiee, Prog. Polym. Sci., 2011, 36, 1754–1765 CrossRef CAS.
- F. Motasemi and F. N. Ani, Renewable Sustainable Energy Rev., 2012, 16, 4719–4733 CrossRef CAS.
- B. Sajjadi, A. R. Abdul Aziz and S. Ibrahim, Renewable Sustainable Energy Rev., 2014, 37, 762–777 CrossRef CAS.
- J. Li, J. Dai, G. Liu, H. Zhang, Z. Gao, J. Fu, Y. He and Y. Huang, Biomass Bioenergy, 2016, 94, 228–244 CrossRef CAS.
- V. K. Tyagi and S.-L. Lo, Renewable Sustainable Energy Rev., 2013, 18, 288–305 CrossRef CAS.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358 RSC.
- B. F. Fernandez, Renewable Energy Focus, 2015, 16, 156–159 CrossRef.
- A. Richel and N. Jacquet, Biomass Convers. Biorefin., 2015, 5, 115–124 CAS.
- V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2011, 40, 4925–4936 RSC.
- S. Mutyala, C. Fairbridge, J. R. J. Paré, J. M. R. Bélanger, S. Ng and R. Hawkins, Fuel Process. Technol., 2010, 91, 127–135 CrossRef CAS.
- D. A. Jones, T. P. Lelyveld, S. D. Mavrofidis, S. W. Kingman and N. J. Miles, Resour., Conserv. Recycl., 2002, 34, 75–90 CrossRef.
- M. Bhattacharya and T. Basak, Energy, 2016, 97, 306–338 CrossRef.
- Z. Du, Y. Li, X. Wang, Y. Wan, Q. Chen, C. Wang, X. Lin, Y. Liu, P. Chen and R. Ruan, Bioresour. Technol., 2011, 102, 4890–4896 CrossRef CAS PubMed.
- K.-S. Chen, Y.-C. Lin, K.-H. Hsu and H.-K. Wang, Energy, 2012, 38, 151–156 CrossRef CAS.
- B. Günther, K. Gebauer, R. Barkowski, M. Rosenthal and C.-T. Bues, Eur. J. Wood Wood Prod., 2012, 70, 755–757 CrossRef.
- J. Robinson, C. Dodds, A. Stavrinides, S. Kingman, J. Katrib, Z. Wu, J. Medrano and R. Overend, Energy Fuels, 2015, 29, 1701–1709 CrossRef CAS.
- M. Adam, D. Beneroso, J. Katrib, S. Kingman and J. P. Robinson, Biofuels, Bioprod. Biorefin., 2017 DOI:10.1002/bbb.1780.
- L. Estel, M. Poux, N. Benamara and I. Polaert, Chem. Eng. Process., 2016, 113, 56–64 CrossRef.
- J. Sun, W. Wang, Q. Yue, C. Ma, J. Zhang, X. Zhao and Z. Song, Appl. Energy, 2016, 175, 141–157 CrossRef CAS.
- R. Rosa, P. Veronesi and C. Leonelli, Chem. Eng. Process., 2013, 71, 2–18 CrossRef CAS.
- W. Wang, Z. Liu, J. Sun, Q. Ma, C. Ma and Y. Zhang, AIChE J., 2012, 58, 3852–3857 CrossRef CAS.
- J. Sun, W. Wang, C. Zhao, Y. Zhang, C. Ma and Q. Yue, Ind. Eng. Chem. Res., 2014, 53, 2042–2051 CrossRef CAS.
- X. Zhao, W. Wang, H. Liu, C. Ma and Z. Song, Bioresour. Technol., 2014, 158, 278–285 CrossRef CAS PubMed.
- C. O. Kappe, Angew. Chem., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- C. Oliver Kappe, Chem. Soc. Rev., 2008, 37, 1127–1139 RSC.
- M. Hotta, M. Hayashi, M. T. Lanagan, D. K. Agrawal and K. Nagata, ISIJ Int., 2011, 51, 1766–1772 CrossRef CAS.
- J. E. Atwater and R. R. Wheeler Jr, J. Mater. Sci., 2004, 39, 151–157 CrossRef CAS.
- J. E. Atwater and R. R. Wheeler, Carbon, 2003, 41, 1801–1807 CrossRef CAS.
- J. E. Atwater and J. R. R. Wheeler, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 125–129 CrossRef CAS.
- K. H. Wu, T. H. Ting, G. P. Wang, C. C. Yang and C. W. Tsai, Synth. Met., 2008, 158, 688–694 CrossRef CAS.
- C. A. Pickles, Miner. Eng., 2009, 22, 1102–1111 CrossRef CAS.
- J. A. Menéndez, A. Arenillas, B. Fidalgo, Y. Fernández, L. Zubizarreta, E. G. Calvo and J. M. Bermúdez, Fuel Process. Technol., 2010, 91, 1–8 CrossRef.
- M.-q. Chen, J. Wang, M.-x. Zhang, M.-g. Chen, X.-f. Zhu, F.-f. Min and Z.-c. Tan, J. Anal. Appl. Pyrolysis, 2008, 82, 145–150 CrossRef CAS.
- J. Du, P. Liu, Z.-h. Liu, D.-g. Sun and C.-y. Tao, J. Fuel Chem. Technol., 2010, 38, 554–559 CrossRef CAS.
- E. Lester and S. Kingman, Energy Fuels, 2004, 18, 140–147 CrossRef CAS.
- X.-H. Wang, H.-P. Chen, X.-J. Ding, H.-P. Yang, S.-H. Zhang and Y.-Q. Shen, BioResources, 2009, 4, 946–959 CAS.
- X. Zhang, H. Lei, G. Yadavalli, L. Zhu, Y. Wei and Y. Liu, Fuel, 2015, 144, 33–42 CrossRef CAS.
- X. Zhang, H. Lei, L. Zhu, Y. Wei, Y. Liu, G. Yadavalli, D. Yan, J. Wu and S. Chen, Fuel, 2015, 160, 375–385 CrossRef CAS.
- H. Lei, S. Ren and J. Julson, Energy Fuels, 2009, 23, 3254–3261 CrossRef CAS.
- S. Ren, H. Lei, L. Wang, Q. Bu, S. Chen, J. Wu, J. Julson and R. Ruan, Bioresour. Technol., 2013, 135, 659–664 CrossRef CAS PubMed.
- Z. Abubakar, A. A. Salema and F. N. Ani, Bioresour. Technol., 2013, 128, 578–585 CrossRef CAS PubMed.
- B. A. Mohamed, C. S. Kim, N. Ellis and X. Bi, Bioresour. Technol., 2016, 201, 121–132 CrossRef CAS PubMed.
- Z. Zhang, D. J. Macquarrie, M. De bruyn, V. L. Budarin, A. J. Hunt, M. J. Gronnow, J. Fan, P. S. Shuttleworth, J. H. Clark and A. S. Matharu, Green Chem., 2015, 17, 260–270 RSC.
- F. Yu, S. Deng, P. Chen, Y. Liu, Y. Wan, A. Olson, D. Kittelson and R. Ruan, Appl. Biochem. Biotechnol., 2007, 136–140, 957–970 CrossRef PubMed.
- A. Domínguez, J. A. Menéndez, Y. Fernández, J. J. Pis, J. M. V. Nabais, P. J. M. Carrott and M. M. L. R. Carrott, J. Anal. Appl. Pyrolysis, 2007, 79, 128–135 CrossRef.
- M. Miura, H. Kaga, A. Sakurai, T. Kakuchi and K. Takahashi, J. Anal. Appl. Pyrolysis, 2004, 71, 187–199 CrossRef CAS.
- E. Yagmur, M. Ozmak and Z. Aktas, Fuel, 2008, 87, 3278–3285 CrossRef CAS.
- Y. F. Huang, W. H. Kuan, S. L. Lo and C. F. Lin, Bioresour. Technol., 2010, 101, 1968–1973 CrossRef CAS PubMed.
- A. Dominguez, Y. Fernandez, B. Fidalgo, J. J. Pis and J. A. Menendez, Chemosphere, 2008, 70, 397–403 CrossRef CAS PubMed.
- Y. Fernández, A. Arenillas, M. A. Díez, J. J. Pis and J. A. Menéndez, J. Anal. Appl. Pyrolysis, 2009, 84, 145–150 CrossRef.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87–102 CrossRef CAS.
- W. T. Tsai, M. K. Lee and Y. M. Chang, Bioresour. Technol., 2007, 98, 22–28 CrossRef CAS PubMed.
- E. Cetin, R. Gupta and B. Moghtaderi, Fuel, 2005, 84, 1328–1334 CrossRef CAS.
- B. Zhang, Z. Zhong, Q. Xie, S. Liu and R. Ruan, J. Environ. Sci., 2016, 45, 240–247 CrossRef PubMed.
- F. C. Borges, Z. Du, Q. Xie, J. O. Trierweiler, Y. Cheng, Y. Wan, Y. Liu, R. Zhu, X. Lin, P. Chen and R. Ruan, Bioresour. Technol., 2014, 156, 267–274 CrossRef CAS PubMed.
- F. C. Borges, Q. Xie, M. Min, L. A. Muniz, M. Farenzena, J. O. Trierweiler, P. Chen and R. Ruan, Bioresour. Technol., 2014, 166, 518–526 CrossRef CAS PubMed.
- D. Beneroso, J. M. Bermudez, A. Arenillas and J. A. Menendez, Bioresour. Technol., 2013, 144, 240–246 CrossRef CAS PubMed.
- Y.-F. Huang, P.-T. Chiueh, W.-H. Kuan and S.-L. Lo, Energy, 2016, 100, 137–144 CrossRef CAS.
- S. Ren, H. Lei, L. Wang, Q. Bu, S. Chen, J. Wu, J. Julson and R. Ruan, J. Anal. Appl. Pyrolysis, 2012, 94, 163–169 CrossRef CAS.
- Q. Xie, M. Addy, S. Liu, B. Zhang, Y. Cheng, Y. Wan, Y. Li, Y. Liu, X. Lin, P. Chen and R. Ruan, Fuel, 2015, 160, 577–582 CrossRef CAS.
- N. Saifuddin, P. Priatharsini and S. B. Hakim, Am. J. Appl. Sci., 2016, 13, 511–521 CrossRef.
- Y.-F. Huang, C.-H. Shih, P.-T. Chiueh and S.-L. Lo, Energy, 2015, 87, 638–644 CrossRef CAS.
- B. Zhang, Z. Zhong, P. Chen and R. Ruan, Bioresour. Technol., 2015, 197, 79–84 CrossRef CAS PubMed.
- Y.-C. Lin, S.-C. Chen, C.-E. Chen, P.-M. Yang and S.-R. Jhang, Fuel, 2014, 135, 435–442 CrossRef CAS.
- Q. Bu, H. Lei, S. Ren, L. Wang, Q. Zhang, J. Tang and R. Ruan, Bioresour. Technol., 2012, 108, 274–279 CrossRef CAS PubMed.
- X. Zhang, H. Lei, L. Zhu, M. Qian, J. C. Chan, X. Zhu, Y. Liu, G. Yadavalli, D. Yan, L. Wang, Q. Bu, Y. Wei, J. Wu and S. Chen, Catal. Sci. Technol., 2016, 6, 4210–4220 CAS.
- X. Zhang, H. Lei, L. Wang, L. Zhu, Y. Wei, Y. Liu and G. Yadavalli, 2014 ASABE – CSBE/SCGAB Annual International Meeting Paper, 2014, Paper No. 141894632, pp. 1–13 Search PubMed.
- Q. Xie, F. C. Borges, Y. Cheng, Y. Wan, Y. Li, X. Lin, Y. Liu, F. Hussain, P. Chen and R. Ruan, Bioresour. Technol., 2014, 156, 291–296 CrossRef CAS PubMed.
- B. Zhang, G. Tan, Z. Zhong and R. Ruan, J. Anal. Appl. Pyrolysis, 2017, 123, 92–98 CrossRef CAS.
- L. Wang, H. Lei, Q. Bu, S. Ren, Y. Wei, L. Zhu, X. Zhang, Y. Liu, G. Yadavalli, J. Lee, S. Chen and J. Tang, Fuel, 2014, 129, 78–85 CrossRef CAS.
- X. Zhang, H. Lei, L. Wang, L. Zhu, Y. Wei, Y. Liu, G. Yadavalli and D. Yan, Green Chem., 2015, 17, 4029–4036 RSC.
- S. Liu, Y. Zhang, L. Fan, N. Zhou, G. Tian, X. Zhu, Y. Cheng, Y. Wang, Y. Liu, P. Chen and R. Ruan, Fuel, 2017, 196, 261–268 CrossRef CAS.
- X. Zhang, H. Lei, L. Zhu, J. Wu and S. Chen, Green Chem., 2015, 17, 4736–4747 RSC.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- L. Wang, H. Lei, S. Ren, Q. Bu, J. Liang, Y. Wei, Y. Liu, G.-S. J. Lee, S. Chen, J. Tang, Q. Zhang and R. Ruan, J. Anal. Appl. Pyrolysis, 2012, 98, 194–200 CrossRef CAS.
- W. Yunpu, D. A. I. Leilei, F. A. N. Liangliang, S. Shaoqi, L. I. U. Yuhuan and R. Roger, J. Anal. Appl. Pyrolysis, 2016, 119, 104–113 CrossRef.
- W. Yao, J. Li, Y. Feng, W. Wang, X. Zhang, Q. Chen, S. Komarneni and Y. Wang, RSC Adv., 2015, 5, 30485–30494 RSC.
- Y. Xue, A. Kelkar and X. Bai, Fuel, 2016, 166, 227–236 CrossRef CAS.
- Y.-T. Cheng and G. W. Huber, Green Chem., 2012, 14, 3114 RSC.
- C. Dorado, C. A. Mullen and A. A. Boateng, Appl. Catal., B, 2015, 162, 338–345 CrossRef CAS.
- H. Zhang, R. Xiao, J. Nie, B. Jin, S. Shao and G. Xiao, Bioresour. Technol., 2015, 192, 68–74 CrossRef CAS PubMed.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- N. Nikbin, P. T. Do, S. Caratzoulas, R. F. Lobo, P. J. Dauenhauer and D. G. Vlachos, J. Catal., 2013, 297, 35–43 CrossRef CAS.
- H. Zhang, T. R. Carlson, R. Xiao and G. W. Huber, Green Chem., 2012, 14, 98–110 RSC.
- H. Zhang, J. Zheng, R. Xiao, D. Shen, B. Jin, G. Xiao and R. Chen, RSC Adv., 2013, 3, 5769 RSC.
- R. French and S. Czernik, Fuel Process. Technol., 2010, 91, 25–32 CrossRef CAS.
- L. Fan, P. Chen, Y. Zhang, S. Liu, Y. Liu, Y. Wang, L. Dai and R. Ruan, Bioresour. Technol., 2016, 225, 199–205 CrossRef PubMed.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Chem. Eng. J., 2012, 207–208, 27–34 CrossRef CAS.
- M. Artetxe, G. Lopez, G. Elordi, M. Amutio, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2012, 51, 13915–13923 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and G. San Miguel, J. Anal. Appl. Pyrolysis, 2005, 74, 370–378 CrossRef CAS.
- A. Lopez-Urionabarrenechea, I. de Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, J. Anal. Appl. Pyrolysis, 2012, 96, 54–62 CrossRef CAS.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 CAS.
- C. A. Mullen and A. A. Boateng, Fuel Process. Technol., 2010, 91, 1446–1458 CrossRef CAS.
- L. Zhou, Y. Jia, T.-H. Nguyen, A. A. Adesina and Z. Liu, Fuel Process. Technol., 2013, 116, 149–157 CrossRef CAS.
- H. Shang, R.-R. Lu, L. Shang and W.-H. Zhang, Fuel Process. Technol., 2015, 131, 167–174 CrossRef CAS.
- X. Zhao, M. Wang, H. Liu, C. Zhao, C. Ma and Z. Song, J. Anal. Appl. Pyrolysis, 2013, 100, 49–55 CrossRef CAS.
- Y. Wan, P. Chen, B. Zhang, C. Yang, Y. Liu, X. Lin and R. Ruan, J. Anal. Appl. Pyrolysis, 2009, 86, 161–167 CrossRef CAS.
- T. Dickerson and J. Soria, Energies, 2013, 6, 514–538 CrossRef CAS.
- Y. Yu, J. Yu, B. Sun and Z. Yan, J. Anal. Appl. Pyrolysis, 2014, 106, 86–91 CrossRef CAS.
- W. H. Kuan, Y. F. Huang, C. C. Chang and S. L. Lo, Bioresour. Technol., 2013, 146, 324–329 CrossRef CAS PubMed.
- Y. F. Huang, W. H. Kuan, C. C. Chang and Y. M. Tzou, Bioresour. Technol., 2013, 131, 274–280 CrossRef CAS PubMed.
- K. Murata, Y. Liu, M. Inaba and I. Takahara, J. Anal. Appl. Pyrolysis, 2012, 94, 75–82 CrossRef CAS.
- J. Sun and Y. Wang, ACS Catal., 2014, 4, 1078–1090 CrossRef CAS.
- B. Zhang, Z. Zhong, K. Ding and Z. Song, Fuel, 2015, 139, 622–628 CrossRef CAS.
- J. Wang, Z. Zhong, Z. Song, K. Ding and A. Deng, Energy Convers. Manage., 2016, 123, 29–34 CrossRef CAS.
- J. F. Haw, W. Song, D. M. Marcus and J. Nicholas, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS PubMed.
- D. Lesthaeghe, V. S. Speybroeck and M. Waroquier, Phys. Chem. Chem. Phys., 2009, 11, 2794–2798 RSC.
- I. M. Hill, S. A. Hashimi and A. Bhan, J. Catal., 2012, 285, 115–123 CrossRef CAS.
- W.-L. Fanchiang and Y.-C. Lin, Appl. Catal., A, 2012, 419–420, 102–110 CrossRef CAS.
- P. Matias, J. M. Lopes, S. Laforge, P. Magnoux, M. Guisnet and F. Ramôa Ribeiro, Appl. Catal., A, 2008, 351, 174–183 CrossRef CAS.
- A. Galadima and O. Muraza, J. Ind. Eng. Chem., 2015, 31, 1–14 CrossRef CAS.
- B. Zhang, Z. Zhong, Z. Song, K. Ding, P. Chen and R. Ruan, J. Power Sources, 2015, 300, 87–94 CrossRef CAS.
- Q. Bu, H. Lei, S. Ren, L. Wang, J. Holladay, Q. Zhang, J. Tang and R. Ruan, Bioresour. Technol., 2011, 102, 7004–7007 CrossRef CAS PubMed.
- Z. Hu, X. Ma and C. Chen, Bioresour. Technol., 2012, 107, 487–493 CrossRef CAS PubMed.
- Q. Bu, H. Lei, L. Wang, G. Yadavalli, Y. Wei, X. Zhang, L. Zhu and Y. Liu, J. Anal. Appl. Pyrolysis, 2015, 112, 74–79 CrossRef CAS.
- Y. F. Huang, W. H. Kuan, S. L. Lo and C. F. Lin, Bioresour. Technol., 2008, 99, 8252–8258 CrossRef CAS PubMed.
- Y. F. Huang, P. T. Chiueh, W. H. Kuan and S. L. Lo, Bioresour. Technol., 2013, 142, 620–624 CrossRef CAS PubMed.
- H. L. Lei, S. Ren and J. Julson, Energy Fuels, 2009, 23, 3254–3261 CrossRef CAS.
- C. Wu, V. L. Budarin, M. J. Gronnow, M. De Bruyn, J. A. Onwudili, J. H. Clark and P. T. Williams, J. Anal. Appl. Pyrolysis, 2014, 107, 276–283 CrossRef CAS.
- F. Mushtaq, T. A. Abdullah, R. Mat and F. N. Ani, Bioresour. Technol., 2015, 190, 442–450 CrossRef CAS PubMed.
- A. A. Salema and F. N. Ani, Bioresour. Technol., 2012, 125, 102–107 CrossRef CAS PubMed.
- R. Zhou, H. Lei and J. Julson, J. Anal. Appl. Pyrolysis, 2013, 101, 172–176 CrossRef CAS.
- D. V. Suriapparao, N. Pradeep and R. Vinu, Energy Fuels, 2015, 29, 2571–2581 CrossRef CAS.
- M. Bartoli, L. Rosi, A. Giovannelli, P. Frediani and M. Frediani, J. Anal. Appl. Pyrolysis, 2016, 122, 479–489 CrossRef CAS.
- D. C. Elliott, T. R. Hart, A. J. Schmidt, G. G. Neuenschwander, L. J. Rotness, M. V. Olarte, A. H. Zacher, K. O. Albrecht, R. T. Hallen and J. E. Holladay, Algal Res., 2013, 2, 445–454 CrossRef.
- W. T. Chen, Y. Zhang, J. Zhang, G. Yu, L. C. Schideman, P. Zhang and M. Minarick, Bioresour. Technol., 2014, 152, 130–139 CrossRef CAS PubMed.
- X. Wang, H. Chen, K. Luo, J. Shao and H. Yang, Energy Fuels, 2008, 22, 67–74 CrossRef CAS.
- V. L. Budarin, J. H. Clark, B. A. Lanigan, P. Shuttleworth, S. W. Breeden, A. J. Wilson, D. J. Macquarrie, K. Milkowski, J. Jones, T. Bridgeman and A. Ross, Bioresour. Technol., 2009, 100, 6064–6068 CrossRef CAS PubMed.
- M. A. Hossain, J. Jewaratnam, P. Ganesan, J. N. Sahu, S. Ramesh and S. C. Poh, Energy Convers. Manage., 2016, 115, 232–243 CrossRef CAS.
- Y.-C. Lin, T.-Y. Wu, W.-Y. Liu and Y.-H. Hsiao, Fuel, 2014, 119, 21–26 CrossRef CAS.
- J. M. Bermúdez, M. Francavilla, E. G. Calvo, A. Arenillas, M. Franchi, J. A. Menéndez and R. Luque, RSC Adv., 2014, 4, 38144 RSC.
- S. Ren, H. Lei, L. Wang, Q. Bu, S. Chen and J. Wu, RSC Adv., 2014, 4, 10731 RSC.
- A. Mamaeva, A. Tahmasebi, L. Tian and J. Yu, Bioresour. Technol., 2016, 211, 382–389 CrossRef CAS PubMed.
- L. Dai, L. Fan, Y. Liu, R. Ruan, Y. Wang, Y. Zhou, Y. Zhao and Z. Yu, Bioresour. Technol., 2016, 225, 1–8 CrossRef PubMed.
- R. Zhang, L. Li, D. Tong and C. Hu, Bioresour. Technol., 2016, 212, 311–317 CrossRef CAS PubMed.
- Q. Bu, H. Lei, L. Wang, Y. Wei, L. Zhu, Y. Liu, J. Liang and J. Tang, Bioresour. Technol., 2013, 142, 546–552 CrossRef CAS PubMed.
- S. Zhang, Q. Dong, L. Zhang and Y. Xiong, Bioresour. Technol., 2015, 191, 17–23 CrossRef CAS PubMed.
- N. M. Mubarak, A. Kundu, J. N. Sahu, E. C. Abdullah and N. S. Jayakumar, Biomass Bioenergy, 2014, 61, 265–275 CrossRef CAS.
- N. M. Mubarak, J. N. Sahu, E. C. Abdullah and N. S. Jayakumar, J. Anal. Appl. Pyrolysis, 2016, 120, 521–528 CrossRef CAS.
- M. N. Noraini, E. C. Abdullah, R. Othman and N. M. Mubarak, Mater. Lett., 2016, 184, 315–319 CrossRef CAS.
- B. A. Mohamed, N. Ellis, C. S. Kim, X. Bi and R. Emam Ael, Sci. Total Environ., 2016, 566–567, 387–397 CrossRef CAS PubMed.
- Y. Wang, L. Dai, L. Fan, D. Duan, Y. Liu, R. Ruan, Z. Yu, Y. Liu and L. Jiang, J. Anal. Appl. Pyrolysis, 2017, 123, 224–228 CrossRef CAS.
- Q. Dong and Y. Xiong, Bioresour. Technol., 2014, 171, 127–131 CrossRef CAS PubMed.
- A. Dominguez, J. A. Menendez, M. Inguanzo and J. J. Pis, Bioresour. Technol., 2006, 97, 1185–1193 CrossRef CAS PubMed.
- A. Marcilla, L. Catalá, J. C. García-Quesada, F. J. Valdés and M. R. Hernández, Renewable Sustainable Energy Rev., 2013, 27, 11–19 CrossRef CAS.
- M. Stals, R. Carleer, G. Reggers, S. Schreurs and J. Yperman, J. Anal. Appl. Pyrolysis, 2010, 89, 22–29 CrossRef CAS.
- H. Lei, https://labs.wsu.edu/lei/documents/2016/09/processes-biofuels-and-bioproducts.ppt, 2016.
- L. Estel, M. Poux, N. Benamara and I. Polaert, Chem. Eng. Process., 2017, 113, 56–64 CrossRef CAS.
- D. Beneroso, T. Monti, E. T. Kostas and J. Robinson, Chem. Eng. J., 2017, 316, 481–498 CrossRef CAS.
- D. Beneroso, A. Albero-Ortiz, J. Monzó-Cabrera, A. Díaz-Morcillo, A. Arenillas and J. A. Menéndez, Fuel, 2016, 172, 146–152 CrossRef CAS.
- S. Karnjanakom, A. Bayu, X. Hao, S. Kongparakul, C. Samart, A. Abudula and G. Guan, J. Mol. Catal. A: Chem., 2016, 421, 235–244 CrossRef CAS.
- E. R. Umeki, C. F. de Oliveira, R. B. Torres and R. G. d. Santos, Fuel, 2016, 185, 236–242 CrossRef CAS.
- W. B. Widayatno, G. Guan, J. Rizkiana, J. Yang, X. Hao, A. Tsutsumi and A. Abudula, Appl. Catal., B, 2016, 186, 166–172 CrossRef CAS.
- Q. Bu, H. M. Morgan Jr, J. Liang, H. Lei and R. Ruan, Advances in Bioenergy, 2016, vol. 1, pp. 69–123 Search PubMed.
- S. S. Lam, A. D. Russell, C. L. Lee and H. A. Chase, Fuel, 2012, 92, 327–339 CrossRef CAS.
- J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794 RSC.
- D. R. Godwin, S. J. Lawton, J. D. Moseley, M. J. Welham and N. P. Weston, Energy Fuels, 2010, 24, 5446–5453 CrossRef CAS.
- J. P. Robinson, S. W. Kingman, C. E. Snape and H. Shang, 2007.
- J. P. Robinson, S. W. Kingman, R. Barranco, C. E. Snape and H. Al-Sayegh, Ind. Eng. Chem. Res., 2010, 49, 459–463 CrossRef CAS.
- A. Bridgwater, J. Anal. Appl. Pyrolysis, 1999, 51, 3–22 CrossRef CAS.
- A. Demirbas, Fuel Process. Technol., 2007, 88, 591–597 CrossRef CAS.
- A. Anca-Couce, Prog. Energy Combust. Sci., 2016, 53, 41–79 CrossRef.
- R. Ruan, P. Chen, R. Hemmingsen, V. Morey and D. Tiffany, Int. J. Agric. Biol. Eng., 2008, 1, 64–68 Search PubMed.
- J. P. Robinson, S. W. Kingman, C. E. Snape, S. M. Bradshaw, M. S. A. Bradley, H. Shang and R. Barranco, Chem. Eng. Res. Des., 2010, 88, 146–154 CrossRef CAS.
- L. Wang, H. Lei and R. Ruan, Production of Biofuels and Chemicals with Microwave, 2015, pp. 251–263 Search PubMed.
- C. Xin, M. M. Addy, J. Zhao, Y. Cheng, S. Cheng, D. Mu, Y. Liu, R. Ding, P. Chen and R. Ruan, Bioresour. Technol., 2016, 211, 584–593 CrossRef CAS PubMed.
- R. Xing, A. V. Subrahmanyam, H. Olcay, W. Qi, G. P. van Walsum, H. Pendse and G. W. Huber, Green Chem., 2010, 12, 1933 RSC.
- L. M. Balster, E. Corporan, M. J. DeWitt, J. T. Edwards, J. S. Ervin, J. L. Graham, S.-Y. Lee, S. Pal, D. K. Phelps, L. R. Rudnick, R. J. Santoro, H. H. Schobert, L. M. Shafer, R. C. Striebich, Z. J. West, G. R. Wilson, R. Woodward and S. Zabarnick, Fuel Process. Technol., 2008, 89, 364–378 CrossRef CAS.
- H. A. Meylemans, R. L. Quintana, B. R. Goldsmith and B. G. Harvey, ChemSusChem, 2011, 4, 465–469 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- X. Zhang and H. Lei, RSC Adv., 2016, 6, 6154–6163 RSC.
- C. J. Chuck and J. Donnelly, Appl. Energy, 2014, 118, 83–91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |