
Direct synthesis of a carbon nanotube interpenetrated doped porous carbon alloy as a durable Pt-free electrocatalyst for the oxygen reduction reaction in an alkaline medium†
Sreekuttan M.
Unni
 a,
Gopinathan M.
Anilkumar
bd,
Masashi
Matsumoto
cd,
Takanori
Tamaki
ad,
Hideto
Imai
cd and
Takeo
Yamaguchi
*ad
a,
Gopinathan M.
Anilkumar
bd,
Masashi
Matsumoto
cd,
Takanori
Tamaki
ad,
Hideto
Imai
cd and
Takeo
Yamaguchi
*ad
aLaboratory for Chemistry and Life Sciences, Tokyo Institute of Technology, R1-17, 4259 Nagatsuta, Midori-ku, Yokohama 226-850, Japan. E-mail: yamag@res.titech.ac.jp
bR&D Centre, Noritake Co., Ltd., 300 Higashiyama, Miyochi-cho, Miyoshi 470-0293, Japan
cDevice Fuctional Analysis Department, Nissan Arc, Ltd., Yokosuka, Kanagawa 237-0061, Japan
dCore Research for Evolutionary Science and Technology, Japan Science and Technology Agency (JST-CREST), Japan
First published on 21st June 2017
Abstract
Direct synthesis of highly durable carbon nanotube interpenetrated porous carbon alloy electrocatalysts for the oxygen reduction reaction (ORR) from a single precursor, trimetallic zeolitic imidazole framework (t-ZIF), is reported. The use of a single precursor improves the uniform distribution of active reaction centres which is crucial for ORR catalysts. The t-ZIF has Fe, Co and Zn metal centres and 2-methylimidazole as a ligand. Carbonisation of the t-ZIF under an inert atmosphere produces nitrogen and Fe/Co–Nx doped carbon/carbon nanotubes alloyed with metal/metal oxide particles encased inside the carbon structures (FeCo-NCZ). The presence of Zn in the t-ZIF induces porosity in carbon during the carbonisation process. The peculiar morphology with a reasonably high surface area provides efficient mass transport and interpenetrated carbon nanotube assisted fast electron transport in the catalyst. X-ray photoelectron spectroscopy reveals that FeCo-NCZ is enriched with different possible active reaction centres such as pyridinic, graphitic and Fe/Co–Nx type nitrogen coordination on the catalyst surface. The ORR activity of FeCo-NCZ in oxygen saturated 0.1 M KOH was comparable to/higher than that of the reference Pt/C catalyst. The displayed onset potential (1.04 V vs. the RHE) and half-wave potential (0.91 V vs. the RHE) of FeCo-NCZ are more positive compared to those of Pt/C and other control-samples. It is noteworthy that the dioxygen reduction kinetics of FeCo-NCZ are comparable to those of Pt/C as evident from the Tafel slope and oxygen reduction follows a four electron pathway. More interestingly, FeCo-NCZ shows better fuel tolerance and electrochemical stability even at 60 °C compared to Pt/C under alkaline conditions.
Introduction
Development of Pt-free electrocatalysts for the oxygen reduction reaction (ORR) is an emerging research area for cost-effective low-temperature fuel cell and metal–air batteries.1,2 Among the various electrocatalysts being developed, low-cost metal (Fe, Co, etc.) and non-metal (N, S, B, P, etc.) doped carbon morphologies gained more attention as electrocatalysts for the oxygen reduction reaction (ORR).3–5 Since the active reaction site density on the doped carbon plays a major role in the efficient reduction of oxygen, many synthetic strategies are being adopted to improve its active site densities.4,6–8 The general method for the catalyst preparation comprises the high-temperature treatment of the mixture of carbon, metal precursor and heteroatom sources.9 However, such a method of preparation leads to the fabrication of electrocatalysts with an unequal distribution of active centres on them.10 This may ultimately be reflected in the electrocatalytic activity towards the ORR and electrochemical stability of electrocatalysts. Carbonisation of a single precursor which contains all the prerequisites in a preferred chemically linked ordered structure will be an efficient strategy for the development of electrocatalysts with uniform active site density on them.11Metal organic framework (MOF) materials have recently gained great interest, as a precursor to the development of ORR electrocatalysts due to their peculiar morphology, high surface area, well-defined pore structure, and preferred metal–ligand interaction.12–14 Among different MOFs, zeolitic imidazole frameworks (ZIFs) are the most studied candidate for the development of Pt-free ORR catalysts.14–20 Dodelet et al., used ZIF-8 (zinc based imidazole framework) as the carbon and nitrogen source for the electrocatalyst preparation.21 There are reports on the synthesis of electrocatalysts from ZIF-67 (cobalt-based imidazole framework).22 However, additional metal or heteroatom sources are necessary to improve the ORR activity of derived carbon.19 Furthermore, David Lou et al. reported a variety of modifications on ZIF-8 and ZIF-67 for different applications including the ORR.23–27 Later, bimetallic ZIFs (both Zn and Co as metal centres) were introduced to improve the ORR activity.28–33 High-temperature annealing of such bimetallic ZIFs results in improved activity under alkaline or acidic conditions towards the ORR.34 During high-temperature annealing, Zn can evaporate and generate porosity in the final carbon.28 This will lead to the improvement in the active reaction site density. Synthesis of such monometallic and bimetallic ZIFs usually follows a traditional time consuming, low yield synthesis process and impedes bulk synthesis of the electrocatalysts. Recently, Gross et al. synthesised ZIF-8 and ZIF-67 through a low-temperature aqueous phase approach and produced ZIFs with high yields.35 However, the carbonised product from such ZIFs showed poor ORR activity in alkaline media.36 Therefore, an appropriate synthetic strategy is required to achieve tailored carbon alloy catalysts with higher/better electrochemical properties.
The fast synthesis of ZIF-8/ZIF-67 through the aqueous room temperature process produces nanometre-sized ZIFs and can accommodate foreign metal ions which makes the formation of ZIFs practically difficult. Thus, adopting a fast room temperature aqueous synthesis procedure of ZIFs can deliver the following benefits: (1) combination of different metal centres, (2) high yields and (3) nano-metre sized particles. Shui et al. reported that the preferred iron-based coordination compound is necessary to tune ZIF-8 derived catalysts to exhibit better ORR activity.37,38 This can be easily achieved by adopting a fast aqueous synthesis approach where the ZIF can efficiently host the in situ formed metal–ligand complex.39 Here, we report the synthesis of carbon electrocatalysts from the Zn, Fe and Co based trimetallic ZIF (t-ZIF) using the fast synthesis procedure. According to Zelenay et al., the combination of Co and Fe in the non-Pt catalysts improves the electrochemical reduction of dioxygen.40 So the carbonisation of the t-ZIF can lead to such preferred coordinations of both iron and cobalt. By adopting the fast synthesis procedure, iron metal ions coordinated with ligand molecules during the synthesis of the ZIF can occupy the pores of the ZIF and deliver efficient active reaction centres without any further addition of iron coordinated molecules. Moreover, the presence of zinc will assist in situ formation of pores during high-temperature annealing through its evaporation at high temperatures. The t-ZIF derived carbon catalyst (FeCo-NCZ) has a morphology of carbon nanotube interpenetrated porous carbon. It shows improved ORR activity in an alkaline medium which is comparable to that of commercial Pt/C.
Results and discussion
Trimetallic (Zn, Co and Fe) ZIF was prepared using the procedure reported by Gross et al.35 Aqueous solutions of the corresponding metal salts and imidazole/triethylamine solution were mixed and stirred for 30 min to prepare the t-ZIF. Carbonisation of the as-made t-ZIF was carried out at high temperatures in a N2 atmosphere. After the carbonisation process, to remove surface metal oxide impurities, the carbon was further subjected to acid wash. A second annealing at 910 °C was performed on the acid washed carbon to get the final catalyst (Scheme 1) (the detailed experimental procedure is given in the Experimental section). The as-made t-ZIF (from precursors of Co, Fe and Zn) was named FeCoZn-ZIF, and the carbon alloy catalyst derived from the above ZIF was denoted as FeCo-NCZ. Control samples were also prepared by changing the metal content during ZIF synthesis. The carbon alloy derived from Co-ZIF (ZIF-containing only Co), CoZn-ZIF (ZIF-containing Co and Zn) and FeZn-ZIF (ZIF-containing Fe and Zn) were named Co-NC, Co-NCZ and Fe-NCZ, respectively.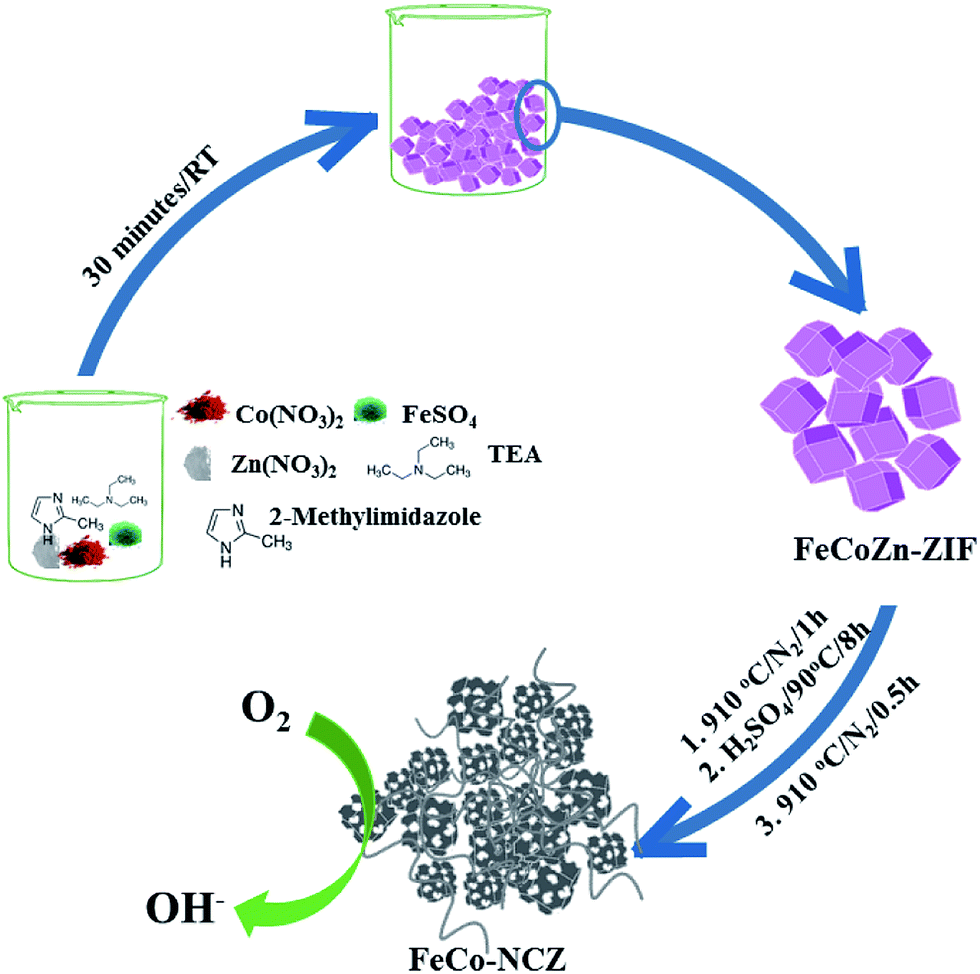 | ||
| Scheme 1 Schematic representation of the synthesis of carbon nanotube interpenetrated porous carbon alloy (FeCo-NCZ) electrocatalysts. | ||
X-ray diffraction (XRD) patterns (Fig. 1(a)) of Co-ZIF and CoZn-ZIF show similar peaks, indicating that the structural feature in the ZIF is the same. The XRD peaks of FeCoZn-ZIF and FeZn-ZIF show similar features to those of Co-ZIF and CoZn-ZIF. This is a clear indication that the addition of iron is not affecting the crystalline features of Co-ZIF and CoZn-ZIF. During the ZIF synthesis, the imidazole coordinated iron is well packed in the available pores of the ZIF without altering its crystalline nature. The XRD patterns reveal a sodalite type crystal structure for all prepared ZIFs.35 After high-temperature annealing, Co-NC and Co-NCZ show diffraction peaks corresponding to those of metallic cobalt, carbide and oxide phases of cobalt (Fig. 1(b)). The peak at 2θ ∼ 26° corresponds to the (002) plane of graphitic carbon. The peaks at 2θ ∼ 31.2, 36.9, 59.5 and 65.4° indicate the presence of the crystalline Co3O4 phase of cobalt.41,42 The peaks at 2θ ∼ 43.46° and 44.3° correspond to the carbide and metallic form of cobalt, respectively. The XRD pattern of Fe-NCZ also shows the presence of oxide (2θ ∼ 30.1, 35.4, 57.3, and 62.9° for Fe3O4), carbide (2θ ∼ 43.3°) and metallic iron (2θ ∼ 44.8°) phases, along with the (002) plane of graphitic carbon.43 More interestingly, the XRD pattern of FeCo-NCZ displays a more intense peak of the metallic phases of Fe/Co (2θ ∼ 44.8°). But the peak intensities of oxide phases of Co (2θ ∼ 65.4°) and Fe (2θ ∼ 35.4, 57.3, and 62.9°) are lower compared to those of metallic phases. This indicates the presence of more crystalline metallic forms of Fe/Co in the FeCo-NCZ.
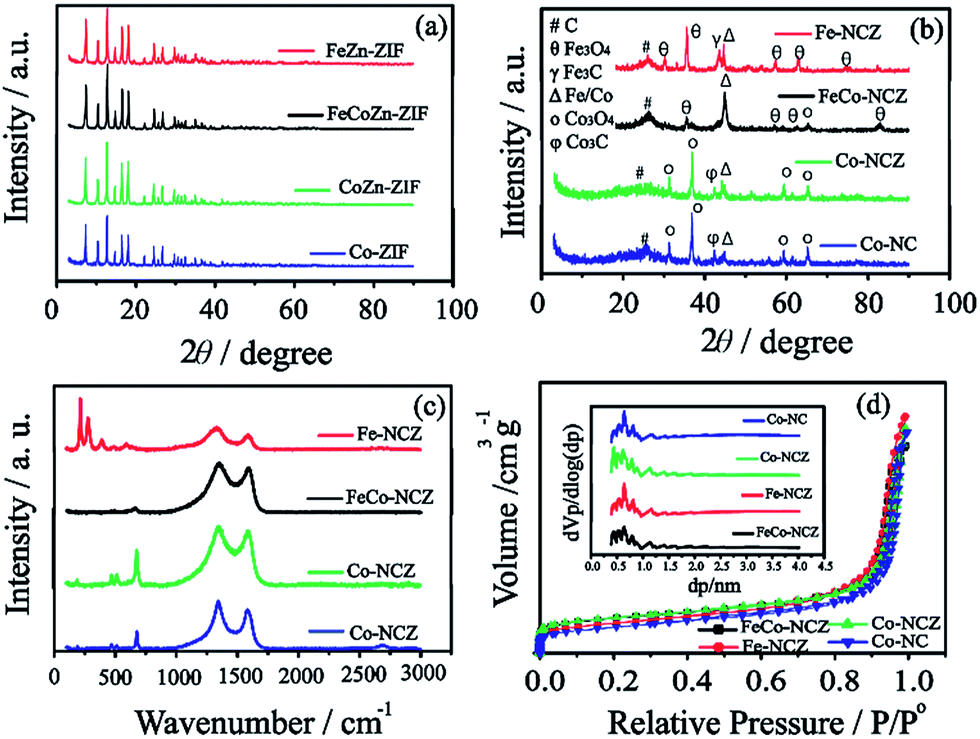 | ||
| Fig. 1 XRD patterns of the prepared ZIFs (a) and ZIF derived carbon (b), (c) Raman spectra of ZIF derived catalysts and (d) nitrogen adsorption–desorption isotherm of different carbon catalysts (inset: the pore size distribution of different carbon catalysts). | ||
Raman spectra of the annealed ZIF show (Fig. 1c) two dominant peaks at 1339.54 and 1585.30 cm−1 corresponding to D and G bands of the carbon. The G band represents the graphitic form of carbon (graphitic lattice vibrational mode with E2g symmetry), and the D band represents the disordered graphite (graphitic lattice vibration mode with A1g symmetry) form of carbon.44 In the annealed catalyst, the D band intensity is higher compared to that of the G band which indicates the presence of more disordered carbon in the annealed catalyst. The graphitization degree was calculated using the intensity ratio of D and G bands. The ID/IG ratio of the catalyst is in the order of Fe-NCZ (1.55) > Co-NC (1.21) > Co-NCZ (1.15) > FeCo-NCZ (1.11). This clearly indicates that the FeCo-NCZ has a better graphitic phase compared to the other prepared catalyst systems. The ID/IG ratio assists in calculating the in-plane crystallite size (La) of the annealed carbon.45 The La of the carbon catalyst is in the order of FeCo-NCZ (17.3 nm) > Co-NCZ (16.7 nm) > Co-NC (15.9 nm) > Fe-NCZ (12.4 nm). The improved crystallite size of FeCo-NCZ represents the reduced resistivity, which is one of the prerequisites for an efficient electrocatalyst. The in situ formed carbon nanotubes in FeCo-NCZ also contribute to the enhancement of the graphitization and lead to better electrical conductivity of the material.46,47 Apart from G and D bands, the peaks corresponding to the transverse and longitudinal optical phonon mode vibration of the oxides of Co (for Co-NCZ and Co-NC) and Fe (for Fe-NCZ) are observed in between 150 and 750 cm−1. This indicates the presence of the oxides of cobalt and iron on the surface of annealed catalysts.48 The less intense peaks of the oxides of cobalt and iron in the case of FeCo-NCZ suggest that these nanoparticles are well encased in the carbon layers.
The surface area of the prepared electrocatalyst was analysed using nitrogen adsorption–desorption isotherms using the Brunauer–Emmet–Teller (BET) method at 77 K. Nitrogen adsorption–desorption isotherms of electrocatalysts resemble a type I isotherm with more microporous features (Fig. 1(d)). The surface area of the electrocatalysts calculated from BET is in the order of Co-NCZ (470 m2 g−1) > FeCo-NCZ (463 m2 g−1) > Fe-NCZ (393 m2 g−1) > Co-NC (340 m2 g−1). Co-NCZ and FeCo-NCZ show almost similar surface areas. As expected, the Co-NC has a lower surface area as compared to the other ZIF derived carbon. A slight decrease in the surface area of Fe-NCZ in comparison to those of FeCo-NCZ and Co-NCZ is mainly due to the difference in the nature of carbon derived from the FeZn-ZIF compared to the carbon from FeCoZn-ZIF and CoZn-ZIF.49 The pore-size distribution measured using the Horvath–Kawazoe (HK) method shows that the pore diameter of all catalysts is in between 0.5 and 1 nm (inset of Fig. 1(d)). The pore volume of the electrocatalyst (calculated using the BJH method) is in the order of Fe-NCZ (1.24 cm3 g−1) > Co-NC (1.15 cm3 g−1) > Co-NCZ (1.13 cm3 g−1) > FeCo-NCZ (1.05 cm3 g−1). Since micropores play a major role towards the improvement in the active reaction site density, the micropore surface area was calculated. FeCo-NCZ (262 m2 g−1) and Co-NCZ (280 m2 g−1) display high micropore surface areas compared to Fe-NCZ and Co-NCZ.
Scanning electron microscopy (SEM) images of FeCoZn-ZIF and Co-ZIF show similar morphological features with an average particle size of 60 nm (Fig. S1†). However, after carbonisation, morphological features of the ZIFs collapsed. More evidence about the morphology of annealed carbon from different ZIFs was obtained from transmission electron microscopy images (TEM). As seen in Fig. 2, the FeCo-NCZ has a morphology of carbon nanotube interpenetrated porous carbon with metal/metal oxide particles embedded in it. The porosity can be created during the acid leaching of metal/metal oxide nanoparticles in addition to the evaporation of Zn from the ZIF. Disordered carbon fringes on the carbon mass clearly indicate the effective doping of heteroatoms.50 The metal/metal oxide nanoparticles are well encased in the carbon shells. The nanometer thick graphene type carbon shell around the metal/metal oxide nanoparticles prevents them from leaching out during the acid washing process. As reported by Gewirth et al.,51 these metal nanoparticles are also capable of activating the doped carbon which covered the nanoparticles to act as an active centre for oxygen reduction. This possibility is theoretically explained by Deng et al.52
 | ||
| Fig. 2 TEM images of FeCo-NCZ at different magnifications ((a) 100 nm, (b) 20 nm, and (c) 5 nm). (d) STEM image of FeCo-NCZ and (e) represents the elemental mapping of FeCo-NCZ. | ||
Along with the porous carbon mass, TEM images also reveal several micrometre long bamboo shaped carbon nanotubes, which are uniformly distributed throughout the carbon. Since Fe catalyses the nanotube formation from small organic molecules, the nanotubes in the present case are formed from the small organic molecules derived from the ZIF at high temperatures.43 The uniform distribution of nanotubes in the carbon matrix further improves the effective electron transport through it. The elemental mapping (Fig. 2e) clearly shows that all elements are well dispersed in the carbon matrix of FeCo-NCZ. The mapping of Fe and Co clearly depicts the uniform distribution over the surface, and the positions of these metal centres are exactly matching on the carbon matrix resembling an alloy type feature. The uniform distribution of these metal centres on the carbon surface assists the formation of improved active reaction centres and makes FeCo-NCZ a better electrocatalyst for the ORR. Similar morphological features were also observed for the Fe-NCZ (Fig. S2†). However, the morphology of Fe-NCZ resembles a densely packed carbon mass and carbon nanotubes. This may be the reason for the reduced surface area of Fe-NCZ compared to those of FeCo-NCZ and Co-NCZ. The HRTEM images of Co-NC (Fig. S3†) and Co-NCZ (Fig. S4†) show the porous nature of carbon morphology with nanoparticles dispersed in them. More interestingly, the bamboo shaped nanotubes were absent in these carbon structures. This indicates that the Co particles in the present case are not producing carbon nanotubes during the carbonisation process. Interestingly, most of the nanoparticles are well encased in the carbon shells.
Different elemental compositions and their chemical environment were analysed using X-ray photoelectron spectroscopy (XPS). All annealed ZIF samples show the presence of carbon, nitrogen, and oxygen. Cobalt is present in FeCo-NCZ, Co-NCZ and Co-NC. XPS demonstrates the presence of iron in FeCo-NCZ and Fe-NCZ. More interestingly, FeCo-NCZ and Co-NCZ show trace amounts of zinc in them. The existence of residual zinc indicates incomplete evaporation. The deconvoluted XPS spectrum of Zn 2p (Fig. S5†) shows the presence of three different types of zinc in FeCo-NCZ and Co-NCZ. They are Zn–O (1021.73 eV), Zn–N (1022.97 eV), and metallic Zn (1020.50 eV).53 Atomic percentages (at%) of all elements present in the prepared electrocatalysts are given in Table S1.† The deconvoluted N 1s spectra (Fig. 3) of all catalysts display six different types of nitrogen coordination such as pyridinic (∼398.22 eV), M–Nx (∼399.30 eV), pyrrolic (400.30 eV), graphitic (401.10 eV), pyridine N-oxide (∼402.61 eV) and π–π* shake-up/N2 (∼403.90 eV).54 The pyridinic and graphitic nitrogen coordination in doped carbon materials improves the reduction of oxygen by improving the onset potential and reduction current.55,56 The at% of pyridinic nitrogen in the catalyst is in the order of Co-NCZ (41.4%) > FeCo-NCZ (41.0%) > Fe-NCZ (34.4%) > Co-NC (29.3%). FeCo-NCZ and Co-NCZ have comparable amounts of pyridinic nitrogen. Similarly, at% of the graphitic nitrogen is in the order of Co-NC (25.0%) > Fe-NCZ (23.6%) > FeCo-NCZ (20.9%) > Co-NCZ (14.3%). However, the sum of both pyridinic and graphitic nitrogen contents is higher for FeCo-NCZ and is expected to improve the reaction site density. The companied effect of both coordinations is reflected on the enhancement of the ORR of FeCo-NCZ in comparison with other catalysts. The M–Nx interactions in Co-NCZ and Co-NC are found to be 15.7 and 17.8 at% respectively. Since a trace amount of Zn is also present in the Co-NCZ, Zn–Nx can also contribute to the total M–Nx of Co-NCZ. The Fe–Nx interaction in Fe-CNZ is calculated to be 16.6 at%. The at% of nitrogen in M–Nx type interaction in FeCo-NCZ is 11.62. The ratio of surface nitrogen to carbon (N/C) is higher for the FeCo-NCZ. The N/C ratio of the electrocatalyst is in the order of FeCo-NCZ (0.065) > Co-NCZ (0.061) > Fe-NCZ (0.057) > Co-NC (0.023). The higher N/C ratio of FeCo-NCZ indicates that the surface of FeCo-NCZ is enriched with preferred nitrogen coordination to improve the active reaction site density. The at% of different nitrogen coordinations is given in Table 1.
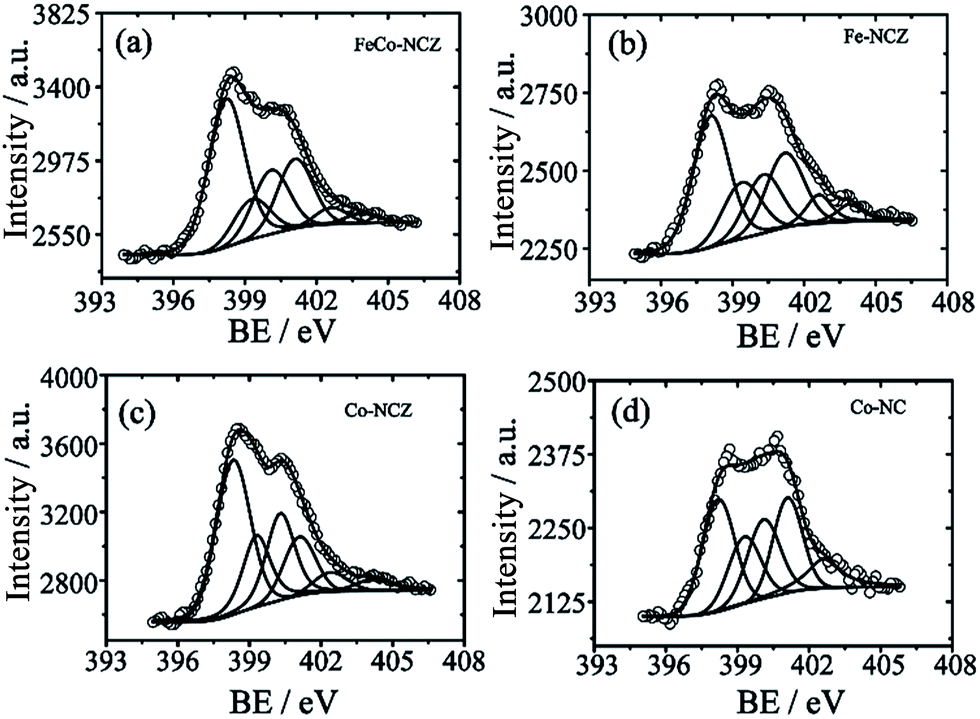 | ||
| Fig. 3 Deconvoluted XPS N 1s spectra of (a) FeCo-NCZ, (b) Fe-NCZ, (c) Co-NCZ and (d) Co-NC. | ||
| FeCo-NCZ | Fe-NCZ | Co-NCZ | Co-NC | |
|---|---|---|---|---|
| Pyridinic | 41.0 | 34.4 | 41.4 | 29.3 |
| M–Nx | 11.7 | 16.6 | 15.7 | 17.8 |
| Pyrrolic | 20.3 | 17.2 | 21.3 | 22.7 |
| Graphitic | 20.9 | 23.6 | 14.4 | 25.0 |
| Pyridine N-oxide | 3.8 | 5.5 | 4.9 | 5.2 |
| π–π* shake-up/N2 | 2.3 | 2.7 | 2.3 | 0.0 |
| Total nitrogen content | 5.65 | 3.32 | 5.22 | 2.10 |
The deconvoluted XPS spectrum of cobalt (Fig. S6†) shows the presence of metallic Co (778.39 eV), Co–Nx (783.93 eV) and oxide of cobalt (779.83 eV) in Co-NC, Co-NCZ and FeCo-NCZ.57 Among these three different coordinations, Co–Nx plays a crucial role as the ORR active centre.58 The at% of cobalt in Co–Nx type coordination is almost similar (25.6%) in FeCo-NCZ, Co-NCZ and Co-NC. The uniform distribution of these Co metal centres may alter the physical properties associated with the ORR of the electrocatalysts. Similarly, the deconvoluted Fe 2p spectrum also indicates the presence of Feo, FeII, and FeIII forms of iron. In Fig. S7,† the peaks at 710.10 and 711.92 eV correspond to the 2p3/2 orbitals of Fe(II) and Fe(III), respectively. The peaks at 723.57 and 725.81 eV correspond to 2p1/2 orbitals of Fe(II) and Fe(III), respectively.59 Fitted Fe 2p spectra of Fe-CNZ also show similar kinds of peaks. Two additional peaks at 707.72 and 714.46 eV corresponding to FeO and Fe–Nx respectively were also observed in FeCo-NCZ and Fe-NCZ.43 Among different iron coordinations, Fe–Nx contributes as an active centre to improve the ORR activity of the electrocatalysts.60 XPS analysis provides clear evidence that all the preferred coordinations which can enhance the electrocatalytic reduction of oxygen are available for FeCo-NCZ. Deconvoluted C 1s spectra (Fig. S8†) of all synthesised electrocatalysts show four distinct peaks corresponding to binding energies of 284.3 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 285.4 eV (C–N), 286.7 eV (C–O), and 288.5 (CO).61
C), 285.4 eV (C–N), 286.7 eV (C–O), and 288.5 (CO).61
The electrocatalytic activity of the carbon alloy catalysts towards the reduction of dioxygen molecules was analysed using the linear sweep voltammetry (LSV) technique in oxygen saturated 0.1 M KOH at a scan rate of 10 mV s−1. Hg/HgO and Pt were used as a reference electrode and counter electrode, respectively. Measured potential is represented in the reversible hydrogen electrode (RHE) by calibrating Hg/HgO in hydrogen saturated 0.1 M KOH (Fig. S9†). Fig. 4a shows the LSV of the electrocatalyst. From the figure, it is clear that FeCo-NCZ displays improved ORR activity compared to Pt/C and other control-samples. FeCo-NCZ shows an onset potential of 1.04 V and a half-wave potential at 0.91 V. More interestingly, Pt/C experiences 10 mV higher overpotential compared to FeCo-NCZ in onset potential and half-wave potential. Among different electrocatalysts synthesised, Co-NC displays poor ORR activity in terms of onset (0.92 V) and half-wave potential (0.82 V). Fe-NCZ and Co-NCZ display almost a similar performance and Co-NCZ displays an overpotential of 10 mV in half-wave potential compared to Fe-NCZ. Fe-NCZ and Co-NCZ display an overpotential of 50 mV and 60 mV respectively in half-wave potential compared to FeCo-NCZ. The Tafel plot (the plot of E vs. log|−jk|) of the FeCo-NCZ and Pt/C shows a Tafel slope of 87.4 mV per decade and 84.9 mV per decades respectively (Fig. 4b). This indicates that the dioxygen reduction mechanism of both Pt/C and FeCo-NCZ is almost similar in 0.1 M KOH solution. Furthermore, the mass activity of the FeCo-NCZ and Pt/C (mass of both carbon and Pt) was measured at the potential of 0.95 V, and it is found that the mass activity of FeCo-NCZ is 1.6 times lower compared to that of Pt/C. Further modification in the catalyst to improve its mass activity is one of the ongoing research studies in our group.
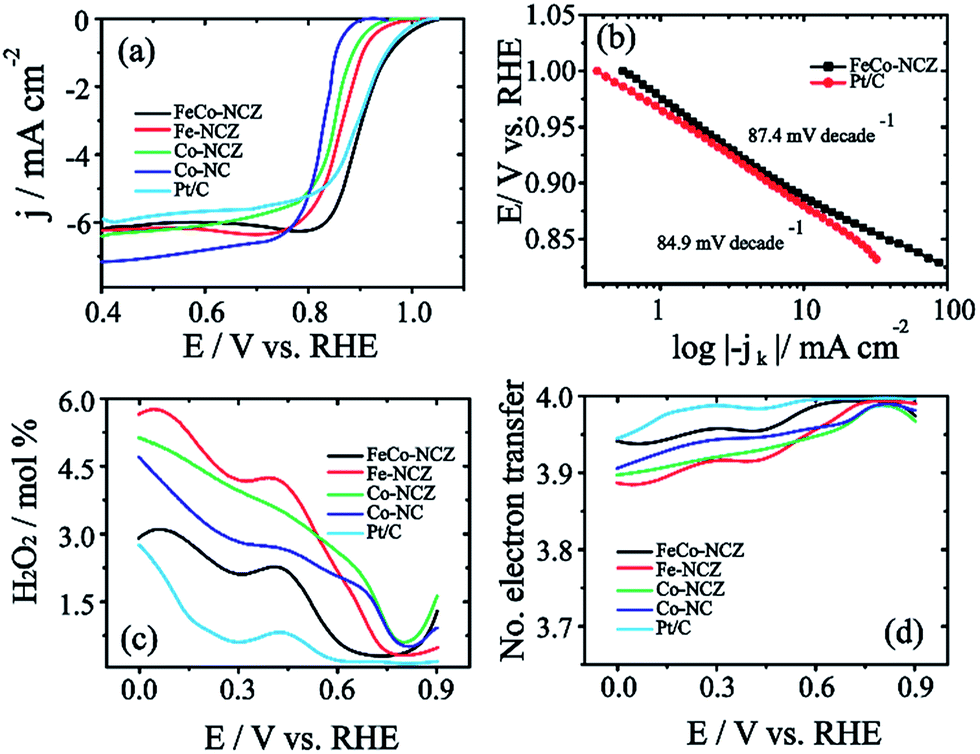 | ||
| Fig. 4 (a) Linear sweep voltammograms (LSV) of all electrocatalysts in oxygen saturated 0.1 M KOH at an electrode rotation rate of 1600 rpm with a scan rate of 10 mV s−1. The anodic scan is used for the measurement of voltammograms. The disc current is normalised to the active electrode area (0.196 cm−2) of the glassy carbon electrode. (b) The Tafel plot of FeCo-NCZ and Pt/C in the low overpotential region, (c) peroxide yield and (d) electron transfer number of the electrocatalysts at different electrode potentials. | ||
The hydrogen peroxide yield (mol%) and the electron transfer number during the ORR were further measured using the RRDE.
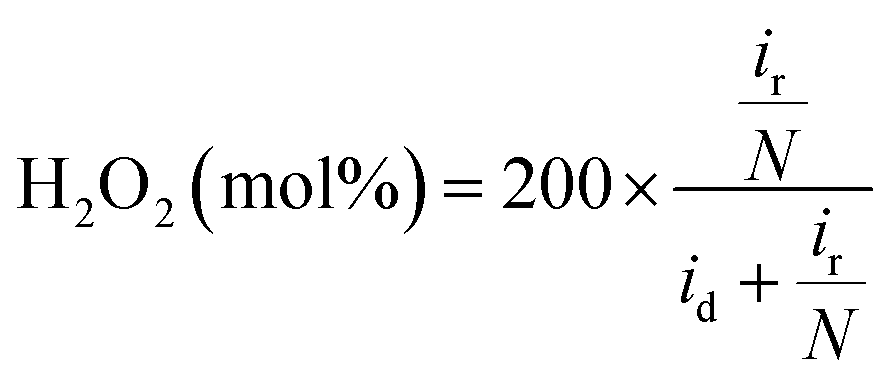 | (1) |
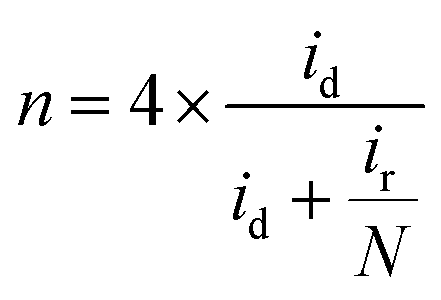 | (2) |
Excellent stability of electrocatalysts at fuel cell working temperatures is one of the significant concerns in the catalyst development. Electrochemical stability of FeCo-NCZ was measured by potential cycling (load cycle) between 0.6 V and 1 V for 10k cycles at a temperature of 60 °C in nitrogen saturated 0.1 M KOH. LSV was taken before and after potential cycling in oxygen saturated 0.1 M KOH at room temperature. This cycling process assists in accelerating the degradation of the carbon catalyst. The cycling stability of Pt/C was also evaluated under similar experimental conditions. FeCo-NCZ showed a 37 mV negative shift in half-wave potential after 10k potential cycles; however, in the case of commercial Pt/C, the negative shift was 67 mV after 10k potential cycles (Fig. 5). This clearly demonstrates that FeCo-NCZ is highly durable compared to Pt/C even at a temperature of 60 °C. Analysis of the TEM images (Fig. S10†) performed after the durability analysis of FeCo-NCZ shows that FeCo-NCZ is free from severe morphological changes even after 10k potential cycles. The excellent durability at 60 °C may be attributed to the presence of high density of active reaction centres on carbon surfaces. But in the case of Pt/C, sintering or dissolution of Pt nanoparticles may occur during the potential cycling which causes the reduction in the catalyst performance.62 The fuel tolerance of the non-Pt electrocatalyst was also studied in oxygen saturated 0.1 M KOH with and without methanol. Fig. S11a† shows that the LSV of FeCo-NCZ is unaltered with the addition of methanol. This indicates that the carbon alloy catalyst (non-Pt) shows high tolerance towards methanol. This shows that the fuel like methanol cannot affect the ORR activity of the FeCo-NCZ catalyst. But the ORR activity is drastically changed with the addition of methanol in the case of Pt/C. This further validates the applicability of FeCo-NCZ in direct methanol alkaline fuel cells.
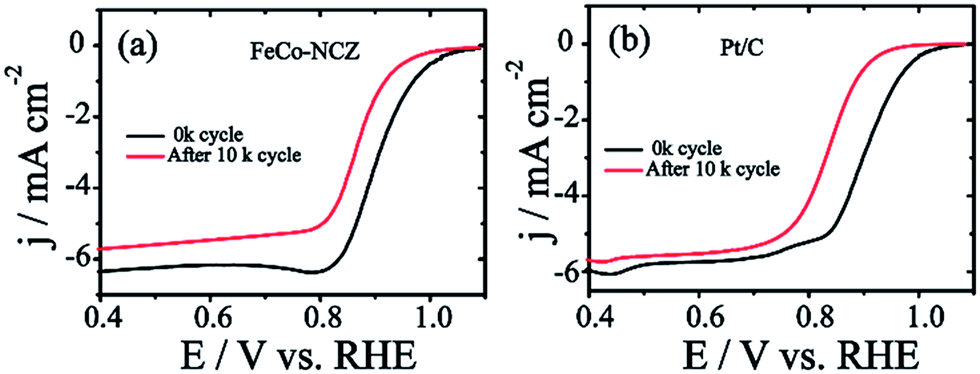 | ||
Fig. 5 LSVs of (a) FeCo-NCZ and (b) Pt/C, before and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles in oxygen saturated 0.1 M KOH at an electrode rotation rate of 1600 rpm with the scan rate of 10 mV s−1 (for the durability test, 10000 load cycles in between 1 V and 0.6 V were performed in nitrogen saturated 0.1 M KOH at 60 °C). 000 potential cycles in oxygen saturated 0.1 M KOH at an electrode rotation rate of 1600 rpm with the scan rate of 10 mV s−1 (for the durability test, 10000 load cycles in between 1 V and 0.6 V were performed in nitrogen saturated 0.1 M KOH at 60 °C). | ||
The more positive onset potential, as well as the half-wave potential of FeCo-NCZ, compared to Pt/C and other control-carbon catalysts is mainly due to the enriched possible active reaction centres such as graphitic, pyridinic and Fe/Co–Nx type coordination on the surface of FeCo-NCZ. The uniform distribution of these active reaction centres is achieved only through adopting a single precursor for the preparation of the electrocatalyst using a high-temperature annealing process. The reasonably high micropore surface area of FeCo-NCZ improves the distribution of active reaction centres on the carbon surfaces and assists in the reduction of dioxygen more efficiently. This high density of active reaction centres on the surface of FeCo-NCZ together with the reduced defective sites (<ID/IG) further helps to improve the electrochemical stability of FeCo-NCZ. This is clearly evidenced by the 37 mV negative shift in half-wave potential after 10k potential cycles in comparison with Pt/C. The peculiar morphology of the FeCo-NCZ plays a significant role in improving the catalytic activity. The carbon nanotube interpenetrated porous carbon assists the efficient mass transport as well as the electron transport throughout the catalyst. The high crystallite size of the FeCo-NCZ indicates low resistivity in FeCo-NCZ. These results emphasise that the FeCo-NCZ is a better electrocatalyst for oxygen reduction in solid alkaline fuel cells.
Experimental
Materials
2-Methylimidazole was purchased from Sigma Aldrich. Iron sulphate heptahydrate (99%), cobalt nitrate hexahydrate (99.5%), zinc nitrate pentahydrate (99%), and triethylamine were purchased from Wako pure chemical Industries (Japan). All chemicals were used as received.Synthesis of carbon alloy electrocatalysts
In a typical experiment, 3.242 g of 2-methyl imidazole was dissolved in 50 ml DI water, and 4 g triethyl ammine (TEA) was added into the imidazole solution. The resulting mixture was kept for stirring for 5 min. 0.55 g zinc nitrate, 0.089 g cobalt nitrate, and 0.085 g iron sulphate were dissolved separately in 50 ml DI water and added to the imidazole–TEA mixture with constant stirring. The stirring was continued for 30 min at room temperature. The resulting precipitate was centrifuged and washed several times with water. The prepared ZIF was kept for drying in an oven at a temperature of 90 °C for 24 h. The ZIF contains 75% Zn and 25% Fe and Co in a ratio of 1:1. The electrocatalysts from the above-synthesised ZIF were prepared by carbonising it at a high temperature (910 °C) for one hour in an inert atmosphere (nitrogen). After carbonisation, the samples were subjected to acid wash for 8 h at 80 °C using 2 M H2SO4, followed by washing with DI water several times. The resulting carbon was dried overnight at 90 °C and annealed again at a temperature of 910 °C for 0.5 h in a nitrogen atmosphere to get the final catalysts. For comparison purpose, catalysts from ZIFs with different metal ions were also prepared using a similar synthesis procedure as described above. The details of the samples are given in Table 2.
| Catalyst | ZIF |
|---|---|
| FeCo-NCZ | FeCoZn-ZIF |
| Fe-NCZ | FeZn-ZIF |
| Co-NCZ | CoZn-ZIF |
| Co-NC | Co-ZIF (ZIF-67) |
Materials characterization
XRD data were obtained using an Ultima IV (Rigaku) with a Cu Kα (λ = 1.5406 Å) X-ray source operating at 40 kV and 40 mA and a scan rate of 3° min−1. Raman analysis was performed using a LabRAM HR Evolution Raman spectrometer using a visible laser beam of wavelength 532 nm. A BELSORP-Max (Microtrac BEL Corp. Japan) was used for surface area analysis. The analysis was carried out at 77 K using ultrapure nitrogen gas. The morphology of the catalysts was analysed using a high-resolution transmission electron microscope (HRTEM) (images were taken using a TOPCON EM-002BF-J operated at an accelerating voltage of 200 kV with a Twin EDS facility) and a scanning electron microscope (S-4800, Hitachi High-Technologies Corporation). The surface elemental composition of the carbon catalyst was analysed using a Quantum 2000 (ULVAC-PHI Inc., Japan) using an X-ray source of monochromatic Al Kα (hν = 1486.58 eV) with a photoelectron takeoff angle of 45° and an X-ray irradiation area of 100 μm Φ.Electrochemical characterization
The electrochemical analysis was carried out on an electrochemical measurement system HZ-7000 and a dynamic electrode HR-301 (HD HOKUTO DENKO). The oxygen reduction reaction (ORR) activity of the electrocatalyst was evaluated using linear sweep voltammetry (LSV) with an electrode rotation rate of 1600 rpm at a scan rate of 10 mV s−1 in oxygen saturated 0.1 M KOH solution. Hg/HgO (1 M KOH) and Pt were used as reference and counter electrodes, respectively. The electrocatalyst coated glassy carbon electrode (0.196 cm2) was used as the working electrode. The potential was converted to the reference hydrogen electrode (RHE) and expressed in the RHE. The calibration of Hg/HgO was performed using the method reported by Dai et al.63 For calibration, 0.1 M KOH is saturated with hydrogen by bubbling pure hydrogen for 30 minutes. A Pt disc was used as the working electrode. Hg/HgO and Pt were used as reference and counter electrodes respectively. LSV was performed at a scan rate of 1 mV s−1. The potential was measured where the LSV curve crosses zero current. This potential was used for calibrating Hg/HgO to the RHE. The catalyst slurry was prepared by sonicating a mixture of 20 mg active material, 80 μl 5 wt% Nafion and 2 ml 3:1 water:IPA mixture for 3 h in a bath sonicator. 10 μl of the resulting slurry was drop coated on the glassy carbon electrode. The catalyst loading of non-Pt carbon catalysts is maintained as 0.5 mg cm−2 on the electrode surface. LSV was performed in an anodic scan. To eliminate the capacitive current from the oxygen reduction current, the blank correction was carried out to present the LSV data. For blank correction, LSV in the nitrogen saturated 0.1 M KOH was performed without electrode rotation. For comparison purpose, the ORR activity of Pt/C (46.1 wt%, with Pt loading 30 μg cm−2) was also evaluated. The hydrogen peroxide yield was evaluated using a rotating ring disc electrode (RRDE) in oxygen saturated 0.1 M KOH solution at an electrode rotation rate of 1600 rpm. The ring potential was kept at 0.5 V vs. Hg/HgO (1.39 V vs. the RHE). The collection efficiency of the ring electrode was measured as 0.37 for the peroxide yield and electron transfer calculation.
Electrochemical stability of FeCo-NCZ was measured by potential cycling (load cycle) between 0.6 V and 1 V for 10000 cycles at a temperature of 60 °C in nitrogen saturated 0.1 M KOH. LSV was taken before and after potential cycling in oxygen saturated 0.1 M KOH at room temperature. In one cycle, the electrode was initially kept at 0.6 V for 3 s and then changed to 1 V for another 3 s. The electrolyte solution temperature was kept at 60 °C, and nitrogen gas was purged throughout the cycling process. For methanol tolerance study, LSV was taken before and after the addition of methanol (2 ml) in oxygen saturated 0.1 M KOH (310 ml) at a scan rate of 10 mV s−1 with an electrode rotation rate of 1600 rpm.
Conclusions
We presented a direct synthesis approach for the preparation of a carbon nanotube interpenetrated porous carbon alloy from a single precursor, t-ZIF, for the ORR in an alkaline medium. An aqueous rapid synthesis approach is used for the preparation of the t-ZIF in high yields. The t-ZIF consists of Fe, Co and Zn-based metal centres and 2-methyl imidazole as a ligand. On carbonisation, the t-ZIF is transformed to carbon catalysts. HR-TEM reveals the morphology of carbon nanotube interpenetrated porous carbon with metal/metal oxide nanoparticles well encased in the carbon shells. Furthermore, EDS analysis provides clear evidence for the uniform distribution of metal atoms and nitrogen moieties on the surfaces. Since the positions of Co and Fe in the EDS images are in the same location, this represents alloy type features of the metal centres in the catalysts. XRD analysis shows that FeCo-NCZ has metallic Fe/Co phases along with much less amount of oxide phases. The BET surface area of FeCo-NCZ is found to be 463 m2 g−1 with microporous features. XPS analysis reveals that the pyridinic, graphitic and Fe/Co–Nx type nitrogen coordination dominates in the FeCo-NCZ compared to that in the other control-catalyst prepared by the same process. This nitrogen coordination is important for efficient ORR activity of Pt-free electrocatalysts. The ORR activity of FeCo-NCZ in oxygen saturated 0.1 M KOH is comparable to that of commercial Pt/C. The displayed onset potential (1.04 V vs. the RHE) and half-wave potential (0.91 V vs. the RHE) of FeCo-NCZ are more positive compared to those of Pt/C and other control-samples. The dioxygen reduction kinetics of FeCo-NCZ are comparable to those of Pt/C as evident from the Tafel slope, and oxygen reduction follows a four electron pathway. More interestingly, FeCo-NCZ shows better fuel tolerance and electrochemical stability even at 60 °C compared to Pt/C. With improved ORR activity and better electrochemical stability, FeCo-NCZ can be a cost-effective electrocatalyst for solid alkaline fuel cells.Acknowledgements
The authors acknowledge the financial assistance from the Japan Society for the Promotion of Science (JSPS), Core Research for Evolutionary Science and Technology, Japan Science and Technology Agency (JST-CREST, JPMJCR1543) and Research program of “Five-star Alliance” in “NJRC Mater. & Dev.”. SMU acknowledges JSPS for the research fellowship.Notes and references
- R. Cao, J.-S. Lee, M. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816–829 CrossRef CAS.
- D. C. Higgins and Z. Chen, Can. J. Chem. Eng., 2013, 91, 1881–1895 CrossRef CAS.
- Z. Yang, J. Ren, Z. Zhang, X. Chen, G. Guan, L. Qiu, Y. Zhang and H. Peng, Chem. Rev., 2015, 115, 5159–5223 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841–2857 RSC.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- H. Chung, G. Wu, D. Higgins, P. Zamani, Z. Chen and P. Zelenay, in Electrochemistry of N4 Macrocyclic Metal Complexes: Volume 1: Energy, ed. J. H. Zagal and F. Bedioui, Springer International Publishing, Cham, 2016, pp. 41–68, DOI:10.1007/978-3-319-31172-2_2.
- Z. Li, G. Li, L. Jiang, J. Li, G. Sun, C. Xia and F. Li, Angew. Chem., Int. Ed., 2015, 54, 1494–1498 CrossRef CAS PubMed.
- K. Chen, Y. Hao, M. Zhang, D. Zhou, Y. Cao, Y. Wang and L. Feng, RSC Adv., 2017, 7, 5782–5789 RSC.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- Q.-L. Zhu, W. Xia, T. Akita, R. Zou and Q. Xu, Adv. Mater., 2016, 28, 6391–6398 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- H. M. Barkholtz and D.-J. Liu, Mater. Horiz., 2017, 4, 20–37 RSC.
- T. Palaniselvam, B. P. Biswal, R. Banerjee and S. Kurungot, Chem.–Eur. J., 2013, 19, 9335–9342 CrossRef CAS PubMed.
- P. Zhang, F. Sun, Z. Xiang, Z. Shen, J. Yun and D. Cao, Energy Environ. Sci., 2014, 7, 442–450 CAS.
- D. Kim, D. W. Kim, W. G. Hong and A. Coskun, J. Mater. Chem. A, 2016, 4, 7710–7717 CAS.
- M. Thomas, R. Illathvalappil, S. Kurungot, B. N. Nair, A. A. P. Mohamed, G. M. Anilkumar, T. Yamaguchi and U. S. Hareesh, ACS Appl. Mater. Interfaces, 2016, 8, 29373–29382 CAS.
- S. Pandiaraj, H. B. Aiyappa, R. Banerjee and S. Kurungot, Chem. Commun., 2014, 50, 3363–3366 RSC.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- W. Xia, J. Zhu, W. Guo, L. An, D. Xia and R. Zou, J. Mater. Chem. A, 2014, 2, 11606–11613 CAS.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- H. Hu, L. Han, M. Yu, Z. Wang and X. W. Lou, Energy Environ. Sci., 2016, 9, 107–111 CAS.
- B. Y. Guan, L. Yu and X. W. Lou, Energy Environ. Sci., 2016, 9, 3092–3096 CAS.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- L. Zhang, H. B. Wu and X. W. Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- M. Wang, T. Qian, J. Zhou and C. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 5213–5221 CAS.
- J. Tang, R. R. Salunkhe, H. Zhang, V. Malgras, T. Ahamad, S. M. Alshehri, N. Kobayashi, S. Tominaka, Y. Ide, J. H. Kim and Y. Yamauchi, Sci. Rep., 2016, 6, 30295 CrossRef CAS PubMed.
- S. Gadipelli, T. Zhao, S. A. Shevlin and Z. Guo, Energy Environ. Sci., 2016, 9, 1661–1667 CAS.
- X. Wang, H. Zhang, H. Lin, S. Gupta, C. Wang, Z. Tao, H. Fu, T. Wang, J. Zheng, G. Wu and X. Li, Nano Energy, 2016, 25, 110–119 CrossRef.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- L. Chong, G. A. Goenaga, K. Williams, H. M. Barkholtz, L. R. Grabstanowicz, J. A. Brooksbank, A. B. Papandrew, R. Elzein, R. Schlaf, T. A. Zawodzinski, J. Zou, S. Ma and D.-J. Liu, ChemElectroChem, 2016, 3, 1541–1545 CrossRef CAS.
- A. F. Gross, E. Sherman and J. J. Vajo, Dalton Trans., 2012, 41, 5458–5460 RSC.
- M. Jiang, X. Cao, D. Zhu, Y. Duan and J. Zhang, Electrochim. Acta, 2016, 196, 699–707 CrossRef CAS.
- J. Shui, C. Chen, L. Grabstanowicz, D. Zhao and D.-J. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10629–10634 CrossRef CAS PubMed.
- J. Tian, A. Morozan, M. T. Sougrati, M. Lefèvre, R. Chenitz, J.-P. Dodelet, D. Jones and F. Jaouen, Angew. Chem., Int. Ed., 2013, 52, 6867–6870 CrossRef CAS PubMed.
- Q. Lai, L. Zheng, Y. Liang, J. He, J. Zhao and J. Chen, ACS Catal., 2017, 7, 1655–1663 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- L. Tan, Y.-D. Yang, N. Li, S. Chen and Z.-Q. Liu, Catal. Sci. Technol., 2017, 7, 1315–1323 CAS.
- S. M. Unni, R. Illathvalappil, S. N. Bhange, H. Puthenpediakkal and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 24256–24264 CAS.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 442–451 CAS.
- S. M. Unni, S. Devulapally, N. Karjule and S. Kurungot, J. Mater. Chem., 2012, 22, 23506–23513 RSC.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- S. Dou, X. Li, L. Tao, J. Huo and S. Wang, Chem. Commun., 2016, 52, 9727–9730 RSC.
- B. Li, X. Ge, F. W. T. Goh, T. S. A. Hor, D. Geng, G. Du, Z. Liu, J. Zhang, X. Liu and Y. Zong, Nanoscale, 2015, 7, 1830–1838 RSC.
- H. Zhong, Y. Luo, S. He, P. Tang, D. Li, N. Alonso-Vante and Y. Feng, ACS Appl. Mater. Interfaces, 2017, 9, 2541–2549 CAS.
- T. Palaniselvam, R. Kannan and S. Kurungot, Chem. Commun., 2011, 47, 2910–2912 RSC.
- J. A. Varnell, E. C. M. Tse, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Nat. Commun., 2016, 7, 12582 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- Y. Yuan, L. Yang, B. He, E. Pervaiz, Z.-G. Shao and M. Yang, Nanoscale, 2017, 9, 6259–6263 RSC.
- M. E. M. Buan, N. Muthuswamy, J. C. Walmsley, D. Chen and M. Rønning, ChemCatChem, 2017, 9, 1663–1674 CrossRef CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- X. Cui, S. Yang, X. Yan, J. Leng, S. Shuang, P. M. Ajayan and Z. Zhang, Adv. Funct. Mater., 2016, 26, 5708–5717 CrossRef CAS.
- P. Chen, K. Xu, Y. Tong, X. Li, S. Tao, Z. Fang, W. Chu, X. Wu and C. Wu, Inorg. Chem. Front., 2016, 3, 236–242 RSC.
- J. Wei, Y. Hu, Z. Wu, Y. Liang, S. Leong, B. Kong, X. Zhang, D. Zhao, G. P. Simon and H. Wang, J. Mater. Chem. A, 2015, 3, 16867–16873 CAS.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- G. Panomsuwan, N. Saito and T. Ishizaki, Phys. Chem. Chem. Phys., 2015, 17, 6227–6232 RSC.
- Y. Zhang, S. Chen, Y. Wang, W. Ding, R. Wu, L. Li, X. Qi and Z. Wei, J. Power Sources, 2015, 273, 62–69 CrossRef CAS.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of the prepared ZIF, TEM images of Fe-NCZ, Co-NCZ and Co-NC, deconvoluted XPS spectrum of C, Fe, Co and Zn in different catalyst systems. See DOI: 10.1039/c7se00249a |
| This journal is © The Royal Society of Chemistry 2017 |