
Thermal charging of supercapacitors: a perspective
Ayar
Al-zubaidi
 ,
Xixi
Ji
and
Jie
Yu
*
,
Xixi
Ji
and
Jie
Yu
*
Shenzhen Engineering Lab for Supercapacitor Materials, Shenzhen Key Laboratory for Advanced Materials, Department of Material Science and Engineering, Shenzhen Graduate School, Harbin Institute of Technology, University Town, Shenzhen 518055, China. E-mail: jyu@hit.edu.cn
First published on 12th June 2017
Abstract
Thermally-induced self-charging of electrochemical capacitors is a recently reported phenomenon, whereby a change in the temperature of a supercapacitor can lead to the generation of a voltage difference across the device. The temperature change is induced for all the device or only some of its components, unaided by or in combination with other voltage-inducing effects. This phenomenon is based on old and known physical concepts, whose use for energy generation became possible due to the advent of nanomaterials. The purpose of this article is to present the research conducted on this phenomenon, and offer a prospective direction for further progress in the field. First, we briefly introduce the existing heat-to-electricity conversion technologies, and their underlying principles. Then, we examine the main thermally-induced phenomena occurring in the environment of an ionic electrolyte, and/or a solid–liquid interface. After that, we review the studies conducted on thermally-induced self-charging in electrochemical capacitors, and the performance factors investigated so far. Finally, we present the future prospects of this field in the form of questions to address, additional factors to inspect, and materials of potential benefit for the design of thermally-chargeable supercapacitors.
 Ayar Al-zubaidi | Ayar Al-zubaidi received the B.Sc. degrees in Chemical Engineering from the University of Technology, Iraq, in 1998, then received the M.Sc. degree from the same university in 2007. As the recipient of the Japanese Government (MEXT) Scholarship, she completed her PhD in Materials Science and Engineering at Nagoya Institute of Technology, Japan, in 2014. She is currently a postdoctoral fellow at the Department of Materials Science and Engineering, Harbin Institute of Technology Shenzhen Graduate School, China, under the direction of Professor Jie Yu. Her current research interests include developing self-charging strategies for supercapacitors, electrochemical heat-to-electricity conversion, and thermally-responsive materials. |
 Xixi Ji | Xixi Ji received the BS degree from the School of Metallurgical Engineering, Hunan University of Technology in 2012, then received the Master's degree from School of Materials Science and Engineering, Harbin Institute of Technology Shenzhen Graduate School, in 2015. She is currently a PhD student at Harbin Institute of Technology, studying under the supervision of Professor Jie Yu. Her current research is focused on the synthesis of two-dimensional functional materials, and their use in bio-sensing applications. |
 Jie Yu | Jie Yu is a professor at Shenzhen Graduate School, Harbin Institute of Technology. He received his bachelor degree (1988) from Central South University, Master (1990) and PhD degree (1997) from the Institute of Metal Research, Chinese Academy of Sciences (CAS). After that, he worked as a postdoctoral fellow in Institute of Physics, CAS. Then he worked in Nanyang Technological University in Singapore as a Research Fellow, National Institute for Materials Science in Japan as a STA postdoctoral Research Fellow, and Chinese University of Hong Kong as a research associate. His research interests include supercapacitors, electrocatalysts for fuel cells, chemical vapor deposition of thin film materials, and electrospinning. |
1. Low-grade heat
The terms “energy efficiency” and “fuel efficiency” are the mathematical way of stating that an energy conversion process does not fully transform the energy of input sources (for example, fuel or hot steam) into theoretically predicted output (travel distance in a vehicle, or work produced in a turbine). For each process involving energy conversion, and depending on the nature of the energy source and the complexity of the conversion system, a portion of the energy input is not used, and is often released to the surroundings in the form of waste heat. This heat is normally classified as low-grade, which means that it is available with a temperature of less than 100–200 °C, and is generated as a byproduct from diverse industrial machinery, such as heat exchanges, boilers, economizers, reactors, kilns, dryers, heat pumps, and so on. It can also be released from non-industrial sources, such as domestic cooling and refrigeration processes, automobile engines, diverse electronic components and microchips, and even from human metabolism.Recovering waste heat serves several purposes, as it (1) improves the energy and fuel efficiency of relevant processes, (2) prevents heat-induced failure in machinery parts, and hence reduces their operational cost, and (3) provides a means of tapping into additional and abundant energy sources, like solar thermal energy, geothermal energy, and ocean thermal energy, to support the continuously depleting fossil fuels.
2. The evolution of heat to electricity
In this section, we address the most common heat-to-electricity technologies, and their key concepts. The full progress of these technologies lies beyond the scope and objective of the present review, so we will try to remain brief in our discussion, while allowing each topic due explanations where considered necessary or of relevance to the topic of our review.2.1. Heat engines
A heat engine is the most common scheme for converting heat into electricity. A heat engine operates through a thermodynamic cycle that transforms heat into mechanical work, through the variation of temperature and pressure. Thermodynamic cycles employed in a heat engine can vary in their complexity and the number of processes they include, but they all share a common feature, which is the use of thermal energy of a fluid while it is being transported between a heat source of higher temperature (TH), to a heat sink of a lower temperature (TL). The values of TH and TL set the maximum efficiency of energy conversion in any heat engine, known as the Carnot efficiency, and defined as:1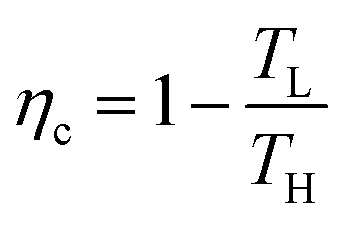 | (1) |
The Carnot efficiency represents the upper limit to which the efficiency of any heat-to-work conversion process is compared.
More straightforward conversion processes convert heat directly into electrical current, using thermoelectric engines, which also work between two heat reservoirs that differ in temperature, but operate based on a different concept known as the Seebeck effect, which will be presented below.
2.2. Solid-state thermoelectrics
There are three main phenomena that portray the interrelation between heat and electricity in a material, and are often referred to as the “three thermoelectric effects”. The first is the establishment of a voltage difference along a conductor when its two ends are kept at two different temperatures, which is referred to as the “Seebeck effect”. The electrons on the hot end, gaining kinetic energy proportional to the increase in temperature, travel in the direction of the cold side, creating a difference in electron density, until an electric field is established to impede further flow of electrons. If two different conductors are joined and their two junctions are placed along a temperature gradient, due to the inherent difference between the electron energies in the two materials, the electron energy levels in each conductor will shift differently, creating a voltage difference between the junctions. On the other hand, charge carriers passing through the conductor would carry along energy (heat), leading to a temperature difference between the opposite ends of the conductor, which is the second thermoelectric effect known as the “Peltier effect”. The third thermoelectric phenomenon is the co-occurrence of a temperature gradient and an electric current, and is called the “Thomson effect”.A thermoelectric material is one with free electrons or holes that carry both charge and heat. When subjected to a temperature gradient, positive charge (and positive potential) builds on the cold side in a thermoelectric material with positive free carriers (P-type thermoelectric), and on the hot side in a material with negative free carriers (N-type thermoelectric).2 A thermoelectric generator (TEG) is a solid-state device that takes advantage of the Seebeck effect, to convert heat directly into electrical energy, when subjected to a heat source of higher temperature on one side, and a heat sink of a lower one on the other. The performance of a thermoelectric material is given by its dimensionless figure of merit, ZT, defined as:3
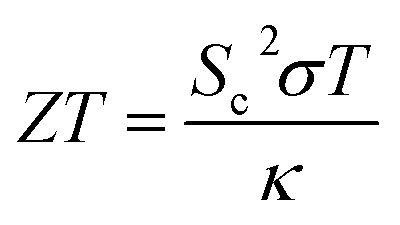 | (2) |
2.3. Thermogalvanic cells
Thermogalvanic cells rely on an electrochemical version of the Seebeck effect. Basically, a potential difference across an electrochemical cell is generated due to the temperature dependence of the redox potential of electrochemically active species in the cell's electrolytic medium. The change in temperature causes a change in the entropy of the redox reaction. Therefore, when a system (cell) containing a redox-active electrolyte is placed at a temperature difference, it will generate voltage proportional to the difference between redox reaction entropies in the electrolyte near the hot and cold electrode, following the equation: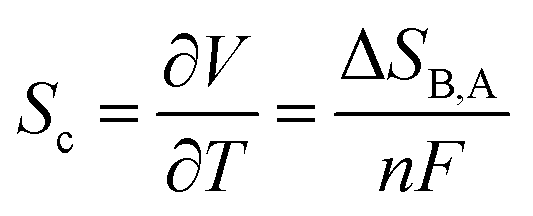 | (3) |
Thermogalvanic cells differ from their solid-state siblings in two main aspects. The first is that thermogalvanic cells involve a net transfer of material, which does not occur in thermoelectric devices. The second is that instead of kinetic energy of electrons, thermogalvanic cells exploit the temperature-dependence of chemical (redox) reactions that takes place at the interface between the electrode and the electrolyte. This means that while the current in a conductor is electronic (charge is carried entirely by electrons/holes), charge in a thermogalvanic cell is electronic along the electrode body, then becomes ionic (carried by the ions of the electrolyte) at the electrode/electrolyte interface. The main implication of this fact is that unlike the flow of electronic current that leaves the state of the conductor unchanged, the passage of ionic current may cause changes in the chemical states in the system.21 Further, the change in temperature influences not only the charge carriers, but the solvent molecules as well, which influences diverse solute–solvent interactions, like viscosity-induced resistance to ion motion, structure breaking of ions from their solvation shell, and solvent molecule reorientation. The redox reaction kinetics (and generated voltage) will therefore be influenced by the choice of the solvent medium, which can be classified as aqueous, non-aqueous, or molten salt-based.
Aqueous thermogalvanic cells have been the most commonly investigated, and among the active materials employed, ferricyanide/ferrocyanide is a typical redox couple, used due to its reversible electron exchange, high Seebeck coefficient (1.4 mV K−1 as reported by Hu et al.),22 resistance to electrode poisoning,23 and high exchange current density that not only increases the current withdrawn from the cell, but also reduces the activation overpotential that tends to reduce the efficiency of the cells under load. The I−/I3− redox couple has also been investigated,24 achieving a Seebeck coefficient of 0.26 mV K−1 with platinum electrodes, and 2.1 mV K−1 with stainless steel electrodes.
Some studies tackled non-aqueous electrolytes such as methanol, dimethylsulfoxide (DMSO), acetone, dimethylformamide (DMF), ethylene glycol (EG), or solid-state ion exchange phenolic membranes,20 reporting Seebeck coefficient values in the range of 0.2–1.19. Recently, Bonetti et al.25 reported a high Seebeck coefficient of 7 mV K−1, using 0.1 M TBAN in 1-dodecanol.
Solvent-based thermogalvanic cells suffer from inherent disadvantages, such as the limitations imposed by the boiling point of the solvent, the reduced ionic charge carrier concentration (and hence electrolyte conductivity), and the heat carried by the solvent from the hot to the cold side of the device.23,26 These disadvantages reduce the Sc/κ ratio, and the figure of merit and power efficiency, compared to those realized in solid-sate thermoelectrics.23,25 Molten salts were therefore explored to overcome these solvent-related limitations. Molten salts are composed of either liquid-state salts, or metals/alloys dissolved in their salt (or eutectic salt mixture) melts. The high ion concentration in molten salts results in electrical conductivity an order of magnitude higher than that in aqueous solutions, and two-to-three orders of magnitude higher than that in organic solutions with similar metal ionic species (see, for example, Table 4 in ref. 27). Also, their high melting points allow the temperature window of the device,20 and hence the power production in the cell, to be expanded. The reported values of the Seebeck coefficient for diverse molten salts were in the range of 0.4 to 2.25 mV K−1,26,27 with a comparable range of the figure of merit. However, molten salts also cause some operational problems, such as electrode embrittlement and cell degradation, leading to high-cost issues associated with the refractory materials needed for salt containment.23,28,29 Ionic liquids (ILs), a category of molten salts with melting points below 100 °C, are considered promising candidates, with high thermal, chemical, and electrochemical stability, and lower thermal conductivity compared to solid-state thermoelectric materials.30 Studies using ionic liquids have reported Seebeck coefficient values in the range of 0.03–0.26 mV K−1 for the I−/I3− redox couple,31 and 2.0 ± 0.02 mV K−1 for the CoII/III tris(bipyridyl) redox couple.32
Finally, gel-based electrolytes were recently reported as the working medium in a solid-state thermogalvanic device used to harvest body heat, achieving a Seebeck coefficient of 1.21 mV K−1, and a maximum output power of about 0.3 μW.33
The electrode material in thermogalvanic devices influences the device power output, through increasing the surface area exposed to the redox species in the electrolyte. Using scrolls of high surface area multiwalled carbon nanotube buckypaper was reported to result in 33% and 77% higher current density per unit temperature gradient, compared to platinum and graphite electrodes, respectively.22 Similar results were obtained through the electrodeposition of Pt black onto Pt disk electrodes in a device using DMSO/IL mixed solvent.32
3. Heat-to-electricity in electrochemical capacitors
Rechargeable energy storage devices, such as lithium ion batteries and supercapacitors, are devices that can reversibly store and deliver electric current, using electrochemical mechanisms. Lithium ion batteries rely on the intercalation reaction of lithium ions in the structure of porous host materials. Supercapacitors, on the other hand, store charge through one or a combination of two possible mechanisms: the first is the reversible physical adsorption of the electrolyte ions onto the surface of a porous electrode material, to form what is known as an “electric double-layer capacitor”, or EDLC, as shown in Fig. 1. The second mechanism stores charge by supplying energy for fast, reversible chemical reactions of the electrolyte ions with chemically active entities on the surface of the electrode, giving rise to so-called “pseudocapacitance”.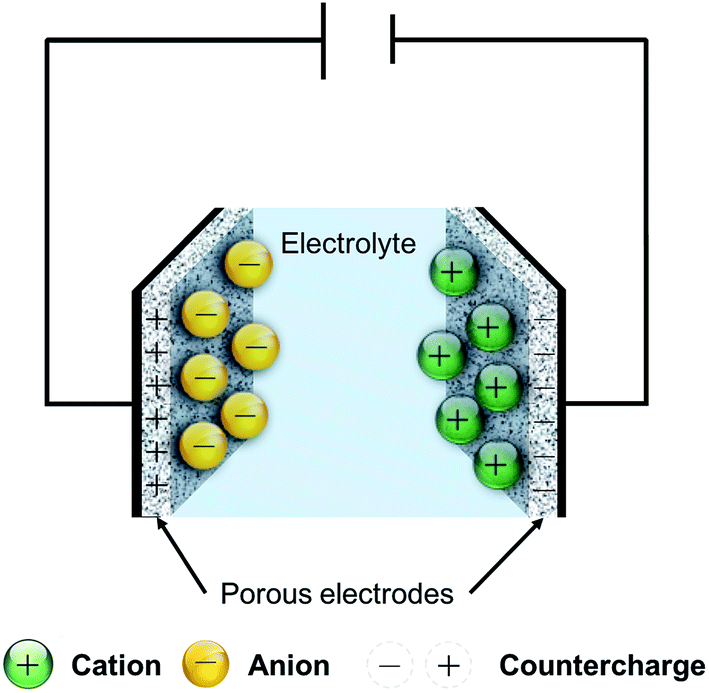 | ||
| Fig. 1 A schematic of an electric double-layer capacitor. | ||
Due to their charge storage mechanism, supercapacitors are capable of faster delivery of stored charge than lithium ion batteries, whose power performance is limited by the slow kinetics of the lithium intercalation reaction. Supercapacitors are therefore characterized by high power density and long cycle life, and may either be used to complement lithium ion batteries, which provide on the other hand higher energy density, or the two can be hybridized into a supercapattery,34 to realize both high energy and high power delivery. In parallel with the continuous search for materials and designs to realize high-performance and cost-effective devices, research efforts in the next stage of energy research extended towards the design of standalone units that integrate energy harvesting and storage in one unit. These units can be divided into two categories in terms of their integration level. The first category constitutes “power packs”, which integrate two devices, each performing a single energy harvesting-or-storage function.35–38 The second category comprises “self-charging” devices that integrate two different functions in a single multitasking device.39–46
In the latter category, the self-charging of supercapacitors has recently been reported by several studies, which used low-grade thermal energy to generate voltage between the two electrodes of the device, by relying on physical concepts that differ from the Seebeck effect in thermogalvanic cells.
In principle, the solid–liquid interface and presence of ions in an electrochemical system allow for diverse thermally-induced effects to take place, which means that a heat-to-electricity Seebeck-like outcome may be achieved if the change in any of the temperature-dependent phenomena occurring in the electrochemical system can cause a potential difference across the system. In particular, thermally-induced changes in the ionic charge density at an electrode/electrolyte interface can lead to the “thermocapacitive” effect, a term that we borrow from Härtel et al.,47 and use here to refer to thermally-induced asymmetry in the density of ionic charge, which may arise due to the combined contribution of several factors, and lead to a potential difference across an electrochemical capacitor. Here, we define an “electrochemical capacitor”, as an electrochemical system composed of electrically conductive electrodes and an ionically conductive electrolytic medium, where (as far as the currently available studies on thermal charging are concerned) charge storage is achieved through ion adsorption on the surface of the electrode, and does not rely on redox-couples like those used in thermogalvanic cells, lithium intercalation-like mechanisms, or diffusion-limited reactions.48 We will first introduce the thermally-induced physical changes that occur in an electrochemical environment, then review studies where such changes were used to achieve self-charging of a supercapacitor.
3.1. The thermocapillary effect
Before ion adsorption onto a porous surface is considered, it is important to point out that the process of wetting itself is an energy conversion mechanism, which exploits the interfacial energy of the wetting of a porous solid with a liquid. The surface tension of a fluid drop/bubble (γ) is a function of the fluid composition and temperature. Therefore, when changes in composition or temperature in the fluid lead to local variations in the surface tension, a phenomenon called the “Marangoni effect” takes place, according to which the resulting gradient in surface tension (or surface energy, according to a recent, more generalized treatment)49 triggers the motion in the fluid parallel to its interface with a second (viscous) phase. The particular case when such a gradient is caused by a gradient in temperature is referred to as the “thermocapillary effect”,50,51 through which the fluid may flow toward warmer or colder regions, depending on the sign of the change of surface tension with temperature (∂γ/∂T).51,52 The use of the interfacial energy of wetting as an energy transformation mechanism was first proposed by Eroshenko,53 and then systematically investigated by Laouir et al.,54 who assessed the feasibility of wetting-based heat-work cycles, using the temperature dependence of the liquid surface tension and its contact angle with the solid as the selection criteria of potential solid–liquid couples for the proposed cycles. Such energy absorption systems (EAS), were experimentally investigated through pressure-induced water infiltration into hydrophobic porous silica55 and zeolites,56 and showed that the increase in temperature reduces the pressure needed to induce pore filling, and improves the system recoverability (infiltration/defiltration reversibility), due to the thermal effect on the solid–liquid interfacial tension.The presence of ions in a liquid modifies many of its properties, and hence the manner and extent of its wetting or infiltration to a porous solid surface. First, water molecules arrange themselves around the ion, forming a solvation shell, the size of which is expected to be more sensitive to the pore size than the pure water molecules. Also, the presence of ions increases the interfacial tension, and was reported to increase the infiltration pressure compared to that of water in energy absorption systems based on the infiltration of NaCl, KCl, or CaCl2 solutions into porous zeolites57–59 and silica.60–62 On the other hand, the charge carried by the ions, affects their affinity for the solid surface and may enhance or reduce their interaction with it, depending on the surface chemistry and polarity. These charge-dependent interactions and transport properties influence the electrolyte's response to the change in temperature, and will be discussed next in the Soret effect section.
3.2. The Soret effect
The flow of matter caused by a macroscopic temperature gradient is referred to as “thermodiffusion”. If thermodiffusion occurs in a fluid mixture of two or more different constituents, different species will respond differently to the same temperature gradient, and molecular separation will occur within the mixture, which is known as the Soret (or Ludwig–Soret) effect, or Soret diffusion.63,64When a temperature difference, ΔT, applied between the boundaries of a multicomponent liquid system generates a temperature gradient, ∇T, the mass transfer of molecular species i driven by thermal diffusion is described by a characteristic thermal diffusion coefficient (DT). Diffusion also occurs by mass transfer according to Fick's law, and is governed by the molecule's mass diffusion coefficient (D). The net mass flow rate (Ji) of a particular species i in the mixture is given by:65
| Ji = JDi + JTi | (4) |
| Ji = −D∇c − DT∇T | (5) |
| ST = DT/D | (6) |
The thermal diffusion coefficient is defined as:66
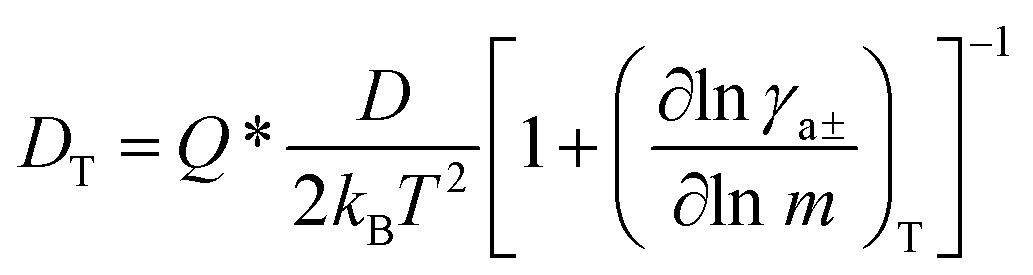 | (7) |
If the liquid contains easily dissociated particles, they will break up into charged ions, which behave as mobile charge carriers surrounded by solvent shells. These ions behave as subcomponents with characteristic diffusion properties, and their interionic forces and ion–solvent interactions are characteristic of their charge. In such systems, and as a temperature gradient is applied on an originally homogeneous solution, several thermally-induced modifications may occur, including:
(1) Changes in the overall thermal motion of the dissolved solute. The motion depends on the thermal heat of transport which, for diluted solutions of monovalent solutes, is an additive function of the individual contributions from the corresponding values of the cationic and anionic species. The relation becomes more complicated for higher valences and solution concentrations.67
(2) Changes in the volume, viscosity, chemical potential, and permittivity of the solvent. Volume changes result from the expansion of the solvent molecules,68 and can be described in terms of the coefficient of thermal expansion (αT), given by:66
∇![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnc = −αT∇T lnc = −αT∇T | (8) |
γa±)/(∂lnm) in eqn (7).
(3) A concentration gradient occurs, due to the local nonhomogeneity in the solute concentration caused by thermal diffusion. This concentration gradient drives mass diffusion in a direction opposite to thermal diffusion, the rate of which depends on the diffusion coefficient of each ion.
(4) Changes occur to the particle–particle interactions. The temperature affects the ion pair-interaction potential which, in addition to temperature, depends on the changes in the chemical potential of the solvent.69,70 Also, the permittivity of the solvent (ε), which tends to decrease with temperature, may affect the diffusion through affecting the ion–ion interaction forces in the solution.71,72
(5) In the particular case of electrolytic solutions, changes occur to the relative motion of positive and negative ions, each migrating at a rate governed by their characteristic coefficient of diffusion (D), thermodiffusion (DT), and the magnitude and sign of their ionic heat of transport 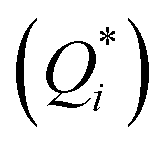 .73
.73
(6) In the vicinity of a neighboring surface, changes occur in the collision frequency between free carriers and the adjacent surface, leading to selective ion attachment to that surface, and breaking the isotropy in the double layer.30
The changes in (5) and (6) necessitate the inclusion of the motion of oppositely charged ions relative to each other, in the equation of diffusion of a single ion. Accordingly, the steady state equation becomes:
| Ji = JDi + JTi + JEi = 0 | (9) |
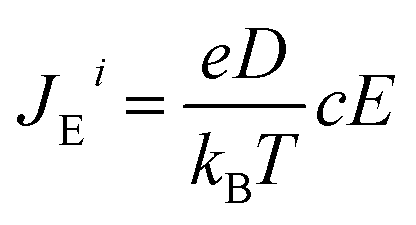 | (10) |
The electric field generated through thermodiffusion is evaluated from:73
| E = −(ψ0/T)∇T | (11) |
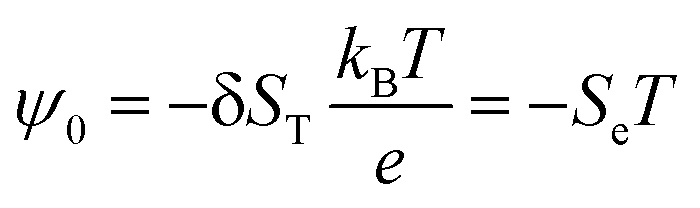 | (12) |
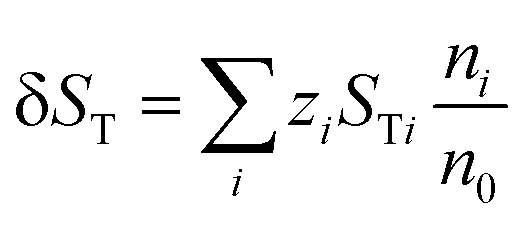 | (13) |
| δST = STC − STA | (14) |
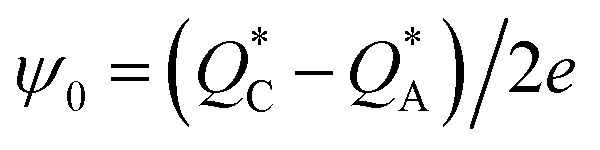 | (15) |
 and
and  being the ionic heat of transport for the cation and anion, respectively.
being the ionic heat of transport for the cation and anion, respectively.
In physical terms, the previous discussion and eqn (9) to (15) can be rephrased as follows: when an electrolytic solution is placed under a temperature difference, and as a result of the characteristic mass and thermal diffusion coefficients, the heats of transport, and the temperature-dependence of the physical properties and interactions of all the components in the solution, a nonhomogeneity in the concentration of ions occurs along the temperature gradient, establishing a difference in the ionic charge carrier concentration at the hot and cold boundaries in the system, and giving rise to a macroscopic electric field. In an electrochemical cell, this electric field manifests as a potential difference, or “thermopotential”, between the hot and cold electrodes of the cell. This thermopotential, which can be viewed as the “ionic” analogue of the electronic Seebeck effect in solid-state thermoelectrics, or as the “physical” version of the chemically-induced Seebeck effect in thermogalvanic cells, is the basic principle behind thermal charging in supercapacitors.
The direction of thermodiffusion of a species can be described by a positive Soret coefficient when the particle migrates from the hot to the cold side (thermophobic behavior), or a negative Soret coefficient in the opposite case (thermophilic behavior).68,73 As a part of a detailed study, Würger73 identified several factors that may govern the direction of motion of particles under a temperature gradient, which we summarize below:
| n± = n0(e±eψ/kBT − 1) | (16) |
In addition to the factors above, one needs to consider in an electrochemical cell, the contribution of the surface of the electrode as well. The surface chemistry and pore structure of the electrode can influence its interaction with the surrounding ions, and might modify their response to the change in temperature. This factor needs to be examined carefully, as it may involve thermally-induced changes to the electrode behavior as well.
The possibility of harvesting the energy of the thermal motion of ions requires that the magnitude of the generated electric field be at least comparable to those achieved using thermoelectric or thermogalvanic approaches. This in fact is not the case, due to the mobile nature of ionic charge carriers in electrolytes,77 and the absence of any chemical reactions to boost the potential difference generated across the electrochemical cell. Put differently, the conversion of heat into electricity in an electrolytic, redox-free electrochemical cell requires design strategies that compensate the said disadvantages, and increase the potential difference generated from the cell.
Recently, several studies managed to realize such ion-based thermal energy harvesting schemes using electrochemical capacitors, by relying on diverse aspects of the Soret effect, and without the use of any redox-active species in the electrolyte. These studies will be reviewed in detail below, and their design parameters and performance indicators are summarized in Table 1. Each study used its own particular expression (thermoelectric coefficient, thermopower, Seebeck coefficient, thermal sensitivity, etc.) to describe the voltage per unit temperature difference (in mV K−1 or mV °C−1) achieved in their set-up. We will use throughout our review the term “thermopotential” instead of the term used in the original publication, for no other reason than the need to use unified terminology and avoid confusion.
| Ref. | Electrode | Electrolyte | Thermopotential (or peak V) | ΔT | Capacitance | Energy density, power density | Remarks |
|---|---|---|---|---|---|---|---|
| a Although the expression “fully charged state” is used in the original publication, a state of full charge may not necessarily be equivalent to reaching the peak value of thermally-induced voltage, and it may not be possible to infer such relation without comparing thermal charging with electrical charging of the same device. | |||||||
| 78 | Nanoporous monel (pore size 480 nm) | NaCl/H2O (26 wt%) | V = 112 mV | 36 °C | — | — | Value of thermopotential varied nonlinearly with ΔT |
| 79 | Working electrode: nanoporous carbon (NC); particle size 50 μm, pore size 1–10 nm, SSA 500 m2 g−1 | NaCl/H2O (26 wt%) | 3.7 mV °C−1 | 35 °C | — | 0.1 J g−1, 0.5 mW g−1 | Energy conversion system constructed from two identical sandwich arrangements, each composed of a NC working electrode, and copper film counter electrode. Working and counter electrodes from the cold side are connected by copper wires with their hot side counterparts |
| Counter electrode: copper film | |||||||
| 80 | (1) Pt sheet | LiCl/H2O (0.1–3.7 M) | 0.75–0.88 mV °C−1 (NC) | 39 °C | 55 F g−1 | 0.1 J g−1, 2.3 mW g−1 | Output voltage is −ve (Pt) or +ve (NC) |
| (2) NC (pore size 2–100 nm, modal value 3 nm, SSA 1200 m2 g−1) | |||||||
| 81 | (1) Pt sheet | EMIMTFSI/AN (2 M), ionic conductivity σ@40 °C = 57.3 ± 0.5 mS cm−1 | −1.7 mV K−1 (Pt), −0.3 mV K−1 (PC) | 20 °C | 18 μF cm−2 (Pt), 3 μF cm−2 (NC) | 0.05 mJ m−2 (Pt) | (1) Vertical cell was used |
| (2) Porous Carbon (PC): SSA 300 m2 g−1, pore size 20 ± 10 nm | (2) 50% fully charged statea reached in 250 s when the cell was heated from the bottom, & 1000 s when heated from the top | ||||||
| (3) Output voltage of both materials had the same −ve sign but different values | |||||||
| 82 | (1) Au | NaOH–polyethyleneoxide (PEO–NaOH) polymer | 10 mV K−1 | 4.5 K | 1.03 mF cm−2 (MWCNT on Au) from EIS results | 1.35 μJ cm−2 (MWCNT on Au) | Output voltage had the same +ve sign and value for both Au and MWCNT@Au |
| (2) MWCNTs on Au | |||||||
| 83 | PANI on graphene/CNT | Solid-state polystyrene sulfonic acid (PSSH) | V = 38 mV | 5.3 K | 120 mF cm−2 | 5.7 mW h g−1, 2.1 W g−1 | (1) Charging time to peak V = 350 s |
| (2) Electrical conductivity of PSSH and thermopotential depended on relative humidity (7.9 mV K−1@70% RH – 5.1 mV K−1@30% RH) | |||||||
| (3) Energy and power density determined from electrical charging | |||||||
| 84 | Activated carbon | Acetonitrile (AN) or propylene carbonate (PC)-based electrolytes | 80–300 mV | ∼41 °C (65-RT) | — | — | (1) Commercial supercapacitor was used |
| (2) Peak V reached in 2 hours | |||||||
| (3) Higher thermopotential achieved with higher number of activation GCD cycles, and lower GCD current density | |||||||
| (4) Achieved voltage decreased with repeated thermal charging cycles | |||||||
| 93 | Working electrode: (1) Ni-coated MWCNTs, (2) Ni-coated NC | NaC2H3O2/FA (1.0 M) | 3.6 mV °C−1 (Ni–MWCNTs), 2 mV °C−1 (Ni–NC) | 45 °C (Ni–MWCNT), 75 °C (Ni–NC) | — | 600 mJ g−1 (Ni–MWCNT), 1800 mJ g−1 (Ni–NC) | Energy densities determined from ΔT ∼ 45–50 °C |
| Counter electrode: NC | |||||||
| 95 | Ion exchange membranes on activated carbon | NaCl/H2O (0.5 M) | 0.097 mV K−1 per membrane | 44 K | — | 2 mJ m−2 | (1) Energy density determined with ΔT = 30 °C |
| (2) Cycle efficiency 0.002% of Carnot efficiency | |||||||
| (3) Peak V = 0.2 mV (at ΔT = 15 °C) reached within 0.5 min | |||||||
| 99 | Activated carbon on graphite | NaCl/H2O (600 mM for charging; 20 mM for discharging) | 80 mV | 50 °C | — | 5.4 J per liter, 40 mW m−2 (130 mW kg−1) | Charging is performed at low temperature (25 °C), and discharging at higher temperature (75 °C) |
| 47 | Activated carbon (SSA = 1200 m2 g−1) | TEA-BF4/AN (1 mol L−1) | 0.6 mV K−1 | 65 °C | 185 mJ g−1 | (1) Commercial supercapacitor (10 F) was used | |
| (2) Peak V (+36 mV) reached after 2 min | |||||||
| (3) Peak V increased to 40 + mV with increasing heating rate | |||||||
| (4) Figure of merit = 0.0039 | |||||||
| (5) Maximum efficiency = 80% of Carnot efficiency | |||||||
| 102 | (1) Pt sheet | NaF, NaCl, NaBr, NaI in H2O (1 M) | 0.53 mV °C−1 (NaF)–NC, 0.63 mV °C−1 (NaCl)–NC, 0.67 mV °C−1 (NaBr)–NC, 0.72 mV °C−1 (NaI)–NC | — | — | — | Thermopotential decreased with increasing anion size for Pt, and increased for NC |
| (2) NC (pore size 3 nm, SSA 1810 m2 g−1) | |||||||
| 103 | (1) Pt sheet | LiCl, NaCl, KCl, CsCl in H2O (0.1–3.7 M) | 0.85 mV °C−1 (LiCl)–NC, 0.52 mV °C−1 (CsCl)–NC | — | — | — | Thermopotential decreased with increasing cation size for NC, but had no clear trend for Pt |
| (2) NC (pore size 1–100 nm, modal value 3 nm) | |||||||
| 104 | (1) Pt sheet | LiCl/DMSO (1 M), LiCl/H2O (1 M), LiCl/FA (1 M) | Pt: −0.3 mV °C−1 (DMSO), −1.0 mV °C−1 (H2O), −5.4 mV °C−1 (FA) | 40 °C | — | — | Thermopotential correlated with dielectric constant for Pt, but had no clear trend for NC |
| (2) NC (pore size 3 nm, SSA 1810 m2 g−1) | NC: 0.15 mV °C−1 (DMSO), 0.68 mV °C−1 (H2O), 0.23 mV °C−1 (FA) | ||||||
| 105 | (1) Pt | LiCl/FA (21 wt%) | 6.5 mV K−1, 4.1 mV K−1, 1.4 mV K−1, 0.4 mV K−1 | ∼50 °C | — | — | Output voltage has −ve sign for all materials (like ref. 104), but thermopotential values are reported with +ve sign |
| (2) Ni | |||||||
| (3) Cu | |||||||
| (4) In | |||||||
4. Thermal charging under temperature difference
The Qiao group78–80 proposed that the electric energy generated from the thermal motion of ions in the double-layer can be increased by using porous nanocarbon electrodes to maximize the ion/electrode exposure area. To verify their hypothesis, they used porous nanocarbon (average pore size of 3 nm and specific surface area (SSA) of 1200 m2 g−1), and performed their experiments using an electrochemical capacitor configuration, albeit for the use of a salt bridge to connect the two compartments where the electrodes were separately kept at two different temperatures.79,80 They referred to their scheme as a “thermally-chargeable supercapacitor”, or TCS. This set-up generated a voltage of about 112 mV in a solution of sodium chloride (NaCl) electrolyte, for a temperature difference of 36 °C.78The first noteworthy observation from the aforementioned studies was the sign of the generated voltage. Comparing nanocarbon versus Pt sheet electrodes, their experiments showed that the detected voltage of the hot Pt electrode decreased below zero (the open-circuit voltage of the device) with an increase in temperature, then instantly reset to zero after connecting the electrodes through a resistor. With porous electrodes, the increase in temperature caused the voltage of the hot electrode to increase to positive values, then decay slowly upon shorting through the resistor. The authors attributed the difference to the influence of negatively-charged functional groups on the surface of nanocarbon, which according to the authors, causes the adsorption of anions (which is favorable on Pt surfaces) to be suppressed on the surface of nanocarbon (concepts illustrated in Fig. 2).80
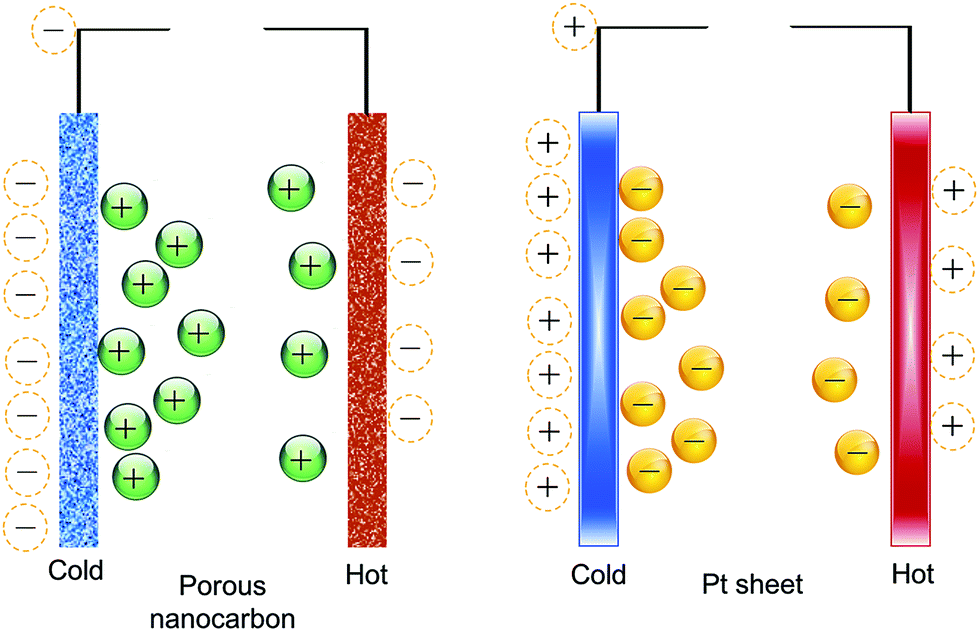 | ||
| Fig. 2 The difference between platinum and nanocarbon electrodes in their ion adsorption and thermal charging behavior, as proposed by ref. 80. | ||
Bonetti et al.81 used a similar scheme, with ionic liquids as the thermal charging medium. Using a vertical supercapacitor cell (electrodes located at the top and bottom of the cell, and no salt bridge used), they performed thermal charging of platinum, and also of porous carbon electrodes with a specific surface area of 300 m2 g−1 and an average pore size of 20 ± 10 nm, in a binary ionic liquid mixture of 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)-imide (EMIMTFSI) in acetonitrile (AN). Unlike the results obtained by the Qiao group, the thermopotential (which they defined using the equation ΔV = −ΛΔT) had the same sign for both electrodes. With a temperature difference of 20 K, this set-up resulted in a thermopotential of −1.7 mV K−1 for Pt, and −0.3 mV K−1 for the porous carbon, the latter value translating into a single-electrode capacitance of 3 μF cm−2, obtained by discharging the nanoporous carbon through external loads with resistance of 1.5 to 10 kΩ. The Pt electrodes had higher areal capacitance (18 μF cm−2), but low Carnot efficiency (0.24 × 10−9). Due to the vertical configuration of the device, the process showed geometry-dependent kinetics; 50% of the fully charged state in Pt electrodes was attained in 250 s when the cell was heated from the bottom, versus 1000 s when the cell was heated from the top, which the authors attributed to the diffusion mechanism being convective in the first case, and diffusive in the second.
Zhao et al.82 used a scheme based on a controlled Soret effect, by using an asymmetric polymer electrolyte prepared from NaOH-treated polyethyleneoxide (PEO–NaOH), to generate thermally-induced voltage in a supercapacitor. The concept was to combine heavy anions with lighter cations, and use the large difference between their respective ionic mobilities to cause selective Soret motion for one of the ionic species in the electrolyte (Na+ cations), versus less or none for the other (alkoxylate and carboxylate anions), as illustrated in Fig. 3. Their set-up, which used electrodes of Au and also of multi-walled carbon nanotubes (MWCNTs) deposited on Au, exhibited yet another behavior distinctly different from that observed in the previous two studies: both electrodes exhibited almost identical voltage-versus-time curves upon heating, and resulted in similar thermopotential values on the order of 10 mV K−1. The areal capacitance of MWCNT/Au was 1.03 mF cm−2, determined from impedance spectroscopy, and corresponding to an energy density of 1.35 μJ cm−2, obtained for a temperature difference of 4.5 K, and by discharging through a load resistance of 20 kΩ.
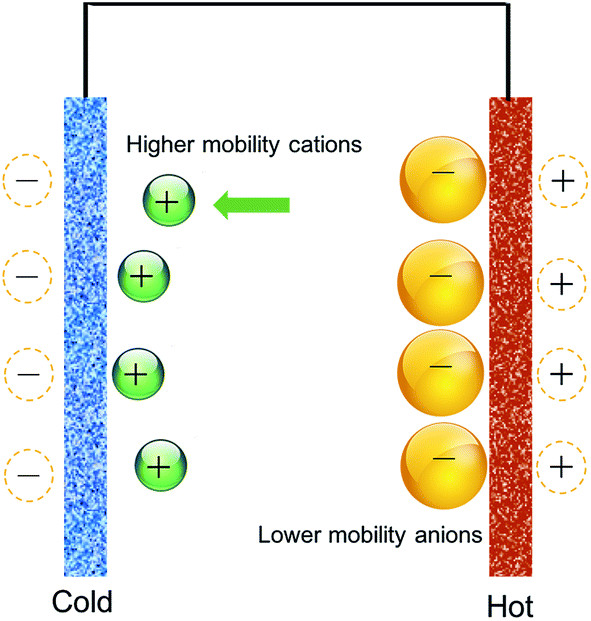 | ||
| Fig. 3 In an asymmetric electrolyte, the increase in temperature selectively causes thermodiffusion of only the ions with higher mobility, leading to charge separation and a potential difference between the two electrodes. | ||
The diversity in the qualitative and quantitative results obtained in different set-ups indicates the necessity to take a closer look at the nature of ion–surface interaction, and include chemical and surface structure factors, as will be addressed in the Future prospects and outlook section.
Kim et al.83 employed a similar asymmetric electrolyte approach to thermally charge a solid-state supercapacitor, using polystyrene sulfonic acid (PSSH) as both a solvent-free electrolyte and a separator film, and polyaniline (PANI)-deposited graphene/nanotube (PANI@CNT–G) composite films as electrodes. The difference from the study done by Zhao et al.82 was the use of PANI@CNT–G as the electrodes, which added the pseudocapacitive contribution of PANI to the double-layer capacitance of graphene and the nanotubes. The thermopotential achieved using PSSH, which is hygroscopic in nature, depended on its relative humidity (RH), varying from about 7.9 mV K−1 at 70% RH, to 6.3 mV K−1 at 50% RH, then 5.1 mV K−1 at 30% RH.
5. Thermal charging in the absence of temperature difference
Creating a temperature difference between the two electrodes in an electrochemical cell is the typical approach to cause a difference in the thermal response at the boundaries in a cell with two identical electrodes. Alternatively, a difference in the thermal response may also be obtained if the two boundaries had inherently dissimilar characteristics, and were subjected to the same increase in temperature. Wang et al.84 reported such an approach as a thermal charging strategy to recover the energy wasted in supercapacitors (charge remaining in the device) after electrical charging and discharging. An electrically charged-then-discharged supercapacitor developed positive voltage when heated under open circuit conditions. This increase in voltage would also occur without heating, albeit on a larger time scale. The phenomenon originates from the difference in kinetics between the fast movement of free electrons in the skeleton of the electrode material, and the slower movement of counter charge at the electrode/electrolyte interface. The latter's slower response to polarization, be it in the form of physical ion adsorption/desorption onto the pores of the electrodes, or chemical reactions of surface functional groups, or both, allows enough time only for the entities (ions/functional groups) with relatively faster kinetics to respond to electric polarization/depolarization upon charge and discharge, leaving the entities with slower kinetics unresponsive, with their associated charge unused. This unused charge would “surface” later and slowly increase the open circuit voltage after the device is discharged. On the other hand, it can be harvested (recovered) more quickly with the application of heat to the device, due to the thermally-induced enhancement in the kinetics of the process involved in charge storage. Fig. 4 shows this concept in a system where electrical charging/discharging takes place at lower (room) temperature (TL) through ion adsorption/desorption on a porous electrode surface. The charge remaining inside the electrode is then thermally harvested at higher temperature (TH). The scheme below assumes that the ions adsorbed on the outer surface of the electrode, or larger-size or shallower pores, would respond more quickly to polarization, compared to those adsorbed inside the smaller-size or deeper ones. This assumption is a reasonable one for porous supercapacitor electrodes, and is consistent with the authors' observation that the thermally-achieved voltage increased when the device was electrically charged to higher voltage or at a slower rate,84 as both conditions provide the force or time for more ions to access as large a portion as possible of the pores in the electrode material, or travel deeper into the pores. This assumption is also consistent with the reported deterioration of the high-rate charge storage capabilities of porous electrodes, due to limited accessibility into the pores.85–92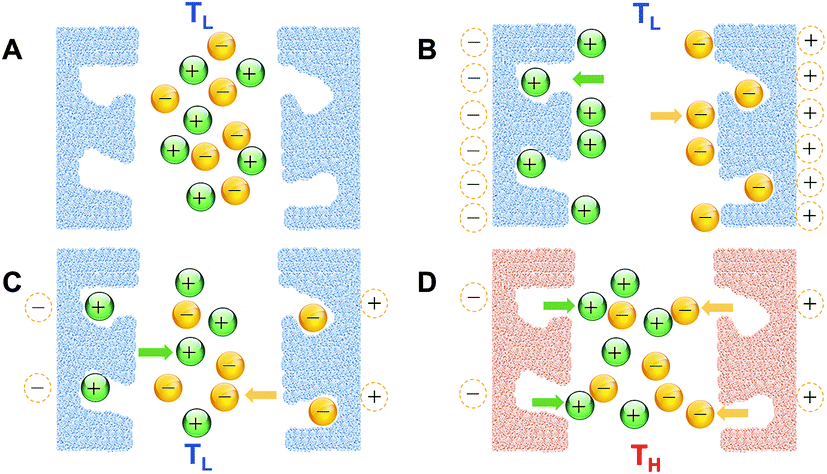 | ||
| Fig. 4 Thermal charging after electrical charging and discharging in the case of charge storage by adsorption/desorption of ions onto the pores of the electrode: (A) device at open-circuit voltage; (B) electrical charging at low T; (C) electrical discharging at low T; (D) thermal charging at high T. | ||
The results obtained above show the possibility of thermal charging without the need to place the cell under a temperature difference, which presents a design advantage since a temperature difference is normally difficult to establish and maintain in a low-grade heat environment.
Lim et al.93 reported a thermal harvesting scheme under a homogeneous temperature field, based on the difference between the working and counter electrode in their surface structure. Using a supercapacitor set-up with 1.0 M sodium acetate/formamide solution, and a counter-electrode of nanoporous carbon, they employed two different types of working electrodes: nickel-coated MWCNTs, or a nickel-coated nanoporous carbon electrode. The thermopotential had a value of 3.6 mV °C−1 for the MWCNT electrode (voltage value of 160 mV for a temperature increase of 45 °C), versus 2 mV °C−1 with the nanocarbon electrode, for which a voltage of 100 mV was generated from a temperature increase as high as 75 °C. The corresponding values of energy density generated in the thermal cycle were 600 and 1800 mJ g−1, attributed to the larger specific surface area of nanocarbon compared to that of the nanotubes.
6. Thermal charging in alternation with electrical charging
The temperature dependence of thermal motion, and hence the possibility of heat-to-electricity conversion, is not limited to devices at rest (a state of zero charge). Thermal effects may also be induced in combination with electric charging/discharging, or other means of energy conversion that the device components are responsive to.In this regard, several studies have reported various variations of a cyclic charging–heating–discharging–cooling (CHDC) scheme (the concept of which is illustrated in Fig. 5), which combines the effect of temperature and concentration on the composition of the double-layer. The cycle, analogous in principle to the Stirling cycle, consists of alternated heating/cooling joined with the energy harvesting technique called “capacitive mixing” (CAPMIX).94 CAPMIX technology involves diverse electrode-based approaches that produce electricity by harvesting “blue energy”, a term that refers to the energy residing in the salinity difference between the salty sea water and fresh river water. The process of extracting that energy involves alternated dipping of porous electrodes in high and low salinity feed solutions. The potential difference across a charged double-layer tends to increase with the decrease in salinity at constant charge, because in lower salinity solutions, ions diffuse away from the electrode, increasing the accumulated electrostatic energy at the expense of the free energy of the solutions, which is known as “capacitive double layer expansion” (CDLE). Accordingly, the potential difference is generated when the electrodes are electrically charged in salty water, then discharged in fresh water.
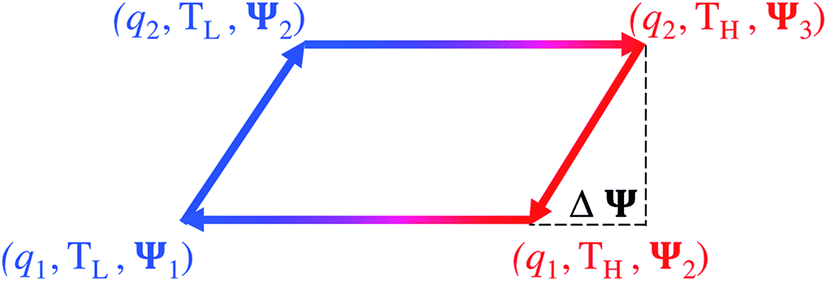 | ||
| Fig. 5 Charging–heating–discharging–cooling (CHDC) cycle. When integrated with the CAPMIX process, charging takes place in the solution of higher salinity (cold sea water), and discharging takes place in the solution of lower salinity (warm river water, typically warmed from mixing the output coolant stream from industrial processes). | ||
Sales et al.95 demonstrated a harvesting scheme that combines CHDC cycles and the phenomenon of thermal membrane potential, according to which if a temperature difference is applied on two half-cells of identical electrolyte concentration and connected through a membrane, an increase in the membrane potential would occur.96,97 This potential difference is caused by the difference in the components' distribution at the membrane–solution interfaces, and also by the thermoelectrodiffusion of ions inside the membrane and in the surrounding solution.98 The system consisted of titanium rods coated with porous electrodes of activated carbon, and sealed by ion exchange membranes, in 0.5 M NaCl solution electrolyte. Two pairs of electrodes were coated with a cation-exchange membrane (CEM) and an anion-exchange membrane (AEM), respectively. Then, the two individual electrodes of each pair were connected through an external load, and alternatingly dipped in two different electrolyte reservoirs: a “cold” reservoir where ion adsorption occurs, and a “warm” one for ion desorption. A temperature difference of ΔT of 44 K between the two reservoirs generated a voltage of about 0.17 mV, which when normalized by the number of membranes used, corresponded to a thermopotential of 0.097 mV K−1. The energy extracted from a 30 °C temperature difference and using a 15 Ω load was about 2 mJ m−2, and the efficiency relative to the Carnot efficiency was 0.002%.
Another variation of this scheme was proposed by Ahualli et al.,99 who combined the thermal effect and salinity effect on the ion kinetics in the double layer of porous electrodes in a joint CHDC–CAPMIX cycle. Their approach was proposed to harvest the temperature difference between the input and output water used as a cooling medium in industrial processes. The authors attributed the increase in the potential of the charged electrodes mainly to the effect of increasing the temperature on the permittivity of the electrolyte (water), according to the equation:99,100
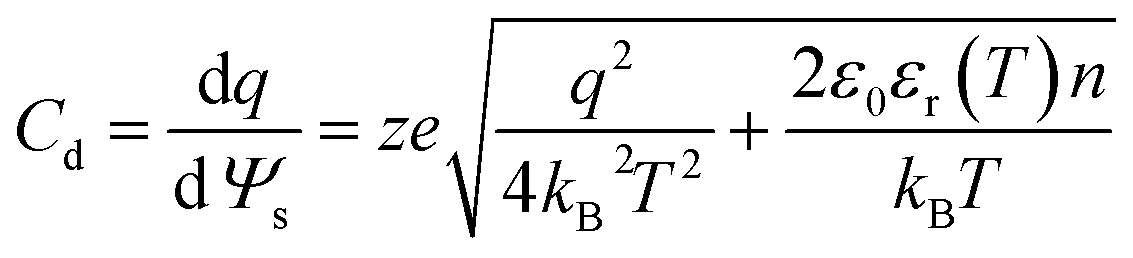 | (17) |
A similar CHDC/CAPMIX combination was separately proposed by Janssen et al.,101 as a means for boosting the efficiency of blue energy harvesting, compared to that of the CAPMIX process alone. Using a planar slit-pore model, they theoretically predicted a two-fold increase in the work extracted from the CAPMIX cycle, if a temperature difference on the order of 10–50 °C is maintained between the charging and discharging solutions, and a decrease in the energetic cost of the sea water desalination process for a temperature difference of 10–20 °C. They also predicted that the required temperature difference becomes smaller with the decrease in pore size.
A simplified scheme was proposed by Härtel et al.,47 who employed only the thermocapacitive CHDC cycle to convert heat into electricity, through intermittent application of a temperature gradient between electrical charging and discharging of a commercial supercapacitor, in which the electrolyte concentration is kept constant. During each cycle, the working electrode is charged, then heated to cause the increase in the thermal motion of adsorbed ions, triggering the increase in the voltage of the device. The harvested voltage would be recovered in the subsequent discharge step at high temperature, after which the electrode is cooled back to its initial temperature before the start of the next cycle. They used a commercially available supercapacitor device, with a capacitance of 10 F, and porous activated carbon electrodes (surface area 1200 m2 g−1), which they heated to 65 °C after charging to 2.5 volts. This set-up achieved a voltage of 36 mV, which was increased to 40 mV by increasing the heating rate (an average thermopotential of 0.6 mV °C−1), corresponding to an energy density of 185 mJ g−1, and a figure of merit of 0.0039, versus a theoretically predicted value of 0.066.
7. Performance parameters
To implement the thermocapacitive effect concept as a practical energy harvesting technology and an alternative to electrical charging, it is important to define the parameters of relevance to the thermal charging efficiency, and map the thermo-electrochemical behavior in relation to those parameters.At present, the only systematic investigation concerned with the parameters involved in thermal charging of supercapacitors was carried out by the Qiao group, which addressed the influence of the size of the electrolytes ions,102,103 the electrolyte concentration,80 the solvent medium,104 and the (metallic) electrode work function.105
7.1. The solute
Comparing electrodes of platinum and nanocarbon with a pore size of 3 nm, Lim et al.102 investigated the influence of anion size on the thermopotential, using an array of aqueous solutions of 1.0 M NaF, NaCl, NaBr, and NaI, and observed that an increase in anion size caused the thermopotential to decrease for Pt electrodes, but increase with nanocarbon electrodes. To assess the influence of the cation,103 they used aqueous solutions of LiCl, NaCl, KCl, or CsCl. They observed that by changing the cation size from 0.180 nm (Li1+) to 0.362 nm (Cs1+), the thermopotential dropped from 0.85 mV °C−1 to about 0.52 mV °C−1 for nanocarbon electrodes, while Pt electrodes did not show a monotonic change with cation size.The opposite trends in the dependence of the thermopotential on the ion size observed for cationic and anionic species on porous electrodes were explained by the authors as being due to a more important role for the cationic species in the thermal charging of the nanocarbon electrode. They rationalized their hypothesis by the negatively-charged chemical functionalities that cause cations to be preferentially adsorbed on the surface, rendering the cation size an influencing factor in the thermopotential. Approximating the adsorption behavior by a Temkin isotherm, they derived an expression for the thermopotential:
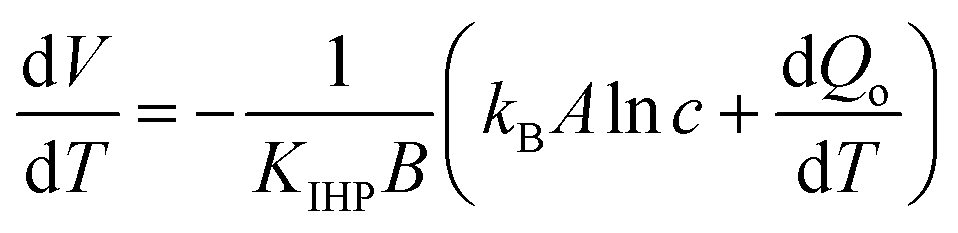 | (18) |
To assess the influence of electrolyte concentration, they used LiCl with concentrations in the range of 0.1 M to 3.7 M, which caused the thermopotential to decrease when Pt electrodes were used. With porous nanocarbon electrodes, no significant influence of the concentration of the electrolyte on the thermopotential was observed within the aforementioned concentration range; the thermopotential varied non-monotonically from 0.86 mV °C−1 at concentration of 0.1 M, to less than 0.75 mV °C−1 for 1.0 M, then 0.88 mV °C−1 for 3.7 M.80
7.2. The solvent
To explore the role of the solvent properties in thermal charging, Lim et al.104 again compared Pt versus porous nanocarbon electrodes, in a 1 M solution of LiCl in DMSO, water, or formamide (FA) as solvent, in order of increasing dielectric constant. For Pt electrodes, the thermopotential correlated positively with the dielectric constant of the solvent, with values of −0.3 mV °C−1, −1.0 mV °C−1, and −5.4 mV °C−1 for DMSO, water, and FA, respectively. With carbon electrodes, the aqueous solution gave the highest thermopotential (0.68 mV °C−1), followed by lower values for FA (0.23 mV °C−1), and DMSO (0.15 mV °C−1). The authors attributed the results to the restriction imposed by the small space inside the nanopores, which causes the size of the solvated ion (and hence that of the solvent molecule) to play a more important role in thermal charging, since the water molecules which gave the highest thermopotential had significantly smaller size (0.25 nm, compared to 0.47 nm for DMSO and 0.62 nm for FA).7.3. The electrode
Whether electrically or thermally induced, ion adsorption is a process that takes place at the interface of two different surfaces: the electrically conductive surface of the electrode, and the ionically conductive electrolyte solution. Thus, the contribution of the electrode properties to the mechanism and efficiency of thermal charging becomes an important factor to be investigated. To incorporate that factor, Lim et al.105 used the contribution of the electrode's work function to the capacitance as described by the Jellium model. This model divides a metal electrode into a core of positively charged ions, and a gas of negatively charged electrons homogenously dispersed on the metal. The positive ions disappear abruptly at one edge of the metal, while the electron gas decays slowly in the form of a tail near the opposite edge, which is known as the “electron spillover effect”. Being the moving part of the metal, the electrons define the metal behavior and its contribution to the capacitance. This contribution will depend on the interaction between these electrons and their positively charged background on the metal (and hence the properties of the metal), and these interactions are captured by the work function of the metal.106By performing thermal charging of four different metallic electrodes (platinum, nickel, copper, or indium), in a solution of LiCl/FA, the authors reported that the thermopotential values for Cu (1.4 mV K−1), Ni (4.1 mV K−1), and Pt 6.5 (mV K−1), correlated linearly with their work function values of 4.8 eV, 5.2 eV, and 5.5 eV, respectively. On the other hand, the data for In (work function 4.1 eV) were scattered and showed a small change in voltage with temperature of 0.4 mV K−1.
Interestingly, the authors reported a nonlinear trend in the relation between the thermopotential and temperature, which they attributed to the enhancement in the adsorption coverage on the electrode at higher temperature. However, one needs to consider the possibility of temperature-induced changes in the electrode properties, contributing to the nonlinearity. It is also important to bear in mind that porous electrodes, which are more relevant to the design of supercapacitors than metallic electrodes, are characteristically different and more complicated in chemical and surface structure compared to metallic electrodes, and require deeper investigation to probe their role in thermal charging.
8. Future prospects and outlook
Despite being based on old physical concepts, thermal charging needs to be addressed as a charging technique for electrochemical capacitors, through defining and controlling the relevant design factors, towards enhancing the generated voltage to a level comparable to that realized through electrical charging. The factors influencing thermal charging may be classified into two categories: factors similar to those involved in electrical charging, and factors that stem from the “dynamic properties” of the materials in the device. Here the word “dynamic” is not used to imply that the properties change with time, but rather with temperature. We propose below some of the factors that can be addressed for a better understanding of the subject:8.1. The electrode side of the story
Apart from the redox reactions of polyaniline-deposited electrodes reported by Kim et al.,83 the currently available studies addressed the changes that occur only on the electrolyte side of the interface. This overlooks any contribution that may stem from the change in the electrode properties with temperature or with accumulation of ions on the surface, which needs to be taken into account as well. For example, the work function of a material, which marks the electrode contribution to the capacitance according to the Jellium model, varies with temperature and with adsorption of ions on the surface as well.107–116 The thermal expansion,117–126 and the electronic structure and electric conductivity,127–131 are also temperature dependent, and may reflect the kinetics of thermally-induced charge storage. It is also important to note that the physical mechanism of thermal charging does not necessarily rule out the possibility of reversible or irreversible temperature-dependent charge transfer between the electrode and electrolyte, which is plausible for porous electrode materials that may contain chemically active functionalities. To monitor the change in the electrode behavior during charging, in situ characterization techniques like Raman spectroscopy,132–143 and Kelvin probe force144,145 and electrochemical force microscopy,146,147 offer valuable tracking tools that can be coupled with thermal charging to probe the local structural and electronic changes occurring on the surface of the electrode during its interaction with other species from its surrounding medium.8.2. The ion-to-pore-size relation
The effect of pore geometry is a critical factor and a common research topic in the field of electrically chargeable supercapacitors,88,148–162 and similar experimental investigation is necessary in thermal charging, for good reasons. First, the current studies did not reveal a universal trend in the sign of the thermopotential with temperature, and the available experimental data are limited and insufficient to draw conclusions. This is because the exact changes occurring for the ion–surface interaction with temperature are yet to be completely clarified, and need to be examined not only in relation to the surface chemistry, but also the feasibility (and fraction) of ion accessibility into the pores. For example, a molecular dynamics study by Vatamanu et al.,163 which investigated the influence of temperature on the differential capacitance of ionic liquids near atomically flat and atomically corrugated (rough) graphene surfaces, showed that at the potential of zero charge, the ion density profile near strongly corrugated graphene surfaces showed a strong preference for the cation to partition to the surface, which in their case was related to size compatibility between the cation and the grooves of the graphitic surface. These effects occurred without the assumption of chemical functionalities attached to the surface, and caused the surface density profile of the cation to change more conspicuously with temperature than that of the anion. In other words, a comprehensive map that elucidates the origin of the magnitude and sign of the thermopotential requires systematic experimental investigation of an expanded array of electrode/electrolyte couples, to include effects from diverse variables on the thermal response of ions near the porous electrode surface.The second reason is related to the performance and efficiency of thermal charging. In addition to the evaluation of a thermally-chargeable capacitor as an energy harvesting device, i.e. by comparing its efficiency to that of the Carnot cycle, it is worthy, from a practical standpoint, to address its efficiency as an energy storage device, by comparing the performance (achieved voltage, rate performance, coulombic efficiency, cyclability, etc.) of thermal charging to that achieved electrically for the same device components. The pore geometry can influence the characteristics of thermodiffusion,164,165 since the pores that act as electrolyte reservoirs, can modify the thermal response of the preadsorbed species, through the interaction of the pore walls with that species.66,166–168 The extent of modification will depend, in addition to the surface chemistry, on the pore geometry.169,170 This suggests that the charge storage mechanism behind thermal charging of porous materials may involve elements characteristically different from those involved in electrical charging.
8.3. Design considerations
It is understandable with research on the subject at its earliest stages, that the currently reported values of generated voltage form only a fraction of those achieved by electrical charging, which necessitates materials and strategies that reduce the gap between the two. To that end, we examine some of the properties that may make a material beneficial for understanding the thermal charging mechanism, or increasing the achieved voltage.Protonic ionic liquids were reported to achieve higher Seebeck coefficient values compared to other ionic liquids used as solvents in thermogalvanic cells,30 and may be interesting to investigate for thermally chargeable capacitors as well, especially considering that they bring along the previously mentioned merits that render all of the ionic liquid family attractive to use in an elevated temperature environment.
Finally, and by trailing the evolution of electrically chargeable supercapacitors, it is possible to further increase the generated voltage by employing redox-additive or redox-active electrolytic solutions174 in a “thermogalvanic supercapacitor” design, in which the electrodes are structurally and chemically designed to combine physical and chemical contributions from both the electrode and the electrolyte, to convert heat to electricity.
Considering the recency of the topic, the current review is not likely to have captured all of the factors that need to be addressed or raised all of the necessary questions that need to be answered with regards to thermal charging in supercapacitors. Further research will obviously raise additional or different questions, or define new elements that determine the direction of research on the subject. Nonetheless, an important purpose of the present review is to attract the attention of researchers from the two fields of energy storage and thermal energy harvesting, through summarizing the strategies that successfully managed to combine the targets of both fields in a single device, and to provide tentative suggestions that may assist and encourage the pursuit of further research to advance this newborn technology.
Acknowledgements
This work was supported by the National Basic Research Program of China (2012CB933003), and the National Natural Science Foundation of China (No. 51272057).References
- S. Percy, C. Knight, S. McGarry, A. Post, T. Moore and K. Cavanagh, in Thermal Energy Harvesting for Application at MEMS Scale, Springer, New York, 2014, pp. 1–6 Search PubMed.
- S. Twaha, J. Zhu, Y. Yan and B. Li, Renewable Sustainable Energy Rev., 2016, 65, 698–726 CrossRef CAS.
- Thermoelectrics Handbook: Macro to Nano, ed. D. M. Rowe, CRC Press, Boca Raton, 1st edn, 2005 Search PubMed.
- X. Shi, L. Chen and C. Uher, Int. Mater. Rev., 2016, 61, 379–415 CrossRef CAS.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16631–16634 CrossRef CAS.
- A. Mehdizadeh Dehkordi, M. Zebarjadi, J. He and T. M. Tritt, Mater. Sci. Eng., R, 2015, 97, 1–22 CrossRef.
- C. Godart, A. P. Gonçalves, E. B. Lopes and B. Villeroy, in Properties and Applications of Thermoelectric Materials, ed. V. Zlatić and A. C. Hewson, Springer, Netherlands, 2009, pp. 19–49 Search PubMed.
- S. V. Faleev and F. Léonard, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 214304 CrossRef.
- W. J. Xie, Y. G. Yan, S. Zhu, M. Zhou, S. Populoh, K. Gałązka, S. J. Poon, A. Weidenkaff, J. He, X. F. Tang and T. M. Tritt, Acta Mater., 2013, 61, 2087–2094 CrossRef CAS.
- C. Gayner and K. K. Kar, Prog. Mater. Sci., 2016, 83, 330–382 CrossRef CAS.
- S. LeBlanc, S. K. Yee, M. L. Scullin, C. Dames and K. E. Goodson, Renewable Sustainable Energy Rev., 2014, 32, 313–327 CrossRef CAS.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS PubMed.
- G. Wang, L. Endicott and C. Uher, Sci. Adv. Mater., 2011, 3, 539–560 CrossRef CAS.
- S. Fan, J. Zhao, J. Guo, Q. Yan, J. Ma and H. H. Hng, Appl. Phys. Lett., 2010, 96, 182104 CrossRef.
- A. Dey, O. P. Bajpai, A. K. Sikder, S. Chattopadhyay and M. A. Shafeeuulla Khan, Renewable Sustainable Energy Rev., 2016, 53, 653–671 CrossRef CAS.
- S. B. Riffat and X. Ma, Appl. Therm. Eng., 2003, 23, 913–935 CrossRef.
- A. Lambrecht, H. Böttner, J. Nurnus, A. Gopinath, T. J. Coutts and J. Luther, AIP Conf. Proc., 2004, 738, 24–32 CrossRef CAS.
- W. Liu, Z. Ren and G. Chen, in Thermoelectric Nanomaterials, ed. K. Koumoto and T. Mori, Springer, Berlin Heidelberg, 2013, pp. 255–285 Search PubMed.
- P. F. Salazar, S. Kumar and B. A. Cola, J. Appl. Electrochem., 2014, 44, 325–336 CrossRef CAS.
- H. L. Chum and R. A. Osteryoung, Review of thermally regenerative electrochemical systems, Solar Energy Research Institute, Golden, Colorado, 1981 Search PubMed.
- J. W. Tester, Evaluation of thermogalvanic cells for the conversion of heat to electricity, MIT Energy Lab, 1992 Search PubMed.
- R. Hu, B. A. Cola, N. Haram, J. N. Barisci, S. Lee, S. Stoughton, G. Wallace, C. Too, M. Thomas, A. Gestos, M. E. dela Cruz, J. P. Ferraris, A. A. Zakhidov and R. H. Baughman, Nano Lett., 2010, 10, 838–846 CrossRef CAS PubMed.
- T. I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CrossRef CAS.
- H. A. H. Alzahrani, J. J. Black, D. Goonetilleke, J. Panchompoo and L. Aldous, Electrochem. Commun., 2015, 58, 76–79 CrossRef CAS.
- M. Bonetti, S. Nakamae, M. Roger and P. Guenoun, J. Chem. Phys., 2011, 134, 114513 CrossRef CAS PubMed.
- K. E. Johnson and S. J. Sime, Electrochim. Acta, 1977, 22, 1043–1046 CrossRef CAS.
- Y. V. Kuzminskii, V. A. Zasukha and G. Y. Kuzminskaya, J. Power Sources, 1994, 52, 231–242 CrossRef CAS.
- I. D. Raistrick, J. Poris and R. A. Huggins, US4315059 A, 1982.
- A. Gunawan, C.-H. Lin, D. A. Buttry, V. Mujica, R. A. Taylor, R. S. Prasher and P. E. Phelan, Nanoscale Microscale Thermophys. Eng., 2013, 17, 304–323 CrossRef CAS.
- E. Laux, S. Uhl, T. Journot, J. Brossard, L. Jeandupeux and H. Keppner, J. Electron. Mater., 2016, 45, 3383–3389 CrossRef CAS.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Chem. Commun., 2011, 47, 6260–6262 RSC.
- M. A. Lazar, D. Al-Masri, D. R. MacFarlane and J. M. Pringle, Phys. Chem. Chem. Phys., 2016, 18, 1404–1410 RSC.
- P. Yang, K. Liu, Q. Chen, X. Mo, Y. Zhou, S. Li, G. Feng and J. Zhou, Angew. Chem,. Int. Ed., 2016, 55, 12050–12053 CrossRef CAS PubMed.
- G. Z. Chen, Int. Mater. Rev., 2017, 62, 173–202 CrossRef CAS.
- Z. Zhang, X. Chen, P. Chen, G. Guan, L. Qiu, H. Lin, Z. Yang, W. Bai, Y. Luo and H. Peng, Adv. Mater., 2014, 26, 466–470 CrossRef CAS PubMed.
- J. Maeng, C. Meng and P. P. Irazoqui, Biomed. Microdevices, 2015, 17, 7 CrossRef PubMed.
- J. Luo, W. Tang, F. R. Fan, C. Liu, Y. Pang, G. Cao and Z. L. Wang, ACS Nano, 2016, 10, 8078–8086 CrossRef CAS PubMed.
- J. Luo, F. R. Fan, T. Jiang, Z. Wang, W. Tang, C. Zhang, M. Liu, G. Cao and Z. L. Wang, Nano Res., 2015, 8, 3934–3943 CrossRef.
- J. Pörhönen, S. Rajala, S. Lehtimäki and S. Tuukkanen, IEEE Trans. Electron Devices, 2014, 61, 3303–3308 CrossRef.
- N. Bagheri, A. Aghaei, M. Y. Ghotbi, E. Marzbanrad, N. Vlachopoulos, L. Häggman, M. Wang, G. Boschloo, A. Hagfeldt, M. Skunik-Nuckowska and P. J. Kulesza, Electrochim. Acta, 2014, 143, 390–397 CrossRef CAS.
- F. Zhou, Z. Ren, Y. Zhao, X. Shen, A. Wang, Y. Y. Li, C. Surya and Y. Chai, ACS Nano, 2016, 10, 5900–5908 CrossRef CAS PubMed.
- Z. Yang, L. Li, Y. Luo, R. He, L. Qiu, H. Lin and H. Peng, J. Mater. Chem. A, 2012, 1, 954–958 RSC.
- G. Wee, T. Salim, Y. M. Lam, S. G. Mhaisalkar and M. Srinivasan, Energy Environ. Sci., 2011, 4, 413–416 CAS.
- A. Ramadoss, B. Saravanakumar, S. W. Lee, Y.-S. Kim, S. J. Kim and Z. L. Wang, ACS Nano, 2015, 9, 4337–4345 CrossRef CAS PubMed.
- T. N. Murakami, N. Kawashima and T. Miyasaka, Chem. Commun., 2005, 3346–3348 RSC.
- T. Miyasaka and T. N. Murakami, Appl. Phys. Lett., 2004, 85, 3932–3934 CrossRef CAS.
- A. Härtel, M. Janssen, D. Weingarth, V. Presser and R. van Roij, Energy Environ. Sci., 2015, 8, 2396–2401 Search PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- R. Tadmor, J. Colloid Interface Sci., 2009, 332, 451–454 CrossRef CAS PubMed.
- L. E. Scriven and C. V. Sternling, Nature, 1960, 187, 186–188 CrossRef.
- V. Pratap, N. Moumen and R. S. Subramanian, Langmuir, 2008, 24, 5185–5193 CrossRef CAS PubMed.
- J.-P. Delville, M. R. d. S. Vincent, R. D. Schroll, H. Chraïbi, B. Issenmann, R. Wunenburger, D. Lasseux, W. W. Zhang and E. Brasselet, J. Opt. A: Pure Appl. Opt., 2009, 11, 34015 CrossRef.
- V. A. Eroshenko, Cycle of Transformation of Thermal Energy into Mechanical Energy and Devices to Achieve It, Soviet Patent No. 1,254,811, 1981.
- A. Laouir, L. Luo, D. Tondeur, T. Cachot and P. Le Goff, AIChE J., 2003, 49, 764–781 CrossRef CAS.
- X. Kong and Y. Qiao, Philos. Mag. Lett., 2005, 85, 331–337 CrossRef CAS.
- Y. Qiao, V. K. Punyamurtula, A. Han, X. Kong and F. B. Surani, Appl. Phys. Lett., 2006, 89, 251905 CrossRef.
- A. Han and Y. Qiao, Appl. Phys. Lett., 2007, 91, 173123 CrossRef.
- A. Han, W. Lu, T. Kim, V. K. Punyamurtula and Y. Qiao, Smart Mater. Struct., 2009, 18, 24005 CrossRef.
- B. Xu, Y. Qiao, T. Park, M. Tak, Q. Zhou and X. Chen, Energy Environ. Sci., 2011, 4, 3632–3639 CAS.
- X. Kong, F. B. Surani and Y. Qiao, Phys. Scr., 2006, 74, 531 CrossRef CAS.
- A. Han, X. Chen and Y. Qiao, Langmuir, 2008, 24, 7044–7047 CrossRef CAS PubMed.
- V. D. Borman, A. A. Belogorlov, G. V. Lisichkin, V. N. Tronin and V. I. Troyan, J. Exp. Theor. Phys., 2009, 108, 389–410 CrossRef CAS.
- C. F. W. Ludwig, Sitzungsberichte Akad. Wiss. Wien Math., Nat. Sci., 1856, 20, 539 Search PubMed.
- C. Soret, Arch. Sci. Phys. Nat., 1879, 2, 48–61 Search PubMed.
- P. Geelhoed, J. Westerweel, S. Kjelstrup and D. Bedeaux, in Encyclopedia of Microfluidics and Nanofluidics, ed. D. Li, Springer, US, 2008, pp. 2061–2064 Search PubMed.
- J. N. Agar and J. C. R. Turner, Proc. R. Soc. London, Ser. A, 1960, 255, 307–330 CrossRef.
- P. N. Snowdon and J. C. R. Turner, Trans. Faraday Soc., 1960, 56, 1812–1819 RSC.
- D. R. Caldwell, J. Phys. Chem., 1973, 77, 2004–2008 CrossRef CAS.
- J. K. G. Dhont, J. Chem. Phys., 2004, 120, 1632–1641 CrossRef CAS PubMed.
- J. K. G. Dhont, J. Chem. Phys., 2004, 120, 1642–1653 CrossRef CAS PubMed.
- M. R. Wright, An Introduction to Aqueous Electrolyte Solutions, John Wiley & Sons, 2007 Search PubMed.
- E. Helfand and J. G. Kirkwood, J. Chem. Phys., 1960, 32, 857–866 CrossRef CAS.
- A. Würger, Rep. Prog. Phys., 2010, 73, 126601 CrossRef.
- A. Würger, Phys. Rev. Lett., 2008, 101, 108302 CrossRef PubMed.
- S. A. Putnam and D. G. Cahill, Langmuir, 2005, 21, 5317–5323 CrossRef CAS PubMed.
- A. Yoshimori, Condens. Matter Phys., 2007, 10, 563–571 CrossRef.
- I. Chikina, V. Shikin and A. A. Varlamov, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 11505 CrossRef CAS PubMed.
- Y. Qiao, V. K. Punyamurtula and A. Han, Appl. Phys. Lett., 2007, 91, 153102 CrossRef.
- Y. Qiao, V. K. Punyamurtual, A. Han and H. Lim, J. Power Sources, 2008, 183, 403–405 CrossRef CAS.
- H. Lim, W. Lu, X. Chen and Y. Qiao, Nanotechnology, 2013, 24, 465401 CrossRef PubMed.
- M. Bonetti, S. Nakamae, B. T. Huang, T. J. Salez, C. Wiertel-Gasquet and M. Roger, J. Chem. Phys., 2015, 142, 244708 CrossRef PubMed.
- D. Zhao, H. Wang, Z. U. Khan, J. Chen, R. Karlsson, M. P. Jonsson, M. Berggren and X. Crispin, Energy Environ. Sci., 2016, 9, 1450–1457 CAS.
- S. L. Kim, H. T. Lin and C. Yu, Adv. Energy Mater., 2016, 6, 1600546 CrossRef.
- J. Wang, S.-P. Feng, Y. Yang, N. Y. Hau, M. Munro, E. Ferreira-Yang and G. Chen, Nano Lett., 2015, 15, 5784–5790 CrossRef CAS PubMed.
- R. de Levie, Electrochim. Acta, 1963, 8, 751–780 CrossRef.
- W. G. Pell, B. E. Conway, W. A. Adams and J. de Oliveira, J. Power Sources, 1999, 80, 134–141 CrossRef CAS.
- W. G. Pell, B. E. Conway and N. Marincic, J. Electroanal. Chem., 2000, 491, 9–21 CrossRef CAS.
- B. E. Conway and W. G. Pell, J. Power Sources, 2002, 105, 169–181 CrossRef CAS.
- M. J. Bleda-Martínez, D. Lozano-Castelló, D. Cazorla-Amorós and E. Morallón, Energy Fuels, 2010, 24, 3378–3384 CrossRef.
- B. Daffos, P.-L. Taberna, Y. Gogotsi and P. Simon, Fuel Cells, 2010, 10, 819–824 CrossRef CAS.
- A. Al-zubaidi, T. Inoue, T. Matsushita, Y. Ishii and S. Kawasaki, Phys. Chem. Chem. Phys., 2012, 14, 16055–16061 RSC.
- T. M. Arruda, M. Heon, V. Presser, P. C. Hillesheim, S. Dai, Y. Gogotsi, S. V. Kalinin and N. Balke, Energy Environ. Sci., 2012, 6, 225–231 Search PubMed.
- H. Lim, Y. Shi and Y. Qiao, Appl. Phys. A, 2016, 122, 443 CrossRef.
- D. Brogioli, R. Ziano, R. A. Rica, D. Salerno and F. Mantegazza, J. Colloid Interface Sci., 2013, 407, 457–466 CrossRef CAS PubMed.
- B. B. Sales, O. S. Burheim, S. Porada, V. Presser, C. J. N. Buisman and H. V. M. Hamelers, Environ. Sci. Technol. Lett., 2014, 1, 356–360 CrossRef CAS.
- G. J. Hills, P. W. M. Jacobs and N. Lakshiminarayanaiah, Nature, 1957, 179, 96–97 CrossRef CAS.
- T. Ikeda, J. Chem. Phys., 1958, 28, 166–167 CrossRef CAS.
- M. Jokinen, J. A. Manzanares, K. Kontturi and L. Murtomäki, J. Membr. Sci., 2016, 499, 234–244 CrossRef CAS.
- S. Ahualli, M. M. Fernández, G. Iglesias, Á. V. Delgado and M. L. Jiménez, Environ. Sci. Technol., 2014, 48, 12378–12385 CrossRef CAS PubMed.
- A. de Keizer, in Fundamentals of Interface and Colloid Science, ed. J. Lyklema, Academic Press, 1995, vol. 2, pp. 3.1–3.232. Search PubMed.
- M. Janssen, A. Härtel and R. van Roij, Phys. Rev. Lett., 2014, 113, 268501 CrossRef PubMed.
- H. Lim, W. Lu, X. Chen and Y. Qiao, Int. J. Electrochem. Sci., 2012, 7, 2577–2583 CAS.
- H. Lim, W. Lu and Y. Qiao, Appl. Phys. Lett., 2012, 101, 63902 CrossRef.
- H. Lim, C. Zhao and Y. Qiao, Phys. Chem. Chem. Phys., 2014, 16, 12728–12730 RSC.
- H. Lim, Y. Shi, M. Wang and Y. Qiao, Appl. Phys. Lett., 2015, 106, 223901 CrossRef.
- J. O. Bockris, A. K. N. Reddy and M. E. Gamboa-Aldeco, Modern Electrochemistry 2A – Fundamentals of Electrodics, J. O'. M. Bockris, Springer, 2000 Search PubMed.
- S. Trasatti, Surf. Sci., 1995, 335, 1–9 CrossRef CAS.
- S. Halas and T. Durakiewicz, Vacuum, 2010, 85, 486–488 CrossRef CAS.
- C. G. Vayenas, S. Bebelis, I. V. Yentekakis, P. Tsiakaras and H. Karasali, Platinum Met. Rev., 1990, 122–130 CAS.
- F. Gossenberger, T. Roman, K. Forster-Tonigold and A. Groß, Beilstein J. Nanotechnol., 2014, 5, 152–161 CrossRef PubMed.
- F. Silva, M. J. Sottomayor and A. Martins, J. Chem. Soc., Faraday Trans., 1996, 92, 3693–3699 RSC.
- R. Rahemi and D. Li, Scr. Mater., 2015, 99, 41–44 CrossRef CAS.
- I. Riess and C. G. Vayenas, Solid State Ioics., 2003, 159, 313–329 CrossRef CAS.
- T. Neubrand, S. Günther, A. Fenske and R. Imbihl, Phys. Chem. Chem. Phys., 2004, 6, 3569–3575 RSC.
- S. van Reenen, S. Kouijzer, R. A. J. Janssen, M. M. Wienk and M. Kemerink, Adv. Mater. Interfaces, 2014, 1, 1400189 CrossRef.
- D. Tsiplakides and C. G. Vayenas, J. Electrochem. Soc., 2001, 148, E189–E202 CrossRef CAS.
- F. Huang, K. T. Yue, P. Tan, S.-L. Zhang, Z. Shi, X. Zhou and Z. Gu, J. Appl. Phys., 1998, 84, 4022–4024 CrossRef CAS.
- H. D. Li, K. T. Yue, Z. L. Lian, Y. Zhan, L. X. Zhou, S. L. Zhang, Z. J. Shi, Z. N. Gu, B. B. Liu, R. S. Yang, H. B. Yang, G. T. Zou, Y. Zhang and S. Iijima, Appl. Phys. Lett., 2000, 76, 2053–2055 CrossRef CAS.
- T. Uchida, M. Tachibana, S. Kurita and K. Kojima, Chem. Phys. Lett., 2004, 400, 341–346 CrossRef CAS.
- H. Jiang, B. Liu, Y. Huang and K. C. Hwang, J. Eng. Mater. Technol., 2004, 126, 265–270 CrossRef CAS.
- L. Deng, R. J. Young, I. A. Kinloch, R. Sun, G. Zhang, L. Noé and M. Monthioux, Appl. Phys. Lett., 2014, 104, 51907 CrossRef.
- A. V. Dolbin, V. B. Esel'son, V. G. Gavrilko, V. G. Manzhelii, N. A. Vinnikov and S. N. Popov, Low Temp. Phys., 2008, 34, 678–679 CrossRef CAS.
- S. Sahoo, A. P. S. Gaur, M. Ahmadi, M. J.-F. Guinel and R. S. Katiyar, J. Phys. Chem. C, 2013, 117, 9042–9047 CAS.
- A. Kondo and K. Maeda, J. Solid State Chem., 2015, 221, 126–131 CrossRef CAS.
- C. Yang, X. Wang and M. A. Omary, Angew. Chem,. Int. Ed., 2009, 48, 2500–2505 CrossRef CAS PubMed.
- W. Gao and R. Huang, J. Mech. Phys. Solids, 2014, 66, 42–58 CrossRef CAS.
- S. B. Cronin, Y. Yin, A. Walsh, R. B. Capaz, A. Stolyarov, P. Tangney, M. L. Cohen, S. G. Louie, A. K. Swan, M. S. Ünlü, B. B. Goldberg and M. Tinkham, Phys. Rev. Lett., 2006, 96, 127403 CrossRef CAS PubMed.
- N. R. Raravikar, P. Keblinski, A. M. Rao, M. S. Dresselhaus, L. S. Schadler and P. M. Ajayan, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 235424 CrossRef.
- P. M. Rafailov, M. Monev, R. Bretzler, S. Evtimova, B. Arnaudov, C. Thomsen, U. Dettlaff-Weglikowska and S. Roth, Phys. Status Solidi B, 2010, 247, 2801–2804 CrossRef CAS.
- L. Zhang, Z. Jia, L. Huang, S. O'Brien and Z. Yu, J. Phys. Chem. C, 2008, 112, 13893–13900 CAS.
- M. Gleeson, B. Kasemo and D. Chakarov, Surf. Sci., 2003, 524, L77–L83 CrossRef CAS.
- Z. Komínková and M. Kalbáč, Phys. Status Solidi B, 2016, 253, 2331–2335 CrossRef.
- M. Bouša, O. Frank, I. Jirka and L. Kavan, Phys. Status Solidi B, 2013, 250, 2662–2667 CrossRef.
- P. M. Rafailov, M. Stoll, J. Maultzsch and C. Thomsen, AIP Conf. Proc., 2004, 723, 153–156 CrossRef CAS.
- M. Kalbac, L. Kavan, M. Zukalova and L. Dunsch, Phys. Status Solidi B, 2007, 244, 4086–4091 CrossRef CAS.
- M. Stoll, P. M. Rafailov, W. Frenzel and C. Thomsen, Chem. Phys. Lett., 2003, 375, 625–631 CrossRef CAS.
- P. Corio, A. Jorio, N. Demir and M. S. Dresselhaus, Chem. Phys. Lett., 2004, 392, 396–402 CrossRef CAS.
- A. Al-zubaidi, Y. Ishii, S. Yamada, T. Matsushita and S. Kawasaki, Phys. Chem. Chem. Phys., 2013, 15, 20672–20678 RSC.
- L. J. Hardwick, M. Hahn, P. Ruch, M. Holzapfel, W. Scheifele, H. Buqa, F. Krumeich, P. Novák and R. Kötz, Electrochim. Acta, 2006, 52, 675–680 CrossRef CAS.
- J. Tarábek, L. Kavan, L. Dunsch and M. Kalbac, J. Phys. Chem. C, 2008, 112, 13856–13861 Search PubMed.
- M. Kalbac, L. Kavan, L. Dunsch and M. S. Dresselhaus, Nano Lett., 2008, 8, 1257–1264 CrossRef CAS PubMed.
- M. Kalbac, L. Kavan and L. Dunsch, J. Phys. Chem. C, 2009, 113, 1340–1345 CAS.
- P. Corio, P. S. Santos, V. W. Brar, G. G. Samsonidze, S. G. Chou and M. S. Dresselhaus, Chem. Phys. Lett., 2003, 370, 675–682 CrossRef CAS.
- D. Tsiplakides, D. Archonta and C. G. Vayenas, Top. Catal., 2007, 44, 469–479 CrossRef CAS.
- L. Collins, J. I. Kilpatrick, I. V. Vlassiouk, A. Tselev, S. A. L. Weber, S. Jesse, S. V. Kalinin and B. J. Rodrigue, Appl. Phys. Lett., 2014, 104, 133103 CrossRef.
- L. Collins, S. Jesse, J. I. Kilpatrick, A. Tselev, O. Varenyk, M. B. Okatan, S. A. L. Weber, A. Kumar, N. Balke, S. V. Kalinin and B. J. Rodriguez, Nat. Commun., 2014, 5, 3871 CAS.
- L. Collins, S. Jesse, J. I. Kilpatrick, A. Tselev, M. B. Okatan, S. V. Kalinin and B. J. Rodriguez, Beilstein J. Nanotechnol., 2015, 6, 201–214 CrossRef CAS PubMed.
- D. Lozano-Castelló, D. Cazorla-Amorós, A. Linares-Solano, S. Shiraishi, H. Kurihara and A. Oya, Carbon, 2003, 41, 1765–1775 CrossRef.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498–2507 CrossRef.
- R. Lin, P. L. Taberna, J. Chmiola, D. Guay, Y. Gogotsi and P. Simon, J. Electrochem. Soc., 2009, 156, A7–A12 CrossRef CAS.
- S. Kondrat, C. R. Pérez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 CAS.
- T. E. Rufford, D. Hulicova-Jurcakova, Z. Zhu and G. Q. Lu, J. Phys. Chem. C, 2009, 113, 19335–19343 CAS.
- G. Salitra, A. Soffer, L. Eliad, Y. Cohen and D. Aurbach, J. Electrochem. Soc., 2000, 147, 2486–2493 CrossRef CAS.
- P. Simon and Y. Gogotsi, Philos. Trans. R. Soc., A, 2010, 368, 3457–3467 CrossRef CAS PubMed.
- H.-K. Song, Y.-H. Jung, K.-H. Lee and L. H. Dao, Electrochim. Acta, 1999, 44, 3513–3519 CrossRef CAS.
- P. Wu, J. Huang, V. Meunier, B. G. Sumpter and R. Qiao, J. Phys. Chem. Lett., 2012, 3, 1732–1737 CrossRef CAS PubMed.
- H. Yamada, H. Nakamura, F. Nakahara, I. Moriguchi and T. Kudo, J. Phys. Chem. C, 2007, 111, 227–233 CAS.
- C. M. Yang, H. J. Jung and Y. J. Kim, J. Colloid Interface Sci., 2015, 446, 208–212 CrossRef CAS PubMed.
- S. M. Mahurin, E. Mamontov, M. W. Thompson, P. Zhang, C. H. Turner, P. T. Cummings and S. Dai, Appl. Phys. Lett., 2016, 109, 143111 CrossRef.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem,. Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- J. Vatamanu, L. Xing, W. Li and D. Bedrov, Phys. Chem. Chem. Phys., 2014, 16, 5174–5182 RSC.
- H. A. Maier, M. J. Hampe and P. A. Bopp, Chem. Phys. Lett., 2011, 518, 55–60 CrossRef CAS.
- S. Yeganegi and E. Pak, Chem. Phys., 2007, 333, 69–76 CrossRef CAS.
- J. Colombani, G. Galliéro, B. Duguay, J.-P. Caltagirone, F. Montel and P. A. Bopp, Phys. Chem. Chem. Phys., 2002, 4, 313–321 RSC.
- G. Galliéro, J. Colombani, P. A. Bopp, B. Duguay, J.-P. Caltagirone and F. Montel, Phys. A, 2006, 361, 494–510 CrossRef.
- D. Niether, D. Afanasenkau, J. K. G. Dhont and S. Wiegand, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4272–4277 CrossRef CAS PubMed.
- J. Colombani, G. Galliéro, B. Duguay, J.-P. Caltagirone, F. Montel and P. A. Bopp, Philos. Mag., 2003, 83, 2087–2095 CrossRef CAS.
- H. Davarzani, M. Marcoux and M. Quintard, Int. J. Heat Mass Transfer, 2010, 53, 1514–1528 CrossRef.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Chen, C. Li and G. Shi, J. Phys. Chem. Lett., 2013, 4, 1244–1253 CrossRef CAS PubMed.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, J. Mater. Chem. A, 2013, 1, 12386–12394 CAS.
| This journal is © The Royal Society of Chemistry 2017 |