
A novel method to introduce acidic and basic bi-functional sites in graphitic carbon nitride for sustainable catalysis: cycloaddition, esterification, and transesterification reactions†
Subhajyoti
Samanta
 and
Rajendra
Srivastava
*
and
Rajendra
Srivastava
*
Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab-140001, India. E-mail: rajendra@iitrpr.ac.in; Fax: +91-1881-223395; Tel: +91-1881-242175
First published on 9th June 2017
Abstract
Graphitic carbon nitride was prepared using urea, thiourea, and a mixture of urea–thiourea. Functional carbon nitride was prepared by reacting g-C3N4 with aqueous H2SO4. Characterizations revealed that bi-functional acidic (–SO3H) and basic (–NH2) sites were introduced after the aqueous H2SO4 treatment. The characterizations also revealed that the surface acidity and basicity were influenced by the concentration of aqueous H2SO4 solution. A detailed characterization of the catalyst was made using a complementary combination of powder X-ray diffraction, N2 adsorption–desorption, scanning/transmission electron microscopy, thermogravimetric analysis, Fourier transform infrared spectroscopy, elemental analysis, and X-ray photoelectron spectroscopy. Furthermore, the bi-functional nature (acidity/basicity) of the catalyst was investigated with NH3 and CO2 temperature-programmed desorption techniques. The activation and utilization of CO2 in the synthesis of cyclic carbonates and quinazoline-2,4(1H,3H)-dione over bi-functional graphitic carbon nitride was investigated. Kinetic and thermodynamic parameters (such as k, Ea, ΔH, ΔG, and ΔS) were calculated for the cycloaddition reaction of CO2 to epichlorohydrin by varying the reaction parameters. Graphitic carbon nitride (prepared with urea–thiourea) treated with 60 wt% aqueous H2SO4 exhibited the highest catalytic activity and selectivity in the synthesis of cyclic carbonates and quinazoline-2,4(1H,3H)-dione. The catalyst was easily recovered and recycled with negligible loss of activity. In addition, the catalyst exhibited significantly high activity in the transesterification reaction of cyclic carbonate with methanol and the esterification reaction of oleic acid with methanol. The uniqueness of this study is the introduction of bi-functional (acid–base) sites by treating g-C3N4 with aqueous sulfuric acid, which was confirmed using NH3 and CO2 temperature-programmed desorption techniques and the various other physico-chemical characterization techniques mentioned above. Due to its simple and economical synthesis procedure, tuneable functional sites, efficient recyclability, and diverse catalytic activity, this catalyst is highly attractive for sustainable catalysis.
1. Introduction
The composition of the Earth's atmosphere can be classified into constant constituents and variable constituents. Human beings and other living organisms are affected by troposphere and atmosphere constituents that have predominantly high concentrations of N2 and O2. CO2 is present in the gaseous form in very low concentrations in the troposphere; however, it is present on earth as carbonate and other forms in water bodies and soil. Human activity, enzymatic degradation, industrial development, and energy production lead to the production of large concentrations of CO2 in the atmosphere.1 Most alternative energy resources that are being developed using biomass also contribute to the increase in CO2 concentration in the atmosphere because the end product of their consumption is CO2. On its own, CO2 is not harmful; however, its accumulation in the troposphere enhances global warming, which is dangerous to living organisms and the planet Earth.2,3 Therefore, it is important to eliminate excess concentrations of CO2 from the atmosphere through proper management.4 In this direction, the development of catalytic processes for the conversion of CO2 to useful chemicals is one of the most attractive ways to utilize CO2.5–12 Although CO2 is non-reactive, it has various modes of activation, which makes it an abundant, economical, and non-toxic carbon (C1) source. It also has the capacity to replace chemical processes based on toxic chemicals (such as phosgene, isocyanates, or carbon monoxide) with environmentally friendly catalytic processes. Only a few industrial processes based on CO2 are in practice; these include synthesis of urea, salicylic acid, para-hydroxy benzoic acid, cyclic and polycarbonate, and methanol.12Insertion of CO2 as a C1 source produces a wide range of important chemicals. Among these reactions, the cycloaddition of CO2 to epoxide (e.g. propylene oxide) for the synthesis of cyclic carbonates (e.g. propylene carbonate) and the cycloaddition of CO2 to 2-aminobenzonitrile for the synthesis of quinazoline-2,4(1H,3H)-dione are of significant interest.13–34 Cyclic carbonates/organic carbonates are among the most important feedstocks; they are extensively used as aprotic polar solvents, electrolytes in lithium ion batteries, precursors for polycarbonates, and intermediates in organic synthesis. Quinazoline-2,4(1H,3H)-dione acts as a key building block for the synthesis of several useful and valuable medicines.
Metal catalysts have been proven to be efficient for the synthesis of organic carbonates; however, their use generally has some drawbacks, especially the presence of undesirable metal residue in the final product. Therefore, organocatalysts have been developed, such as halide salts, porphyrin, phthalocyanine, Schiff's bases and several similar homogenous complexes.15–17 Because CO2 is an acidic molecule, it can be activated with the aid of a basic catalyst.18–32 The synthesis of cyclic carbonate is one of the early catalytic examples of metal organic framework-based catalytic reactions.33–37 Most of these catalysts are prepared using a wide range of costly chemicals by multi-step synthesis processes. Most homogenous complexes and heterogeneous catalyst-based catalytic processes involve large quantities of solvents and co-catalysts to catalyze this reaction.38–40 Therefore, the development of economical and solvent-free syntheses of cyclic carbonate is highly desirable.
Carbon-based materials are inexpensive and can be easily prepared on a large scale. Among carbon-based materials, graphitic carbon nitride (g-C3N4) is emerging as a metal-free heterogeneous catalyst because of the various active centres in its structure, its electron conductivity, and its high thermal and chemical stability.41 Due to its versatile physicochemical properties, g-C3N4 has demonstrated promising applications in numerous fields, especially photocatalysis and heterogeneous catalysis.41–43 g-C3N4 possesses a two-dimensional structure with π-conjugated planar layers that are composed of tris-triazine moieties bridged by nitrogen atoms. Due to the presence of uncondensed amino groups (in the forms of –NH2 and –NH– groups) at the edges of the graphitic sheets, it can be used in several base-catalyzed reactions. A few efforts have been made to exfoliate g-C3N4 with H2SO4 to generate more functional sites.44–47 In an early study, it was reported that sulphur was introduced by the treatment of g-C3N4 with different concentrations of H2SO4.44 However, the sulphur concentration decreased with increasing activation temperature. In another study, it was reported that –SO3H groups were introduced in the g-C3N4 structure by treatment with 6 N H2SO4.45 The resulting –SO3H-functionalized g-C3N4 was used to prepare Au-nanoparticles-supported g-C3N4 for photocatalytic water reduction.45 Since then, a few more attempts have been made to prepare functional g-C3N4 by treatment with H2SO4, and the resulting materials have been used in photocatalysis.46,47 Very recently, it was reported that H2SO4-treated g-C3N4 afforded –NH2-functionalized g-C3N4.48 Because g-C3N4 contains amine functionalities, efforts have also been made to use it as a catalyst in the activation of CO2, especially in cyclic carbonate synthesis.48–52 In order to improve its catalytic activity in cyclic carbonate synthesis, several active components have been incorporated in the structure of g-C3N4. Metallophthalocyanine–carbon nitride hybrid catalysts, alkali metal hydroxide-incorporated g-C3N4 materials, alkali bromide-incorporated g-C3N4 materials and several other additives have been incorporated in the structure of g-C3N4 to improve its catalytic activity.49–52
For the activation and utilization of CO2 in cycloaddition reactions, bi-functional catalysts (acid–base) are preferred. In this study, g-C3N4 was prepared using urea, thiourea, and urea–thiourea as precursors. The unique aspect of this study is the introduction of bi-functional (acid–basic) sites by treating g-C3N4 with aqueous sulfuric acid at different temperatures, which was thoroughly confirmed using NH3 and CO2 temperature-programmed desorption techniques and various other physico-chemical characterization techniques. Although attempts have been made in the past to functionalize g-C3N4 with aqueous sulfuric acid, the simultaneous incorporation of –SO3H and –NH2 groups in g-C3N4 prepared with two different precursors has not been reported. The catalytic activities of the parent and H2SO4-treated g-C3N4 were investigated in the activation and utilization of CO2 as a C1 source in the atom-economic synthesis of a cyclic carbonate and quinazoline-2,4(1H,3H)-dione. The effectiveness of this catalyst was extended to the transesterification of cyclic carbonate with methanol and the esterification of oleic acid with methanol to demonstrate the basic and acidic properties of the catalyst.
2. Materials and methods
All the materials used in this study were AR grade and were used as received without further purification. Urea, thiourea, and H2SO4 were purchased from SD Fine Chemicals India Pvt. Limited. Epoxides, solvents, and carbonates were procured from Spectrochem India Pvt. Limited. 2-Aminobenzonitrile was obtained from Sigma-Aldrich. Oleic acid and methanol were purchased from Loba. Chemi. India. Deionized water obtained from a Millipore Milli-Q system was used in the synthesis. High purity CO2 gas was obtained from Sigma Gas Service, India.2.1 Material synthesis
For comparative study, conventional acidic zeolite beta53 and basic catalysts (MgO and Mg/Al hydrotalcite)54,55 were synthesized by following reported procedures. Na–Y was kindly donated by Sud-Chemi India Pvt. Ltd.
2.2 Materials characterization
X-ray diffraction (XRD) patterns were recorded in the 2θ range of 5° to 80° with a scan rate of 2° min−1 on a PAN analytical X’PERT PRO diffractometer, Netherlands, using Cu Kα radiation (λ = 0.1542 nm, 40 kV, 45 mA). Thermogravimetric analysis was carried out using a TGA/DSC 1 STARe Mettler Toledo instrument, Switzerland, in the temperature range of 100 °C to 900 °C under an air flow of 10 mL min−1. Nitrogen adsorption–desorption measurements were performed at −200 °C using a Quantachrome Instruments Autosorb-IQ volumetric adsorption analyzer, USA. The sample was out-gassed at 150 °C for 3 h in the degas port of the adsorption apparatus. The specific surface areas of the materials were calculated from the adsorption data points obtained at P/P0 between 0.05 and 0.3 using the Brunauer–Emmett–Teller (BET) equation. The pore diameter was estimated using the Barret–Joyner–Halenda (BJH) method. The morphologies of the materials were investigated using a scanning electron microscope (SEM, JEOL JSM-6610LV, Japan) at an accelerating voltage of 5 kV. The nanostructures were investigated using a high resolution transmission electron microscope (TECHNAI, FEI-G2) at an accelerating voltage of 300 kV. X-ray photoelectron spectroscopy (XPS) was carried out using a PHI 5000 VersaProbeII XPS system (ULVAC-PHI, INC, Japan) with a microfocused (100 μm, 25 W, 15 kV) monochromatic Al-Kα source (hν = 1486.6 eV), a hemispherical analyzer and a multichannel detector. Elemental analysis (CHNS) was performed using a Fischer Thermo Scientific instrument. NH3-TPD and CO2-TPD were performed using a CHEMBET™ TPR/TPD instrument, Quantachrome, USA. In a typical experiment, 100 mg of sample was placed in a U-shaped flow-through quartz sample tube. Before the TPD experiments, the catalyst was pre-treated in He (30 mL min−1) at 450 °C for 1 h. After cooling to 50 °C, ammonia (partial pressure 100 Torr) (10% NH3 in He)/CO2 (99.99%) was adsorbed on the samples for 1 h. The sample was subsequently flushed with a He stream (30 mL min−1) at 50 °C for 1 h to remove physisorbed ammonia/CO2. The TPD experiments were carried out in the range of 50 °C to 400 °C at a heating rate of 10 °C min−1. The ammonia/CO2 concentration in the effluent was monitored using a gold-plated filament thermal conductivity detector. FT-IR measurements were performed on a Bruker Ternsor-F27 instrument.2.3 Catalytic reaction procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). The reaction mixture was analyzed by GC (Yonglin 6100; BP-5; 30 m × 0.25 mm × 0.25 μm) and the product was confirmed using GC-MS (Schimadzu GCMS-QP 2010 Ultra; Rtx-5 Sil Ms; 30 m × 0.25 mm × 0.25 μm).
000 rpm). Crushed ice was added to the reaction mixture, and the flask was maintained in a refrigerator for 10 to 12 h to precipitate the product. The precipitate was filtered, washed with cold water, and then dried under vacuum. The product was confirmed by its 1H NMR and FT-IR spectra, which matched well with the reported literature.56
000 rpm). The reaction mixture was analyzed by GC (Yonglin 6100; BP-5; 30 m × 0.25 mm × 0.25 μm) and the product was confirmed using GC-MS (Schimadzu GCMS-QP 2010 Ultra; Rtx-5 Sil Ms; 30 m × 0.25 mm × 0.25 μm).
000 rpm). Crushed ice was added to the reaction mixture, and the flask was maintained in a refrigerator for 10 to 12 h to precipitate the product. The precipitate was filtered, washed with cold water, and then dried under vacuum. The product was confirmed by its 1H NMR and FT-IR spectra, which matched well with the reported literature.56
3. Results and discussion
In this study, carbon nitride (CN) was prepared with different source materials: urea, thiourea and a mixture of [(1:1) wt%] urea–thiourea. Prepared CN was treated with different concentrations of H2SO4, which resulted in the formation of –SO3H and –NH2-functionalized bi-functional catalysts. Details of the physicochemical characterization and catalytic activities of the catalysts are provided in the following section.
3.1 Physicochemical characterization
The structures and phase purities of the synthesized materials were confirmed by powder XRD (Fig. 1a). In all cases, CN exhibited two reflections at 12.6° and 27.4° that can be indexed to the 100 and 002 graphitic planes, corresponding to the in-plane structural packing motif and interlayer stacking peak of the aromatic systems, respectively.50 The d-spacings for these two planes are very close to 0.698 nm and 0.325 nm, which matches well with g-C3N4 (Fig. 1a).58 The H2SO4-treated samples exhibited more intense 002 reflections. With increasing H2SO4 content, the intensity of the 002 reflection increased (Fig. 1b). Close inspection of the XRD patterns shows a marginal shift in the 2θ value (from 27.4 to 27.8) for the H2SO4-treated samples (Fig. 1b). From the changes in the intensity and 2θ value, one can conclude the following: (a) H2SO4 treatment can exfoliate g-C3N4 layers; (b) H2SO4 treatment can disintegrate the framework structure and generate more exposed g-C3N4 units, which is responsible for the increase in the intensity. With further increase in the H2SO4 content (for example, 80% H2SO4), the framework structure collapsed, and the resulting material did not exhibit any reflection corresponding to the g-C3N4 framework unit (Fig. S1, ESI†). | ||
| Fig. 1 XRD patterns of (a) carbon nitrides (CN(U), CN(TU), CN(UTU)) prepared using various precursors, including urea, thiourea and urea–thiourea; (b) H2SO4-treated CN(UTU) samples. | ||
The nitrogen adsorption–desorption isotherms of the CN materials exhibited type I/IV isotherms with H3 hysteresis loops (Fig. S2†). The studies showed that the material prepared with urea exhibited the highest adsorbed volume, whereas the material prepared with thiourea exhibited the lowest adsorbed volume. The BJH pore size distribution shows that the materials do not exhibit ordered pores. The textural properties of the various carbon nitride materials synthesized in this study are provided in Table 1. The surface area and total pore volume of CN(U) are the highest among the various CN materials investigated in this study. The isotherm shows that the H2SO4-treated materials exhibited low adsorbed volumes compared to the parent material, CN(UTU). The highest adsorbed volume and the most porous structure were observed for S-CN(UTU)-60. The surface area and total pore volume of the S-CN(UTU)-60 material are the highest among the various H2SO4-treated CN(UTU) materials prepared in this study. The reasons for the enhanced textural properties of S-CN(UTU)-60 are provided in the next paragraph.
| Physico-chemical properties | |||||
|---|---|---|---|---|---|
| Catalyst | Surface area (m2 g−1) | External surface area (m2 g−1) | Total pore volume (cm3 g−1) | NH3-TPD (mmol g−1) | CO2-TPD (mmol g−1) |
| a Recycled catalyst. | |||||
| CN(U) | 91.274 | 81.237 | 0.370 | 2.20 × 10−2 | 5.34 × 10−3 |
| CN(TU) | 28.493 | 28.103 | 0.075 | 1.81 × 10−2 | 8.04 × 10−3 |
| CN(UTU) | 32.053 | 32.053 | 0.147 | 3.12 × 10−2 | 5.09 × 10−3 |
| S-CN(UTU)-20 | 6.937 | 3.831 | 0.019 | 4.07 × 10−1 | 6.62 × 10−1 |
| S-CN(UTU)-40 | 4.021 | 0.004 | 0.015 | 9.02 × 10−1 | 9.13 × 10−1 |
| S-CN(UTU)-60 | 10.20 | 0 | 0.065 | 9.12 × 10−1 | 9.72 × 10−1 |
| S-CN(UTU)-60a | 10.31 | 0 | 0.066 | — | — |
| Elemental analysis data | |||||
|---|---|---|---|---|---|
| Catalyst | C (%) | N (%) | S (%) | H (%) | C/N |
| CN(UTU) | 40.01 | 57.12 | — | 1.78 | 0.70 |
| S-CN(HTU)-20 | 36.86 | 56.12 | 2.55 | 1.19 | 0.65 |
| S-CN(HTU)-40 | 32.51 | 52.11 | 5.82 | 2.89 | 0.62 |
| S-CN(HTU)-60 | 30.21 | 49.88 | 7.85 | 3.96 | 0.61 |
| S-CN(HTU)-60a | 30.75 | 49.80 | 7.89 | 4.04 | 0.61 |
The microstructures of the CN materials were investigated by scanning electron microscopy (SEM). All the CN materials exhibited irregular morphologies. Urea-derived CN(U) exhibited an irregular aggregated morphology with small oligomeric units supported on a cross-linked polymeric microstructure with cracks on its surface (Fig. S3†). A highly cross-linked smooth surface (with some wrinkling on the surface, marked as the yellow domain) was observed for CN(TU), which confirms that the thiourea-facilitated condensation and polymerization process was more efficient than that of urea. A different morphology evolved when the two different precursors were mixed and heated together. Due to the differences in the rates of condensation and polymerisation of the two precursors and the difference in the rates of evolution of CO2 and SO2 during the heat treatment process, different domains evolved which provided separate aggregated porous micro-structures, as shown in the SEM images (Fig. S3†). Elemental analysis of CN(UTU) obtained by energy dispersive X-ray analysis confirms the presence of C and N in the sample (Fig. S4†). The morphologies of the H2SO4-treated samples were different from that of the parent CN(UTU) material (Fig. 2). When CN(UTU) was added to the aqueous H2SO4 solution, dissolution occurred. With increasing H2SO4 concentration, the dissolution increased, as shown in the photographs recorded during the sample preparation (Fig. 3). During the dissolution process, exfoliation along with disintegration of the structure can occur. After the treatment with H2SO4, the reaction mixture was added to ice-cold water. During this process, reintegration took place and the microstructure re-evolved (Scheme S1†). Due to the different extents of disintegration with different H2SO4 concentrations and the differences in the reintegration process due to the differences in the acidity of the medium, different microstructures evolved (Fig. 2). Photographs recorded during the sample preparation clearly show that with increasing H2SO4 concentration, dissolution of CN was facilitated (Fig. 3). At the beginning, a yellowish coagulant was observed when CN(UTU) was treated with aq. H2SO4 (60% aqueous solution). After 2 h of treatment at 80 °C, complete dissolution occurred. In contrast, when CN(UTU) was treated with 40% and 20% aqueous H2SO4, complete dissolution did not occur (Fig. 3). The extent of dissolution varied in the order of H2SO4 (60%) > H2SO4 (40%) > H2SO4 (20%) (Fig. 3). After 2 h of treatment at 80 °C, the reaction mixture was cooled to ambient temperature and was then added to ice-cold water for re-precipitation. At higher concentrations of H2SO4, the polymeric carbon nitride disintegrated into oligomeric carbon nitride units (which were dissolved in 60% aqueous H2SO4 solution). When the dissolved solution was added to ice-cold water, reintegration of oligomeric units occurred and the structure re-evolved. The difference in the XRD peak intensity before and after H2SO4 treatment shows that the 002 plane was more exposed after reintegration. At a very high concentration of H2SO4 (80% aqueous solution) the polymeric carbon nitride disintegrated into very small oligomeric carbon nitride units which could not reintegrate into the original polymeric carbon nitride, as confirmed from the XRD investigation (Fig. S1†). During the growth process, the material attempted to lower its energy and favourably evolve in the form of a spheroid morphology, which is evident from the SEM images of the S-CN(UTU)-60 sample (Fig. 2). Elemental analysis of S-CN(UTU)-60 obtained from the energy dispersive X-ray analysis confirms the presence of C, N, and S in the sample (Fig. S3†). This further confirms that S was incorporated after the H2SO4 treatment. In the case of S-CN(UTU)-60, re-integration of highly dispersed oligomeric units of CN took place, which resulted in a material with a high surface area compared with S-CN(UTU)-40 and S-CN(UTU)-20. In contrast, in the cases of S-CN(UTU)-20 and S-CN(UTU)-40, partial dissolution of the CN structure occurred, and the dissolved CN oligomeric units re-integrated on the external surface and inter-crystalline porous structure to provide materials with low surface areas. Because the dis-integration and re-integration process occurred in aqueous acidic solution in which no gas evolved, their surface areas were low compared to that of their parent material.
 | ||
| Fig. 2 SEM images of (a) S-CN(UTU)-20, (b) S-CN(UTU)-40, (c) and (d) S-CN(UTU)-60. | ||
 | ||
| Fig. 3 Photographs recorded at different stages of sample preparation during the treatment of CN(UTU) with different concentrations of H2SO4. | ||
The micro-nanostructures were also evaluated with the aid of transmission electron microscopy (TEM). The low magnification TEM images show a spheroid-like morphology similar to that observed in the SEM images (Fig. 4a and S5†). The higher magnification TEM image shows that the spheroid morphology is composed of a highly connected spider web-like sheet morphology (Fig. 4b). The sheet-like morphology has fewer cross-linked and dense cross-linked domains (Fig. 4b). This cross-linked sheet-like structure also creates porous domains with different dimensions. This nanocrystal morphology can be observed if the disintegration and reintegration processes are not uniform. The HRTEM image confirms that the sheet-like domains are highly ordered, as shown by the lattice fringes (Fig. 4c and S5†). The inter-planar distance between two adjacent planes of S-CN(UTU)-60 was found to be 0.326 nm (as obtained from the HRTEM images), which is identical to the standard 002 plane of graphitic C3N4.43
 | ||
| Fig. 4 (a and b) TEM images and (c) HRTEM image showing the lattice fringes of the highly active S-CN(UTU)-60 catalyst. | ||
The thermal stabilities of the materials were investigated by thermogravimetric analysis (Fig. 5). The thermograms of the CN materials show that the thermal stability varies with the source material. CN(U) prepared with urea decomposed more readily in air compared to CN(TU), as revealed from the TGA analysis. It may be noted that the yield of CN(TU) was higher than that of CN(U), which confirms that the sulphur groups present in TU facilitated the condensation process and produced g-C3N4 material in high yield. It may further be noted that no sulphur was observed in the elemental analysis, which confirms that during the condensation and polymerization process, all sulphur escaped in the form of SO2. All the H2SO4-treated samples decomposed at lower temperatures compared to the parent material (Fig. 5). The thermal stabilities of the acid-treated materials follows the trend S-CN(UTU)-20 > S-CN(UTU)-40 > S-CN(UTU)-60. This confirms that during the H2SO4 treatment, exfoliation and framework disintegration occurred, which could generate several functional groups (such as –OH and –SO3H, along with more uncondensed –NH2 groups); this facilitated the thermal decomposition of the material in air. The presence of these functional groups can be easily confirmed by FT-IR and elemental analysis.
 | ||
| Fig. 5 Thermograms of the parent and H2SO4-treated carbon nitride samples investigated in this study. | ||
Detailed elemental analysis of the materials was performed using a CHNS analyzer to quantify the contents of carbon, nitrogen, and sulphur present in the samples. The weight percentages of all the elements present in the samples are given in Table 1. Elemental analysis shows that the parent CN(UTU) contains C, N, and H elements with a C/N ratio of 0.7. For the H2SO4-treated samples, the presence of C, N, S, and H elements were confirmed, with C/N ratios less than 0.7. CHNS analysis clearly shows that more S was incorporated in S-CN(UTU)-60, which was treated with 60% aqueous H2SO4 solution. The elemental analysis data correlates well with the results obtained from the XPS analysis (discussed in the following section).
FT-IR is a useful technique to investigate the presence of various functional groups in a material. The broad band appearing in the range of 3000 to 3300 cm−1 is mainly due to the N–H stretching of the amine groups (Fig. 6). The FT-IR band at 1637 cm−1 can be assigned to the C–N stretching vibration (Fig. 6). Several vibrations in the range of 1400 to 1550 cm−1 are observed; these can be attributed to the presence of C–N heterocycles of the melamine unit (Fig. 6). The FT-IR peaks at 1240 cm−1 and 1310 cm−1 are attributed to sp2 hybridized C–(NH) units. A sharp peak at 807 cm−1 is ascribed to the tris-s-triazine of the melamine unit in all the materials (Fig. 6). The FT-IR spectra of the S-CN(UTU) samples show that several new functional groups were introduced after H2SO4 treatment. Very distinct bands in the range of 3000 to 3400 cm−1 can be assigned to the uncondensed/free –NH2 and –OH groups. Appearance of a strong peak at 650 cm−1 (strong S–O bond) confirms the presence of the –SO3H functionality in all the H2SO4-treated materials. This is further supported by the presence of two additional peaks at 1350 cm−1 and 1150 cm−1, which can be attributed to S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretching and symmetric stretching vibrations, respectively. It is interesting to note that these three peaks are absent in the parent CN(UTU) and the other two CN(U) and CN(TU) materials (Fig. 6). The presence of –NH2, –OH, and –SO3H groups in the H2SO4-treated samples shows that g-C3N4 was exfoliated; some melamine units were then cleaved and produced these new functional groups.
O asymmetric stretching and symmetric stretching vibrations, respectively. It is interesting to note that these three peaks are absent in the parent CN(UTU) and the other two CN(U) and CN(TU) materials (Fig. 6). The presence of –NH2, –OH, and –SO3H groups in the H2SO4-treated samples shows that g-C3N4 was exfoliated; some melamine units were then cleaved and produced these new functional groups.
 | ||
| Fig. 6 FT-IR spectra of the parent and H2SO4-treated carbon nitride samples investigated in this study. | ||
X-ray photoelectron spectroscopy was carried out in order to determine the presence of various elements (and their oxidation states) in the highly active S-CN(UTU)-60 catalyst and its parent material, CN(UTU). Full survey scans of the parent material CN(UTU) and the H2SO4-treated material S-CN(UTU)-60 are presented in Fig. 7a. C, N, and O appeared in CN(UTU), whereas C, N, S, and O are found in S-CN(UTU)-60. If one compares the relative intensities of the constituent elements, it is very clear that the O 1s intensity increased after the H2SO4 treatment, which confirms that greater numbers and more types of surface –OH species are formed compared to the parent material. Similarly, the C 1s intensity decreased after the H2SO4 treatment, which confirms that the loss of carbon atoms occurred during the H2SO4 treatment. The high resolution spectra of C 1s, N 1s and O 1s (Fig. 7b–d) exhibit no obvious changes in their binding energies, which further confirms that the oxidation state of all the constituent elements remained unchanged after the H2SO4 treatment. Although the binding energy did not change, the deconvolution spectra for S-CN(UTU)-60 are different from those of the parent material, which signifies that the surface nature of the constituent elements changed after the H2SO4 treatment. The high resolution spectra of C 1s in the CN(UTU) and S-CN(UTU)-60 samples exhibit two characteristic peaks at 288.4 eV and 284.3 eV (Fig. 7b) which can be attributed to the sp2 hybridized carbon atom of g-C3N4 and the skeleton carbon of the melamine unit, respectively.59 Both the CN(UTU) and S-CN(UTU)-60 samples exhibit three peaks for N 1s at 397.5 eV, 399.1 eV, and 400.3 eV (Fig. 7c), which can be assigned to an sp2 hybridized N atom bonded to a C atom and a tertiary bond to the carbon atom in the form of (N–C3) and (C–N–H) in the amine functional groups, respectively.59 Furthermore, the high resolution XPS spectra for both the samples indicate that O 1s also has two peaks at 530.2 eV and 528.3 eV (Fig. 7d) which are due to the hydroxyl groups present in the sample. The high resolution XPS spectrum of elemental S 2p indicates that it has three maxima at 164 eV, 165.4 eV, and 169.3 eV (Fig. 7e). The peak at 164 eV is ascribed to the C–S bond, and the peak at 165.4 eV is due to the –N–S bond in S-CN(UTU)-60. In addition, the peak at 169.3 eV is characteristic of the –SO group, which further confirms that –SO3H has been incorporated in the sample.60 It is noteworthy that the S 2p peaks are absent in CN(UTU), which implies that after H2SO4 treatment, the –SO3H group was incorporated in the sample, resulting in the formation of a bi-functional catalyst. The XPS results correlate well with the CHNS results, which confirms that sulphur was incorporated after the H2SO4 treatment. These results are in agreement with the FT-IR studies, which shows that the –SO3H group appeared after H2SO4 treatment.
 | ||
| Fig. 7 (a) Surface survey XPS spectra of CN(UTU) and S-CN(UTU)-60, (b) high resolution XPS spectra of C 1s of CN(UTU) and S-CN(UTU)-60, (c) high resolution XPS N 1s spectra of CN(UTU) and S-CN(UTU)-60, (d) high resolution O 1s XPS spectra of CN(UTU) and S-CN(UTU)-60, and (e) high resolution S 2p XPS spectrum of S-CN(UTU)-60. | ||
As revealed by FT-IR and XPS analysis, the acid-treated samples contain –SO3H and –NH2 functionalities. Therefore, it was important to evaluate their acidities and basicities. The acidities and basicities of the material were evaluated using NH3/CO2-TPD analysis. The basicity was studied by TPD of CO2 molecules adsorbed on the CN materials (Fig. 8a). CO2 is an acidic probe molecule that can be adsorbed on the basic sites of a catalyst. CO2 was adsorbed at 50 °C on the catalyst surface and then desorbed in the range of 50 °C to 300 °C. CO2 was weakly adsorbed on the parent CN samples. The desorption profile clearly shows that among the various untreated samples, CN(UTU) exhibited marginally higher CO2 adsorption. The desorption profile clearly shows that CO2 was adsorbed more on the H2SO4-treated samples. Furthermore, the desorption temperature shifted to a higher temperature as the concentration of H2SO4 increased, which shows that sites with higher basic strength are present in S-CN(UTU)-60 compared to the other H2SO4-treated samples. The amounts of CO2 desorbed from CN(U), CN(TU), CN(UTU), S-CN(UTU)-20, S-CN(UTU)-40, and S-CN(UTU)-60 were found to be 5.34 × 10−3, 8.04 × 10−3, 5.09 × 10−3, 6.62 × 10−1, 9.13 × 10−1, and 9.72 × 10−1 mmol g−1, respectively. This analysis confirms that after the H2SO4 treatment, uncondensed –NH2 sites with different numbers and types were produced and are responsible for the varied basicities of the CN samples.
 | ||
| Fig. 8 (a) CO2-TPD profiles and (b) NH3-TPD profiles of various materials investigated in this study. Inset: TPD profiles of parent carbon nitrides are shown here for better resolution. | ||
NH3 is a basic probe molecule that can be adsorbed on the acid sites of a catalyst. NH3 was adsorbed at 50 °C on the catalyst surface and then desorbed in the range of 50 °C to 350 °C (Fig. 8b). NH3 was weakly adsorbed on the parent CN samples. The desorption profile clearly shows that among various untreated samples, CN(UTU) exhibited marginally higher NH3 desorption. The desorption profile also shows that CN(TU) has two types of acid sites, whereas CN(U) possesses only one type of acid site. The desorption profiles clearly show that NH3 was adsorbed more on the H2SO4-treated samples. Furthermore, the desorption temperature shifted to higher temperatures as the concentration of H2SO4 increased, which shows that sites with higher acid strength are present in S-CN(UTU)-60 compared to the other H2SO4-treated samples. The desorption profile also shows that S-CN(UTU) possesses four different types of acid sites. The amounts of NH3 desorbed from CN(U), CN(TU), CN(UTU), S-CN(UTU)-20, S-CN(UTU)-40, and S-CN(UTU)-60 were found to be 2.20 × 10−2, 1.81 × 10−2, 3.12 × 10−2, 4.07 × 10−1, 9.02 × 10−1 and 9.12 × 10−1 mmol g−1, respectively. This analysis confirmed that after the H2SO4 treatment, –SO3H sites with different numbers and types were produced on the surface of CN(UTU) and are responsible for the varied acidities of the CN samples.
3.2 Evaluation of the catalytic activity
Literature reports suggest that amine-functionalized bases are important to activate and catalyze the cycloaddition of CO2 to epoxides to form cyclic carbonates. We also learned from the literature that this reaction takes place efficiently when basic functional sites and acid sites both are present concomitantly in the catalyst system/reaction vessel. CN is a mildly basic catalyst.43 CN(UTU) was treated with different concentrations of H2SO4 to obtain acid sites in the catalyst. The various physico-chemical characterizations described above clearly show that the resulting S-doped CN materials are bi-functional catalysts. The importance of bi-functional sites in the cycloaddition of CO2 to epoxides can be judged from the catalytic activity obtained from this material. The basic sites present in the material activate the CO2 molecules, whereas the acid sites in the material activate and polarize the epoxide ring. The simultaneous activation of two reactant molecules present in the same material at close proximity facilitates the insertion of activated CO2 in the polarized C–O bond of the epoxide to produce the cyclic carbonate.The catalysts prepared in this study were investigated in the synthesis of cyclic carbonates by the cycloaddition reaction of CO2 to epoxides (Table 2). Epichlorohydrin (ECH) was chosen as a model substrate in the reaction. The reaction did not take place in the absence of a catalyst. Only a very low ECH conversion of 10.1% was obtained using CN(U) (Table 2). However, using CN(UTU), the conversion of ECH increased to 21.3%; this suggests that more basic sites were introduced (Table 2). The order of reactivity is consistent with the order of basicity obtained from the CO2 TPD analysis. It was interesting to observe that H2SO4 treatment influences the reactivity of the catalyst. CN(UTU) treated with 60 wt% aqueous H2SO4 exhibited the highest cyclic carbonate yield (Table 2). In these cases, minor amounts of 3-chloropropanol or 2-chloropropanol were observed as side products during the GC-MS analysis. More than 90% yield of the product was obtained using S-CN(UTU)-60 after 4 h of reaction at a very mild pressure of 10 bar.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Selectivity (%) |
| a Reaction conditions: epoxide (63 mmol), catalyst (50 mg), temperature (100 °C), time (1 h), CO2 pressure (10 bar). b Epoxide = epichlorohydrin. c epoxide = propylene oxide. d Reaction time (2 h). e Reaction time (3 h). f Reaction time (4 h). g Reaction time (8 h). The values in entries 5 to 13 are presented as mean ± SD (n = 3). | |||
| 1 | CN(U) | < 1b | — |
| 2 | CN(U) | (10.1)b (7.4)c | (93.2)b (95.4)c |
| 3 | CN(TU) | (15.3)b (12.7)c | (97.1)b (96.3)c |
| 4 | CN(UTU) | (21.3)b (16.8)c | (97.2)b (98.1)c |
| 5 | S-CN(UTU)-20 | (31.2 ± 0.6)b (22.4 ± 0.4)c | (98.3 ± 0.1)b (98.1 ± 0.2)c |
| 6 | S-CN(UTU)-40 | (47.1 ± 0.8)b (31.6 ± 0.3)c | (99.2 ± 0.3)b (99.1 ± 0.2)c |
| 7 | S-CN(UTU)-60 | (58.2 ± 1.1)b (35.4 ± 0.5)c | (99.3 ± 0.3)b (99.1 ± 0.1)c |
| 8d | S-CN(UTU)-60 | (69.3 ± 0.9)b (46.7 ± 0.7)c | (99.2 ± 0.2)b (98.9 ± 0.4)c |
| 9e | S-CN(UTU)-60 | (80.3 ± 0.7)b (53.8 ± 0.6)c | (99.1 ± 0.1)b (99.0 ± 0.2)c |
| 10f | S-CN(UTU)-60 | (92.8 ± 0.5)b (61.5 ± 1.0)c | (99.2 ± 0.3)b (99.3 ± 0.2)c |
| 10g | S-CN(UTU)-60 | (93.5 ± 0.7)c | (99.0 ± 0.4)c |
| 11 | MgO | (2.8 ± 0.1)b,f | (20.5 ± 0.1)b,f |
| 12 | Mg/Al hydrotalcite | (9.2 ± 0.4)b,f | (28.2 ± 0.2)b,f |
| 13 | Zeolite beta | (12.8 ± 0.3)b,f | (64.5 ± 0.4)b,f |

3.3 Reaction kinetic investigation
After determining the best catalyst for the cycloaddition reaction, the reaction parameters were optimized. The cyclic carbonate yield was influenced by the CO2 pressure, and the results are shown in Fig. 9a. With increasing CO2 pressure, the cyclic carbonate yields increased. At a lower pressure of 2 bar, the ECH conversion was very low (close to 20%). However, at an initial CO2 pressure of 10 bar, the conversion was >92%. At a CO2 pressure higher than 10 bar, only a marginal increase in the cyclic carbonate yield was obtained. The following conclusion can be drawn from the CO2 pressure study: (1) CO2 is not involved in the rate determining step in this pressure range; (2) the functional groups obtained after H2SO4 treatment play important roles in the activation of CO2; (3) CO2 has good solubility in the reaction medium in the optimum pressure range. | ||
| Fig. 9 Influence of reaction parameters: (a) CO2 pressure, (b) catalyst amount, (c) temperature, and (d) time for the cycloaddition reaction of CO2 to epichlorohydrin. | ||
The amount of catalyst plays an important role in the cyclic carbonate yield. When the catalyst amount was increased from 10 mg to 50 mg, the cyclic carbonate yield increased (Fig. 9b). With further increase in the catalyst amount, only a marginal increase in the cyclic carbonate yield was obtained. Therefore, 50 mg of catalyst was selected as an optimum catalyst amount for further investigation.
Temperature influenced the cyclic carbonate yield significantly. At lower temperatures (60 °C), the conversion of epoxide was only 10.2%, whereas it reached >90% at 100 °C; this indicates that a larger fraction of molecules attained the activation energy for the ring-opening cycloaddition reaction (Fig. 9c). Only a marginal increase in the catalytic activity was observed above 100 °C, demonstrating that 100 °C was ideal for this reaction.
The influence of reaction time was investigated at 100 °C and 10 bar CO2 pressure. The reaction time had a remarkable influence on the reaction; the conversion of ECH increased sharply within the first 4 h and then remained almost unchanged with high selectivity, which showed that a reaction time of 4 h was optimal for high ECH conversion with good selectivity for the desired product (Fig. 9d).
The kinetics of the cycloaddition was studied using ECH. The general rate formula for cycloaddition is given by eqn (1).
| Rate = −d[ECH]/dt = k (ECH)a(CO2)b(catalyst)c | (1) |
A large excess of CO2 was used; therefore, the CO2 concentration can be kept constant. Furthermore, the catalyst amount was also kept constant under the present investigation for the kinetic investigation; therefore eqn (1) can be simplified to eqn (2).
| Rate = −d[ECH]/dt = kobs(ECH)a | (2) |
Cycloaddition reactions were carried out at 100 °C by varying the ECH concentration from 63 mmol to 252 mmol while keeping the CO2 pressure and catalyst amount constant (Fig. 10a). With increasing ECH concentration, the ECH conversion decreased. In order to determine the influence of ECH concentration on the rate of the cycloaddition reaction, the rate was plotted versus ECH concentration (Fig. 10a). It was found that the rate of the cycloaddition reaction exhibited a 1st order dependence with respect to the ECH concentration. Therefore, eqn (2) can be converted to eqn (3) and then reduced to eqn (4).
| −ln[ECH] = kobst | (3) |
| Rate = kobs(ECH)1 | (4) |
| kobs = Aexp(−Ea/RT) | (5) |
| lnkobs = lnA − Ea/RT | (6) |
 | ||
| Fig. 10 Plots of (a) rate vs. epichlorohydrin concentration and (b) ln(k) vs. 1/T for the cycloaddition reaction of CO2 to epichlorohydrin. | ||
Moreover, the activation energy for the cycloaddition reaction was determined using the Arrhenius equation (eqn (5)) based on the calculated rate constants in the temperature range of 333 to 383 K, where R is the universal gas constant (8.314 J mol−1 K−1), T is the absolute temperature (K), and A and Ea are the pre-exponential factor (s−1) and activation energy (kJ mol−1), respectively. The Arrhenius plot was used to determine the activation enthalpy (ΔH = Ea − RT). The free energy of activation (ΔG) was determined using the equation ΔG = −RTln(NAhk/RT),61 where R is the gas constant, NA is Avogadro's number, h is Planck's constant, k is the rate constant and T is the optimum reaction temperature (100 °C). Finally, the activation of entropy was calculated using the formula ΔS = (ΔH − ΔG)/T. The values of Ea, ΔH, ΔG, and ΔS were calculated to be 55.33 kJ mol−1, 52.23 kJ mol−1, 30.54 kJ mol−1, and 0.216 kJ mol−1, respectively.
Propylene oxide was successfully converted to propylene carbonate. Details of the investigation of the conversion of propylene oxide to propylene carbonate are provided in Table 2. In this case, a relatively longer reaction time was required to obtain good yields compared to ECH. Therefore, it may be concluded that epichlorohydrin was more active than propylene oxide. The recyclability of the catalyst was examined under the optimum conditions (Fig. 10). After the reaction, acetonitrile was added to the reaction mixture. The catalyst was recovered using a centrifuge, followed by rinsing with acetonitrile and drying in an oven at 80 °C for 8 h. The recovered catalyst was reused in the next cycle. S-CN(UTU)-60 was recycled, and the results are shown in Fig. 11. The recycling study shows that the conversion and selectivity were almost constant during five cycles of use. These results strongly suggest that this catalyst is stable and can be used for multiple cycles. The recovered catalyst was subjected to XRD (Fig. S6†), BET (Table 1), and SEM analysis (Fig. S7 and S8†). The XRD pattern shows no change in the crystallinity after the recycling study (Fig. S6†). Only a marginal change in the textural properties was observed for the recovered catalyst compared to the fresh catalyst (Table 1). CHNS analysis of the recovered catalyst shows that the N and S contents were similar to those of the fresh catalyst. The catalytic activity of the best catalyst reported in this study, S-CN(UTU)-60, was compared with those of selected catalysts reported in the literature, such as zeolite, MOF, base-functionalized silica-based catalyst, and metal complexes (Table S1†). The comparative catalytic activity shows that S-CN(UTU)-60 exhibited better or similar catalytic activity to catalysts reported in the literature. Furthermore, the cycloaddition reaction of CO2 to epichlorohydrin was performed using conventional acidic (zeolite beta) and basic (MgO and Mg/Al hydrotalcite) catalysts under the optimized reaction conditions (Table 2). Comparison of the catalytic activity shows that S-CN(UTU)-60 exhibited much higher activity and selectivity than conventional acid and base catalysts.
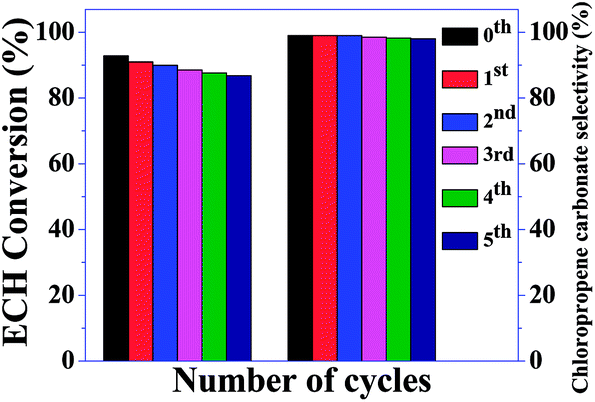 | ||
| Fig. 11 Catalytic activity data obtained during the recycling of S-CN(UTU)-60 in the cycloaddition of CO2 to epichlorohydrin (ECH). | ||
Based on the catalytic results and kinetic investigation, a plausible mechanism for the formation of cyclic carbonate by the cycloaddition of epoxide and CO2 is presented in Scheme S2.† The synergistic participation of the bi-functional sites present in the catalyst is responsible for the high catalytic activity. Simultaneous activation of CO2 by the basic sites (amines present in the catalyst) and the epoxide ring by the acid sites (–SO3H groups present in the catalyst) occurs. The oxygen anion of the epoxide reacts with the CO group of the activated CO2 through intramolecular nucleophilic cyclization, which forms the cyclic carbonate product and regenerates the catalyst.
Having found the best catalyst for the cycloaddition of CO2 to epoxide, the application of this catalyst was further investigated in the synthesis of quinazoline-2,4(1H,3H)-dione by the reaction of 2-aminobenzonitrile and CO2. In this case, only one product, quinazoline-2,4(1H,3H)-dione, was obtained. The influence of solvent was found to be crucial; the catalytic activity data obtained with different solvents is presented in Table 3. In contrast to the above CO2 cycloaddition reaction with epoxide, this reaction did not proceed in solvent-free conditions. It was found that the reaction proceeded well in DMF or DMSO. The reaction did not take place in non-polar solvents such as toluene. The reaction did not take place in the aprotic polar solvent acetonitrile, whereas only a low yield of the product was obtained in a polar protic solvent, methanol. Moderate yield of the product was obtained when the reaction was performed in H2O. The facile recovery and high yield of the product in DMF encouraged us to choose this solvent for further catalytic investigation. The influence of the reaction parameters was studied to optimize the reaction conditions; the comparative catalytic activities with the various catalysts prepared in this study are presented in Table 3. The reaction conditions were optimized as follows: 2-aminobenzonitrile (2 mmol), DMF (5 mL), temperature (130 °C), catalyst (100 mg), time (12 h), and CO2 pressure (25 bar). The catalytic activity data shows that the reaction proceeded efficiently when the catalyst was treated with high concentrations of H2SO4. The order of catalytic activity of the various catalysts investigated in this study is the same as the order obtained in the case of cyclic carbonate synthesis. Furthermore, the cycloaddition reaction of CO2 to 2-aminobenzonitrile was performed using conventional acidic (zeolite beta) and basic catalysts (MgO) under the optimized reaction conditions. A comparison of the catalytic activity shows that S-CN(UTU)-60 exhibited much higher activity and selectivity than conventional acid and base catalysts (Table 3).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Product yield (%) |
| a Reaction conditions: 2-aminobenzonitrile (2 mmol), solvent (5 mL), catalyst (100 mg), time (12 h), temperature (130 °C), CO2 (25 bar). The values in entries 3 to 14 are presented as mean ± SD (n = 3). | |||
| 1 | CN(U) | DMF | 16.4 |
| 2 | CN(TU) | DMF | 26.6 |
| 3 | CN(UTU) | DMF | 39.5 ± 0.5 |
| 4 | S-CN(UTU)-20 | DMF | 74.2 ± 0.9 |
| 5 | S-CN(UTU)-40 | DMF | 80.3 ± 0.6 |
| 6 | S-CN(UTU)-60 | DMF | 90.2 ± 0.8 |
| 7 | S-CN(UTU)-60 | DMSO | 93.2 ± 1.1 |
| 8 | S-CN(UTU)-60 | Water | 40.8 ± 1.0 |
| 9 | S-CN(UTU)-60 | Methanol | 7.4 ± 0.2 |
| 10 | S-CN(UTU)-60 | Acetonitrile | 0 |
| 11 | S-CN(UTU)-60 | Toluene | 0 |
| 12 | S-CN(UTU)-60 | Neat | 0 |
| 13 | MgO | DMF | 27.4 ± 0.3 |
| 14 | Zeolite beta | DMF | 7.8 ± 0.4 |

These results clearly show that CO2 adsorption and reaction with 2-aminobenzonitrile was facilitated in the presence of S-CN(UTU)-60. The basic sites present in the catalyst play a dual role in the synthesis of quinazoline-2,4(1H,3H)-dione. The basic sites activate the CO2 molecules and also abstract acidic protons from the NH2 group of 2-aminobenzonitrile due to the electron withdrawing group (–CN) attached to the benzene ring, leading to the formation of intermediate 1a (Scheme S3†). In the second step, intermediate 1a undergoes nucleophilic attack from a CO2 molecule and thereby generates intermediate 1b. Then, the oxygen anion in intermediate 1b reacts with the –CN group and generates cyclic intermediate 1c. Ring opening of intermediate 1c leads to the formation of isocyanate intermediate 1d, which undergoes intramolecular cyclization leading to the formation of intermediate 1e. Tautomerization of 1e leads to the formation of the stable cyclic amide quinazoline-2,4(1H,3H)-dione product and regenerates the catalyst.
Cyclic carbonates can be converted to di-alkyl carbonates by transesterification reactions. The importance of the bi-functional (acid and base) nature of the catalyst can also be determined from transesterification and esterification reactions. In general, transesterification is facilitated in the presence of base catalyst, whereas esterification is facilitated in the presence of acid catalyst. The catalysts investigated in this study were evaluated in the transesterification of ethylene carbonate and propylene carbonate with methanol. The reaction parameters were optimized, and the catalytic activities of various catalysts under the optimized reaction conditions is shown in Table 4. Reactions were conducted at 150 °C using a cyclic carbonate: methanol ratio of 1:10. Similar to the above reaction, in this reaction, methanol was used in great excess; thus, the reaction follows a pseudo-first-order rate law. Quantitative yield of the dialkyl carbonate was obtained after 8 h over the highly active catalyst. The transesterification reaction is directly proportional to the number and strength of the basic sites present in the material. The order of activity follows the trend S-CN(UTU)-60 > S-CN(UTU)-40 > S-CN(UTU)-20, which matches well with the basicity order obtained from the TPD analysis. The advantage of S-CN(UTU)-60 over the other catalysts is evident from the di-alkyl carbonate yield obtained from the transesterification of cyclic carbonate with methanol. The high yield of di-alkyl carbonate in the case of S-CN(UTU)-60 correlates well with the basicity obtained from the CO2 TPD investigation. Furthermore, the transesterification reaction of ethylene carbonate and methanol was performed using conventional acidic (zeolite beta) and basic catalysts (MgO and Na–Y) under the optimized reaction conditions. A comparison of the catalytic activity shows that S-CN(UTU)-60 exhibited much higher activity and selectivity than the conventional acid and base catalysts (Table 4). This result shows that S-CN(UTU)-60 has good basicity and can be applied in various transesterification reactions.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Selectivity (%) |
| a Reaction conditions: cyclic carbonate (50 mmol), methanol (500 mmol), catalyst (50 mg), temperature (150 °C), time (6 h). b Cyclic carbonate = ethylene carbonate. c Cyclic carbonate = propylene carbonate. d Reaction time (8 h). e Reaction time (8 h) and temperature (170 °C). The values in entries 3 to 11 are presented as mean ± SD (n = 3). | |||
| 1 | CN(U) | (60.2)b (11.4)c | (83.1)b (76.3)c |
| 2 | CN(TU) | (64.3)b (17.6)c | (85.3)b (79.5)c |
| 3 | CN(UTU) | (71.5 ± 0.7)b (21.8 ± 0.8)c | (89.1 ± 0.4)b (81.2 ± 0.2)c |
| 4 | S-CN(UTU)-20 | (78.2 ± 0.5)b (30.4 ± 0.6)c | (91.2 ± 0.3)b (84.3 ± 0.1)c |
| 5 | S-CN(UTU)-40 | (82.1 ± 1.0)b (35.6 ± 0.8)c | (94.6 ± 0.3)b (85.5 ± 0.2)c |
| 6 | S-CN(UTU)-60 | (88.4 ± 0.8)b (43.4 ± 0.9)c | (98.3 ± 0.2)b (89.4 ± 0.4)c |
| 7d | S-CN(UTU)-60 | (98.9 ± 1.1)b (63.4 ± 1.1)c | (99.3 ± 0.2)b (89.7 ± 0.2)c |
| 8e | S-CN(UTU)-60 | (93.4 ± 1.3)c | (99.3 ± 0.4)e (90.3 ± 0.2)c |
| 9 | MgO | (38.6 ± 0.7)b | (97.6 ± 0.2)e |
| 10 | Na–Y | (26.7 ± 0.4)b | (96.3 ± 0.4)e |
| 11 | Zeolite beta | (15.4 ± 0.2) | (86.1 ± 0.5)e |
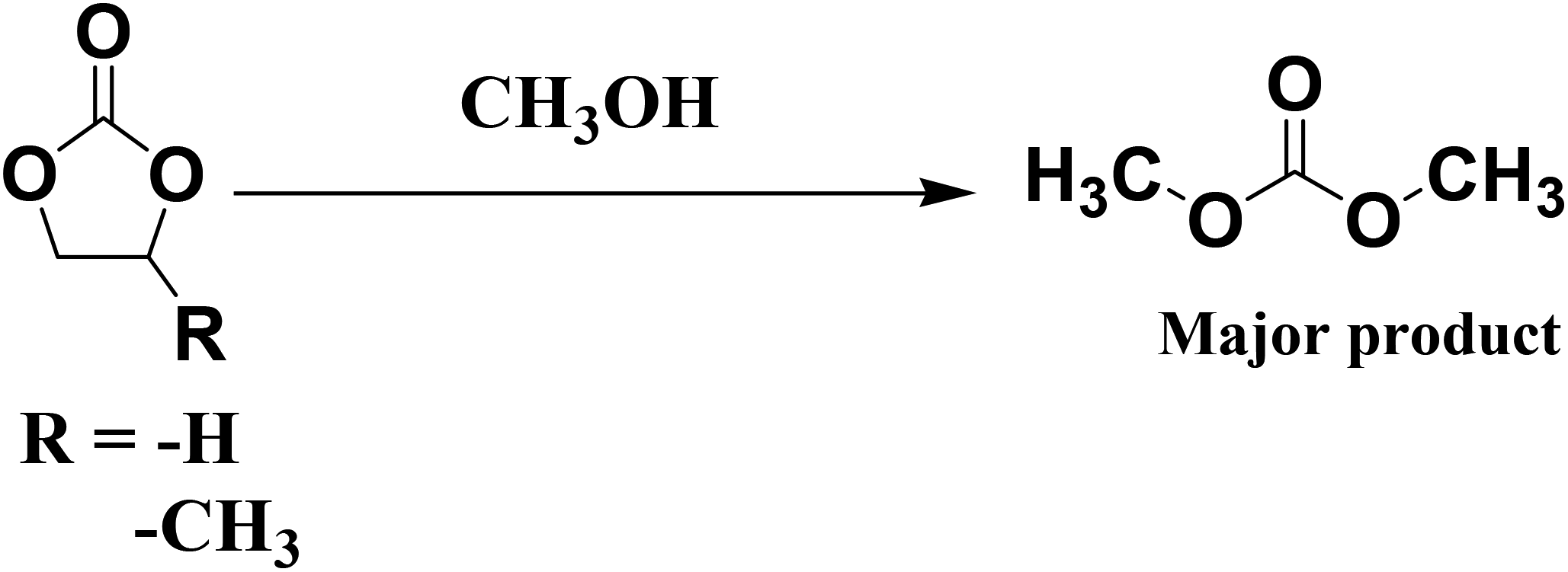
The highly active catalyst S-CN(UTU)-60 was further investigated in the esterification of oleic acid with methanol. Esterification reactions also demonstrate the efficiency of an acid catalyst. The reaction parameters were optimized, and the catalytic activity of S-CN(UTU)-60 under the optimized reaction conditions at different time intervals is shown in Fig. 12a. The reaction was conducted at 100 °C using an oleic acid: methanol ratio of 1:10. Quantitative yield of the methyl oleate (1H NMR spectrum, Fig. S9†) was obtained after 4.5 h over the highly active catalyst S-CN(UTU)-60. The esterification reaction is directly proportional to the amount and strength of the acid sites present in the material. The order of activity follows the trend S-CN(UTU)-60 > S-CN(UTU)-40 > S-CN(UTU)-20, which matches well with the acidity order obtained from TPD analysis (Fig. 12b). The advantage of S-CN(UTU)-60 over the other catalysts is clearly evident from the methyl oleate yield obtained from the esterification of oleic acid with methanol. The high yield of methyl oleate in the case of S-CN(UTU)-60 correlates well with the acidity obtained from the NH3 TPD investigation. Furthermore, the esterification reaction of oleic acid and methanol was performed using the acidic catalyst zeolite beta. A methyl oleate yield of 20.9% was obtained under our optimized reaction conditions. This result shows that S-CN(UTU)-60 has good acidity and can be applied in various esterification reactions.
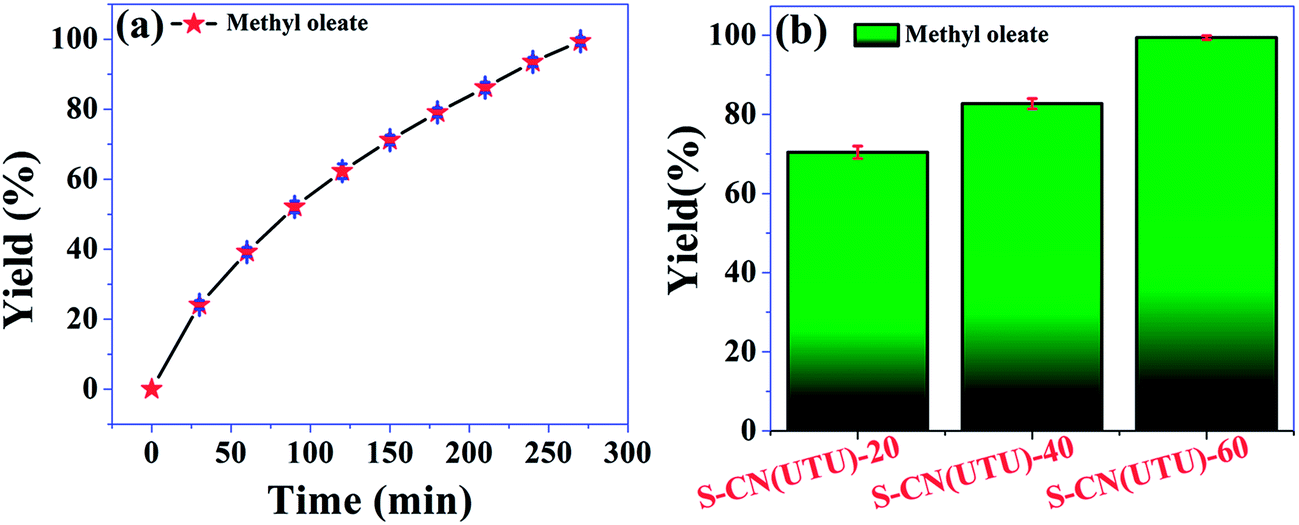 | ||
| Fig. 12 (a) Methyl oleate yield determined by alkali titration at different time intervals, (b) comparison of the catalytic activity of the H2SO4-treated catalysts in the esterification reaction after 4.5 h. Error bars in both graphs indicate mean ± SD (n = 3). | ||
4. Conclusions
Carbon nitride was treated with aqueous H2SO4 to introduce bi-functional acidic and basic sites in the material. Elemental analysis, EDAX, and XPS investigations showed that sulphur was incorporated in the material after H2SO4 treatment. FT-IR and XPS analysis confirmed that –SO3H, –NH2, and –OH functionalities were present in the H2SO4-treated samples. SEM and TEM investigations confirmed that during the H2SO4 treatment, the carbon nitride structure disintegrated and was reintegrated under ice-cold water conditions to produce carbon nitride materials with different morphologies when compared to the parent material. However, the primary framework of carbon nitride did not change, as supported by the XRD and TEM investigations. CO2-TPD and NH3-TPD analysis confirmed that the sample treated with 60% aqueous H2SO4 exhibited the highest acidity and basicity. The 60% aqueous H2SO4-treated sample exhibited the highest activity in the activation and utilization of CO2 in the synthesis of cyclic carbonates and quinazoline-2,4(1H,3H)-dione. The activation energy for the synthesis of cyclic carbonate by the cycloaddition of CO2 to epichlorohydrin was calculated to be 55.33 kJ mol−1. The catalyst was found to exhibit the highest activity for the synthesis of cyclic carbonate without using any solvent or promoters/co-catalysts. The catalyst exhibited no loss in activity or catalyst functionality even after five cycles of use. Its high activity correlated well with the acidity/basicity of the catalyst obtained from the TPD investigation. The merit of the bi-functional catalyst prepared in this study was demonstrated in the cycloaddition of CO2 to epoxide. Basic sites present in the catalyst activated CO2 molecules, whereas acid sites present in the catalyst activated the epoxide ring and polarized the C–O bond to facilitate CO2 insertion to form the cyclic carbonate. Simultaneous activation of CO2 and epoxide/2-aminobenzonitrile by the synergistic participation of the bi-functional sites present in the carbon nitride catalyst was responsible for the high catalytic activity. The bi-functional nature of the catalyst was also demonstrated with the aid of transesterification and esterification reactions. The application of these catalysts was successfully demonstrated in the transesterification of cyclic carbonate with methanol to produce dialkyl carbonate. The 60% aqueous H2SO4-treated sample showed the highest activity, which correlated well with the results obtained from CO2-TPD. The application of these catalysts was successfully demonstrated in the esterification of oleic acid with methanol. The 60% aqueous H2SO4-treated sample showed the highest activity, which correlated well with the results obtained from NH3-TPD. The process using the present catalyst system avoids hazardous substances such as phosgene or isocyanate and is performed at low temperature and pressure. The facile economical synthesis, tuneable acid–base properties, and efficient recyclability of the catalysts have encouraged us to use them in various catalytic applications.Acknowledgements
This work is financially supported by Nano Mission, Department of Science and Technology, New Delhi (SR/NM/NS-1054/2015). S. S is grateful to MHRD, New Delhi for providing SRF. The authors thank the DST FIST-funded XPS facility at the Department of Physics, IIT Kharagpur. The authors are grateful to SAIF, IIT Bombay for HRTEM analysis. The authors are also thankful to the Director, IIT Ropar for providing TPD facilities through an interdisciplinary project.Notes and references
- T. M. Mcdonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- F. Jin, X. Zeng, Z. Jing and H. Enomoto, Ind. Eng. Chem. Res., 2012, 51, 9921–9937 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- P. T. Anastas, Green Chem., 2003, 5, G29–G34 RSC.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS.
- Z. Z. Yang, L. N. He, J. Gao, A. H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 CAS.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. A. Cantat, Angew. Chem., Int. Ed., 2012, 5, 187–190 CrossRef PubMed.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef CAS.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- F. Solymosi and H. Knözinger, J. Catal., 1990, 122, 166–177 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 89, 81–85 CrossRef CAS.
- X. B. Lu, Y. J. Zhang, K. Jin, L. M. Luo and H. Wang, J. Catal., 2004, 227, 537–541 CrossRef CAS.
- X. B. Lu, H. Wang and R. He, J. Mol. Catal., 2002, 186, 33–42 CrossRef CAS.
- S. Ravi, D.-H. Kang, R. Roshan, J. Tharun, A. C. Kathalikkattil and D. W. Park, Catal. Sci. Technol., 2015, 5, 1580–1587 CAS.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 91, 133–139 CrossRef CAS.
- R. R. Kuruppathparambil, R. Babu, H. M. Jeong, G. Y. Hwang, G. S. Jeong, M. Kim and D. W. Park, Green Chem., 2016, 18, 6349–6356 RSC.
- S. Roy, B. Banerjee, A. Bhaumik and S. M. Islam, RSC Adv., 2016, 6, 31153–31160 RSC.
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS.
- S. I. Fujita, B. M. Bhanage, Y. Ikushima and M. Arai, Green Chem., 2001, 3, 87–91 RSC.
- R. A. Watile, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 1051–1055 CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- S. Wang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 2308–2320 CrossRef CAS PubMed.
- H. S. Kim, J. J. Kim, H. N. Kwon, M. J. Chung, B. G. Lee and H. G. Jang, J. Catal., 2002, 205, 226–229 CrossRef CAS.
- S. I. Fujita, M. Tanaka and M. Arai, Catal. Sci. Technol., 2014, 4, 1563–1569 CAS.
- D. B. Nale, S. Rana, K. Parida and B. M. Bhanage, Catal. Sci. Technol., 2014, 4, 1608–1614 CAS.
- W. Lu, J. Ma, J. Hu, J. Song, Z. Zhang, G. Yang and B. Han, Green Chem., 2014, 16, 221–225 RSC.
- B. Sarmah, B. Satpati and R. Srivastava, J. Colloid Interface Sci., 2017, 493, 307–316 CrossRef CAS PubMed.
- Z. Xie, M. Zhu, A. Nambo, J. B. Jasinski and M. A. Carreon, Dalton Trans., 2013, 42, 6732–6735 RSC.
- M. A. Carreon, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2012, 51, 1306–1314 Search PubMed.
- O. Zalomaeva, A. Chibiryaev, K. Kovalenko, O. Kholdeeva, B. Balzhinimaev and P. V. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS.
- R. Babu, A. C. Kathalikkattil, R. Roshan, J. Tharun, D. Kim and D. Park, Green Chem., 2016, 18, 232–242 RSC.
- P. Rani, P. F. Siril and R. Srivastava, Mol. Catal., 2017, 433, 100–110 CrossRef.
- J. A. Castro-Osma, C. Alonso-Moreno, A. Lara-Sanchez, J. Martinez, M. North and A. Otero, Catal. Sci. Technol., 2014, 4, 1674–1684 CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1–15 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2006, 90, 314–326 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- S. Verma, R. N. B. Baig, M. N. Nadagouda and R. S. Verma, Chem. Commun., 2015, 51, 15554–15557 RSC.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- H. Yan, Y. Chen and S. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CrossRef CAS.
- S. Patnaik, S. Martha, G. Madras and K. Parida, Phys. Chem. Chem. Phys., 2016, 18, 28502–28514 RSC.
- F. Cheng, H. Wang and X. Dong, Chem. Commun., 2015, 51, 7176–7179 RSC.
- R. C. Pawar, S. Kang, J. H. Park, J.-H. Kim, S. Ahn and C. S. Lee, Sci. Rep., 2016, 6, 31147, DOI:10.1038/srep31147.
- Z. Huang, F. Li, B. Chen and G. Yuan, Catal. Sci. Technol., 2016, 6, 2942–2948 CAS.
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 CAS.
- T. Zhang, X. Wang, X. Huang, Y. Liao and J. Chen, RSC Adv., 2016, 6, 2810–2818 RSC.
- J. Xu, F. Wu, Q. Jiang and Y. X. Li, Catal. Sci. Technol., 2015, 5, 447–454 CAS.
- J. Xu, J. K. Shang, Q. Jiang, Y. Wang and Y. X. Li, RSC Adv., 2016, 6, 55382–55392 RSC.
- R. Kore, B. Satpati and R. Srivastava, Chem.–Eur. J., 2011, 17, 14360–14365 CrossRef CAS PubMed.
- Y. Ding, G. Zhang, H. Wu, B. Hai, L. Wang and Y. Qian, Chem. Mater., 2001, 13, 435–440 CrossRef CAS.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS.
- Y. Zhao, B. Yu, Z. Yang, H. Zhang, L. Hao, X. Gao and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 5922–5925 CrossRef CAS PubMed.
- L. Espinosa, A. Gevers, B. Woldt, M. Graß and M. Meier, Green Chem., 2014, 16, 1883–1892 RSC.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, W. Sun and W.-K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS.
- S. Martha, A. Nashim and K. Parida, J. Mater. Chem. A, 2013, 1, 7816–7824 CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CrossRef CAS.
- A. Jönoson, E. Wehtje, P. Adlercreutz and B. Mattiasson, Biochim. Biophys. Acta, 1999, 1430, 313–322 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 show the XRD patterns, N2 adsorption–desorption isotherms, SEM images, and TEM images; Fig. S6–S8 show the XRD pattern, SEM images, and EDAX spectrum of the recycled catalyst; Fig. S9 shows the 1H NMR spectrum of methyl oleate. Scheme S1 shows a schematic of the morphology evolution after H2SO4 treatment, and Schemes S2 and S3 show the proposed mechanism for the cycloaddition reaction of CO2 to epoxide and 2-aminobenzonitrile. See DOI: 10.1039/c7se00223h |
| This journal is © The Royal Society of Chemistry 2017 |