Drastic improvement in the photocatalytic activity of Ga2O3 modified with Mg–Al layered double hydroxide for the conversion of CO2 in water†
Received
17th April 2017
, Accepted 27th June 2017
First published on 28th June 2017
Abstract
Photocatalytic conversion of CO2 to useful chemicals is a promising countermeasure against increasing concentrations of CO2 in the atmosphere as photocatalytic reactions can drive a thermodynamically uphill reaction (so-called “artificial photosynthesis”). Our group has earlier reported that Ga2O3 photocatalysts loaded with a Ag cocatalyst exhibit high activity for the conversion of CO2 to CO in water, and further modification of the catalysts with ZnGa2O4 improves the selectivity for CO over the other reduction products by suppressing H2. In this study, Ga2O3 modified with a Mg–Al layered double hydroxide (LDH) considerably enhanced not only the amount of CO evolved but also the selectivity toward CO evolution. By using 1.0 g of a Mg–Al LDH/Ga2O3 composite photocatalyst loaded with 0.25 wt% of a Ag cocatalyst, the CO formation rate and selectivity for CO were 211.7 μmol h−1 and 61.7%, respectively. Moreover, the turnover number (TON) of electrons utilized to reduce CO2 to CO was 75.4 per Ag atom during 5 h of photoirradiation.
Introduction
Photocatalysis has been increasingly attracting attention because it can be used to convert light energy to chemical energy, which can be stored. This conversion has implications for the current critical environmental issues. The photocatalytic splitting of H2O to H2 and O2 under solar light illumination has been already reported.1–3 H2 can be used as an energy source for various applications, such as fuel cells and hydrogen internal combustion engines. Photocatalytic artificial photosynthesis involves photoabsorption (light harvesting), charge separation, and H2O oxidation, as well as the consumption of the photogenerated electrons for reduction.4,5 Because it is important to remove CO2 (a greenhouse gas) from the atmosphere, a photocatalytic reaction that can reduce CO2via an uphill process (ΔG > 0) can be a promising technique.5–7 Previously, our group has reported the photocatalytic activity of Ga2O3, Zn-modified Ga2O3, and ZnGa2O4 for the conversion of CO2 to CO in an aqueous NaHCO3 solution in the presence of a Ag cocatalyst.8–10 Although the Ag-loaded Ga2O3 photocatalyst exhibited high performance, the selectivity toward CO evolution among the reduction products was extremely low, and H2 was predominantly formed.8 As the process for H2 formation competes for the electrons required for CO2 reduction, further optimization is crucial for suppressing this process. In this regard, a Ga2O3 photocatalyst modified with a Zn species has been reported to drastically increase the selectivity toward CO evolution by suppressing H2 evolution.10 X-ray diffraction (XRD) and X-ray absorption fine structure (XAFS) analyses have revealed that after calcination at 1223 K, ZnGa2O4 is formed on the surface of Ga2O3 modified with Zn species.8,10 The ZnGa2O4 thus formed, which itself can act as a photocatalyst,10 clearly suppresses H2 production and improves the selectivity toward CO evolution among the reduction products. The CO evolution rate (108 μmol h−1 for 1.0 g of Ag/ZnGa2O4/Ga2O3) was deemed insufficient, while the selectivity toward CO evolution was improved by modification with ZnGa2O4.
Modification of a photocatalyst with a solid base material, which can provide adsorption sites for CO2 molecules, is promising to regulate the selectivity for the photocatalytic conversion of CO2 in water. For this purpose, MgO-modified TiO2,11,12 magnesium/aluminum layered double oxide (LDO)-grafted TiO2,13 and self-assembled carbon nitride (C3N4), as well as different types of LDHs (LDH-C3N4),14 have been reported to exhibit photocatalytic activity for CO2 conversion. Moreover, our group has reported that a Ta2O5 photocatalyst modified with alkaline earth metal oxides, particularly SrO, drastically improves the selectivity toward CO evolution for CO2 conversion in an aqueous NaHCO3 solution.15 Layered double hydroxides (LDHs) are known as anionic clays, consisting of highly ordered two-dimensional hydroxide sheets and charge-compensating interlayer anions.16,17 The general formula of LDHs is [M1−α2+Mα3+(OH)2]α+(Aα/nn−)·mH2O, where M2+ and M3+ represent the divalent and trivalent cations, respectively; α represents the molar ratio of M3+/(M2+ + M3+), and An− is the interlayer anion with a valence of n.18 Mg–Al LDH (Mg6Al2(OH)16(CO3)·4H2O), the most well-known LDH, is widely used as a solid base material for the removal of various anions from wastewater,19–21 for base-catalyzed reactions,22–24 and for CO2 capture and storage.25 Moreover, Ebitani et al. have reported that Mg–Al LDH catalyzes the aldol condensation of carbonyl compounds in an aqueous solution,26 indicating that the Mg–Al LDH and its derivatives can be used as solid base catalysts in the presence of water because their surface basic sites exhibit high water tolerance.
Meanwhile, the photocatalytic conversion of CO2 in water using a series of LDH photocatalysts, whose surface properties can drive the selective reduction of CO2 to CO by suppressing H2 evolution, has also been reported by our group.27–29 In this study, drastic changes in the amount of CO evolved and the selectivity toward CO by the modification of a Ga2O3 photocatalyst with Mg–Al LDH were investigated.
Experimental section
Preparation of Mg–Al LDH-modified Ga2O3
Gallium oxide (Ga2O3, 99.99%, Kojundo Chemical Laboratory Co., Ltd.) was used as received. First, an aqueous solution of metal components (5.80 g of MgCl2·6H2O (Kojundo Chemical Laboratory Co., Ltd.) and 1.27 g of AlCl3 (Wako Pure Chemical Industries, Ltd.), total volume 333 mL) was slowly dropped into 0.06 M aqueous Na2CO3 (Wako Pure Chemical Industries, Ltd.) solution containing the required amount of Ga2O3 powder while simultaneously maintaining the pH between 9.9 and 10.1 by the addition of aqueous NaOH solution. The Mg/Al ratio in Mg–Al LDH was maintained constant at 3. The resulting suspension was aged at 333 K for 22 h at ambient pressure with vigorous stirring. The solid precipitate was washed with ultrapure water and collected by suction filtration. The cake was dried at 333 K under vacuum. The concentration of modified Mg–Al LDH was varied from 30 to 95 mol% in accordance with eqn (1).| | | x (mol%) = 100 × (AMg + AAl)/(AMg + AAl + AGa). | (1) |
Here, AMg, AAl, and AGa represent the amount of Mg atoms, Al atoms, and Ga2O3 in the prepared photocatalyst. For example, x = 50 implies that the molar ratio of (Mg + Al)/Ga2O3 = 1.0. Hereafter, x-MgAl/Ga2O3 represents Mg–Al LDH-modified Ga2O3 with a molar ratio of x. Reference composite samples of x-Mg/Ga2O3 and x-Al/Ga2O3, corresponding to Mg(OH)2- and Al(OH)3-modified Ga2O3, respectively, were prepared by the same procedure as that used to prepare x-MgAl/Ga2O3.
Loading of the Ag cocatalyst on the photocatalysts
The Ag cocatalyst was loaded on the photocatalysts by impregnation. The required amount of the as-synthesized x-MgAl/Ga2O3 powder was dispersed in an aqueous solution containing the required amount of AgNO3 (Wako Pure Chemical Industries, Ltd.) and evaporated at 353 K. The resulting powder was calcined at 723 K for 2 h under air. In this study, yAg/x-MgAl/Ga2O3 represents a Ag cocatalyst (y wt%) loaded on a x-MgAl/Ga2O3 photocatalyst.
Preparation of the reference photocatalysts
Ag cocatalyst-loaded Mg–Al LDH (Ag/MgAl).
Mg–Al LDH was synthesized by co-precipitation as described above with the exception of using Ga2O3 powder in aqueous Na2CO3. The Ag cocatalyst (1.0 wt%) was loaded on the prepared Mg–Al LDH by impregnation as described above.
Ag cocatalyst-loaded bare Ga2O3 (yAg/Ga2O3).
The Ag cocatalyst (y wt%) was loaded on Ga2O3 by the impregnation method as described above.
Mg–Al LDH-modified yAg/Ga2O3 (x-MgAl/yAg/Ga2O3).
An aqueous solution of precursors (MgCl2·6H2O and AlCl3) was slowly dropped into aqueous Na2CO3, which contains the yAg/Ga2O3 powder, by the same procedure as that utilized for x-MgAl/Ga2O3. A Ag cocatalyst was primarily loaded on a Ga2O3 photocatalyst for this reference material, and then Mg–Al LDH was prepared on it, while the Ag cocatalyst was deposited on Mg–Al LDH-modified Ga2O3 finally using the normal procedure.
Physical mixture of Mg–Al LDH and yAg/Ga2O3 (x-MgAl + yAg/Ga2O3).
Calculated amounts of the prepared Mg–Al LDH and yAg/Ga2O3 powders were sufficiently mixed in an agate mortar.
Ag cocatalyst-loaded SiO2-modified Ga2O3 (yAg/x-Si/Ga2O3).
The required amounts of Ga2O3 powder, tetraethyl orthosilicate (TEOS, Wako Pure Chemical Industries, Ltd.), and urea (Wako Pure Chemical Industries, Ltd.) were mixed in ultrapure water, and the resulting suspension was aged at 373 K for 24 h under hydrothermal conditions in a stainless-steel autoclave equipped with an inner Teflon vessel. The impregnation of the Ag cocatalyst and calcination at 723 K afforded the yAg/x-Si/Ga2O3 composite.
Catalyst characterization
Powder X-ray diffraction (XRD) patterns of the prepared samples were recorded on an XRD instrument (Ultima IV, Rigaku) using Cu Kα radiation (λ = 0.154 nm). The Brunauer–Emmett–Teller (BET) specific surface areas of the samples were estimated from their N2 adsorption isotherms at 77 K (liquid N2 temperature) using a volumetric adsorption measurement instrument (BELSORP-miniII, MicrotracBEL). Prior to the measurement, the powder sample was evacuated at 373 K for 1 h using a pretreatment instrument (BELPREP-vacII, MicrotracBEL). Scanning electron microscopy (SEM) images of the synthesized samples were recorded on a field-emission scanning electron microscopy instrument (FE-SEM, SU-8220, Hitachi High-Technologies) at an accelerating voltage of 3.0 kV. Transmission electron microscopy (TEM) images were recorded on a JEM-2100F TEM system (Japan Electron Optics Laboratory; JEOL) using an accelerating voltage of 200 kV. Further characterizations such as UV/Vis diffuse reflectance spectra (DRS), surface atomic ratios of metal components based on X-ray photoelectron spectroscopy (XPS) measurements, Ag 3d XPS measurements, TEM images of the photocatalysts, and transient photocurrent measurements are available in the ESI.†
Photocatalytic conversion of CO2 in an aqueous solution
The photocatalytic activity for the conversion of CO2 in an aqueous solution was evaluated using an inner-irradiation type reaction vessel in a quasi-flowing batch system. First, photocatalyst powder was dispersed in 1.0 L of an aqueous NaHCO3 solution, and the dissolved air in this suspension was degassed by a flow of high-purity CO2 (99.999%). CO2 was continuously bubbled into the reaction solution at a flow rate of 30 mL min−1. The suspension was irradiated using a 400 W high-pressure Hg lamp with a quartz filter equipped with a cooling water system. The gaseous products thus obtained, e.g., CO, H2, and O2, were analyzed by gas chromatography (GC-2010 Tracera, Shimadzu Corp.) using a MICROPACKED ST column equipped with an on-line barrier discharge ionization detector. The formation rates (μmol h−1) of these products were calculated using a 1.0 mL volumetric pipe. The selectivity toward CO evolution in the reduction products and the balance between the consumed electrons (e−) and holes (h+) were expressed by eqn (2) and (3), respectively.| | | Selectivity toward CO evolution (%) = 100 × RCO/(RCO + RH2). | (2) |
| | | Consumed e−/h+ = (RCO + RH2) × 2/(RO2 × 4). | (3) |
Here, RCO, RH2, and RO2 represent the formation rates of CO, H2, and O2, respectively.
Results and discussion
Fig. 1 shows the XRD patterns of the synthesized composite photocatalysts (A and B) and reference samples (C and D). Bare Ga2O3 (D) exhibited a typical reflection pattern corresponding to β-phase Ga2O3.30 In the XRD pattern of bare Mg–Al LDH (C), sharp diffraction peaks, corresponding to the reflections of the (003), (006), and (009) planes, were observed at 11.5°, 23.2°, and 34.8°, respectively.16 The positions of these peaks should be determined by the interlayer distance between each hydroxide sheet; a previous study has reported that carbonate ions (CO32−) are mainly intercalated in the interlayer space.31 Moreover, two diffraction peaks were observed at around 60°, corresponding to the reflections of the (110) and (113) planes, respectively. As compared to bare Ga2O3 (D) and bare Mg–Al LDH (C), the XRD pattern of 95-MgAl/Ga2O3 (B) showed a mixed phase comprising Ga2O3 and Mg–Al LDH. The peak position corresponding to the reflection of the (003) plane was the same as that observed for bare Mg–Al LDH, indicating that CO32− intercalated Mg–Al LDH was formed in the 95-MgAl/Ga2O3 composite. With the incorporation of Ga3+ into the hydroxide sheets within the LDH structure, the peak position corresponding to the reflection of the (110) plane is expected to be shifted to a low angle, because the ionic radii of six-fold coordinated Al3+ and Ga3+ are 53.5 and 62.0 pm, respectively.32 Based on this consideration, the peak corresponding to the reflection of the (110) plane slightly shifted from 60.7° to 60.4°, suggesting that the LDH structure contains marginal amounts of Ga3+ derived from Ga2O3. In other words, the results from XRD analysis indicated that Mg–Al LDH primarily consists of Mg2+, Al3+, and a small amount of Ga3+ within the hydroxide sheets, and a chemical connection is formed between Ga2O3 and Mg–Al LDH through these hydroxide sheets. The absorption edge of the 95-MgAl/Ga2O3 composite photocatalyst in the UV/Vis diffuse reflectance spectra (Fig. S1†) was almost the same as that of bare Ga2O3, indicating that the modification of Ga2O3 with Mg–Al LDH does not affect its absorption properties. With the Ag cocatalyst loading of 0.25 wt% on the 95-MgAl/Ga2O3 composite, a broad absorption shoulder was observed at approximately 280–330 nm, corresponding to the plasmonic absorption of Ag metal particles or the d–d transitions of Ag2O. Fig. 2 shows the SEM and TEM images of the composite photocatalysts. Several micrometer-sized pillar-shaped particles with several cracks and pore structures were observed in the SEM images of Ga2O3 (A). In the SEM images of the composite photocatalyst (B), almost all of the Ga2O3 particles were covered by platelet-like particles, which is characteristic of Mg–Al LDHs. Moreover, a core–shell structure (Ga2O3 core and Mg–Al LDH shell) was clearly observed in the TEM image (C). Small black circles observed in the magnified view of the TEM image (D) were expected to be Ag-cocatalyst particles, with a diameter of several nanometers. Fig. 3 shows the formation rate of the gas evolved, the selectivity toward CO evolution, and the consumed e−/h+ ratio in the photocatalytic conversion of CO2 in water under UV light irradiation. 0.25Ag/95-MgAl/Ga2O3 (A) exhibited excellent activity for the photocatalytic conversion of CO2 to CO with a high selectivity of greater than 60%. CO2 is thought to be reduced by the photogenerated electrons in preference to proton (H2O). On the other hand, 0.25Ag/Ga2O3 (D) mainly produced H2 as the reduction product. The modification with Mg–Al LDH significantly improved the activity and selectivity for the photocatalytic conversion of CO2; nevertheless, Ag-loaded bare Mg–Al LDH (C) did not exhibit activity. The consumed e−/h+ ratio was approximately 1.0 for (A), (B), and (D), indicating that stoichiometric “reduction” and “oxidation” successfully occur.
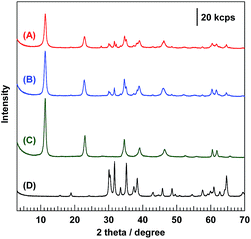 |
| | Fig. 1 XRD patterns of (A) 0.25Ag/95-MgAl/Ga2O3, (B) 95-MgAl/Ga2O3, (C) Mg–Al LDH, and (D) Ga2O3. | |
 |
| | Fig. 2 SEM images of (A) Ga2O3 and (B) 95-MgAl/Ga2O3, and TEM images of (C) 95-MgAl/Ga2O3 and (D) 1.0Ag/95-MgAl/Ga2O3. | |
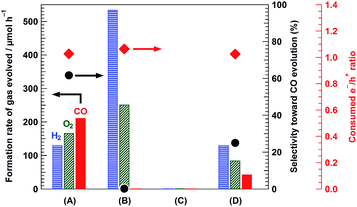 |
| | Fig. 3 Formation rate of the products evolved (bar graphs), selectivity toward CO evolution (black circle), and the consumed e−/h+ ratio (red diamond) in the photocatalytic conversion of CO2 in water using (A) 0.25Ag/95-MgAl/Ga2O3, (B) 95-MgAl/Ga2O3, (C) 1.0Ag/Mg–Al LDH, and (D) 0.25Ag/Ga2O3. Red: CO, green: O2 and blue: H2. Weight of Ga2O3 in the photocatalysts (A, B, and D) 0.19 g; weight of the photocatalyst (C) 0.5 g; reaction solution: 1.0 L of an aqueous NaHCO3 solution (0.1 M); CO2 supply: 30 mL min−1; light source: a 400 W high-pressure Hg lamp (through a quartz glass jacket); photoirradiation time: 1 h. | |
Fig. 4 shows the XRD patterns of 1.0Ag/x-MgAl/Ga2O3 (where x = 0, 50, 70, 85, and 95). The (003) plane reflection was not observed for 1.0Ag/0-MgAl/Ga2O3 (G) and 1.0Ag/50-MgAl/Ga2O3 (F), whereas an XRD pattern was observed for 1.0Ag/x-MgAl/Ga2O3 (x = 70, 85, and 95), with diffraction peaks corresponding to the LDH layer structure despite performing calcination at 723 K during the loading of the Ag cocatalyst. However, when bare Mg–Al LDH is calcined at 723 K, it is known to decompose to an amorphous mixed oxide, accompanied with the collapse of the layered structure, as shown in (A).16 This result indicates that the thermal stability of Mg–Al LDH in the 1.0Ag/x-MgAl/Ga2O3 composite photocatalyst is better than that of bare Mg–Al LDH, probably because of the chemical interaction between LDH and Ga2O3. Table 1 summarizes the differences in the specific surface areas and photocatalytic activities with different molar ratios of Mg–Al LDH. Only Ga2O3 was considered to serve as a photocatalyst in this composite material because Ag-loaded bare Mg–Al LDH did not exhibit any photocatalytic activity. Therefore, the amount of photocatalyst that can generate photoexcited electrons and holes should vary with the x value; for example, 500 mg of the 50-MgAl/Ga2O3 composite contains 420 mg of the Ga2O3 photocatalyst and 80 mg of Mg–Al LDH. Ag-Loaded bare Ga2O3 (x = 0) produced H2, O2, and CO in the stoichiometric balance of e− to h+ (e−/h+ = 1.03). The selectivity for CO over the other reduction products was 29.9% for bare Ga2O3, indicating that the photogenerated electrons are considerably consumed for the reduction of protons (H+) to H2. Meanwhile, with the increase of x (x = 30 and 50), the total photocatalytic activity (determined from the amount of evolved O2) improved despite the decrease in the amount of Ga2O3. The balance of e−/h+ was maintained approximately stoichiometric by varying the x value. With respect to the reduction products, the modification of Ga2O3 with Mg–Al LDH led to the increase in the formation rate of CO evolved from the reduction of CO2. In particular, the formation rate of CO was 149.7 μmol h−1 for x = 50; this value is more than twice as high as that observed for bare Ga2O3. With the increase of x to greater than 50, the photocatalytic activity, and hence, the formation rate of CO, gradually decreased with the increase in the Mg–Al LDH content (i.e., x). For example, the formation rate of O2 for x = 95, reflecting the total photocatalytic activity, was less than half of that for bare Ga2O3. In contrast, the selectivity for CO increased with increasing x. The suppression of H2 is considered to be particularly important because the reduction of H+ to H2 easily occurs for the photocatalytic conversion of CO2 in water. The 1.0Ag/95-MgAl/Ga2O3 composite photocatalyst exhibited 71.2% selectivity for CO. In other words, the Mg–Al LDH modification of Ga2O3 effectively suppresses H2 evolution. Based on the result obtained for bare Mg–Al LDH, which itself was not active for this reaction, the composite photocatalyst comprising Ga2O3 and Mg–Al LDH is crucial for the selective reduction of CO2 to CO. As shown in Fig. S2(a–c),† the particle size of the Ag cocatalyst loaded on the photocatalyst was dispersed by the modification of Ga2O3 with Mg–Al LDH. The CO evolution with high selectivity in the case of the 1.0Ag/95-MgAl/Ga2O3 composite photocatalyst might be caused by not only the alteration of surface properties but also the existence of highly-dispersed Ag cocatalyst particles. Moreover, a small shift to high binding energy in the Ag 3d X-ray photoelectron spectrum (Fig. S3(d)†) indicated that the as-synthesized Ag cocatalyst particle, which was mainly composed of Ag metal, was slightly oxidized by the use in the photocatalytic conversion of CO2 in water. The atomic ratio of Ga on the surface of the composite photocatalysts clearly decreased with the increasing molar ratio of Mg–Al LDH (Fig. S4†), reflecting that the shielding effect of Mg–Al LDH also drastically decreases the total photocatalytic activity. Accordingly, the anodic photocurrent of the Ga2O3 photocatalyst in the presence of methanol as a sacrificial reagent remarkably decreased by the modification with Mg–Al LDH (Fig. S5†). SEM and TEM images (Fig. 2), photocatalytic activities (Table 1), the surface atomic ratio (Fig. S4†), and photocurrent values (Fig. S5†) clearly revealed that the modification of Ga2O3 with Mg–Al LDH improves the selectivity for CO, while the total photocatalytic activity is suppressed by inactive Mg–Al LDH.
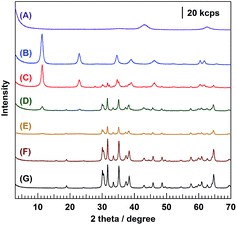 |
| | Fig. 4 XRD patterns of (A) Mg–Al LDH calcined at 723 K, (B) Mg–Al LDH, (C) 1.0Ag/95-MgAl/Ga2O3, (D) 1.0Ag/85-MgAl/Ga2O3, (E) 1.0Ag/75-MgAl/Ga2O3, (F) 1.0Ag/50-MgAl/Ga2O3, and (G) 1.0Ag/Ga2O3. | |
Table 1 Specific surface area (SBET) and the results from the conversion of CO2 in water for the x-MgAl/Ga2O3 composite photocatalyst (x = 30, 50, 70, 75, and 95), bare Ga2O3, and bare Mg–Al LDH. The weight of the Ag cocatalyst: 1.0 wt%. The total weight of the photocatalyst was fixed at 0.5 g. Reaction solution: 1.0 L of an aqueous NaHCO3 solution (0.1 M). CO2 supply: 30 mL min−1. Light source: a 400 W high-pressure Hg lamp (through a quartz glass jacket). Photoirradiation time: 1 h
| Photocatalyst |
S
BET/m2 g−1 |
Weight/g |
Formation rate of products evolved/μmol h−1 |
Selectivity toward CO evolution (%) |
Consumed e−/h+ ratio |
| Total |
Ga2O3 |
H2 |
O2 |
CO |
| Bare Ga2O3 |
13 |
0.50 |
0.50 |
147.3 |
101.6 |
62.9 |
29.9 |
1.03 |
| 30-MgAl/Ga2O3 |
16 |
0.50 |
0.46 |
167.5 |
137.5 |
116.9 |
41.1 |
1.03 |
| 50-MgAl/Ga2O3 |
22 |
0.50 |
0.42 |
200.7 |
168.7 |
149.7 |
42.7 |
1.04 |
| 70-MgAl/Ga2O3 |
35 |
0.50 |
0.35 |
169.7 |
149.9 |
145.5 |
46.2 |
1.05 |
| 75-MgAl/Ga2O3 |
47 |
0.50 |
0.30 |
107.6 |
125.0 |
145.3 |
57.5 |
1.01 |
| 95-MgAl/Ga2O3 |
92 |
0.50 |
0.09 |
21.7 |
36.9 |
53.7 |
71.2 |
1.02 |
| Bare Mg–Al LDH |
89 |
0.50 |
0.00 |
Trace |
Trace |
Trace |
— |
— |
Fig. 5 shows the result obtained from control experiments for the photocatalytic conversion of CO2 in an aqueous NaHCO3 solution using the 95-MgAl/Ga2O3 composite photocatalyst. The effects of the presence of the photocatalyst, photoirradiation, addition of NaHCO3 into the reaction solution, flow of CO2 gas, and loading of the Ag cocatalyst on 95-MgAl/Ga2O3 were investigated. A photocatalytic reaction did not occur in the absence of the photocatalyst (A) and in the absence of photoirradiation (B). With the irradiation of UV light, stoichiometric amounts of H2 and O2 were formed from the photocatalytic splitting of pure water (C). In this experiment, H2 was mainly produced as the reduction product despite the supply of CO2 to the system and the stable reaction solution pH of 6.0, which is probably not suitable for the reduction of CO2 to CO. Under He gas flow (D), water splitting to H2 and O2 was accompanied with marginal amounts of formed CO, presumably owing to the presence of the NaHCO3 additive. Photocatalytic activity for CO2 conversion was not observed in the absence of the Ag cocatalyst (E). These results indicated that all of the above-discussed factors are crucial to the conversion of CO2 to CO using the 95-MgAl/Ga2O3 composite photocatalyst. We previously reported that bidentate HCO3–Ga derived by reacting the dissolved CO2 molecule in water with the hydroxyl group anchored on the Ga atoms is the intermediate species for the photocatalytic conversion of CO2 in water over the Ga2O3-based photocatalyst.33 In this sense, the selectivity toward CO evolution was changed with the concentration of dissolved CO2 molecules in the reaction solution, which should be altered by the above listed factors and the pH value.
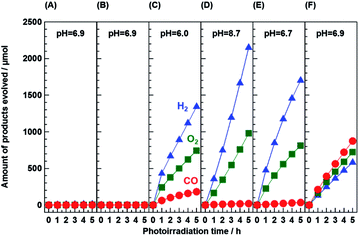 |
| | Fig. 5 Amount of products evolved from various control experiments for the conversion of CO2 in water using the 0.25Ag/95-MgAl/Ga2O3 composite photocatalyst. Red circle: CO, green square: O2, blue triangle: H2. (A) Without photocatalyst powder, (B) without irradiation (dark conditions), (C) without the NaHCO3 additive, (D) without the CO2 gas flow (He gas flow), (E) without the Ag cocatalyst, and (F) normal conditions. Photocatalyst weight: 1.0 g, reaction solution: 1.0 L of an aqueous NaHCO3 solution (0.1 M), CO2 supply: 30 mL min−1, light source: a 400 W high-pressure Hg lamp (through a quartz glass jacket), and photoirradiation time: 5 h. | |
Fig. 6 shows the formation rates of the products evolved from the conversion of CO2 in an aqueous NaHCO3 solution using the 95-MgAl/Ga2O3 composite photocatalyst with various amounts of the Ag cocatalyst. The 95-MgAl/Ga2O3 photocatalyst did not exhibit activity for the conversion of CO2 to CO in the absence of the loaded Ag cocatalyst. However, the introduction of 0.1 wt% Ag led to considerable enhancement of CO evolution. The formation rate of CO was 213.9 μmol h−1 for the 95-MgAl/Ga2O3 photocatalyst with the Ag cocatalyst loading of 0.1 wt%, whereas it was only 1.4 μmol h−1 for the 95-MgAl/Ga2O3 photocatalyst without any Ag cocatalyst. From this result, it can be concluded that the presence of the Ag cocatalyst is crucial for the conversion of CO2 to CO in this photocatalytic system, as described above. Furthermore, with increasing Ag cocatalyst loading on the 95-MgAl/Ga2O3 photocatalyst, the selectivity toward CO evolution was improved. However, the actual amount of the evolved products drastically decreased, and the formation rates of H2, O2, and CO were 26.5, 45.1, and 53.3 μmol h−1, respectively, with a Ag cocatalyst loading of 1.0 wt%. The sintered Ag particles, which were observed in the TEM images of 1.0Ag/95-MgAl/Ga2O3 and 3.0Ag/95-MgAl/Ga2O3 (Fig. S2(e) and (f)†), might induce a recombination of the photogenerated charges. In contrast, the conspicuous Ag particles were not found in the TEM image of the 0.25Ag photocatalyst (Fig. S2(d)†), indicating that the highly-dispersed Ag cocatalyst, which exhibits supreme activity for the selective conversion of CO2, was loaded on it. By considering the selectivity and formation rate of the product, the 0.25Ag/95-MgAl/Ga2O3 composite photocatalyst is the most suitable catalyst for the conversion of CO2 to CO under UV light irradiation. The formation rate of CO using the catalyst loaded with 0.25 wt% Ag was 211.7 μmol h−1. This value reflects remarkably high activity as the actual amount of the reduction products of CO2 (211.7 μmol h−1 per 0.19 g of Ga2O3) is greater than that reported previously.6 Typically, the amount of the products evolved in the photocatalytic reaction is often expressed as unreal amounts, which is normalized by the photocatalyst weight, photoirradiation area, or light source intensity (for example, the normalized activity with the photocatalyst weight for 0.25Ag/95-MgAl/Ga2O3 was 1114 μmol h−1 gGa2O3−1, gGa2O3: weight of Ga2O3 in the composite photocatalyst). In this case, the turnover number (TON) of the consumed electrons per Ag atom for CO evolution was 75.4 during 5 h of photoirradiation. Moreover, the ratio of the consumed electrons to holes, e−/h+, was 1.01 after 5 h of photoirradiation, indicating that this photocatalytic system reduces CO2 into CO, accompanied with the stoichiometric oxidation of H2O into O2.
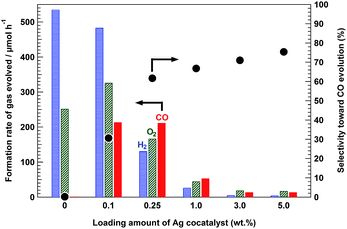 |
| | Fig. 6 Effect of the amount of the Ag cocatalyst on the formation rates of products evolved and the selectivity toward CO evolution in the photocatalytic conversion of CO2 in water. Red: CO, green: O2, and blue: H2. Black circle: selectivity toward CO evolution (right axis). Photocatalyst weight: 1.0 g, reaction solution: 1.0 L of an aqueous NaHCO3 solution (0.1 M), CO2 supply: 30 mL min−1, light source: a 400 W high-pressure Hg lamp (through a quartz glass jacket), and photoirradiation time: 1 h. | |
Fig. 7 shows the formation rates of products and the selectivity for CO over bare Ga2O3 (A), 95-MgAl/Ga2O3 (B), 95-Al/Ga2O3 (C), 95-Mg/Ga2O3 (D), and 95-Si/Ga2O3 (E) with a Ag cocatalyst loading of 0.25 wt%. The photocatalytic activity of 95-Si/Ga2O3, which has a sufficiently large specific surface area, was significantly less than that of bare Ga2O3, and CO evolution was completely suppressed by SiO2 modification. Hence, the increase in the specific surface area is not the only reason for the improved selectivity toward CO evolution by the modification of Ga2O3 with Mg–Al LDH. Modification with Al(OH)3 led to the increased specific surface area and improved selectivity toward CO evolution as compared with those observed for bare Ga2O3. Al2O3 (or Al(OH)3) has been reported to serve as an adsorbate for capturing CO2, resulting in the desorption of CO2 at 473–673 K in CO2-temperature programmed desorption (TPD) measurements; in other words, i.e., medium-strength basic sites are mainly present.34 In contrast, the specific surface area of the 95-Mg/Ga2O3 composite was not altered by modification with Mg(OH)2 because of its low specific surface area; hence, it does not exhibit improved selectivity toward CO evolution despite the high ability of MgO or Mg(OH)2 as a CO2-capturing material.35,36 Hence, the high activity as well as selectivity toward CO evolution for the 95-MgAl/Ga2O3 composite photocatalyst are related to the high specific surface area, resulting from Mg–Al LDH modification, and the excellent ability of Mg–Al LDH as an adsorbent for CO2,37,38 which helps in preserving the CO2 molecules near the Ag cocatalyst. As shown above, the 95-MgAl/Ga2O3 composite exhibited high activity for the stoichiometric splitting of water into H2 and O2 in the absence of the Ag cocatalyst, and a Ag cocatalyst loading of 0.25 wt% was beneficial for the formation of a considerable amount of CO as the reduction product of CO2, with high selectivity toward CO evolution. Interestingly, 95-MgAl/0.25Ag/Ga2O3, which was prepared by a procedure slightly different from that used to prepare 0.25Ag/95-MgAl/Ga2O3, exhibited a lower CO formation rate and selectivity for CO as compared with those observed for 0.25Ag/95-MgAl/Ga2O3 (Table S1†). For the 95-MgAl/0.25Ag/Ga2O3 composite, all Ag particles were considered to be present on the Ga2O3 surface, i.e., Ag/Ga2O3 should be covered with Mg–Al LDH, whereas almost the entire Ag cocatalyst is impregnated on Mg–Al LDH for 0.25Ag/95-MgAl/Ga2O3, which covers Ga2O3. Hence, the transfer of the photogenerated electrons from the conduction band of the Ga2O3 photocatalyst to the Ag particles via Mg–Al LDH permits the selective reduction of CO2 into CO. Moreover, the direct transfer of the photogenerated electrons from Ga2O3 to the Ag cocatalyst may preferentially cause the reduction of H+ into H2. Furthermore, the physical mixture of Mg–Al LDH and Ag/Ga2O3 did not exhibit any remarkable improvement in the amount of CO evolved as compared with that observed for Ag/Ga2O3, but only exhibited enhanced water splitting to H2 and O2, indicating that the selectivity toward CO evolution decreases. The chemical connection between Ga2O3 and Mg–Al LDH, as described in the part for XRD patterns, was estimated to play a crucial role in the improved selectivity toward CO evolution (see Fig. S6† for the schematic of these photocatalysts).
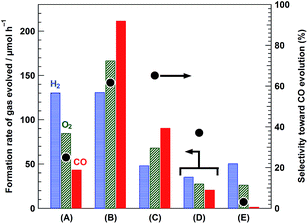 |
| | Fig. 7 Formation rates of the products evolved and the selectivity toward CO evolution in the photocatalytic conversion of CO2 in water using (A) 0.25Ag/Ga2O3 (without Mg–Al LDH modification), (B) 0.25Ag/95-MgAl/Ga2O3, (C) 0.25Ag/95-Al/Ga2O3 (Al modification), (D) 0.25Ag/95-Mg/Ga2O3 (Mg modification), and (E) 0.25Ag/95-Si/Ga2O3 (Si modification). Red: CO, green: O2, and blue: H2. Black circle: selectivity toward CO evolution (right axis). Photocatalyst weight: 0.19 g (A), 1.0 g (B–E), reaction solution: 1.0 L of an aqueous NaHCO3 solution (0.1 M), CO2 supply: 30 mL min−1, light source: a 400 W high-pressure Hg lamp (through a quartz glass jacket), and photoirradiation time: 1 h. | |
Fig. 8 shows the results obtained from isotopic experiments, which were conducted to confirm the carbon source in the CO evolved from the conversion of 13C-labeled CO2 (13CO2) in an aqueous NaHCO3 solution using the 0.25Ag/95-MgAl/Ga2O3 photocatalyst. The GC-MS profile was obtained by monitoring ion currents of m/z = 28 and 29 for the gas-phase products, which were initially separated using the GC system, accompanied by chromatograms recorded using a GC system equipped with a thermal conductivity detector (TCD). Three peaks were observed at approximately 100, 150, and 500 s in the GC/TCD chromatogram, corresponding to H2, O2, and CO, respectively. Another peak was observed at m/z = 29 at the same position as the peak corresponding to CO in the GC chromatogram, indicating that this photocatalytic system produces 13C-labeled CO (13CO, m/z = 29) as the reduction product of 13CO2. Meanwhile, a peak was not observed at m/z = 28 (unlabeled CO was not formed in the gas phase). Moreover, the amount of 13CO evolved, as evaluated from the peak area in the m/z = 29 profile, was in agreement with that quantified using the flame ionization detection GC equipped with a methanizer. This result clearly indicated that the CO evolved in this reaction originates from CO2 substrates and that the decomposition of the NaHCO3 additive or contaminated organic residues does not affect the photocatalytic activity for CO2 conversion. Moreover, the selectivity toward CO evolution was gradually decreased with repeating the photocatalytic reaction, as shown in Fig. S7.† Ag 3d XPS analysis (Fig. S3†) revealed the formation of Ag+ during the photocatalytic reaction, and TEM images (Fig. S8†) clearly elucidated that the aggregation of Ag cocatalyst particles accompanied with a slight change in the oxidation state was a fundamental factor of the deactivation of photocatalytic activity for the conversion of CO2 in water. The enhancement in the durability of the photocatalyst should be promoted in the near future for making a contribution to our sustainable society.
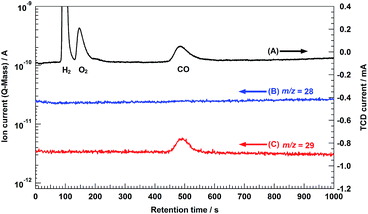 |
| | Fig. 8 GC-MS profiles for the isotopic experiments for the conversion of CO2 in water using the 0.25Ag/95-MgAl/Ga2O3 composite photocatalyst. (A) GC/TCD chromatogram, (B) Q-mass profile of m/z = 28, and (C) Q-mass profile of m/z = 29. Photocatalyst weight: 1.0 g, reaction solution: 1.0 L of an aqueous NaHCO3 solution (0.1 M), 13CO2 supply: 30 mL min−1, and light source: a 400 W high-pressure Hg lamp (through a quartz glass jacket). | |
Conclusions
In this study, a Mg–Al LDH/Ga2O3 composite photocatalyst modified with a Ag cocatalyst exhibited high activity for CO2 conversion in an aqueous NaHCO3 solution under UV light irradiation. The CO formation rate for the 95-MgAl/Ga2O3 photocatalyst loaded with 0.25 wt% Ag was 211.7 μmol h−1. In addition, the selectivity toward CO evolution among the reduction products was improved by the modification of the Ga2O3 photocatalyst with Mg–Al LDH, indicating that the transfer of the photogenerated electrons from the Ga2O3 photocatalyst to the Ag particles via Mg–Al LDH facilitates the selective reduction of CO2 into CO. Control experiments and isotopic labeling studies revealed that CO2 molecules dissolved in the reaction solution form CO by the conversion of CO2 using the Ag-loaded composite photocatalyst. In addition, the stoichiometric formation of O2 as the oxidation product of H2O suggested that H2O serves as the electron donor and a proton source. Hence, the increase in the specific surface area by the modification with Mg–Al LDH and the high ability of Mg–Al LDH as an adsorbent for CO2, which concentrates CO2 molecules near the Ag cocatalyst, improve the selective conversion of CO2 into CO using H2O as the reductant under UV light irradiation. The strategic improvement in the photocatalytic activity for the CO2 conversion, which is based on providing the CO2 adsorption sites, was reasonably demonstrated in this study.
Acknowledgements
This study was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “All Nippon Artificial Photosynthesis Project for Living Earth” (No. 2406) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan; the Precursory Research for Embryonic Science and Technology (PRESTO), supported by the Japan Science and Technology Agency (JST); and the Program for Elements Strategy Initiative for Catalysts & Batteries (ESICB), commissioned by the MEXT of Japan. Shoji Iguchi is grateful for the JSPS Research Fellowships for Young Scientists.
References
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406–13413 CrossRef CAS PubMed.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
- K. Teramura, Z. Wang, S. Hosokawa, Y. Sakata and T. Tanaka, Chem.–Eur. J., 2014, 20, 9906–9909 CrossRef CAS PubMed.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, J. Mater. Chem. A, 2015, 3, 11313–11319 CAS.
- Z. Wang, K. Teramura, Z. Huang, S. Hosokawa, Y. Sakata and T. Tanaka, Catal. Sci. Technol., 2015, 6, 1025–1032 Search PubMed.
- Q. Li, L. Zong, C. Li and J. Yang, Appl. Surf. Sci., 2014, 314, 458–463 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS.
- L. Liu, C. Zhao, J. Xu and Y. Li, Appl. Catal., B, 2015, 179, 489–499 CrossRef CAS.
- J. Hong, W. Zhang, Y. Wang, T. Zhou and R. Xu, ChemCatChem, 2014, 6, 2315–2321 CrossRef CAS.
- K. Teramura, H. Tatsumi, Z. Wang, S. Hosokawa and T. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 431–437 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- C. Delhoyo, Appl. Clay Sci., 2007, 36, 103–121 CrossRef CAS.
- Z. P. Xu, J. Zhang, M. O. Adebajo, H. Zhang and C. Zhou, Appl. Clay Sci., 2011, 53, 139–150 CrossRef CAS.
- R. L. Goswamee, P. Sengupta, K. G. Bhattacharyya and D. K. Dutta, Appl. Clay Sci., 1998, 13, 21–34 CrossRef CAS.
- J. Das, B. S. Patra, N. Baliarsingh and K. M. Parida, Appl. Clay Sci., 2006, 32, 252–260 CrossRef CAS.
- K. H. Goh, T. T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev., 2001, 43, 443–488 CAS.
- S. Abelló, F. Medina, D. Tichit, J. Pérez-Ramírez, J. C. Groen, J. E. Sueiras, P. Salagre and Y. Cesteros, Chem.–Eur. J., 2005, 11, 728–739 CrossRef PubMed.
- C. Xu, Y. Gao, X. Liu, R. Xin and Z. Wang, RSC Adv., 2013, 3, 793–801 RSC.
- M. K. Ram Reddy, Z. P. Xu, G. Q. Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504–7509 CrossRef CAS.
- K. Ebitani, K. Motokura, K. Mori, T. Mizugaki and K. Kaneda, J. Org. Chem., 2006, 71, 5440–5447 CrossRef CAS PubMed.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- S. Iguchi, K. Teramura, S. Hosokawa and T. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 17995–18003 RSC.
- S. Iguchi, K. Teramura, S. Hosokawa and T. Tanaka, Catal. Today, 2015, 251, 140–144 CrossRef CAS.
- X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089–13092 CrossRef CAS PubMed.
- S. Miyata, Clays Clay Miner., 1983, 31, 305–311 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- K. Teramura, K. Hori, Y. Terao, Z. Huang, S. Iguchi, Z. Wang, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2017, 121, 8711–8721 CAS.
- Y. Bang, S. J. Han, S. Kwon, V. Hiremath, I. K. Song and J. G. Seo, J. Nanosci. Nanotechnol., 2014, 14, 8531–8538 CrossRef CAS PubMed.
- R. Philipp and K. Fujimoto, J. Phys. Chem., 1992, 96, 9035–9038 CrossRef CAS.
- S. C. Lee, H. J. Chae, S. J. Lee, B. Y. Choi, C. K. Yi, J. B. Lee, C. K. Ryu and J. C. Kim, Environ. Sci. Technol., 2008, 42, 2736–2741 CrossRef CAS PubMed.
- Z. Yong, V. Mata and A. r. E. Rodrigues, Sep. Purif. Technol., 2002, 26, 195–205 CrossRef CAS.
- Z. Yong and A. r. E. Rodrigues, Energy Convers. Manage., 2002, 43, 1865–1876 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis DR spectra, TEM images, Ag 3d XPS, surface atomic ratios of the photocatalysts, photoelectrochemical measurements, photocatalytic activities of reference photocatalysts, durability of the photocatalytic activity, and schematic illustrations of photocatalysts. See DOI: 10.1039/c7se00204a |
|
| This journal is © The Royal Society of Chemistry 2017 |

 *ab,
Shotaro
Kidera
a,
Soichi
Kikkawa
a,
Saburo
Hosokawa
*ab,
Shotaro
Kidera
a,
Soichi
Kikkawa
a,
Saburo
Hosokawa







