
An investigation of Cu–Re–ZnO catalysts for the hydrogenolysis of glycerol under continuous flow conditions†
Mzamo L.
Shozi
 a,
Venkata D. B. C.
Dasireddy
a,
Sooboo
Singh
a,
Pheladi
Mohlala
b,
David J.
Morgan
c,
Sarwat
Iqbal
c and
Holger B.
Friedrich
*a
a,
Venkata D. B. C.
Dasireddy
a,
Sooboo
Singh
a,
Pheladi
Mohlala
b,
David J.
Morgan
c,
Sarwat
Iqbal
c and
Holger B.
Friedrich
*a
aSchool of Chemistry and Physics, University of KwaZulu-Natal, Durban, 4000, South Africa. E-mail: friedric@ukzn.ac.za
bSasol South Africa (Pty) Ltd, Group Technology R&T, 1 Klasie Havenga Street, Sasolburg 1974, South Africa
cCardiff Catalysis Institute, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
First published on 28th June 2017
Abstract
Cu and Re monometallic and bimetallic catalysts supported on ZnO were synthesized via wet impregnation. The catalysts were characterized using XRD, TPR, Pulse TPD, TEM, SEM, XPS and BET surface area. TPR results showed that the presence of rhenium increases the reduction temperature of the catalysts and TPD showed that the presence of copper decreases the Brønsted acidity of the catalysts. SEM showed an improved distribution of metal oxide on the support after the incorporation of rhenium. These catalysts were evaluated in the hydrogenolysis of glycerol in a continuous flow fixed bed reactor in a temperature range of 150–250 °C and a H2 pressure of 60 bar. All catalysts were active, with activity being higher over the rhenium containing catalysts. At the lowest temperature (150 °C), 1,2-propanediol had the highest selectivity which decreased with increase in temperature. Subsequently, the selectivity to lower alcohols, such as methanol, ethanol and 1-propanol, and ethylene glycol increased with temperature as 1,2-propanediol reacted further to these products due to C–C bond cleavage. This was also observed when the hydrogen content was increased at constant temperature (250 °C). All catalysts were found to be stable in terms of activity and selectivity to lower alcohols over a period of at least 24 hours at 250 °C and 60 bar H2 pressure.
1. Introduction
Glycerol can be obtained from plant cellulose and is also a major by-product of the biodiesel production process. Its availability is expected to increase due to the remarkable growth of biodiesel production worldwide.1 Since the existing supply and demand market cannot accommodate the excess amounts of glycerol generated, the conversion of glycerol to value-added products has become important.2 One of the processes that converts glycerol to value-added products is hydrogenolysis, a catalytic reaction involving the cleavage of a chemical bond with the simultaneous addition of a hydrogen atom, where glycerol is converted to mono-alcohols and diols.3–5 Mono-alcohols such as ethanol and 1-propanol are important, as ethanol is used as fuel and a fuel additive and 1-propanol is used as a multipurpose solvent and chemical feedstock for organic compounds.6 Diols obtained are usually 1,2-propanediol (1,2-PDO) and ethylene glycol (EG), which find important uses in the pharmaceutical industry and in antifreeze.7Hydrogenolysis of glycerol over supported copper catalysts has been extensively researched over the last decade.8–34 Chaminand et al.8 reported the use of CuO/ZnO in the hydrogenolysis of glycerol and obtained a selectivity of 100% to 1,2-PDO with a conversion of glycerol of 19%. Using tungstic acid as an additive had little effect on the conversion, however, the selectivity to 1,2-PDO dropped to 81% as it fragmented due to C–C cleavage. When investigating the use of Cu–ZnO catalysts with varying Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn weight ratios, the highest conversion (37%) was obtained when the ratio was 50:50 with 92% selectivity to 1,2-propanediol.12 CuO/ZnO catalysts prepared by the oxalate gel (OG) and co-precipitation (CP) methods were compared and the former found to give a higher conversion of glycerol due to higher surface area. A selectivity of ∼87% to 1,2-PDO was obtained over both catalysts. A comparison of supported Cu catalysts prepared via incipient wetness (IW) and ion exchange (IE) methods was later reported,16 where the hydrogenolysis of glycerol was performed in the both the liquid and gas phase. The highest conversion (52%) was obtained for the catalysts with a composition of 60 wt% Cu and 40 wt% ZnO, with a selectivity to 1,2-PDO of 98% in the liquid phase. In the gas phase, however, the highest conversion (92%) was obtained for Cu/SiO2 prepared via IE with a Cu concentration of 0.05 mol L−1, with the selectivity to acetol and 1,2-PDO being 69% and 20%, respectively.
:Zn weight ratios, the highest conversion (37%) was obtained when the ratio was 50:50 with 92% selectivity to 1,2-propanediol.12 CuO/ZnO catalysts prepared by the oxalate gel (OG) and co-precipitation (CP) methods were compared and the former found to give a higher conversion of glycerol due to higher surface area. A selectivity of ∼87% to 1,2-PDO was obtained over both catalysts. A comparison of supported Cu catalysts prepared via incipient wetness (IW) and ion exchange (IE) methods was later reported,16 where the hydrogenolysis of glycerol was performed in the both the liquid and gas phase. The highest conversion (52%) was obtained for the catalysts with a composition of 60 wt% Cu and 40 wt% ZnO, with a selectivity to 1,2-PDO of 98% in the liquid phase. In the gas phase, however, the highest conversion (92%) was obtained for Cu/SiO2 prepared via IE with a Cu concentration of 0.05 mol L−1, with the selectivity to acetol and 1,2-PDO being 69% and 20%, respectively.
When Feng et al. investigated the use of Cu/ZnO/MOx (MOx = Al2O3, TiO2, ZrO2) catalysts prepared via CP, they found these to be highly active in the gas phase hydrogenolysis of glycerol.18 All catalysts showed high conversion (>90%) over a temperature range of 240–300 °C with selectivity to both acetol and 1,2-PDO found to decrease with an increase in temperature, due to subsequent lower alcohol formation. Furthermore, the gas phase hydrogenolysis of glycerol was reported over a temperature range of 180–300 °C, using Cu/MOx (MOx = Al2O3, TiO2, ZnO) catalysts prepared by impregnation.23 The conversion of glycerol was consistent with the acidity of the catalysts, with the Cu/Al2O3 catalyst showing the highest conversion over the temperature range. The selectivity to 1,2-PDO was also highest for this catalyst at 87.9% at lower temperatures, which then decreased due to subsequent formation of propanols and ethanol. The Cu/ZnO catalyst showed the highest selectivity (86.5–91.2%) to acetol over the temperature range of 180–270 °C.
Huang et al.10 prepared CuO/SiO2 catalysts and investigated the effect of residual sodium. The highest conversion (32.7%) was obtained when the amount of sodium was at its lowest with a selectivity to 1,2-PDO of 98.7%. They later reported the use of Cu/SiO2 catalysts prepared by homogeneous and heterogeneous deposition-precipitation (DP).20 For the catalyst prepared via homogeneous-DP, a conversion of 22.1% was obtained with selectivity to 1,2-PDO being 98%. Catalyst prepared via heterogeneous-DP, gave a conversion of 35% and selectivity of 93.7% to 1,2-PDO.
Guo et al.11 investigated the effect of the supports (Al2O3, HY zeolite, 13X zeolite, HZSM-5 zeolite, Hβ zeolite) on some Cu catalysts. The catalysts showed low glycerol conversion with the exception of the alumina catalyst, which achieved a conversion of 34.6% and selectivity to propanediols of 93.9%. The effect of temperature and glycerol concentration was further investigated for this catalyst. The conversion of glycerol increased to a maximum of 52.2% over a temperature range of 180–240 °C, while the selectivity to propanediols decreased due to the formation of methanol and ethanol via C–C cleavage. The conversion of glycerol and the yield of propanediols was found to decrease at glycerol concentrations of below 50% due to the formation of water as a by-product. This is contradictory to what was reported by Dasari et al. who observed a decrease in selectivity for glycerol concentrations above 80% due to polymerisation of the reaction products.35
The effect of Cr as a promoter for Cu catalysts in glycerol hydrogenolysis has also been reported.13 The catalyst containing a Cu:Cr ratio of 1:3 gave the best catalytic performance with a glycerol conversion of 80.3% and a selectivity to 1,2-PDO of 83.9%. Mane et al.17 found the role of promoters in copper chromite catalysts to be important for the activity and selectivity in glycerol hydrogenolysis. They investigated the effect of Al, Zn and Ba as promoters. The catalyst with Ba as a promoter showed the highest conversions of 34% and 65% in batch and continuous operations respectively. A selectivity of 85% to 1,2-PDO was obtained for the batch operation. CuO/MgO catalysts prepared by CP and impregnation with various Cu loadings have also been investigated in the hydrogenolysis of glycerol.15 For both methods, the catalyst with a Cu loading of 15 wt% gave the highest conversion. Selectivity to 1,2-PDO was 95.8% for the catalyst prepared via co-precipitation and 92.8% for the catalyst prepared via impregnation.
The use of rhenium as a catalyst promoter in the hydrogenolysis of glycerol has received much attention.36–47 Ma and co-workers have, over the years, reported various influences of rhenium in glycerol hydrogenolysis,36,37,42,45,46 with the reactions being carried out in an autoclave. When they used Re2(CO)10 as a promoter on supported Ru monometallic catalysts, the conversion and selectivity to 1,2-PDO and 1,3-PDO increased. They also reported the use of monometallic Ru and bimetallic supported Ru–Re catalysts. The conversion was found to increase when Re was incorporated due to a synergistic effect between Re and Ru. Re was reported to improve the dispersion of Ru on the support surface and prevent Ru metal aggregation during hydrogenolysis. An investigation of the Re/Ru ratio in another study revealed that an increase in the amount of Re led to an increase in conversion due to Re increasing the acidity of the catalyst. Acid sites are involved in the dehydration step which also increases the selectivity to the diols.48,49
In this study we report for the first time the effect of reaction parameters such as temperature, H2:glycerol ratio and time on stream on the continuous flow hydrogenolysis of glycerol over Re and Cu monometallic and Re–Cu bimetallic catalysts supported on ZnO. These catalysts were chosen as copper has affinity to produce diols, while rhenium enhances catalytic activity and selectivity in glycerol hydrogenolysis.
2. Experimental
2.1. Catalyst preparation
The supported Cu and Re catalysts were prepared by impregnating ZnO (Sigma-Aldrich, 99.9%) with an aqueous solution of Cu(NO3)2·3H2O (Sigma-Aldrich, 97%) and NH4ReO4 (Aldrich, 99%) to obtain a nominal loading of 30 and 1 wt% respectively. After impregnation, the catalysts were dried at 110 °C for 12 hours, followed by calcination in air at 500 °C for 8 hours for Cu/ZnO and 500 °C for 3 hours for Re/ZnO. The Re–Cu catalyst was prepared by impregnating the calcined Cu/ZnO catalyst with an aqueous solution of NH4ReO4 (Aldrich, 99%) to obtain a nominal loading amount of 1 wt% Re. The catalyst was calcined in air at 500 °C for 8 hours after drying at 110 °C for 12 hours.2.2. Catalyst characterization
The BET surface area and pore volume measurements were carried out using a Micrometrics Tristar II Surface area and Porosity Analyzer. Prior to analysis, samples were weighed and degassed at 200 °C overnight under constant flow of nitrogen gas. Inductively coupled plasma analysis was performed using a Perkin Elmer Optical Emission Spectrometer Optima 5300 DV. Prior to analysis, the prepared samples were digested in acid and then diluted accordingly. Powder X-ray diffraction (XRD) was conducted on a Bruker D8 Advance diffractometer with Cu (Kα, λ = 1.5406 Å) as the radiation source.Temperature programmed reduction (TPR) analyses were carried out on a Micromeritics Autochem 2920 chemisorption analyser. Prior to reduction approximately 50 mg of the sample was placed in a U-shaped quartz tube. The sample was dried by heating at 5 °C min−1 under helium flow (50 mL min−1) to 120 °C and kept at this temperature for 10 minutes. The sample was then cooled to room temperature and subsequently heated again at 10 °C min−1 under 10% H2/Ar flow (50 mL min−1) to 1000 °C and kept at this temperature for 10 minutes. The water formed during reduction was trapped using a dry ice/isopropanol bath. The amount of hydrogen consumed during reduction was measured with the TCD. Surface concentrations of Brønsted acidic sites on the prepared catalyst were determined by temperature programmed desorption of propylene. Pulse chemisorption-TPD-MS experiments were carried out on a Micromeritics Autochem 2920 chemisorption analyser coupled with Cirrus Mass Spectrometer. Approximately 50 mg of sample was placed in a quartz U-tube containing quartz wool, fitted with a thermocouple for continuous temperature measurements. Before the TPD experiments the calcined samples were outgassed at 550 °C for 60 minutes in a flow of helium (50 mL min−1) at 10 °C min−1. Subsequently the sample was cooled down to 100 °C, the adsorption temperature. The activation of the samples was followed by pulse chemisorption where 30 injections of propylamine vapour were dosed onto the catalyst by means of an inert gas, helium, flowing through a 1 cm3 loop. The last part of the analysis involved a temperature-programmed desorption (TPD), which was carried out with a linear heating rate (10 °C min−1) in a flow of helium (50 mL min−1) from 100 °C to 550 °C for 60 minutes. At this step in the analysis, the mass spectrometer (MS) began scanning for propylene, the product of interest.
The surface morphology of the catalysts was observed using a Zeiss Ultra plus Field Emission Gun microscope. The images were captured using SmartSEM software. Prior to SEM analysis, the samples were mounted on aluminium stubs using double-sided carbon tape and subsequently gold spluttered using the Polaron SC500 coating unit. Transmission electron microscopy (TEM) images were viewed using a Jeol JEM-1010 Electron Microscope. The images were captured and analysed using iTEM software. Prior to analysis, the samples were sonicated in ethanol after which they were placed on a copper grid. XPS data was acquired on a Thermo-Fisher Scientific K-Alpha+ X-ray photoelectron spectrometer, utilizing monochromatic Al Kα radiation operating at a power of 72 W (6 mA × 12 kV). High resolution scans were performed at a pass energy of 40 eV, with a 0.1 eV step, whilst survey spectra were acquired at a pass energy of 150 eV and a step size of 1 eV. Charge neutralization was achieved using a combination of low energy electrons and argon ions which gave a reproducible C(1s) binding energy of 284.8 eV for all samples.
2.3. Catalytic testing
Catalytic testing was carried out using a continuous flow fixed-bed reactor comprising of a reactor tube with a length of 250 mm and an internal diameter of 20 mm. The catalysts were pelletized and sieved to a mesh size range of 300–600 μm. A catalyst volume of 3 mL was mixed with an equal amount of 24-grit carborundum and loaded in the reactor tube. The GHSV of hydrogen was maintained at 1860 h−1 and the LHSV of glycerol at 10 h−1 for all reactions. Reactions were carried out between 250–325 °C at a hydrogen pressure of 60 bar. Prior to testing, the catalysts were activated for 12 hours under hydrogen (50% in N2); Re/ZnO at 250 °C, Cu/ZnO at 280 °C and Re–Cu/ZnO at 400 °C; after which the reactor was cooled to operating temperatures. The liquid products and unreacted glycerol were collected in a catchpot cooled to −10 °C and analysed on a Perkin Elmer Clarus 500 GC equipped with an FID and a PONA column. The gas samples were collected and analysed for methane and carbon oxides on a Perkin Elmer Clarus 400 GC equipped with a TCD. Mass balances were 100 ± 5% on a carbon basis. All data points were obtained in duplicate within an error of ±2%.3. Results and discussion
3.1. Catalyst characterization
| Catalyst | Metal content/wt% | SBET/m2 g−1 | Pore volume/cm3 g−1 | |
|---|---|---|---|---|
| Cu | Re | |||
| ZnO | — | — | 5.4 | 0.011 |
| CuO/ZnO | 29.71 | — | 2.2 | 0.0036 |
| ReOx/ZnO | — | 1.29 | 5.1 | 0.0051 |
| Re–CuO/ZnO | 29.46 | 1.10 | 2.0 | 0.0029 |
 | ||
| Fig. 1 XRD diffractograms of (a) ReOx/ZnO, (b) CuO/ZnO and (c) Re–CuO/ZnO. | ||
 | ||
| Fig. 2 TPR profiles of ZnO support, Cu/ZnO, Re/ZnO and Re–Cu/ZnO catalysts. | ||
| Catalyst | C | Cu | Re | O | Zn |
|---|---|---|---|---|---|
| CuO/ZnO | 3.1 | 15.4 | — | 44.8 | 36.7 |
| ReOx/ZnO | 7.8 | — | 1.8 | 45.1 | 45.3 |
| Re–CuO/ZnO | 2.4 | 15.3 | 0.9 | 46.1 | 35.3 |
3.2. Catalytic testing
Fig. 3 shows the effect of temperature on the activity of the catalysts in the hydrogenolysis of glycerol. For all catalysts, the conversion was found to increase with temperature, as would be expected. The Cu/ZnO catalyst had the lowest activity which tripled when rhenium was added, likely due to the promoting effect (increasing acidity) that rhenium has in the hydrogenolysis of glycerol.36–38 This also explains why Re/ZnO is also more active than Cu/ZnO.
 | ||
| Fig. 3 Conversion of glycerol with temperature over the different catalysts. Reaction conditions: 60 wt% glycerol/H2O, 3 mL catalyst bed, 60 bar H2, H2:glycerol ratio = 1:1. | ||
Fig. 4a–c show the effect of temperature on the product selectivities over the different catalysts. With increase in temperature, the selectivity towards 1,2-PDO decreased over all the catalysts and the selectivity towards EG, ethanol, methanol and 1- and 2-propanol increased. This could be due to the hydrogenolysis of 1,2-PDO at higher temperatures to these products as it is reported that at temperatures above 200 °C, 1,2-PDO is susceptible to further hydrogenolysis to lower alcohols.4,18,35,66
 | ||
| Fig. 4 Effect of temperature on product selectivity over (a) Re/ZnO, (b) Cu/ZnO and (c) Re–Cu/ZnO. Reaction conditions: 60 wt% glycerol/H2O, 3 mL catalyst bed, 60 bar H2, H2:glycerol ratio = 1:1. Other products: acetol, methane, ethane, propane, CO, CO2, unknowns. | ||
The different product selectivities observed over the three catalysts can be related to metal-support interactions and the difference in acidity of the support. The selectivity to 1,2-PDO (96%) was highest over Re/ZnO at a temperature of 150 °C, due to the higher concentration of Brønsted acid sites which assists in the dehydration step which leads to the formation of 1,2-PDO.
The selectivity to EG was higher over the Cu containing catalysts, reaching 45% over Re–Cu/ZnO. The formation of EG from glycerol is believed to follow a reaction pathway previously reported.67Scheme 1 shows the possible routes to obtaining EG from glycerol over metal catalysts. Glycerol is dehydrogenated to glyceraldehyde which undergoes decarbonylation to form EG and carbon monoxide. Glyceraldehyde may also go through retro-aldol condensation to form glycolaldehyde and formic acid, which are subsequently hydrogenated to EG and methanol. High EG selectivity can also be due to the higher propensity for C–C cleavage over copper containing catalysts and it has been reported that Cu has a strong ability to break C–C bonds to form lower alcohols.9,23 Another trend observed is that the selectivity towards methanol, ethanol and the propanols increased with decrease in selectivity to 1,2-PDO. The likely mechanism of formation of these lower alcohols from 1,2-PDO has also been reported.64,65,67 Amongst other products formed, some unknowns were observed which are believed to be condensation products as previously reported.68
 | ||
| Scheme 1 Proposed reaction pathways for the formation of EG from glycerol.67 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :glycerol ratio.
Fig. 5a–c shows the conversion of glycerol with respect to the hydrogen to glycerol molar ratio at a reaction temperature of 250 °C at a constant hydrogen pressure of 60 bar. An increase in the conversion was seen with an increase of the hydrogen to glycerol ratio. With the increase in the hydrogen to glycerol ratio, the selectivity towards 1,2-PDO decreased with increase in the selectivity towards EG and mono-alcohols. This is likely due to the hydrogenolysis of 1,2-PDO to further products at higher hydrogen to glycerol ratios. Especially notable is that the bimetallic catalyst, unlike the monometallic equivalent, gives predominantly EG and that the selectivity to this product reaches 73% with increasing H2:glycerol ratio. Doping with rhenium thus enhances the C–C bond cleavage ability of Cu.
:glycerol ratio.
Fig. 5a–c shows the conversion of glycerol with respect to the hydrogen to glycerol molar ratio at a reaction temperature of 250 °C at a constant hydrogen pressure of 60 bar. An increase in the conversion was seen with an increase of the hydrogen to glycerol ratio. With the increase in the hydrogen to glycerol ratio, the selectivity towards 1,2-PDO decreased with increase in the selectivity towards EG and mono-alcohols. This is likely due to the hydrogenolysis of 1,2-PDO to further products at higher hydrogen to glycerol ratios. Especially notable is that the bimetallic catalyst, unlike the monometallic equivalent, gives predominantly EG and that the selectivity to this product reaches 73% with increasing H2:glycerol ratio. Doping with rhenium thus enhances the C–C bond cleavage ability of Cu.
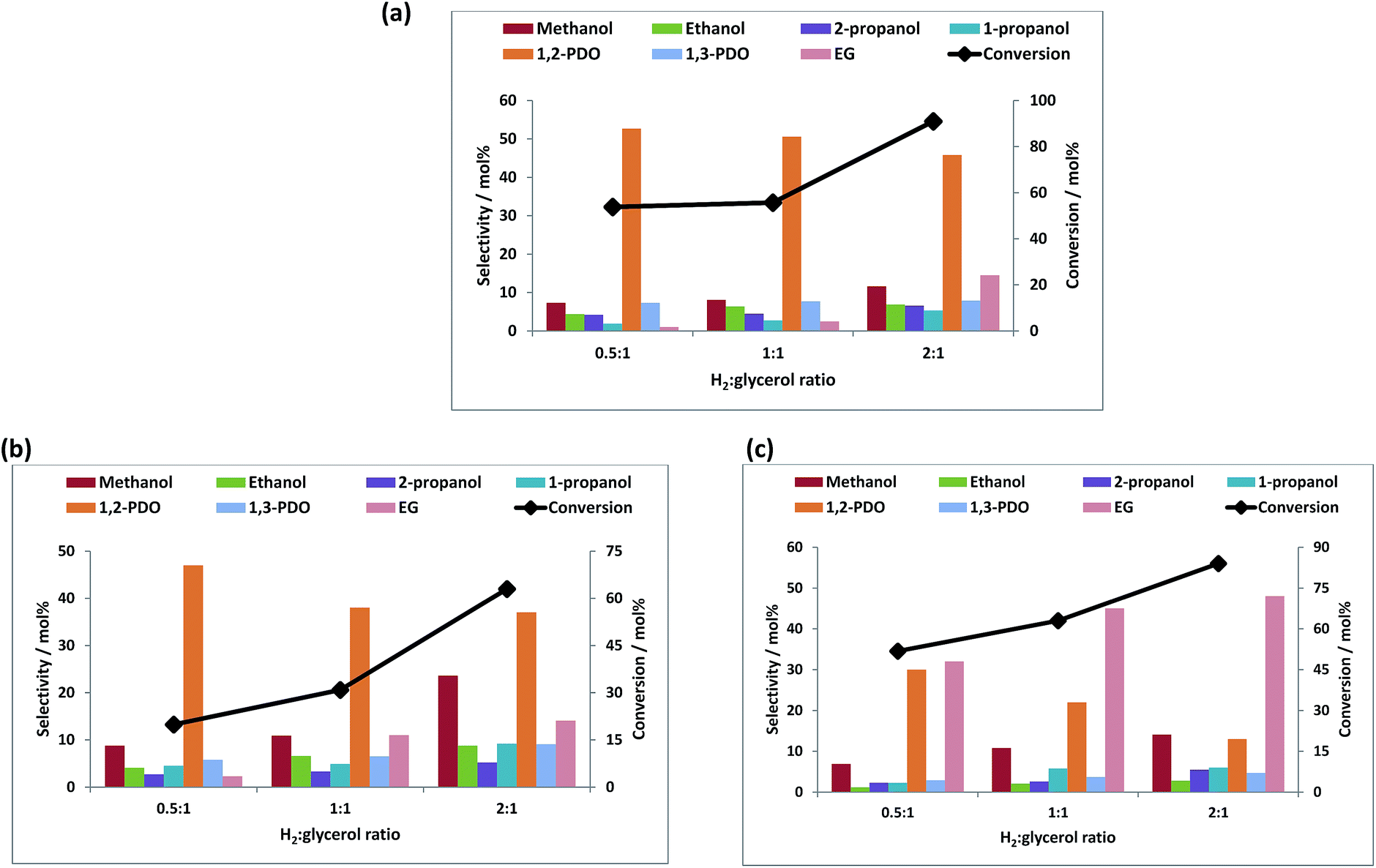 | ||
| Fig. 5 Effect of H2:glycerol ratio on conversion and product selectivity over (a) Re/ZnO, (b) Cu/ZnO and (c) Re–Cu/ZnO. Reaction conditions: 60 wt% glycerol/H2O, 3 mL catalyst bed, 60 bar H2, H2:glycerol ratio = 1:1. Other products: acetol, methane, ethane, propane, CO, CO2, unknowns. | ||
:glycerol ratio of 1:1. All showed a steady state conversion of glycerol and stable selectivity towards products over a period of at least 24 hours (Fig. 6a–c). Amongst all the catalysts, Re–Cu/ZnO showed highest conversion (63%) and selectivity (45%) towards EG. The Cu/ZnO and Re/ZnO catalysts showed higher selectivity towards 1,2-PDO. There was no change in the catalysts' structure after 24 hours as can be seen by the XRD patterns of the used catalysts (Fig. S6†).
 | ||
| Fig. 6 Effect of time-on-steam on conversion and product selectivity over (a) Re/ZnO, (b) Cu/ZnO and (c) Re–Cu/ZnO. Reaction conditions: 60 wt% glycerol/H2O, 3 mL catalyst bed, 60 bar H2, H2:glycerol ratio = 1:1. Other products: acetol, methane, ethane, propane, CO, CO2, unknowns. | ||
4. Conclusions
The Cu and Re supported on ZnO catalysts are effective in the hydrogenolysis of glycerol. An increase in temperature led to the expected increase in the conversion of glycerol. The rhenium containing catalysts were found to be the most active at all temperatures. The temperature was seen to have an influence in the selectivity to the various products, where an increase in temperature favoured the formation of lower alcohols. At 250 °C, where the activity was the highest over all catalysts, the highest selectivity to the lower alcohols was also obtained. Increasing the H2:glycerol ratio enhanced the activity over all catalysts as well as the selectivity to the lower alcohols. All catalysts were stable for a period of at least 24 hours. Rhenium plays an important role as a promoter in the hydrogenolysis of glycerol as it was observed that even in small quantities (∼1 wt%), it is able to enhance the activity of the catalysts, as well as the selectivity to lower alcohols due to its acidic nature. Indeed, the synergistic effect of Re–Cu is seen by the bimetallic catalyst forming dominantly EG, while the monometallic equivalent formed dominantly 1,2-PDO.
Acknowledgements
The authors would like to thank the National Research Foundation for financial support, the Microscopy and Microanalysis Unit (University of KwaZulu-Natal) for SEM and TEM analysis, Sasol Technology R&T for TPR and TPD analysis and the Cardiff Catalysis Institute for XPS analysis.References
- O. Valerio, T. Horvath, C. Pond, M. Manjusri and A. Mohanty, Ind. Crops Prod., 2015, 78, 141–147 CrossRef CAS.
- Z. Gholami, A. Z. Abdullah and K.-T. Lee, Renewable Sustainable Energy Rev., 2014, 39, 327–341 CrossRef CAS.
- Y. Wang, J. Zhou and X. Guo, RSC Adv., 2015, 5, 74611–74628 RSC.
- M. L. Shozi, V. D. B. C. Dasireddy, S. Singh, P. Mohlala, D. J. Morgan and H. B. Friedrich, ACS Sustainable Chem. Eng., 2016, 4, 5752–5760 CrossRef CAS.
- M. R. Nanda, Z. Yuan, W. Qin and C. Xu, Catal. Rev.: Sci. Eng., 2016, 58, 309–336 CAS.
- N. Rahmat, A. Z. Abdullah and A. R. Mohamed, Renewable Sustainable Energy Rev., 2010, 14, 987–1000 CrossRef CAS.
- M. Wang, H. Yang, Y. Xie, X. Wu, C. Chen, W. Ma, Q. Dong and Z. Hou, RSC Adv., 2016, 6, 29769–29778 RSC.
- J. Chaminand, L. a. Djakovitch, P. Gallezot, P. Marion, C. Pinel and C. Rosier, Green Chem., 2004, 6, 359–361 RSC.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, R. B. N. Prasad and N. Lingaiah, Catal. Lett., 2008, 126, 119–124 CrossRef CAS.
- Z. Huang, F. Cui, H. Kang, J. Chen and C. Xia, Appl. Catal., A, 2009, 366, 288–298 CrossRef CAS.
- L. Guo, J. Zhou, J. Mao, X. Guo and S. Zhang, Appl. Catal., A, 2009, 367, 93–98 CrossRef CAS.
- A. Bienholz, F. Schwab and P. Claus, Green Chem., 2010, 12, 290–295 RSC.
- N. Kim, S. Oh, J. Joo, K. Jung and J. Yi, Top. Catal., 2010, 53, 517–522 CrossRef CAS.
- Z. Zhou, X. Li, T. Zeng, W. Hong, Z. Cheng and W. Yuan, Chin. J. Chem. Eng., 2010, 18, 384–390 CrossRef CAS.
- Z. Yuan, J. Wang, L. Wang, W. Xie, P. Chen, Z. Hou and X. Zheng, Bioresour. Technol., 2010, 101, 7088–7092 CrossRef CAS PubMed.
- A. Bienholz, H. Hofmann and P. Claus, Appl. Catal., A, 2011, 391, 153–157 CrossRef CAS.
- R. B. Mane, A. A. Ghalwadkar, A. M. Hengne, Y. R. Suryawanshi and C. V. Rode, Catal. Today, 2011, 164, 447–450 CrossRef CAS.
- Y. Feng, H. Yin, A. Wang, L. Shen, L. Yu and T. Jiang, Chem. Eng. J., 2011, 168, 403–412 CrossRef CAS.
- E. S. Vasiliadou and A. A. Lemonidou, Appl. Catal., A, 2011, 396, 177–185 CrossRef CAS.
- Z. Huang, F. Cui, J. Xue, J. Zuo, J. Chen and C. Xia, Catal. Today, 2012, 183, 42–51 CrossRef CAS.
- F. Vila, M. López Granados, M. Ojeda, J. L. G. Fierro and R. Mariscal, Catal. Today, 2012, 187, 122–128 CrossRef CAS.
- S. Xia, R. Nie, X. Lu, L. Wang, P. Chen and Z. Hou, J. Catal., 2012, 296, 1–11 CrossRef CAS.
- Y. Feng, H. Yin, L. Shen, A. Wang, Y. Shen and T. Jiang, Chem. Eng. Technol., 2013, 36, 73–82 CrossRef CAS.
- A. Wolosiak-Hnat, E. Milchert and B. Grzmil, Chem. Eng. Technol., 2013, 36, 411–418 CrossRef CAS.
- D. Durán-Martín, M. Ojeda, M. L. Granados, J. L. G. Fierro and R. Mariscal, Catal. Today, 2013, 210, 98–105 CrossRef.
- S. Zhu, X. Gao, Y. Zhu, Y. Zhu, H. Zheng and Y. Li, J. Catal., 2013, 303, 70–79 CrossRef CAS.
- L. Niu, R. Wei, H. Yang, X. Li, F. Jiang and G. Xiao, Chin. J. Catal., 2013, 34, 2230–2235 CrossRef CAS.
- E. S. Vasiliadou, T. M. Eggenhuisen, P. Munnik, P. E. de Jongh, K. P. de Jong and A. A. Lemonidou, Appl. Catal., B, 2014, 145, 108–119 CrossRef CAS.
- S. Xia, W. Du, L. Zheng, P. Chen and Z. Hou, Catal. Sci. Technol., 2014, 4, 912–916 CAS.
- C.-J. Yue, L.-P. Gu, Y. Su and S.-P. Zhu, React. Kinet., Mech. Catal., 2014, 111, 633–645 CrossRef CAS.
- D. Sun, Y. Yamada and S. Sato, Appl. Catal., A, 2014, 475, 63–68 CrossRef CAS.
- R. V. Sharma, P. Kumar and A. K. Dalai, Appl. Catal., A, 2014, 477, 147–156 CrossRef CAS.
- S. Wang and H. Liu, Chin. J. Catal., 2014, 35, 631–643 CrossRef CAS.
- S. Zhu, X. Gao, Y. Zhu and Y. Li, Green Chem., 2016, 18, 782–791 RSC.
- M. A. Dasari, P.-P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231 CrossRef CAS.
- L. Ma, D. He and Z. Li, Catal. Commun., 2008, 9, 2489–2495 CrossRef CAS.
- L. Ma and D. He, Top. Catal., 2009, 52, 834–844 CrossRef CAS.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- Y. Amada, S. Koso, Y. Nakagawa and K. Tomishige, ChemSusChem, 2010, 3, 728–736 CrossRef CAS PubMed.
- Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2010, 94, 318–326 CrossRef CAS.
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 105, 117–127 CrossRef CAS.
- L. Ma, Y. Li and D. He, Chin. J. Catal., 2011, 32, 872–876 CrossRef CAS.
- Y. Amada, H. Watanabe, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, J. Phys. Chem. C, 2012, 116, 23503–23514 CAS.
- S. Marincean, L. Peereboom, Y. Xi, D. J. Miller and J. E. Jackson, Chem. Ind., 2007, 115, 427–436 CAS.
- Y. Li, H. Liu, L. Ma and D. He, RSC Adv., 2014, 4, 5503–5512 RSC.
- L. Ma and D. He, Catal. Today, 2010, 149, 148–156 CrossRef CAS.
- Q. Tong, A. Zong, W. Gong, L. Yu and Y. Fan, RSC Adv., 2016, 6, 86663–86672 RSC.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 318, 244–251 CrossRef CAS.
- M. Balaraju, V. Rekha, P. S. S. Prasad, B. L. A. P. Devi, R. B. N. Prasad and N. Lingaiah, Appl. Catal., A, 2009, 354, 82–87 CrossRef CAS.
- H. Zhu, L. Dong and Y. Chen, J. Colloid Interface Sci., 2011, 357, 497–503 CrossRef CAS PubMed.
- M.-F. Luo, Y.-J. Zhong, X.-X. Yuan and X.-M. Zheng, Appl. Catal., A, 1997, 162, 121–131 CrossRef CAS.
- A. Jones, Tempature-Programmed Reduction for Solid Materials Characterization, Taylor & Francis, 1986 Search PubMed.
- C. Kemball, Catalysis, Royal Society of Chemistry, 1981 Search PubMed.
- F. E. López-Suárez, S. Parres-Esclapez, A. Bueno-López, M. J. Illán-Gómez, B. Ura and J. Trawczynski, Appl. Catal., B, 2009, 93, 82–89 CrossRef.
- H. K. Harold, in Surface Acidity, Elsevier, 1989, vol. 45, ch. 5, pp. 72–90 Search PubMed.
- Y. Feng, H. Yin, A. Wang, L. Shen, L. Yu and T. Jiang, Chem. Eng. J., 2011, 168, 403–412 CrossRef CAS.
- B. Li, X. Hong, J.-J. Lin, G.-S. Hu, Q. Yu, Y.-J. Wang, M.-F. Luo and J.-Q. Lu, Appl. Surf. Sci., 2013, 280, 179–185 CrossRef CAS.
- J. H. Flores and M. I. Pais da Silva, Colloids Surf., A, 2008, 322, 113–123 CrossRef CAS.
- V. Montes, M. Checa, A. Marinas, M. Boutonnet, J. M. Marinas, F. J. Urbano, S. Järas and C. Pinel, Catal. Today, 2014, 223, 129–137 CrossRef CAS.
- D. L. King, L. Zhang, G. Xia, A. M. Karim, D. J. Heldebrant, X. Wang, T. Peterson and Y. Wang, Appl. Catal., B, 2010, 99, 206–213 CrossRef CAS.
- C. B. Rodella and R. Buffon, Appl. Catal., A, 2004, 263, 203–211 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- A. S. Fung, P. A. Tooley, M. J. Kelley, D. C. Koningsberger and B. C. Gates, J. Phys. Chem., 1991, 95, 225–234 CrossRef CAS.
- T. Miyazawa, Y. Kusunoki, K. Kunimori and K. Tomishige, J. Catal., 2006, 240, 213–221 CrossRef CAS.
- I. Gandarias, P. L. Arias, J. Requies, M. B. Gueemez and J. L. G. Fierro, Appl. Catal., B, 2010, 97, 248–256 CrossRef CAS.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Catal. Lett., 2008, 120, 307–311 CrossRef CAS.
- E. Van Ryneveld, A. S. Mahomed, P. S. Van Heerden, M. J. Green, C. Holzapfel and H. B. Friedrich, Catal. Sci. Technol., 2014, 4, 832–837 CAS.
- E. van Ryneveld, A. S. Mahomed, P. S. van Heerden and H. B. Friedrich, Catal. Lett., 2011, 141, 958–967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Deconvolution of TPR profiles, SEM and TEM images, used catalyst XRD and Re(4f) and Cu(2p) core-level XPS spectra for the catalysts. See DOI: 10.1039/c7se00199a |
| This journal is © The Royal Society of Chemistry 2017 |