
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmentally friendly nitrogen-doped carbon quantum dots for next generation solar cells†
Darragh
Carolan
 *ab,
Conor
Rocks
a,
Dilli Babu
Padmanaban
a,
Paul
Maguire
a,
Vladimir
Svrcek
b and
Davide
Mariotti
a
*ab,
Conor
Rocks
a,
Dilli Babu
Padmanaban
a,
Paul
Maguire
a,
Vladimir
Svrcek
b and
Davide
Mariotti
a
aNanotechnology & Integrated Bio-Engineering Centre (NIBEC), Ulster University, Jordanstown, Newtownabbey, Co. Antrim BT37 0QB, UK. E-mail: d.carolan@ulster.ac.uk
bResearch Centre of Photovoltaics, National Institute of Advanced Industrial Science and Technology-AIST, Central 2, Tsukuba, Japan
First published on 9th August 2017
Abstract
Shine on you crazy carbon! In this work, nitrogen-doped carbon quantum dots (N-CQDs) are synthesized using a simple custom atmospheric pressure microplasma. The method is facile, rapid, and environmentally friendly and the N-CQDs can be produced in a few minutes with no need for high temperature, complicated chemical techniques, or surface ligands. The N-CQDs are formed using molecular precursors and can be produced in different solvent mixtures. Material characterization techniques show a high degree of nitrogen doping on the QD surface with the amount of nitrogen depending on initial reaction conditions. The N-CQDs show interesting quantum confined optical properties that depend on the amount of nitrogen incorporation. Importantly, the band energy structure of the N-CQDs is elucidated and they are incorporated into a photovoltaic device as the photoactive layer achieving an extraordinary open-circuit voltage of 1.8 V and a power conversion efficiency of 0.8% (champion device), amongst the highest reported to date for group IV and carbon based quantum dots.
Introduction
Semiconductor nanocrystals (NCs) or quantum dots (QDs) have emerged in the last 25 years as a new class of fluorescence nanomaterial with unique properties.1 Much is now known about how to control the size, shape, composition and surface chemistry of II–VI and III–V QD materials,2–4 allowing fine control of their photophysical and electronic properties.5,6 These insights have paved the way for application in a wide range of areas including biological imaging,7,8 solar cells,9 light emitting devices,10 phosphors11 and field-effect transistors.12However, many of these II–VI and III–V materials contain rare and heavy metals, while their toxicity to cells and the environment may adversely affect their use in “real-world” applications.13–16 Moreover, several national and international regulations (e.g. the European Union's Restriction of Hazardous Substances Directive) severely limits the use of heavy metal cations such as Cd, Pb, and Hg in consumer electronics.17 Hence, it is important to develop alternative QDs with good optical and electrical properties but composed of materials with low toxicities. This has driven development of heavy metal free alternatives including group IV materials18–23 (e.g. Si, Ge, C) and ternary I–III–VI alloys24 (e.g. CuInxGa(1−x)Se2, CIGS).
Carbon nanoparticles have recently emerged as nontoxic, cheap, and biocompatible alternatives to their toxic counterparts.25–30 There has been enormous interest in this material as evidenced by the number of reviews published recently.31–36 Currently, carbon nanoparticles include graphene quantum dots, carbon quantum dots, and carbon nanodots (GQDs/CQDs/CNDs), and clear classification of these different forms can be difficult.37 However, the different varieties of CNDs have quite distinctive optical properties, in combination with related structural features, which are both principally determined by the synthetic route. Due to their low toxicity and promising optical properties, these materials have been used in applications such as sensors, bioimaging, drug delivery, photodynamic therapy, photocatalysis, phosphors, light emitting devices and photovoltaic devices.31,38–42
Normally synthetic routes for CNDs are classified as top-down or bottom-up. Top-down methods43–46 break down a wide range of initial carbon materials, such as carbon nanotubes, nanodiamonds, or graphite. Bottom-up syntheses47–50 are fast and simple methods to produce a wide variety of CNDs, with various surface functionalities and tunable properties. Doping of CNDs with heteroatoms can improve their optical properties due to effective surface passivation and/or incorporation of the element into the nanocrystal core.51 The most common doping atoms are nitrogen52 and sulphur,53,54 however boron, phosphorous and metals like copper and gadolinium have also been used.55–57 Early results suggested the favourable effect of nitrogen on the optical properties of carbon based quantum dots, with high quantum yields observed.58,59 In general, the bottom up synthesis of doped CNDs involves a molecular precursor which provides the carbon framework, and another molecular precursor introduces desired elements into the structure. The most popular combination to date has been citric acid as the carbon source, combined with nitrogen containing molecules, such as ethylenediamine or urea, while the most prominent formation route has been solvothermal processing.60 For example, Peng et al. used glucose and citric acid as the carbon sources and dopamine or 4,7,10-trioxa-1,13-tridecanediamine as the nitrogen sources. Various combinations of these precursors were heated to form carbon dots with different photophysical and electrochemical properties. The carbon dots produced were used as a sensitizer in solar cells.61 Nitrogen-doped carbon quantum dots can be rapidly synthesized on a large scale by simply pyrolyzing ethanolamine in air.62
At present, QD photovoltaic (PV) research is dominated by lead (Pb) based devices. Several groups have reported cells based on these materials with power conversion efficiencies (PCEs) of 5–10%.63,64 The advantage of QD-PVs is to exploit QD-like behaviour to harvest solar energy more efficiently; thus, the individual QD properties must be preserved while allowing effective charge transport. Because of the undesirable toxicity of Pb compounds as mentioned previously, there is an imminent need to investigate alternative quantum dots for use in environmentally friendly PVs. In this regard, carbon based QDs have been used in PVs in combination with polymers,65,66 and to sensitize TiO2.67,68 However, CNDs have never been used as the absorbing layer alone.69 In addition to low-toxicity and compatibility with environmentally sustainable strategies, C-based materials have very high absorption coefficients and offer great opportunities with tunable properties.
Herein, we demonstrate the use of an atmospheric pressure microplasma22,70–73 to successfully produce nitrogen-doped carbon QDs (N-CQDs). Microplasmas have been used to synthesize many different types of NCs/QDs,74,75 however they have rarely been utilized in the preparation of CQDs.76 The N-CQDs were found to be highly crystalline with the degree of nitrogen incorporation depending on the initial precursor concentrations. Their surface contained numerous functional groups, allowing easy dispersability in water. Their optical properties showed a dependence on the amount of nitrogen incorporation. The energy band structure of N-CQDs was determined and they were employed in solar cell devices as the photoactive layer without the need for additional polymer composites or sensitizing materials. An exceptionally high open-circuit voltage (1.8 V) was demonstrated along with a PCE of 0.8% (for our champion device), amongst the highest for group IV and CND materials.
Results and discussion
N-CQDs were synthesized using an atmospheric pressure microplasma in contact with water containing the carbon and nitrogen sources (see ESI† for detailed experimental procedures). Typically, citric acid (CA) and ethylenediamine (EDA) were dissolved in 10 mL of deionized water, and a direct-current (DC) atmospheric-pressure microplasma was applied to the liquid surface (Fig. 1(a)). A digital image of this microplasma–liquid interaction is shown in Fig. 1(b). The microplasma was applied for 30 minutes as the anode, the discharge current was fixed at 6 mA and the distance between the nickel tubing and the liquid surface was adjusted to maintain a discharge voltage of 1.3 kV. During the treatment, the solution changed from colourless to yellow/brown within a few minutes, indicating the rapid formation of N-CQDs; see Fig. 1(c). As the reaction proceeded, the colour of the solution became darker, attributed to the formation of more QDs. No visible sedimentation or aggregation was observed, suggesting that the N-CQDs contained surface moieties making them inherently soluble in water.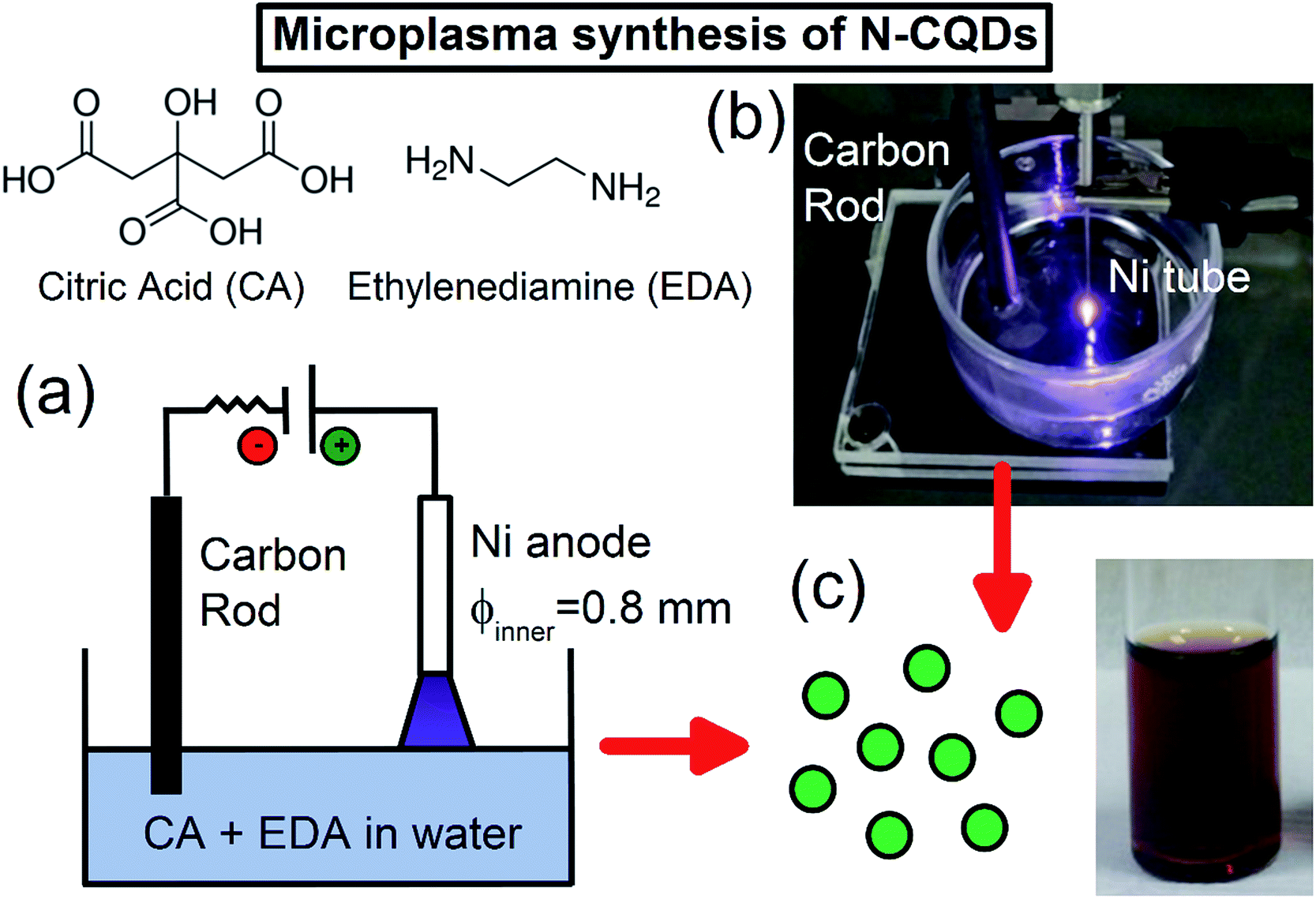 | ||
| Fig. 1 Microplasma synthesis of nitrogen-doped carbon dots (N-CQDs) from molecular precursors, (a) schematic of the setup used, (b) digital photograph of the microplasma–liquid interaction and (c) the N-CQDs produced from this setup with 1.05 g citric acid and 556 μL ethylenediamine dissolved in 10 mL water. 5.6 × 3.4 in; double column, 600 dpi. | ||
The reaction also depended on the ratio of COOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH2 groups and the solution composition in general (see ESI† for discussion on formation mechanism). Importantly, our excitation dependent, crystalline, emissive material is slightly different to other reported excitation dependent amorphous particles50,76 and graphene quantum dots.59 In the following discussion, we analyze samples produced with a fixed amount of CA (1.051 g in 10 mL water) but different amounts of the nitrogen precursor (139–834 μL) to check if this has any effect on the N-CQDs properties.
:NH2 groups and the solution composition in general (see ESI† for discussion on formation mechanism). Importantly, our excitation dependent, crystalline, emissive material is slightly different to other reported excitation dependent amorphous particles50,76 and graphene quantum dots.59 In the following discussion, we analyze samples produced with a fixed amount of CA (1.051 g in 10 mL water) but different amounts of the nitrogen precursor (139–834 μL) to check if this has any effect on the N-CQDs properties.
Transmission electron microscopy (TEM) imaging of N-CQDs produced with 556 μL EDA (1:2 molar ratio COOH:NH2) (Fig. 2(a)) shows that they are highly size and shape monodispersed, with no evidence of aggregation. Fig. 2(b) shows a histogram of N-CQD diameters, determined by analysis of TEM images of 200 QDs at random locations on the grid. Fitting the histogram to a Gaussian model yielded a mean diameter of 5.3 nm, with a standard deviation of 1.4 nm. TEM imaging of the other samples with different EDA concentrations (see Fig. ESI 4(a–c)†) revealed that they contained similarly sized N-CQDs (see histograms), illustrating that changing the amount of nitrogen precursor does not significantly alter the size of the NCs produced. High-resolution TEM (HR-TEM) imaging was used to confirm the crystallinity and establish the crystal phase of the NCs, see Fig. 2(c). HR-TEM imaging (Fig. 2(c)) showed that the NCs form a continuous crystalline phase, without the presence of packing defects. The lattice fringes shown in Fig. 2(c) correspond to a d spacing of 3.2 Å, close to the (002) spacing reported for graphite (3.3 Å).46,77,78 The slight variation in this value is due to turbostratic disorder in the carbon quantum dots.79,80 The fast Fourier transform (FFT) of the N-CQDs (Fig. 2(d)) could also be indexed to graphitic carbon. X-ray diffraction (XRD) was also employed to check the crystal phase of the N-CQDs (556 μL EDA). The XRD pattern of the N-CQDs deposited on glass is shown in Fig. ESI 5† which shows a diffraction peak centred at 2θ = 31.7° in agreement with the (002) plane of graphite.78 The peak at 40° is ascribed to the (012) reflection of rhombohedral graphite which normally coexists with the more common hexagonal phase. The peak at around 78° is attributed to the (110) reflection of graphite.81
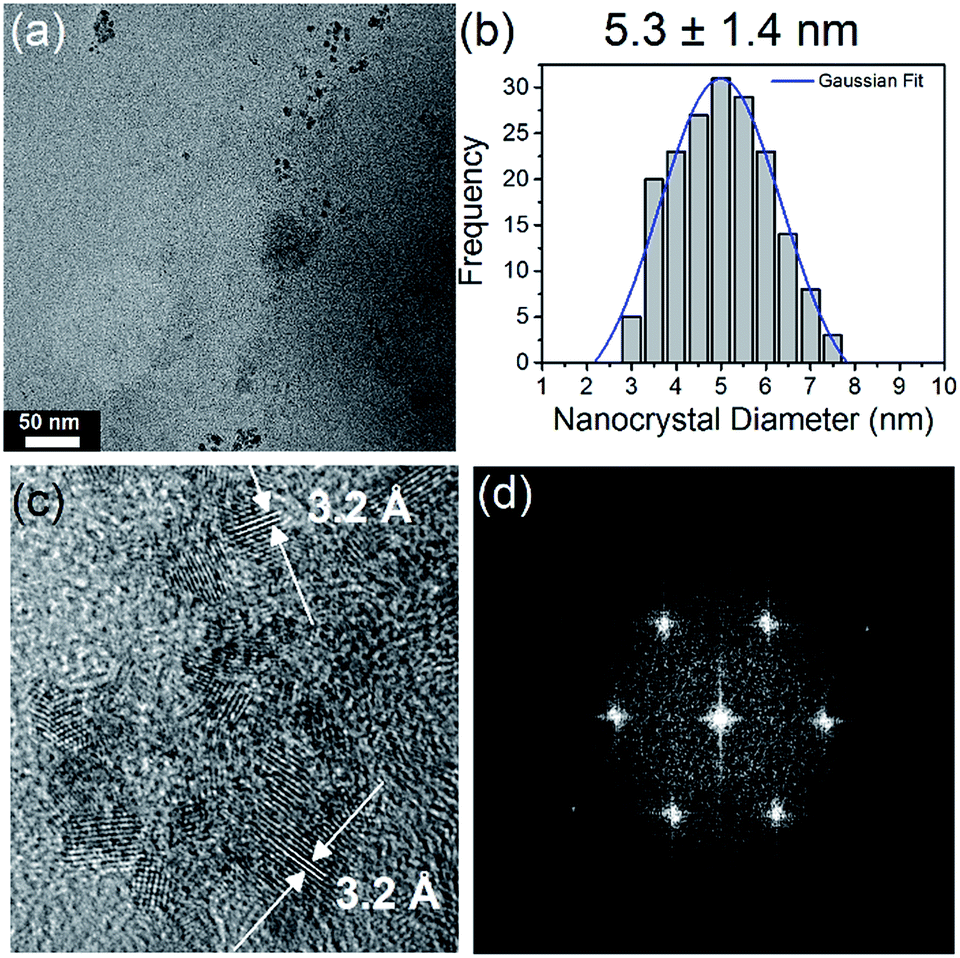 | ||
| Fig. 2 (a) TEM image of the N-CQDs produced with 556 μL EDA, (b) size distribution histogram, (c) HRTEM image with lattice spacing indicated (d) FFT of the N-CQDs. 3.2 × 3.2 in; single column, 600 dpi. | ||
The N-CQD (556 μL EDA) surface chemistry was characterized by Fourier transform infrared (FTIR) spectroscopy, see Fig. 3(a). The broad peak at 3275 cm−1 is a mixture of O–H and NH2 stretching vibrations.82,83 The peaks around 2954 cm−1 are attributed to CH3 and CH2 stretching modes and the peaks at 1775 and 1700 cm−1 are ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations.82,84 The peak at 1650 cm−1 is attributed to the CC stretching vibration. The peak at 1560 cm−1 is assigned to the CN stretching vibration while the peak at 1435 cm−1 is given to C–N vibrations.82 The two peaks at 1400 and 1350 cm−1 are attributed to a partially dissociated carboxylate anion.85 Peaks at 1170 and 880 cm−1 are ascribed to C–C and CC modes, respectively.86 All other samples with different EDA concentrations showed similar features, see Fig. ESI 6.† Interestingly, analysis of the CN peak intensity at 1560 cm−1 showed an increase in the signal as the amount of nitrogen precursor was increased from 139 to 834 μL (see Fig. 3(b), red circles). The opposite trend was seen for the CO peak at 1700 cm−1 (see Fig. 3(b), blue squares) suggesting that as the nitrogen content of the QDs was increased, a simultaneous decrease in the oxygen content also occurred. X-ray photoelectron spectroscopy (XPS) was also carried out to complete the analysis of the chemical composition of the N-CQDs. The XPS survey spectrum for all N-CQD samples (Fig. ESI 7(a)†) show peaks due to the presence of C, N, and O at a binding energy of around 285, 400, and 531 eV, respectively. High resolution spectra of the C 1s and N 1s spectra were analyzed to determine the C and N configurations in the N-CQDs and are shown in Fig. 3(c) and ESI 7(b),† respectively. The C 1s spectrum could be deconvoluted into 5 components corresponding to sp2 CC at 284.5 eV, sp3 C–C/C–H at 285.5 eV, C–O/C–N at 286.0 eV, CN at 287.4 eV, and CO at 287.9 eV.82,83,87 The N 1s spectrum exhibited 4 peaks centred at 398.3, 399.6, 400.1, and 400.6 eV related to CN, NH2, N–C3 and pyrrolic C–N–C.82,83 The deconvoluted C 1s and N 1s spectra for the other samples produced with different nitrogen precursor concentrations are shown in Fig. ESI 8† and are similar for all samples. The atomic percentage of N in each sample was found to increase with increasing EDA used in the synthesis, see Fig. 3(d), red circles. When 139 μL EDA was used, the atomic percentage of nitrogen was found to be 9.4%. Upon increasing the amount of EDA employed to 278, 556, and 834 μL, the nitrogen percentage concomitantly increased to 11.9, 12.7, and 15.5%, respectively (Fig. 3(d)). The atomic percentage of oxygen decreased from 24.6 to 17.3% as the initial amount of nitrogen precursor was increased from 139 to 834 μL, see Fig. 3(d), blue squares. These XPS results are in excellent agreement with the FTIR analysis and indicate that functional groups such as COOH, OH and N groups are present.
O stretching vibrations.82,84 The peak at 1650 cm−1 is attributed to the CC stretching vibration. The peak at 1560 cm−1 is assigned to the CN stretching vibration while the peak at 1435 cm−1 is given to C–N vibrations.82 The two peaks at 1400 and 1350 cm−1 are attributed to a partially dissociated carboxylate anion.85 Peaks at 1170 and 880 cm−1 are ascribed to C–C and CC modes, respectively.86 All other samples with different EDA concentrations showed similar features, see Fig. ESI 6.† Interestingly, analysis of the CN peak intensity at 1560 cm−1 showed an increase in the signal as the amount of nitrogen precursor was increased from 139 to 834 μL (see Fig. 3(b), red circles). The opposite trend was seen for the CO peak at 1700 cm−1 (see Fig. 3(b), blue squares) suggesting that as the nitrogen content of the QDs was increased, a simultaneous decrease in the oxygen content also occurred. X-ray photoelectron spectroscopy (XPS) was also carried out to complete the analysis of the chemical composition of the N-CQDs. The XPS survey spectrum for all N-CQD samples (Fig. ESI 7(a)†) show peaks due to the presence of C, N, and O at a binding energy of around 285, 400, and 531 eV, respectively. High resolution spectra of the C 1s and N 1s spectra were analyzed to determine the C and N configurations in the N-CQDs and are shown in Fig. 3(c) and ESI 7(b),† respectively. The C 1s spectrum could be deconvoluted into 5 components corresponding to sp2 CC at 284.5 eV, sp3 C–C/C–H at 285.5 eV, C–O/C–N at 286.0 eV, CN at 287.4 eV, and CO at 287.9 eV.82,83,87 The N 1s spectrum exhibited 4 peaks centred at 398.3, 399.6, 400.1, and 400.6 eV related to CN, NH2, N–C3 and pyrrolic C–N–C.82,83 The deconvoluted C 1s and N 1s spectra for the other samples produced with different nitrogen precursor concentrations are shown in Fig. ESI 8† and are similar for all samples. The atomic percentage of N in each sample was found to increase with increasing EDA used in the synthesis, see Fig. 3(d), red circles. When 139 μL EDA was used, the atomic percentage of nitrogen was found to be 9.4%. Upon increasing the amount of EDA employed to 278, 556, and 834 μL, the nitrogen percentage concomitantly increased to 11.9, 12.7, and 15.5%, respectively (Fig. 3(d)). The atomic percentage of oxygen decreased from 24.6 to 17.3% as the initial amount of nitrogen precursor was increased from 139 to 834 μL, see Fig. 3(d), blue squares. These XPS results are in excellent agreement with the FTIR analysis and indicate that functional groups such as COOH, OH and N groups are present.
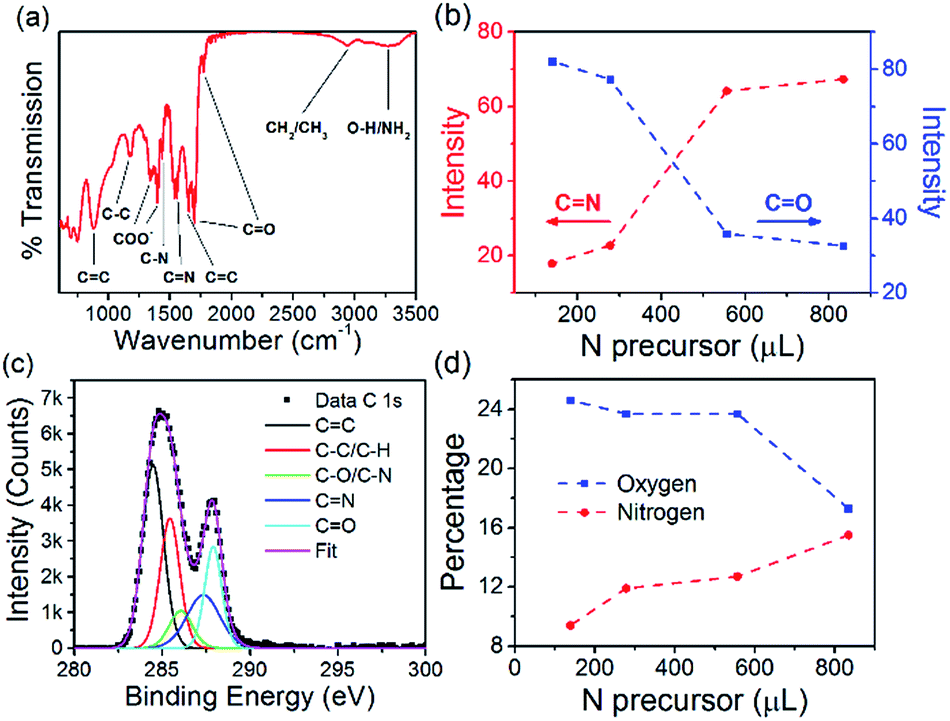 | ||
| Fig. 3 (a) FTIR spectrum of the N-CQDs with 556 μL EDA, (b) variation of the CN (red circles) and CO (blue squares) FTIR peak intensities as a function of the amount of nitrogen precursor used, (c) high resolution scan of the C 1s region, (d) variation of the nitrogen (red circles) and oxygen (blue squares) XPS atomic percentages as a function of the amount of nitrogen precursor used. 3.2 × 2.4 in; single column, 600 dpi. | ||
Taken together, this structural and surface characterization (TEM, XRD, XPS, FTIR) suggests that these QDs are composed of a graphitic like carbon sp2 core with a high degree of nitrogen and oxygen incorporation at the surface which varies as the starting amount of the nitrogen source is altered. Numerous functional groups at the surface impart excellent aqueous solubility to the N-CQDs without the need for further modification. These results also show that the microplasma synthesis technique allows for simple, fine control over the N-CQDs composition by simply varying the ratio of COOH:NH2 groups in the initial reaction mixture.
The optical properties of the N-CQDs were investigated using ultraviolet-visible (UV-Vis) absorbance and photoluminescence (PL) spectroscopy. The UV-Vis spectra of the as-synthesized N-CQD samples in water are shown in Fig. 4(a). The spectra show an onset in the visible region and increasing absorption in the UV. The spectrum for the N-CQDs produced by microplasma treatment of 139 μL EDA (1:0.5 molar ratio of COOH:NH2, 9.4% N) shows an onset of absorption (wavelength position where the absorbance value is equal to or above 0.02) at 495 nm. Increasing the concentration of EDA, and therefore nitrogen content of the QDs, further shifted the onset of absorption to 520 (278 μL EDA, 1:1 molar ratio of COOH:NH2, 11.9% N) and 564 nm (556 μL EDA, 1:2 molar ratio of COOH:NH2, 12.7% N). However, further increasing the EDA concentration to 834 μL (1:3 molar ratio of COOH:NH2, 15.5% N), shifted the onset back to around 495 nm. Peaks and shoulders were clearly seen in all samples. For N-CQDs produced with 139 μL EDA, a peak was observed at 271 nm with a shoulder at 265 nm. Spectra for the other samples showed similar features with a shoulder at ca. 340 nm and peaks at 278 (278 μL EDA), 288 (556 μL EDA) and 281 nm (834 μL EDA). These features are normally attributed to aromatic carbon sp2 core domains (π–π*/σ–π* transitions) and/or surface mediated absorption (n–π* transitions of CN and CO groups at the surface).36,88,89 Clearly, the changing positions of these features suggests that the absorption profiles of the N-CQDs depend on the initial COOH:NH2 ratio and therefore, different surface states (see above discussion). All the materials show absorption tails extending into the visible region of the solar spectrum making them good candidates for solar absorber materials. Tauc plots could be produced and are shown in Fig. 4(b). Fitting the linear part of the spectra (see green dotted lines) and extrapolating to the x-axis produces values between 2.2 and 2.7 eV for the electronic bandgap of each sample. This is also confirmed by analysing the derivative of the absorption coefficient, see Fig. ESI 9.† When the concentration of EDA is increased, the bandgap increases accordingly; 2.2, 2.56, 2.67, and 2.7 eV for samples containing 139, 278, 556, and 834 μL EDA, see inset of Fig. 4(b). Photoluminescence spectra of each sample, taken at an excitation wavelength of 360 nm, are shown in Fig. 5(a). Emission was observed in the visible region and the peak position showed a clear dependence on the nitrogen content. With increasing nitrogen content, the wavelength position of the PL showed a red-shift to longer wavelengths. When the nitrogen content was 9.4% (139 μL EDA) the PL peak position was observed at 441 nm (2.8 eV). Upon increasing the nitrogen content to 11.9% (278 μL EDA), the peak position shifted to 455 nm (2.7 eV). Increasing the nitrogen content further to 12.7% (556 μL EDA) moved the PL to 468 nm (2.6 eV), while a content of 15.5% (834 μL EDA) gave a similar spectrum shifted by only 5 nm to 473 nm (2.6 eV). Considering that TEM analysis showed similar sized QDs in each sample, these results demonstrate that the N-CQDs PL can easily be tuned by precise control over the nitrogen content in the microplasma synthesis. Spectra were also acquired at different excitation wavelengths for each sample, see Fig. 5(b) and ESI 10–13.† PL spectra for 12.7% nitrogen content (556 μL EDA) in Fig. 5(b) were clearly dependent on the excitation wavelength used; the wavelength position of the PL maximum red-shifted from 460 to 580 nm as the excitation wavelength was increased from 230 to 540 nm, see Fig. ESI 10(a) and (b).† This excitation-dependent PL behaviour is common in fluorescent CQDs and may be affected by the particle size distribution (Fig. 2 and ESI 4†) as well as the surface states (Fig. 3, ESI 7(b) and 8†). Increasing the excitation wavelength also resulted in a 20-fold increase in the PL intensity until excitation at 400 nm, whereupon the emission intensity starts to fall off, see Fig. ESI 10(c).† All the other samples showed similar trends (Fig. ESI 11–13†). Interestingly, the excitation wavelength where maximum emission (highest PL intensity) occurs also seemed to depend on the amount nitrogen incorporation. For 9.4% nitrogen (139 μL EDA), the highest emission appeared at an excitation of 340 nm, see Fig. ESI 10(c).† N-CQDs with 11.9% nitrogen (278 μL EDA), showed the best emission at excitation of 360 nm, while samples with 12.7% (556 μL EDA) and 15.5% nitrogen (834 μL EDA) showed the most intense emission at excitation of 400 nm, Fig. ESI 11–13(d).† Owing to similarly sized QDs in each sample, these interesting emission characteristics can be attributed to the formation of different recombination pathways and energy states at the N-CQD surface as the amount of nitrogen is altered, leading to modified emission characteristics. However, the optical properties of CQDs are not clearly explained in the literature to date and future research efforts must focus on a more fundamental understanding of these phenomena. The absolute quantum yield (QY), measured using an integrating sphere, was determined to be 33%. Compared to many reports on the QY of CQDs, this value is quite high and can be attributed to effective passivation of surface trap states by the nitrogen atoms, leading to more efficient radiative recombination.
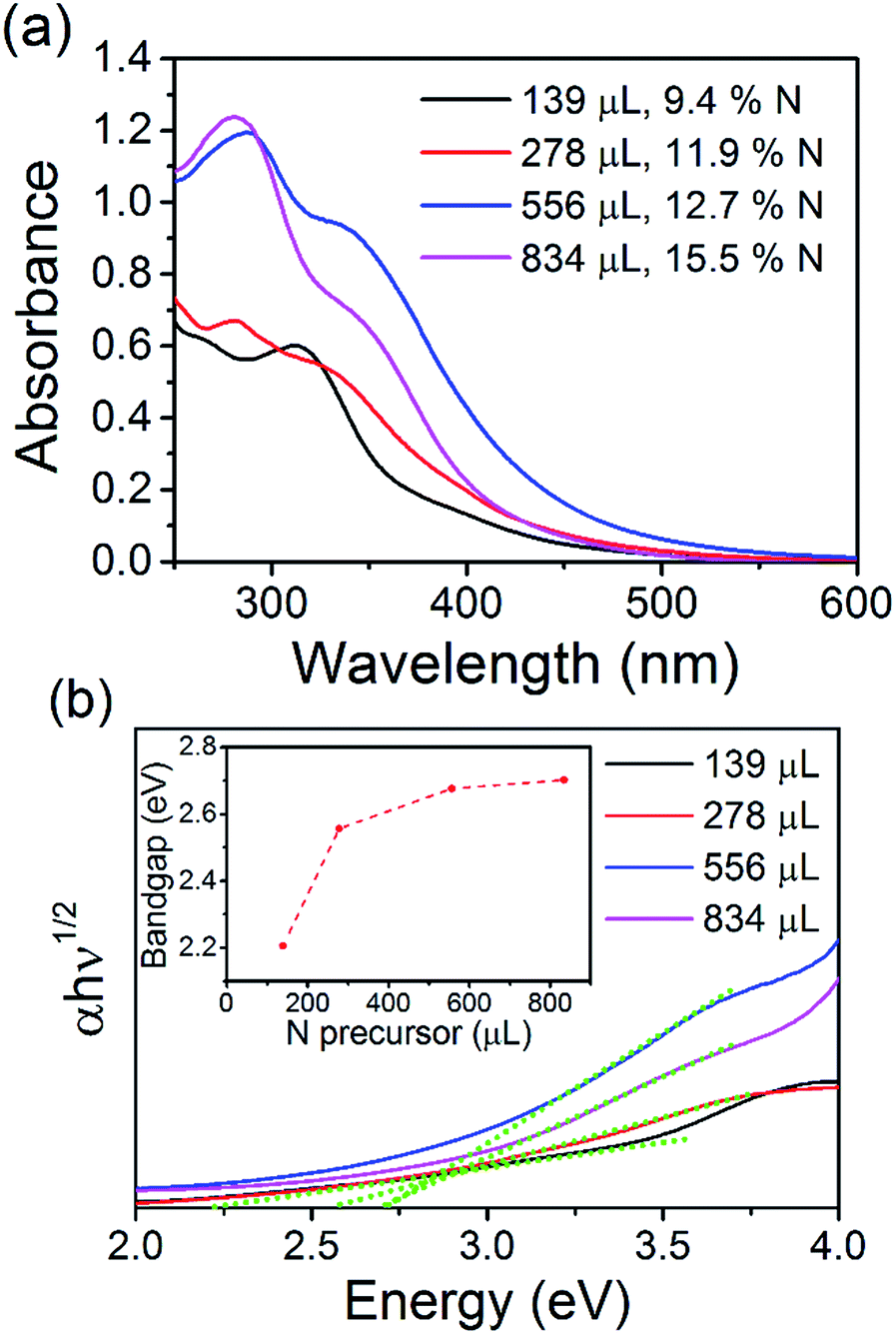 | ||
| Fig. 4 (a) UV-Vis spectra of the N-CQDs in solution, taken inside the integrating sphere, (b) Tauc plots for the N-CQDs, inset: variation of the bandgap with increasing nitrogen incorporation. 3 × 4.6 in; single column, 600 dpi. | ||
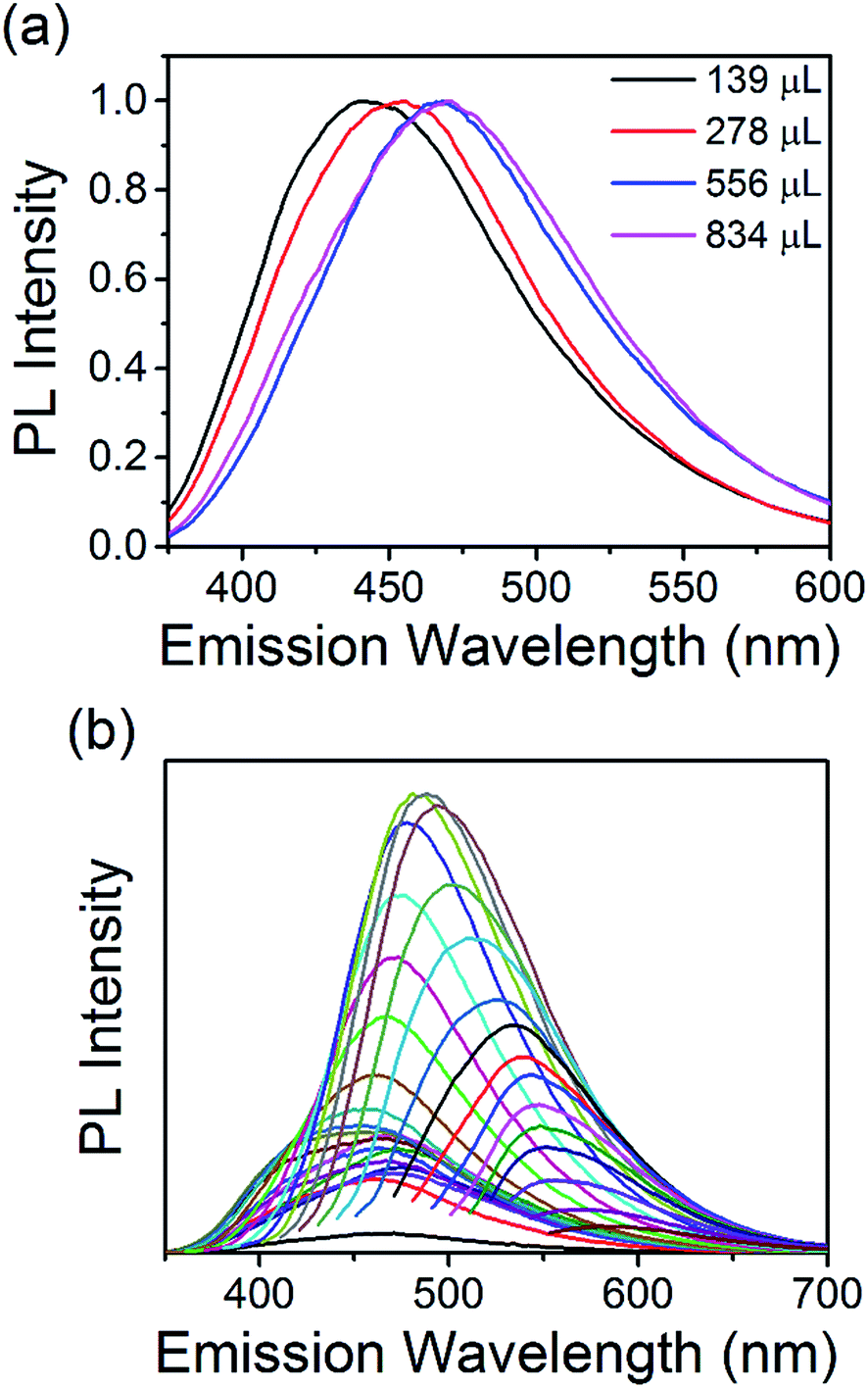 | ||
| Fig. 5 (a) PL spectra of each N-CQD sample taken at an excitation wavelength of 360 nm, (b) PL spectra for the sample containing 12.7% nitrogen (556 μL EDA) taken with different excitation wavelengths from 230 to 540 nm. 3 × 4.8 in; single column, 600 dpi. | ||
To employ the N-CQDs produced here in third generation photovoltaic devices a comprehensive study of their band energy diagram and bandgap must be carried out and we have focused here on N-CQDs with 556 μL EDA (12.7% nitrogen). To do this, we have carried out numerous measurements such as ultraviolet photoelectron spectroscopy (UPS) and Kelvin probe microscopy (KP). The valence band energy (EVB) of the N-CQDs was determined from UPS measurements according to the following equation: EVB = −21.2 + (Ecutoff − Eonset), where Eonset is the onset of photoemission in the low binding energy region, Ecutoff is the inelastic cutoff binding energy and 21.2 eV is the energy of the light source (He 1α). As shown in Fig. 6(a), Eonset was determined to be 2.1 eV from the intersection of linear fits to the baseline signal and the onset of photoemission. In the same fashion, Ecutoff was determined to be 16.7 eV, see Fig. 6(b), thus, EVB = −21.2 + (16.7 − 2.1) = −6.6 eV. From the previously obtained value for the bandgap of the N-CQDs (see Fig. 4(b)), we can calculate the conduction band energy, ECB = EVB ± Eg = −6.6 ± 2.7 = −3.9 eV. The Fermi energy (EF) was determined to be −5.24 ± 0.01 eV from Kelvin probe (KP) measurements, see Fig. 7(c), lying closer to the conduction band than the valence band. This is expected as nitrogen atoms serve as n-type dopants providing excess electrons, which results in an upward shift of the Fermi level. These measurements (UPS, UV-Vis and KP) have therefore allowed the determination of the energy band diagram, Fig. 7(c). This estimation of the energy band diagram of the N-CQDs is a fundamental step towards their application in photovoltaics. Importantly, this application benefits from numerous advantages like low cost, environmentally friendly, nontoxic “green” processes and abundant inorganic materials as well as the ability to tailor the band energies and alignment. The full device structure consists of the transparent conducting oxide indium tin oxide (ITO, 150 nm), a TiO2 hole blocking layer (40 nm), N-CQDs spray-coated from solution as the active layer and gold contacts (340 nm), sputtered directly on top of the N-CQD layer, see Fig. 7(a). The work function of the gold contact was measured by ambient photoemission spectroscopy (APS) and was found to lie at −4.8 eV. Similarly, the work function of the ITO contact was found to lie at −4.7 eV, see Fig. 7(c). Cross-sectional scanning electron microscopy imaging (X-SEM), Fig. 7(b), showed that the N-CQDs formed a compact layer which is 3 μm thick.
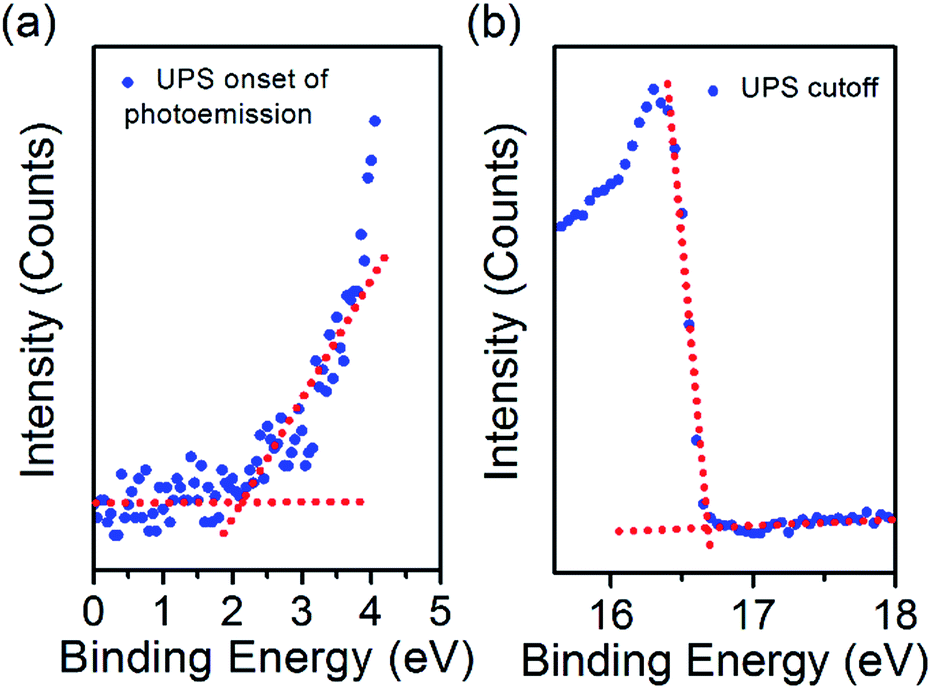 | ||
| Fig. 6 (a) UPS onset of photoemission for the N-CQDs film, (b) UPS cutoff for the N-CQDs film. 3.1 × 2.35 in; single column, 600 dpi. | ||
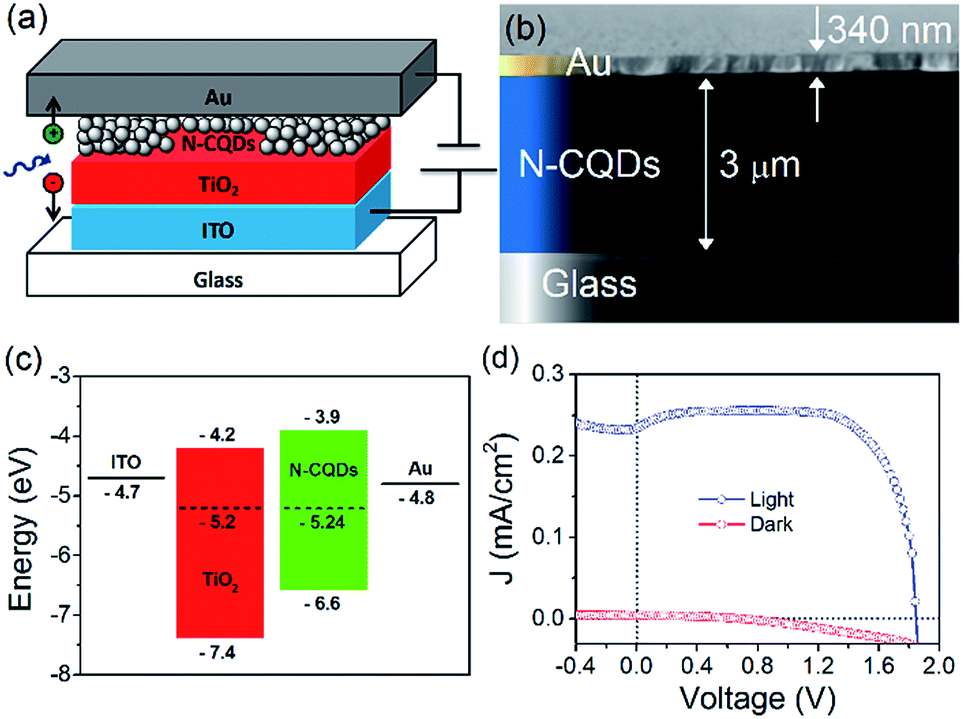 | ||
| Fig. 7 (a) Device structure, (b) X-SEM of the device, (c) energy band structure of the devices fabricated in this work and (d) J–V curve for the best device. 3.2 × 2.4 in; single column, 600 dpi. | ||
The current density–voltage characteristics of the best performing device is shown in Fig. 7(d). The device shows good rectification with a high fill factor (FF) of 0.79, short-circuit current density (JSC) of 0.23 mA cm−2, open-circuit voltage (VOC) of 1.8 V, and power conversion efficiency determined to be 0.8%. This excellent performance can be attributed to efficient alignment of the Fermi level of each material, see Fig. 7(c) as well as excellent charge separation and conduction in the N-CQD film. Other devices showed similar performance, with slightly lower efficiencies (Fig. ESI 14(a)–(d)†). Kwon et al. fabricated a polymer solar cell with CNDs and observed a high VOC of 1.6 eV, although no explanation of the operating mechanism was given.90 The low JSC and efficiency were attributed to the insulating nature of the CND ligands. Zhang et al. studied the effect of CNDs electron transport layers in small molecule solar cells with a champion VOC of 0.904 V reported.91 Devices with CNDs as the ETL exhibited enhanced performance compared to devices without an ETL, and importantly that of devices with LiF as the ETL. Electron mobility measurements showed that more balanced charge transport was achieved with CNDs. The authors found that when CNDs were utilised, the photogenerated excitons were dissociated into free carriers and these were efficiently transported and collected with minimal geminate or bimolecular recombination, leading to improvements in device performance. However, the differences in device structure, materials used, and performance prevents a direct comparison between these works and our results.
The device current is in part determined by the bandgap (>2 eV) and by the thickness of the QD layer (see Fig. 7(b)), which very likely is not matched to the diffusion length of the absorber. Furthermore, collection efficiency may be limited due to Schottky barrier formation with the metal contact.63,92–94 These observations immediately point out to straightforward improvements (e.g. optimizing the thickness of the QD layer,95 introducing a hole extraction layer96,97 and structuring of the underlying layer98), which can significantly impact the short-circuit current and the overall efficiency.
The FF and VOC are already exceptional and there could be various reasons for these results. The work function difference between the electrodes is too small (−0.1 eV) and cannot be the main driving force for charge separation and transfer. The large VOC is essentially relying on favourable band alignment with a negligible barrier between the conduction band of the TiO2 and the N-CQDs, which is critical,99 and allows for efficient injection of electrons to the wide-bandgap semiconductor without sacrificing the open-circuit voltage.
High doping levels in the QDs contribute to a large electron–hole quasi Fermi level splitting, but thermalization losses, photon spontaneous emission, entropy-related losses, incomplete light trapping inside the solar cell and non-radiative exciton recombination can affect the VOC. The VOC can be also affected by recombination rates and it can be argued that the high degree of passivation (demonstrated through PL and QY measurements) drastically reduces bimolecular recombination.100,101 Based on these premises, the VOC can come close to the thermodynamic limit (i.e. the bandgap of the N-CQD) with relatively minor losses.102,103
In general, groups have used either graphene QDs (GQDs) or CQDs in a solar cell devices, and the material was used as a sensitizer or mixed with a light sensitive polymer to improve its performance, see Table ESI 1† for an overview of carbon-based dots used in photovoltaic devices.53,65–68,90,91,104–110 Compared to these reports, our material does not require further functionalization, is not mixed with a polymer, and is not used as a sensitizer. This allows for simple, fast integration in solar cell structures. The N-CQDs are simply spray cast from solution and are used solely as the photoactive layer with no additional pre- or post-treatment, mixing or annealing. To the best of our knowledge, this is the first attempt to employ N-CQDs alone as the active layer in quantum dot photovoltaic devices. A high performance is achieved as evidenced by the values for the VOC, FF, and efficiency, demonstrating the high quality of our as-prepared samples. While the short-circuit current is relatively low, this can be dramatically improved by varying the absorbing layer thickness to the diffusion length and selecting a suitable electron blocking layer.
Conclusions
We have employed a simple, fast, and environmentally friendly method to synthesize nitrogen-doped carbon quantum dots from readily available precursors. By changing the initial amount of the precursors, it was possible to influence the final composition of the QDs. TEM imaging showed that the N-CQDs were well dispersed, highly crystalline and their size did not depend on the starting reaction conditions. Surface analysis showed that the nitrogen incorporation in the N-CQDs increased as the initial ratio of COOH:NH2 was increased. The N-CQDs possessed surface molecular arrangements that allowed them to be readily dispersed in water without any additional reagents or ligands. Optical characterization showed an obvious dependence on the amount of nitrogen doping in the QDs with shifts in the onset of absorption, bandgap, and photoluminescence. The energy band diagram for the N-CQDs was determined which allowed them to be used as the photoactive layer in solar cell devices which showed good charge separation and band alignment with a champion device VOC of 1.8 V and an efficiency of 0.8%. The result clearly show that N-CQDs could offer great opportunities for high efficiency devices where improvement in device fabrication and architecture could lead to much improved current densities while maintaining an exceptionally high open-circuit voltage.
Acknowledgements
This work was supported by EPSRC (EP/M024938/1), Invest NI (PoC-608). The authors also acknowledge fruitful discussion and exchanges supported by EU COST Action TD1208.References
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- G. Schmid, Nanoparticles From Theory to Application, Wiley-VCH, 2011 Search PubMed.
- A. L. Rogach, Semiconductor nanocrystal quantum dots: synthesis, assembly, spectroscopy and applications, Springer, 2008 Search PubMed.
- V. I. Klimov, Semiconductor and metal nanocrystals: synthesis and electronic and optical properties, Marcel Dekker, Inc, 2004 Search PubMed.
- Y. Wang and N. Herron, J. Phys. Chem., 1991, 95, 525–532 CrossRef CAS.
- C. B. Murray, D. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. a. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 933 CrossRef.
- E. Mutlugun, P. L. Hernandez-Martinez, C. Eroglu, Y. Coskun, T. Erdem, V. K. Sharma, E. Unal, S. K. Panda, S. G. Hickey, N. Gaponik, A. Eychmüller and H. V. Demir, Nano Lett., 2012, 12, 3986–3993 CrossRef CAS PubMed.
- J.-S. Lee, M. V Kovalenko, J. Huang, D. S. Chung and D. V Talapin, Nat. Nanotechnol., 2011, 6, 348–352 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039–7050 RSC.
- L. Ye, K. Yong, L. Liu, I. Roy, R. Hu, J. Zhu, H. Cai, W. Law, J. Liu, K. Wang, J. Liu, Y. Liu, Y. Hu, X. Zhang, M. T. Swihart and P. N. Prasad, Nat. Nanotechnol., 2012, 7, 453–458 CrossRef CAS PubMed.
- F. M. Winnik and D. Maysinger, Acc. Chem. Res., 2013, 46, 672 CrossRef CAS PubMed.
- European Council Directive 2002/95/EC, Restriction of the use of certain hazardous substances (RoHS), 2002 Search PubMed.
- K. Linehan and H. Doyle, Small, 2014, 10, 584–590 CrossRef CAS PubMed.
- D. Carolan and H. Doyle, J. Nanopart. Res., 2014, 16, 2721 CrossRef.
- D. Carolan and H. Doyle, J. Nanomater., 2015, 2015, 1–9 CrossRef.
- S. Askari, V. Svrcek, P. Maguire and D. Mariotti, Adv. Mater., 2015, 27, 8011–8016 CrossRef CAS PubMed.
- V. Svrcek, K. Dohnalova, D. Mariotti, M. T. Trinh, R. Limpens, S. Mitra, T. Gregorkiewicz, K. Matsubara and M. Kondo, Adv. Funct. Mater., 2013, 23, 6051–6058 CrossRef CAS.
- D. Carolan and H. Doyle, Part. Part. Syst. Charact., 2017, 34, 1600303 CrossRef.
- H. Zhong, Z. Bai and B. Zou, J. Phys. Chem. Lett., 2012, 3, 3167–3175 CrossRef CAS PubMed.
- S.-T. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J.-H. Liu, Y. Liu, M. Chen, Y. Huang and Y.-P. Sun, J. Phys. Chem. C, 2009, 113, 18110–18114 CAS.
- Y. Hu, J. Yang, J. Tian, L. Jia and J.-S. Yu, RSC Adv., 2015, 5, 15366–15373 RSC.
- J. Zhou, X. Shan, J. Ma, Y. Gu, Z. Qian, J. Chen and H. Feng, RSC Adv., 2014, 4, 5465 RSC.
- D. Sun, R. Ban, P.-H. Zhang, G.-H. Wu, J.-R. Zhang and J.-J. Zhu, Carbon, 2013, 64, 424–434 CrossRef CAS.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS PubMed.
- H. Tao, K. Yang, Z. Ma, J. Wan, Y. Zhang, Z. Kang and Z. Liu, Small, 2012, 8, 281–290 CrossRef CAS PubMed.
- J. Zhang and S. H. Yu, Mater. Today, 2016, 19, 382–393 CrossRef CAS.
- J. Wang and J. Qiu, J. Mater. Sci., 2016, 51, 4728–4738 CrossRef CAS.
- P. Zuo, X. Lu, Z. Sun, Y. Guo and H. He, Microchim. Acta, 2016, 183, 519–542 CrossRef CAS.
- G. E. LeCroy, S.-T. Yang, F. Yang, Y. Liu, K. A. S. Fernando, C. E. Bunker, Y. Hu, P. G. Luo and Y.-P. Sun, Coord. Chem. Rev., 2016, 320, 66–81 CrossRef.
- A.-J. Wang, H. Li, H. Huang, Z.-S. Qian and J.-J. Feng, J. Mater. Chem. C, 2016, 4, 8146–8160 RSC.
- Y. Park, J. Yoo, B. Lim, W. Kwon and S.-W. Rhee, J. Mater. Chem. A, 2016, 4, 11582–11603 CAS.
- A. Cayuela, M. L. Soriano, C. Carrillo-Carrión and M. Valcárcel, Chem. Commun., 2016, 52, 1311–1326 RSC.
- X. Gao, C. Du, Z. Zhuang and W. Chen, J. Mater. Chem. C, 2016, 4, 6927–6945 RSC.
- P. Miao, K. Han, Y. Tang, B. Wang, T. Lin and W. Cheng, Nanoscale, 2015, 7, 1586–1595 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2014, 44, 362–381 RSC.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921 RSC.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, A. Kyle Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- D. Tan, Y. Yamada, S. Zhou, Y. Shimotsuma, K. Miura and J. Qiu, Carbon, 2014, 69, 638–640 CrossRef.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- L. Bao, Z.-L. Zhang, Z.-Q. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D.-W. Pang, Adv. Mater., 2011, 23, 5801–5806 CrossRef CAS PubMed.
- A. M. Schwenke, S. Hoeppener and U. S. Schubert, Adv. Mater., 2015, 27, 4113–4141 CrossRef CAS PubMed.
- K. Wei, J. Li, Z. Ge, Y. You and H. Xu, RSC Adv., 2014, 4, 52230–52234 RSC.
- K. Linehan and H. Doyle, RSC Adv., 2014, 4, 12094 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- F. Wang, S. Pang, L. Wang, Q. Li, M. Kreiter and C. Liu, Chem. Mater., 2010, 22, 4528–4530 CrossRef CAS.
- L. Wang, S.-J. Zhu, H.-Y. Wang, S.-N. Qu, Y.-L. Zhang, J.-H. Zhang, Q.-D. Chen, H.-L. Xu, W. Han, B. Yang and H.-B. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- S. Chandra, P. Patra, S. H. Pathan, S. Roy, S. Mitra, A. Layek, R. Bhar, P. Pramanik and A. Goswami, J. Mater. Chem. B, 2013, 1, 2375–2382 RSC.
- S. K. Bhunia, N. Pradhan and N. R. Jana, ACS Appl. Mater. Interfaces, 2014, 6, 7672–7679 CAS.
- M. K. Barman, B. Jana, S. Bhattacharyya and A. Patra, J. Phys. Chem. C, 2014, 118, 20034–20041 CAS.
- W. Wu, L. Zhan, W. Fan, J. Song, X. Li, Z. Li, R. Wang, J. Zhang, J. Zheng, M. Wu and H. Zeng, Angew. Chem., Int. Ed., 2015, 54, 6540–6544 CrossRef CAS PubMed.
- A. B. Bourlinos, A. Bakandritsos, A. Kouloumpis, D. Gournis, M. Krysmann, E. P. Giannelis, K. Polakova, K. Safarova, K. Hola and R. Zboril, J. Mater. Chem., 2012, 22, 23327–23330 RSC.
- Y. Dong, H. Pang, H. Bin Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- D. Qu, M. Zheng, L. Zhang, H. Zhao, Z. Xie, X. Jing, R. E. Haddad, H. Fan and Z. Sun, Sci. Rep., 2014, 4, 5294 CrossRef CAS PubMed.
- S. Gogoi and N. Karak, Nano-Micro Lett., 2017, 9, 40 CrossRef.
- H. Peng, Y. Li, C. Jiang, C. Luo, R. Qi, R. Huang, C.-G. Duan and J. Travas-Sejdic, Carbon, 2016, 100, 386–394 CrossRef CAS.
- X. Dong, Y. Su, H. Geng, Z. Li, C. Yang, X. Li and Y. Zhang, J. Mater. Chem. C, 2014, 2, 7477 RSC.
- C. Piliego, L. Protesescu, S. Z. Bisri, M. V. Kovalenko and M. A. Loi, Energy Environ. Sci., 2013, 6, 3054–3059 CAS.
- X. Lan, O. Voznyy, F. P. García De Arquer, M. Liu, J. Xu, A. H. Proppe, G. Walters, F. Fan, H. Tan, M. Liu, Z. Yang, S. Hoogland and E. H. Sargent, Nano Lett., 2016, 16, 4630–4634 CrossRef CAS PubMed.
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS PubMed.
- V. Gupta, N. Chaudhary, R. Srivastava, G. D. Sharma, R. Bhardwaj and S. Chand, J. Am. Chem. Soc., 2011, 133, 9960–9963 CrossRef CAS PubMed.
- X. Yan, X. Cui, B. Li and L. S. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS PubMed.
- P. Mirtchev, E. J. Henderson, N. Soheilnia, C. M. Yip and G. A. Ozin, J. Mater. Chem., 2012, 22, 1265–1269 RSC.
- J. B. Essner and G. A. Baker, Environ. Sci.: Nano, 2017, 4, 1216–1263 RSC.
- S. Askari, I. Levchenko, K. Ostrikov, P. Maguire and D. Mariotti, Appl. Phys. Lett., 2014, 104, 163103 CrossRef.
- D. Mariotti, J. Patel, V. Švrček and P. Maguire, Plasma Processes Polym., 2012, 9, 1074–1085 CrossRef CAS.
- J. McKenna, J. Patel, S. Mitra, N. Soin, V. Švrček, P. Maguire and D. Mariotti, Eur. Phys. J.: Appl. Phys., 2011, 56, 24020 CrossRef.
- Q. Chen, J. Li and Y. Li, J. Phys. D: Appl. Phys., 2015, 48, 424005 CrossRef.
- D. Mariotti and R. M. Sankaran, J. Phys. D: Appl. Phys., 2010, 43, 323001 CrossRef.
- T. Velusamy, A. Liguori, M. Macias-Montero, D. B. Padmanaban, D. Carolan, M. Gherardi, V. Colombo, P. Maguire, V. Svrcek and D. Mariotti, Plasma Processes Polym., 2017, e1600224 CrossRef.
- Z. Wang, Y. Lu, H. Yuan, Z. Ren, C. Xu and J. Chen, Nanoscale, 2015, 7, 20743–20748 RSC.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- J. Zhou, C. Booker, R. Li, X. Zhou, T.-K. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef CAS PubMed.
- B. E. Warren, Phys. Rev., 1941, 59, 693–698 CrossRef CAS.
- T. Zheng and J. R. Dahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 3061–3071 CrossRef.
- A. Milev, M. Wilson, G. S. K. Kannangara and N. Tran, Mater. Chem. Phys., 2008, 111, 346–350 CrossRef CAS.
- F. Arcudi, L. Đorđević and M. Prato, Angew. Chem., Int. Ed., 2016, 55, 2107–2112 CrossRef CAS PubMed.
- H. Ding, J.-S. Wei and H.-M. Xiong, Nanoscale, 2014, 6, 13817–13823 RSC.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric identification of organic compounds, Wiley, 1991 Search PubMed.
- G. Socrates, Infrared characteristic group frequencies, Wiley, Chichester; New York, 1980 Search PubMed.
- B. Stuart, Infrared spectroscopy: fundamentals and applications, Wiley, 2004 Search PubMed.
- W. Kwon, S. Do, J.-H. Kim, M. Seok Jeong and S.-W. Rhee, Sci. Rep., 2015, 5, 12604 CrossRef CAS PubMed.
- P. Roy, P.-C. Chen, A. P. Periasamy, Y.-N. Chen and H.-T. Chang, Mater. Today, 2015, 18, 447–458 CrossRef CAS.
- Q. Li, M. Zhou, Q. Yang, Q. Wu, J. Shi, A. Gong and M. Yang, Chem. Mater., 2016, 28, 8221–8227 CrossRef CAS.
- W. Kwon, G. Lee, S. Do, T. Joo and S.-W. Rhee, Small, 2014, 10, 506–513 CrossRef CAS PubMed.
- H. Zhang, Q. Zhang, M. Li, B. Kan, W. Ni, Y. Wang, X. Yang, C. Du, X. Wan and Y. Chen, J. Mater. Chem. C, 2015, 3, 12403–12409 RSC.
- J. P. Clifford, K. W. Johnston, L. Levina and E. H. Sargent, Appl. Phys. Lett., 2007, 91, 253117 CrossRef.
- J. M. Luther, M. Law, M. C. Beard, Q. Song, M. O. Reese, R. J. Ellingson and A. J. Nozik, Nano Lett., 2008, 8, 3488–3492 CrossRef CAS PubMed.
- M.-J. Choi, J. Oh, J.-K. Yoo, J. Choi, D. M. Sim and Y. S. Jung, Energy Environ. Sci., 2014, 7, 3052 CAS.
- X. Lan, S. Masala and E. H. Sargent, Nat. Mater., 2014, 13, 233–240 CrossRef CAS PubMed.
- P. R. Brown, R. R. Lunt, N. Zhao, T. P. Osedach, D. D. Wanger, L.-Y. Chang, M. G. Bawendi and V. Bulović, Nano Lett., 2011, 11, 2955–2961 CrossRef CAS PubMed.
- B.-R. Hyun, J. J. Choi, K. L. Seyler, T. Hanrath and F. W. Wise, ACS Nano, 2013, 7, 10938–10947 CrossRef CAS PubMed.
- D. A. R. Barkhouse, R. Debnath, I. J. Kramer, D. Zhitomirsky, A. G. Pattantyus-Abraham, L. Levina, L. Etgar, M. Grätzel and E. H. Sargent, Adv. Mater., 2011, 23, 3134–3138 CrossRef CAS PubMed.
- C.-H. M. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed.
- C. M. Sutter-Fella, Y. Li, M. Amani, J. W. Ager, F. M. Toma, E. Yablonovitch, I. D. Sharp and A. Javey, Nano Lett., 2016, 16, 800–806 CrossRef CAS PubMed.
- R. T. Ross, J. Chem. Phys., 1967, 46, 4590–4593 CrossRef CAS.
- T. Markvart, Phys. Status Solidi, 2008, 205, 2752–2756 CrossRef CAS.
- L. C. Hirst and N. J. Ekins-Daukes, Prog. Photovoltaics, 2011, 19, 286–293 Search PubMed.
- J. Briscoe, A. Marinovic, M. Sevilla, S. Dunn and M. Titirici, Angew. Chem., Int. Ed., 2015, 54, 4463–4468 CrossRef CAS PubMed.
- Y.-Q. Zhang, D.-K. Ma, Y.-G. Zhang, W. Chen and S.-M. Huang, Nano Energy, 2013, 2, 545–552 CrossRef CAS.
- M. Sun, X. Ma, X. Chen, Y. Sun, X. Cui and Y. Lin, RSC Adv., 2014, 4, 1120–1127 RSC.
- C. Xie, B. Nie, L. Zeng, F.-X. Liang, M.-Z. Wang, L. Luo, M. Feng, Y. Yu, C.-Y. Wu, Y. Wu and S.-H. Yu, ACS Nano, 2014, 8, 4015–4022 CrossRef CAS PubMed.
- J. J. Huang, Z. F. Zhong, M. Z. Rong, X. Zhou, X. D. Chen and M. Q. Zhang, Carbon, 2014, 70, 190–198 CrossRef CAS.
- C. Liu, K. Chang, W. Guo, H. Li, L. Shen, W. Chen and D. Yan, Appl. Phys. Lett., 2014, 105, 73306 CrossRef.
- R. Narayanan, M. Deepa and A. K. Srivastava, J. Mater. Chem. A, 2013, 1, 3907 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Digital images of N-CQDs produced under different conditions, TEM images, XRD, FTIR, XPS and photoluminescence data for N-CQDs with different N contents, performance of several other solar cells prepared in this study and an overview table for other GQDs/CQDs systems used in photovoltaic devices. See DOI: 10.1039/c7se00158d |
| This journal is © The Royal Society of Chemistry 2017 |