
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the mechanism of the Ley–Griffith (TPAP) alcohol oxidation†
Timothy J.
Zerk
a,
Peter W.
Moore
a,
Joshua S.
Harbort
b,
Sharon
Chow
a,
Lindsay
Byrne
c,
George A.
Koutsantonis
 d,
Jeffrey R.
Harmer
b,
Manuel
Martínez
e,
Craig M.
Williams
*a and
Paul V.
Bernhardt
*a
d,
Jeffrey R.
Harmer
b,
Manuel
Martínez
e,
Craig M.
Williams
*a and
Paul V.
Bernhardt
*a
aSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane 4072, Queensland, Australia. E-mail: p.bernhardt@uq.edu.au; c.williams3@uq.edu.au
bCentre for Advanced Imaging, University of Queensland, Brisbane 4072, Australia
cCentre for Microscopy, Characterisation and Analysis, University of Western Australia, Crawley, Western Australia 6009, Australia
dSchool of Molecular Sciences, University of Western Australia, Crawley, Western Australia 6009, Australia
eDepartament de Química Inorgànica I Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès 1-11, E-08028 Barcelona, Spain
First published on 17th October 2017
Abstract
The Ley–Griffith reaction is utilized extensively in the selective oxidation of alcohols to aldehydes or ketones. The central catalyst is commercially available tetra-n-propylammonium perruthenate (TPAP, n-Pr4N[RuO4]) which is used in combination with the co-oxidant N-methylmorpholine N-oxide (NMO). Although this reaction has been employed for more than 30 years, the mechanism remains unknown. Herein we report a comprehensive study of the oxidation of diphenylmethanol using the Ley–Griffith reagents to show that the rate determining step involves a single alcohol molecule, which is oxidised by a single perruthenate anion; NMO does not appear in rate law. A key finding of this study is that when pure n-Pr4N[RuO4] is employed in anhydrous solvent, alcohol oxidation initially proceeds very slowly. After this induction period, water produced by alcohol oxidation leads to partial formation of insoluble RuO2, which dramatically accelerates catalysis via a heterogeneous process. This is particularly relevant in a synthetic context where catalyst degradation is usually problematic. In this case a small amount of n-Pr4N[RuO4] must decompose to RuO2 to facilitate catalysis.
Introduction
The controlled oxidation of a primary alcohol to an aldehyde is a fundamentally important reaction deployed in academic and industrial settings1 to access versatile chemical building blocks, synthetic intermediates, and final targets. Amongst the multitude of reagents and conditions available to perform this functional group transformation,2,3 selectivity (i.e. avoiding over-oxidation) and versatility (i.e. tolerant of other functional groups) are key criteria.4Historically, one-step alcohol oxidations to aldehydes, have relied heavily on chromium reagents (e.g. pyridinium chlorochromate5 (PCC)) activated sulfur protocols (e.g. Swern6,7 and Corey–Kim8) and manganese compounds (e.g. MnO2).9 More recently developed methods include hypervalent iodine compounds (e.g. Dess–Martin periodinane (DMP)10,11 followed by 2-iodoxybenzoic acid (IBX)12–14), and nitroxyl radicals (e.g. (2,2,6,6-tetramethyl-piperidin-1-yl)oxyl (TEMPO)15 followed by 2-azaadamantane N-oxyl (AZADO)16). In addition, molecular oxygen based methods using transition metals17–20 have also appeared in significant numbers. On balance, however, the mainstay protocols that dominate the one-step alcohol oxidation landscape are Swern, IBX/DMP, TEMPO and Ley–Griffith oxidations.21 The Ley–Griffith reaction followed on from early work by Sharpless reporting that ruthenium complexes (e.g. [Ru(PPh3)3Cl2]) catalyzed one-step oxidation of alcohols.22
The Ley–Griffith oxidation21,23–28 utilizes the catalyst n-Pr4N[RuO4] in combination with an excess (1.5 equivalents) of the co-oxidant N-methylmorpholine N-oxide (NMO – Scheme 1), both available commercially. Like many named reactions, modifications of the Ley–Griffith reaction have been reported29–37 (e.g. NMO·TPB38 and DABCOO·TPB39 from our own laboratory), as have numerous alternative applications using TPAP as a catalyst.40–51
 | ||
| Scheme 1 Reaction conditions of the Ley–Griffith alcohol oxidation. | ||
Since the initial report of n-Pr4N[RuO4]-catalyzed alcohol oxidation some 30 years ago only Lee et al.52,53 have attempted to elucidate the mechanism. However, the work of Lee was focused on the sub-stoichiometric n-Pr4N[RuO4] alcohol oxidation (i.e. in the absence of the essential co-oxidant NMO) and was confined to a single spectroscopic method (UV-Vis). Under these conditions n-Pr4N[RuO4] decomposed to insoluble ruthenium dioxide which obscured UV-Vis absorption spectra of both the [RuO4]− chromophore and the oxidized organic product. This work is quoted throughout the literature associated with the Ley–Griffith oxidation,23–25 stating that the mechanism is auto-catalytic and noting that the overall reaction appears to involve a three-electron reduction of n-Pr4N[RuO4].24
Perruthenate exhibits a diversity of reactivity depending primarily on solvation. In aqueous solution it has been proposed to react via a single electron process,54 based on its stoichiometric oxidation of cyclobutanol, which led to ring-opened products. This was evidence of radical formation at the α-carbon, and a corresponding low yield (33%) of cyclobutanone was found. When cyclobutanol was oxidized with n-Pr4N[RuO4]/NMO in dichloromethane Ley and Griffith observed cyclobutanone as the product in a 95% yield, indicating that a clean 2-electron oxidation is mediated by perruthenate under these conditions.21
We recently demonstrated that the role of NMO is to rescue the highly reactive RuV form of the catalyst by rapidly reoxidising it to [RuO4]−, thus sustaining catalysis.55 Others have suggested that NMO is required to access the true catalyst, in forming a perruthenate–NMO adduct.56 Clearly, the catalyst, co-oxidant and the substrate (i.e. n-Pr4N[RuO4], NMO and alcohol) are all essential yet the function of each in the oxidation reaction (producing aldehyde/ketone) has not previously been investigated. In view of the popularity that the Ley–Griffith reaction commands in synthesis, and the lack of mechanistic understanding, we initiated an extensive spectroscopic evaluation, which has resolved important mechanistic details regarding the catalytic cycle of this key reaction.
Experimental
Reagents, equipment and synthetic details are given in the ESI.†UV-Vis monitored reaction kinetics
In a typical experiment, 2.0 mL of a fresh solution of n-Pr4N[RuO4] (0.25 mM) was added to a 1 cm UV-Vis quartz cuvette and thermostatted at 303 K. After 10 minutes, NMO was added from a concentrated stock solution (0.85 M in MeCN) pre-dried over molecular sieves for 16 h. Finally, diphenylmethanol was added from a stock solution (0.1 M in MeCN) to initiate the reaction. The maximum reaction rate (vmax) was obtained in each case from a tangent to the steepest portion of the time-resolved single wavelength profile at 336 nm (ESI Fig. S2†). The slopes of the log–log plots of vmax against the initial concentrations of n-Pr4N[RuO4], NMO and diphenylmethanol revealed the reaction order of each reagent. For alcohol-dependent kinetic measurements the reference cuvette contained a solution of 0.25 mM n-Pr4N[RuO4] in MeCN such that the constant spectrum of the catalyst was subtracted. This allowed a greater range of alcohol concentrations to be exploited without flooding the spectrophotometer detector. Nevertheless the detector was still saturated by absorbance from the benzophenone product for experiments utilizing high initial concentrations of diphenylmethanol, but this did not affect the accurate determination of vmax.Results
Time-resolved UV-Vis spectroscopy
The Ley–Griffith alcohol oxidation reaction was followed using time-resolved UV-Vis spectroscopy (Fig. 1). A large excess of NMO co-oxidant (pre-dried over 4 Å molecular sieves) was added to ensure the regeneration of perruthenate during catalysis. We have recently shown how NMO rescues the unstable reduced RuV catalyst by reoxidation to [RuO4]−.55 Diphenylmethanol was selected as the alcohol substrate because it does not absorb above 280 nm while the corresponding ketone (benzophenone) absorbs at 336 nm (ε 120 M−1 cm−1 – Fig. 1C) and at 280 nm (ε 2910).56 This allows a quantitative and straightforward analysis of product formation in situ without the need for chromatography or mass spectrometry. Additionally, diphenylmethanol is a secondary alcohol so over-oxidation to a carboxylic acid is avoided. | ||
| Fig. 1 (A) Time-resolved UV-Vis spectra following the oxidation of 12.5 mM diphenylmethanol by 0.25 mM n-Pr4N[RuO4] and 67 mM NMO in MeCN (303 K). Spectra are displayed at ten minute intervals over the course of 8 h. Inset – absorbance profile at 336 nm. (B) UV-Vis spectrum of n-Pr4N[RuO4] in MeCN. λmax 316 nm (ε 2430 M−1 cm−1) and 385 nm (ε 2230). (C) UV-Vis spectrum of benzophenone in MeCN. λmax 336 nm (ε 120 M−1 cm−1). | ||
Typical alcohol oxidation experiments proceeded to completion over the course of several hours (Fig. 1A). Although the emerging benzophenone chromophore dominates the profile of Fig. 1A, the distinctive spectral features of the perruthenate anion at 316 nm and 385 nm remain throughout the reaction. This confirms that perruthenate is a catalytic, not stoichiometric, reagent and is at steady state throughout. Subtracting the ‘baseline’ spectrum of perruthenate (Fig. 1B) leads to ESI Fig. S3† which, along with the extinction coefficient of benzophenone at 336 nm, reveals that the alcohol is quantitatively converted to the ketone (Fig. S3† – inset).
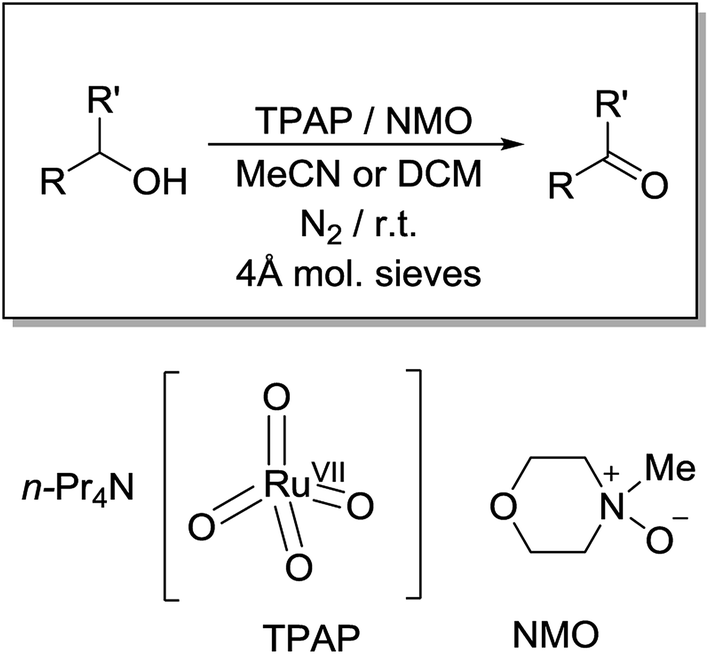
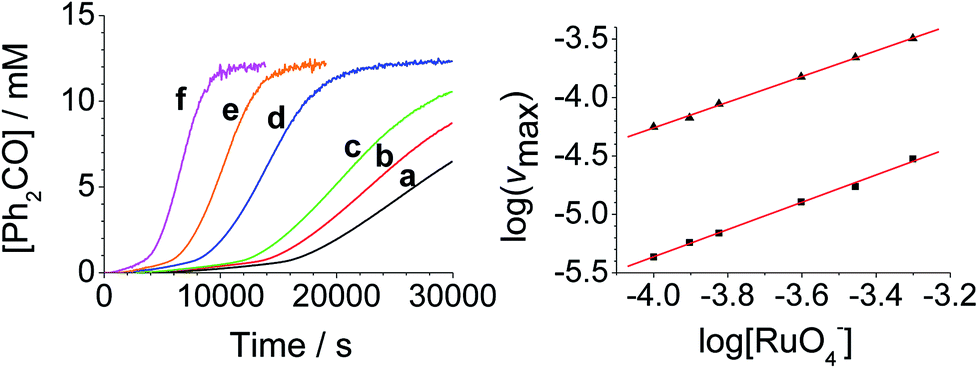
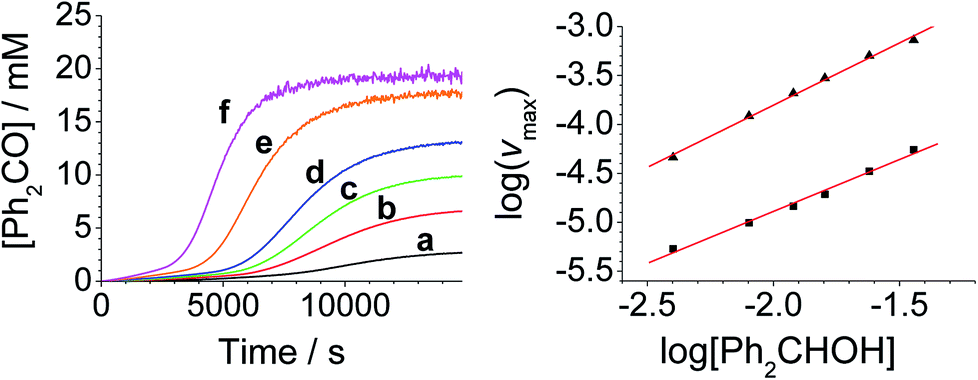
The concentration versus time profile for the oxidation (inset of Fig. 1A) is unusual in showing two distinct linear phases before the reaction is complete at ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (∼2¾ h). The reaction initially proceeds slowly through the first hour where an induction phase is apparent (0–4000 s), and only a small proportion (∼10%) of the product is formed. At ∼5000 s the reaction rate undergoes a ten-fold acceleration and the reaction proceeds rapidly and quantitatively to completion. No satisfactory kinetic model could reproduce the time-resolved absorbance changes. However, the overall reaction could be separated into its slow and fast catalytic phases, which were each analyzed independently in terms of their reagent concentration dependence. The steady state catalytic rate of alcohol oxidation (vmax) was obtained from the slope of the linear portions of the slow and rapid phases (see Fig. S2†). The reaction order with respect to perruthenate, alcohol and NMO was determined by plotting the logarithm of the reaction velocity (during the linear regions of the slow and fast phase) versus the logarithm of reagent concentration. The resulting concentration dependent profiles and log–log plots are shown in Fig. 2–4.
000 s (∼2¾ h). The reaction initially proceeds slowly through the first hour where an induction phase is apparent (0–4000 s), and only a small proportion (∼10%) of the product is formed. At ∼5000 s the reaction rate undergoes a ten-fold acceleration and the reaction proceeds rapidly and quantitatively to completion. No satisfactory kinetic model could reproduce the time-resolved absorbance changes. However, the overall reaction could be separated into its slow and fast catalytic phases, which were each analyzed independently in terms of their reagent concentration dependence. The steady state catalytic rate of alcohol oxidation (vmax) was obtained from the slope of the linear portions of the slow and rapid phases (see Fig. S2†). The reaction order with respect to perruthenate, alcohol and NMO was determined by plotting the logarithm of the reaction velocity (during the linear regions of the slow and fast phase) versus the logarithm of reagent concentration. The resulting concentration dependent profiles and log–log plots are shown in Fig. 2–4.
 | ||
| Fig. 2 [RuO4]−-dependent kinetics (Left). Concentration profile for benzophenone during a reaction between n-Pr4N[RuO4], 150 mM NMO and 12 mM diphenylmethanol in MeCN (T = 303 K) (a) black – 0.100 mM, (b) red – 0.125 mM, (c) green – 0.150 mM, (d) blue – 0.250 mM, (e) orange – 0.350 mM, (f) pink – 0.500 mM n-Pr4N[RuO4]. (Right) log–log plots for the first (■ – slope = 1.1) and second (▲ – slope = 1.0) phases. | ||
 | ||
| Fig. 3 [Diphenylmethanol]-dependent kinetics (Left) concentration profile for benzophenone during a reaction between 0.25 mM n-Pr4N[RuO4], 150 mM NMO and diphenylmethanol in MeCN (T = 303 K). (a) Black – 4.0 mM, (b) red – 8.0 mM, (c) green – 12.0 mM, (d) blue – 16.0 mM, (e) orange – 24.0 mM, (f) pink – 36 mM diphenylmethanol. (Right) log–log plots for the first (■ – slope = 1.1) and second (▲ – slope = 1.2) phases. | ||
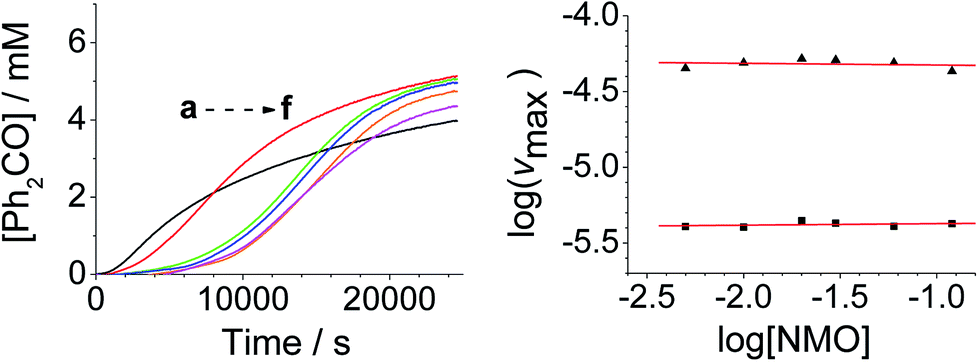 | ||
| Fig. 4 [NMO]-dependent kinetics (Left) concentration profile for benzophenone during a reaction between 0.25 mM n-Pr4N[RuO4], 6.0 mM diphenylmethanol and NMO in MeCN (T = 303 K). (a) Black – 5.0 mM, (b) red – 10.0 mM, (c) green – 20.0 mM, (d) blue – 30.0 mM, (e) orange – 60.0 mM, (f) pink – 120 mM NMO. Note: the different behavior with 5 mM NMO is due to an excess of alcohol versus NMO. (Right) log–log plots for the first (■) and second (▲) phases. | ||
From the data it is apparent that the rate law for both phases of the reaction is identical; first order in perruthenate, first order in alcohol and zero order with respect to NMO (eqn (1a) and (1b)). It has been shown previously that perruthenate is an effective oxidant on its own,21,54,57 which suggests that NMO is not required to access the true catalyst. The zero-order dependence of vmax on the concentration of NMO confirms this.
| vmax = k[RuO4−][ROH] – (induction phase) | (1a) |
| v′max = k′[RuO4−][ROH] – (rapid phase) | (1b) |
These findings are not consistent with the work of Lee et al. who reported the rate law as second order with respect to perruthenate based on its decomposition to RuO2.53 Nevertheless, our reaction mechanism does not explain why there are two distinct phases to the reaction i.e. there are two distinct values for the bimolecular rate constants in eqn (1a) and (1b). The observed behavior suggests that, as the oxidation proceeds, one of the products accelerates the reaction i.e. the reaction is autocatalytic. Indeed when higher initial concentrations of alcohol and perruthenate are employed, an initially faster rate of oxidation leads to a surge of product formation, the induction period being much shorter.
Addition of either benzophenone or N-methyl morpholine at the start of the reaction had no effect on the kinetics; the concentration–time profiles were identical to those shown in Fig. 1–4. The effect of the addition of water was also studied (being a significant factor given the fact that the Ley–Griffith oxidation requires molecular sieves to drive the reaction to completion) by adding different concentrations at the start of the reaction (Fig. 5). The near-zero slope of the log–log plots up to 10 mM H2O indicates that water is also not involved in the rate law. However, when much higher water concentrations were added (>40 mM), the induction period was truncated and the maximum rate was also slightly increased.
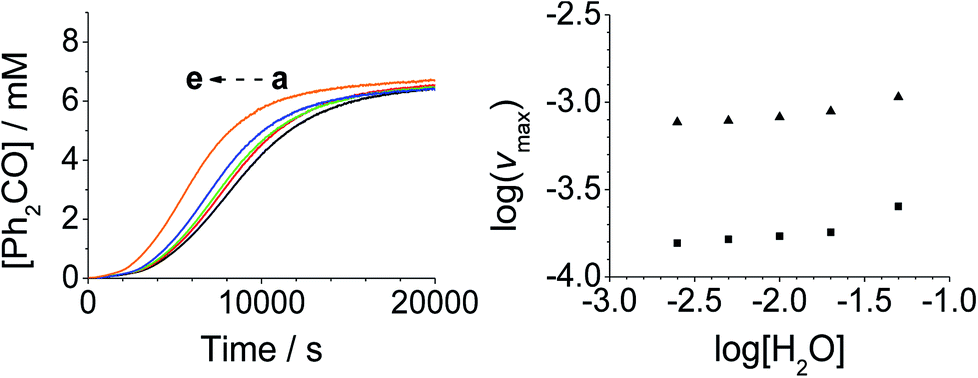 | ||
| Fig. 5 [H2O]0-dependent kinetics (Left) concentration profile for benzophenone during a reaction between 0.25 mM n-Pr4N[RuO4], 6.0 mM diphenylmethanol and 20 mM NMO in MeCN (T = 303 K). (a) Black – 2.5 mM, (b) red – 5.0 mM, (c) green −10.0 mM, (d) blue – 20.0 mM, (e) orange – 50.0 mM H2O added at t0. (Right) log–log plots for the first (■) and second (▲) phases. | ||
EPR spectroscopy
EPR spectroscopy provides a lens through which the paramagnetic catalyst alone is viewed as the organic components do not contribute to the spectra. The continuous wave (CW) EPR spectrum of a frozen solution of n-Pr4N[RuO4] in MeCN was measured at X-band frequency (Fig. 6) and simulated with EPR50F58 to reveal a slightly distorted d1 tetrahedral complex (gx,y = 1.937, gz = 1.910). These g-values (Table 1) are comparable with those measured for other distorted d1 tetraoxidoanions: [CrO4]3−gx = 1.84, gy = 1.85, gz = 1.94;59 [MnO4]2−gx = 1.98, gy = 1.97, gz = 1.94;60 [ReO4]2−gx,y = 1.72, gz = 1.85.61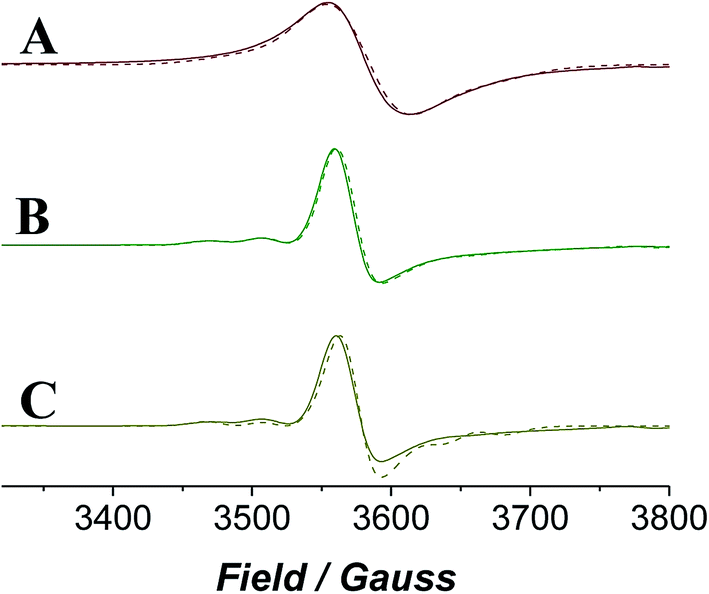 | ||
| Fig. 6 X-band (νav = 9.696 GHz) CW EPR spectra measured for n-Pr4N[RuO4] in acetonitrile, with differing additives. (A) Neat n-Pr4N[RuO4], (B) n-Pr4N[RuO4] + NMO, (C) n-Pr4N[RuO4] + NMO + diphenylmethanol. T = 6 K for all. Experimental spectra are solid lines and simulated spectra are the broken lines. | ||
| Sample | g x | g y | g z | A x | A y | A z (G) |
|---|---|---|---|---|---|---|
| a 101Ru, I = 5/2, abundance 17.06%; 99Ru, I = 5/2, abundance 12.76%; — indicates the value is undetermined. | ||||||
| n-Pr4N[RuO4] | 1.937 | 1.937 | 1.910 | ∼90 | ∼90 | — |
| n-Pr4N[RuO4] + NMO | 1.938 | 1.938 | 1.910 | ∼110 | ∼110 | — |
| n-Pr4N[RuO4] + NMO + Ph2CHOH | 1.939 | 1.936 | 1.918 | ∼110 | ∼110 | — |
When NMO was added to n-Pr4N[RuO4] the EPR signal became slightly more anisotropic and peaks from hyperfine coupling to 99Ru (13%) and 101Ru (17%) (both I = 5/2) were resolved (Fig. 6). However, the spectrum lacked any discernible superhyperfine coupling to the 14N (I = 1) nucleus of NMO. Inner sphere coordination of NMO can be discounted as this would change the geometry and symmetry of the complex and would be accompanied by a more significant effect on the spin Hamiltonian parameters than is observed in Fig. 6. The reluctance of perruthenate to expand its coordination number above four is not unexpected for a tetrahedral oxidoanion and is demonstrated by its persistent optical spectrum in a range of solvents.52,55,62 When NMO was added to [RuO4]− the UV-Vis spectrum was similarly unaffected (Fig. S4†). The subtle changes observed in the EPR spectrum must arise from weak outer-sphere interactions. When diphenylmethanol was added to the mixture of [RuO4]− and NMO (i.e. under Ley–Griffith conditions) then rapidly frozen the CW EPR spectrum did not change either (Fig. 6C); there is no evidence of an alcohol complex of RuVII.
The progress of the Ley–Griffith oxidation was followed by parallel EPR/UV-Vis spectroscopy. At various intervals, a small amount of the reaction mixture in the UV-Vis cell was removed, frozen and the EPR spectrum measured. A lower concentration of alcohol was chosen to slow the reaction so that several measurements could be taken during both the induction and rapid phases (Fig. 7). Throughout the induction period, the only notable change to the EPR was a small decrease in perruthenate signal intensity; furthermore, when the oxidation reached its maximum rate (ca. 8000 s), ∼25% of the perruthenate signal had been lost. No new EPR peaks appeared during this time and the field positions of the existing peaks did not change. Given the fact that both RuVI (d2) and RuV (d3) are EPR-active and adopt different coordination geometries from perruthenate63,64 the results in Fig. 7 indicate the absence of both species. As a whole, these observations indicate that perruthenate is partially converted into an EPR-silent species with a featureless UV-Vis spectrum during the induction period.
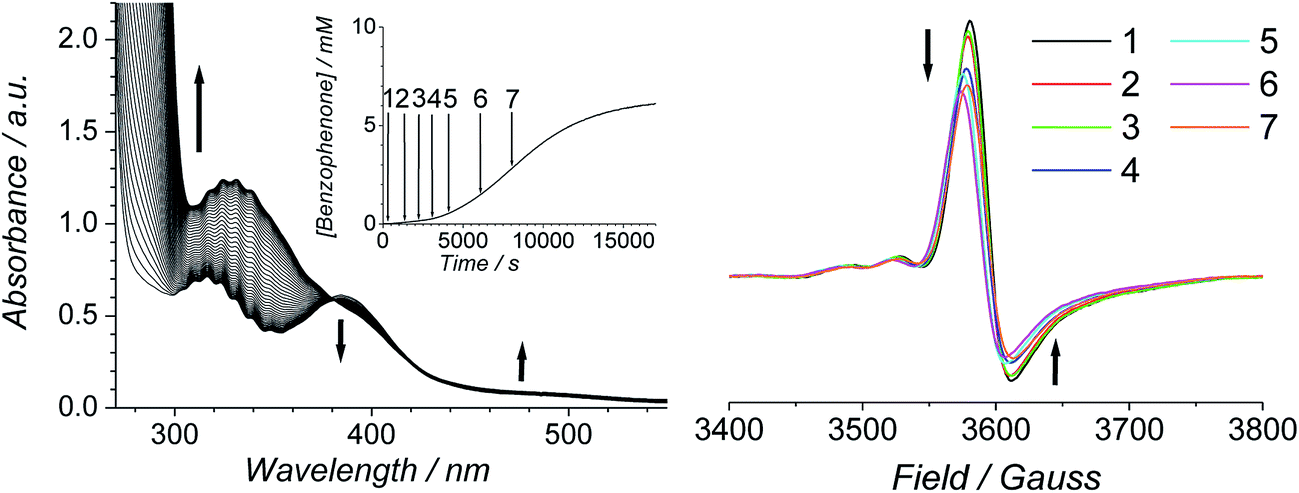 | ||
| Fig. 7 (Left) UV-Vis and concentration profile for benzophenone (inset) during a reaction between 0.25 mM n-Pr4N[RuO4], 6.0 mM diphenylmethanol and 60 mM NMO in MeCN (T = 303 K). (Right) Frozen X-band spectra (νav = 9.7041 GHz) measured at the intervals indicated in the inset (T = 6 K). | ||
In the absence of NMO, perruthenate is irreversibly reduced by alcohols or water and insoluble ruthenium dioxide dihydrate (RuO2·2H2O) is the product.52,53,57,62,65,66 This is observed as a fine black precipitate which produces a featureless, baseline-shifted UV-Vis spectrum; no EPR spectrum of this species has been reported. Indeed when diphenylmethanol was reacted with n-Pr4N[RuO4] in a 1:1 stoichiometric ratio in the absence of NMO the same outcome was achieved. The reaction was followed by EPR spectroscopy and samples of the mixture were taken every minute then frozen before their spectrum was measured. After 2 minutes the initially yellow-green perruthenate solution had become a black suspension and the sample was EPR silent (ESI Fig. S5†).
Therefore, despite the high initial concentration of NMO, an appreciable amount of RuO2·2H2O forms during the induction phase in order to account for the ∼25% decrease in the perruthenate EPR signal (Fig. 7 (right)). The formation of RuO2·2H2O also explains the slight baseline shift observed in the optical spectrum during the reaction and the small decrease in the perruthenate peak at 385 nm (see arrows in Fig. 7 (left) as well as Fig. 1A).
99Ru NMR
We also anticipated that 99Ru NMR could be useful for following the catalyst throughout the oxidation. It was expected that the relatively symmetrical nature of the [RuO4]− anion would provide the opportunity to observe a signal in spite of the low receptivity and quadrupole moment. Unfortunately across an exhaustively searched sweep width we were unable to obtain a discernible 99Ru NMR signal from n-Pr4N[RuO4].RuO2·2H2O is a co-catalyst
The effect of RuO2·2H2O was consequently explored by comparing parallel Ley–Griffith oxidations of diphenylmethanol with and without added ruthenium dioxide (see ESI† for preparative details). The results are shown in Fig. 8. When RuO2·2H2O was added at the start of the reaction (inset) the induction period was bypassed and the oxidation proceeded rapidly and smoothly to completion. The contribution of this additive to the absorption spectrum can be seen by comparing Fig. 1 and 7 (no RuO2·2H2O) with Fig. 8 (added RuO2·2H2O). However, the net change in absorbance at 336 nm (due to benzophenone formation) is equivalent in both cases. Overall, these results indicated that addition of RuO2·2H2O bypasses the slow induction phase, but the yield of product remains the same.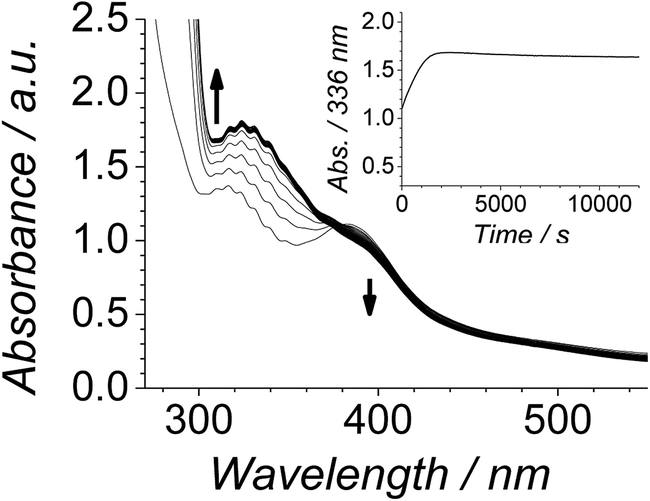 | ||
| Fig. 8 Time-resolved spectra following the oxidation of 6.0 mM diphenylmethanol by 0.25 mM n-Pr4N[RuO4] and 60 mM NMO in MeCN (T = 303 K) with 16 μL of RuO2·2H2O stock solution added at t0. Spectra are displayed at five minute intervals. Inset – single wavelength profile at 336 nm. | ||
18O-enriched alcohol
The possibility of O-atom transfer from perruthenate to the carbonyl product was studied using 18O-labelled piperonol (78% enriched) as a substrate and naturally abundant (16O) perruthenate and NMO (see ESI†). After standard Ley–Griffith oxidation 18O enrichment in the product piperonal was 68% which is not significantly different from the starting material (Fig. S6†). Mass spectroscopy of an aliquot of the crude reaction solution after the oxidation also showed the presence of naturally abundant NMO and [RuO4]− with no incorporation of 18O. These observations rule out any possibility of O-atom transfer from perruthenate to alcohol.Discussion
Notwithstanding the complicated kinetic profile of the Ley–Griffith oxidation (Fig. 1A) its rate law has been established (eqn (1a) and (1b)) and only involves perruthenate and alcohol. Furthermore, the same reaction order persists during both the slow initial phase and the faster second phase. Although ruthenium dioxide accelerates the oxidation, it is not an effective oxidant. Indeed a comprehensive study by Nobuko and Masakatsu has already demonstrated that RuO2 and RuO2·2H2O are ineffective oxidants of unactivated alcohols.67Ruthenium dioxide is indirectly formed by the concerted, two-electron reduction of perruthenate (kred) to the highly unstable RuV which disproportionates (kdisp) to RuO2·2H2O and ruthenate(VI) (Scheme 2). Ruthenate(VI) is also capable of alcohol oxidation and so may undergo a second reaction with alcohol to generate more RuO2·2H2O.54,68 Therefore, any RuV that undergoes disproportionation in the presence of alcohol is quantitatively converted to RuO2·2H2O.
 | (2) |
 | ||
| Scheme 2 Mechanism proposed for the Ley–Griffith alcohol oxidation in acetonitrile. | ||
Evidently ruthenium dioxide provides an active surface upon which oxidation takes place i.e. catalysis moves from a purely homogeneous to a heterogeneous regime. Given the fact that both perruthenate and alcohol appear in the rate law during the accelerated second phase (eqn (1b)), both species must be adsorbed onto the surface of the suspended RuO2·2H2O (i.e. a Langmuir-type mechanism). This is analogous to the oxidations of alcohols and hydrocarbons by permanganate which are catalyzed by colloidal MnO2.69,70
Under Ley–Griffith oxidation conditions, excess NMO limits RuV disproportionation by competitively re-oxidizing RuV to perruthenate (rate constant kox in eqn (2)).55 However, as the reaction proceeds, more water is produced and the NMO concentration decreases. Both factors make RuV disproportionation (kdisp in eqn (2)) more competitive with RuV re-oxidation by NMO (Scheme 2). It is ultimately the balance of these two reactions which determines how quickly the rapid phase of the reaction is accessed. Furthermore, higher alcohol and perruthenate concentrations accelerate the (bimolecular) reaction (kred in eqn (2)) and thus leading to a surge in RuV concentration which, at steady state, is compensated by more rapid disproportionation (last term of eqn (2)) and production of the RuO2·H2O co-catalyst. Accordingly, the rapid catalytic phase occurs sooner at higher concentrations of alcohol and n-Pr4N[RuO4] (Fig. 2 and 3), which is consistent with synthetic scale Ley–Griffith oxidation reactions. Conversely, higher concentrations of NMO prolong the induction period by keeping [RuV] low (Fig. 4).
There are innumerable reports detailing the synthetic application of the Ley–Griffith protocol, but throughout the literature there is a curious lack of any reference to an induction period like that observed here (Fig. 1–5). Synthetic-scale oxidations typically utilize much higher concentrations of reagents ([ROH], [RuO4]− ∼ 0.5 M, a 100-fold concentration increase)25 than those examined here which may explain the disparity; higher concentrations of alcohol and perruthenate being coupled to a short induction period.
However, there is an additional relevant explanation. The overwhelming majority of synthetic studies use commercially available (97% pure) n-Pr4N[RuO4]. When the oxidation of diphenylmethanol was repeated in our study using this commercial catalyst, instead of the pure synthesized product (Fig. S7†), no induction period was observed and the oxidation proceeded smoothly to completion. The baseline shift of the spectrum at t0 indicates that a small amount of insoluble material (ruthenium dioxide) is present in the commercially obtained catalyst (Fig. S8†). It is well known that n-Pr4N[RuO4] is sensitive to moisture and light and must be kept under argon (preferably in a refrigerator). Regardless of these precautions, n-Pr4N[RuO4] eventually degrades to a black solid with no catalytic activity. Even fresh bottles of commercial 97% pure n-Pr4N[RuO4] contain ruthenium dioxide and this is evidently most of the 3% impurity that is present (Fig. S8†). Interestingly, in practical terms, an induction period is undesirable, and by serendipitous fortune the commercial catalyst containing a small (∼3%) amount of impurity bypasses this complication. However, n-Pr4N[RuO4] is still the essential oxidant and complete degradation to inert RuO2 inactivates the catalyst.
Conclusions
A mechanistic study of the oxidation of diphenylmethanol using the Ley–Griffith reagent reveals that the rate determining step involves a single alcohol molecule which is oxidized in a concerted two electron process by a single [RuO4]− anion. NMO is not required to form the active oxidant, its role being stoichiometric as the oxidant which reactivates the RuV form of the catalyst to RuVII. A major finding of this study is that ruthenium dioxide, formed during the reaction, or present initially through degradation of the n-Pr4N[RuO4] catalyst, acts as a heterogeneous co-catalyst for the reaction. When freshly crystallized n-Pr4N[RuO4] is utilized, the oxidation proceeds slowly at the beginning of the reaction until a sufficient concentration of solid ruthenium dioxide is formed by disproportionation of the RuV form of the catalyst. Ironically, slightly impure catalyst performs more predictably, without an induction phase, than the pure catalyst. Nevertheless, the overall yield of product under both conditions is the same. Disproportionation of RuV is accelerated by the presence of water (produced by the reaction), or high catalyst/alcohol concentrations.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge financial support from the Australian Research Council (DP150104672 to PVB) (DP160102887 and FT110100851 to CMW) (FT120100421 to JRH) and from Spanish Ministerio de Economía y Competitividad (CTQ2015-65707C2-1). TJZ, PWM and JSH are grateful for Australian Post-graduate Awards.References
- G. Tojo and M. I. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones, Springer, US, 1st edn, 2006 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations, A Guide to Functional Group Preparations, Wiley, Hoboken, 2010 Search PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms and Structure, Wiley, 2007 Search PubMed.
- L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms, Elsevier Academic Press, 2005 Search PubMed.
- E. J. Corey and J. W. Suggs, Tetrahedron Lett., 1975, 16, 2647–2650 CrossRef.
- K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef CAS.
- A. J. Mancuso, D. S. Brownfain and D. Swern, J. Org. Chem., 1979, 44, 4148–4150 CrossRef CAS.
- E. J. Corey and C. U. Kim, J. Am. Chem. Soc., 1972, 94, 7586–7587 CrossRef CAS.
- R. J. Gritter and T. J. Wallace, J. Org. Chem., 1959, 24, 1051–1056 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251–262 CrossRef CAS PubMed.
- A. Duschek and S. F. Kirsch, Angew. Chem., Int. Ed., 2011, 50, 1524–1552 CrossRef CAS PubMed.
- M. J. Gallen, R. Goumont, T. Clark, F. Terrier and C. M. Williams, Angew. Chem., Int. Ed., 2006, 45, 2929–2934 CrossRef CAS PubMed.
- P. Lucio Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef CAS.
- S. Hamada, T. Furuta, Y. Wada and T. Kawabata, Angew. Chem., Int. Ed., 2013, 52, 8093–8097 CrossRef CAS PubMed.
- M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227–8241 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030–2050 RSC.
- W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 1625–1627 RSC.
- K. B. Sharpless, K. Akashi and K. Oshima, Tetrahedron Lett., 1976, 17, 2503–2506 CrossRef.
- W. P. Griffith and S. V. Ley, Aldrichimica Acta, 1990, 13 CAS.
- W. P. Griffith, Chem. Soc. Rev., 1992, 21, 179–185 RSC.
- S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 1994, 639–666 CrossRef.
- P. Langer and J. Prakt, Chem, 2000, 342, 728–730 CAS.
- M. Pagliaro, S. Campestrini and R. Ciriminna, Chem. Soc. Rev., 2005, 34, 837–845 RSC.
- W. P. Griffith, Ruthenium Oxidation Complexes: Their Uses as Homogenous Organic Catalysts, Springer, Netherlands, 2013 Search PubMed.
- B. Hinzen and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1997, 1907–1908 RSC.
- B. Hinzen, R. Lenz and S. V. Ley, Synthesis, 1998, 1998, 977–979 CrossRef.
- S. V. Ley, I. R. Baxendale, G. Brusotti, M. Caldarelli, A. Massi and M. Nesi, Il Farmaco, 2002, 57, 321–330 CrossRef CAS PubMed.
- A. Bleloch, B. F. G. Johnson, S. V. Ley, A. J. Price, D. S. Shephard and A. W. Thomas, Chem. Commun., 1999, 1907–1908 RSC.
- M. Pagliaro and R. Ciriminna, Tetrahedron Lett., 2001, 42, 4511–4514 CrossRef CAS.
- S. Campestrini, M. Carraro, R. Ciriminna, M. Pagliaro and U. Tonellato, Tetrahedron Lett., 2004, 45, 7283–7286 CrossRef CAS.
- Y. Xie, Z. Zhang, S. Hu, J. Song, W. Li and B. Han, Green Chem., 2008, 10, 278–282 RSC.
- R. Ciriminna, P. Hesemann, J. J. E. Moreau, M. Carraro, S. Campestrini and M. Pagliaro, Chem.–Eur. J., 2006, 12, 5220–5224 CrossRef CAS PubMed.
- C. D. G. Read, P. W. Moore and C. M. Williams, Green Chem., 2015, 17, 4537–4540 RSC.
- P. W. Moore, P. M. Mirzayans and C. M. Williams, Chem.–Eur. J., 2015, 21, 3567–3571 CrossRef CAS PubMed.
- P. W. Moore, Y. Jiao, P. M. Mirzayans, L. N. Q. Sheng, J. P. Hooker and C. M. Williams, Eur. J. Org. Chem., 2016, 2016, 3401–3407 CrossRef CAS.
- A.-K. C. Schmidt and C. B. W. Stark, Org. Lett., 2011, 13, 4164–4167 CrossRef CAS PubMed.
- A. Goti and M. Romani, Tetrahedron Lett., 1994, 35, 6567–6570 CrossRef CAS.
- K. R. Guertin and A. S. Kende, Tetrahedron Lett., 1993, 34, 5369–5372 CrossRef CAS.
- A. Goti, F. De Sarlo and M. Romani, Tetrahedron Lett., 1994, 35, 6571–6574 CrossRef CAS.
- M. H. Yates, Tetrahedron Lett., 1997, 38, 2813–2816 CrossRef CAS.
- I. E. Markó, A. Gautier, M. Tsukazaki, A. Llobet, E. Plantalech-Mir, C. J. Urch and S. M. Brown, Angew. Chem., Int. Ed., 1999, 38, 1960–1962 CrossRef.
- I. E. Markó, P. R. Giles, M. Tsukazaki, I. Chellé-Regnaut, C. J. Urch and S. M. Brown, J. Am. Chem. Soc., 1997, 119, 12661–12662 CrossRef.
- V. Piccialli and T. Caserta, Tetrahedron Lett., 2004, 45, 303–308 CrossRef CAS.
- H. Cheng and C. B. W. Stark, Angew. Chem., Int. Ed., 2010, 49, 1587–1590 CrossRef CAS PubMed.
- A.-K. C. Schmidt and C. B. W. Stark, Org. Lett., 2011, 13, 5788–5791 CrossRef CAS PubMed.
- R. Bloch and C. Brillet, Synlett, 1991, 829–830 CrossRef CAS.
- V. Piccialli, Molecules, 2014, 19, 6534–6582 CrossRef PubMed.
- D. G. Lee, Z. Wang and W. D. Chandler, J. Org. Chem., 1992, 57, 3276–3277 CrossRef CAS.
- W. D. Chandler, Z. Wang and D. G. Lee, Can. J. Chem., 2005, 83, 1212–1221 CrossRef CAS.
- D. G. Lee and L. N. Congson, Can. J. Chem., 1990, 68, 1774–1779 CrossRef CAS.
- T. J. Zerk, P. W. Moore, C. M. Williams and P. V. Bernhardt, Chem. Commun., 2016, 52, 10301–10304 RSC.
- K. J. Tony, V. Mahadevan, J. Rajaram and C. S. Swamy, React. Kinet. Catal. Lett., 1997, 62, 105–116 CrossRef CAS.
- A. C. Dengel, R. A. Hudson and W. P. Griffith, Transition Met. Chem., 1985, 10, 98–99 CrossRef CAS.
- R. A. Martinelli, G. R. Hanson, J. S. Thompson, B. Holmquist, J. R. Pilbrow, D. S. Auld and B. L. Vallee, Biochemistry, 1989, 28, 2251–2258 CrossRef CAS PubMed.
- Y. R. Sekhar and H. Bill, J. Chem. Phys., 1985, 82, 645–647 CrossRef CAS.
- M. Greenblatt and J. H. Pifer, J. Chem. Phys., 1980, 72, 529–534 CrossRef CAS.
- P. McGeehin, B. Henderson and P. C. Benson, Proc. R. Soc. London, Ser. A, 1975, 346, 497–513 CrossRef CAS.
- R. E. Connick and C. R. Hurley, J. Am. Chem. Soc., 1952, 74, 5012–5015 CrossRef CAS.
- A. C. Dengel, W. P. Griffith, C. A. O'Mahoney and D. J. Williams, J. Chem. Soc., Chem. Commun., 1989, 1720–1721 RSC.
- A. C. Dengel, J. F. Gibson and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1991, 2799–2800 RSC.
- R. Lenz and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1997, 3291–3292 RSC.
- M. Hasan, M. Musawir, P. N. Davey and I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2002, 180, 77–84 CrossRef CAS.
- M. Matsumoto and N. Watanabe, J. Org. Chem., 1984, 49, 3435–3436 CrossRef CAS.
- A. J. Bailey, W. P. Griffith, S. I. Mostafa and P. A. Sherwood, Inorg. Chem., 1993, 32, 268–271 CrossRef CAS.
- D. G. Lee and J. F. Perez-Benito, J. Org. Chem., 1988, 53, 5725–5728 CrossRef CAS.
- D. G. Lee and J. F. Perez-Benito, Can. J. Chem., 1985, 63, 1275–1279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04260d |
| This journal is © The Royal Society of Chemistry 2017 |