
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ex situ generation of stoichiometric HCN and its application in the Pd-catalysed cyanation of aryl bromides: evidence for a transmetallation step between two oxidative addition Pd-complexes†
Steffan K.
Kristensen
 a,
Espen Z.
Eikeland
b,
Esben
Taarning
c,
Anders T.
Lindhardt
*d and
Troels
Skrydstrup
*a
a,
Espen Z.
Eikeland
b,
Esben
Taarning
c,
Anders T.
Lindhardt
*d and
Troels
Skrydstrup
*a
aCarbon Dioxide Activation Center (CADIAC), The Interdisciplinary Center (iNANO), Department of Chemistry, Aarhus University, Gustav Wieds Vej 14, 8000 Aarhus, Denmark. E-mail: ts@chem.au.dk
bCenter for Materials Crystallography, The Interdisciplinary Center (iNANO), Department of Chemistry, Aarhus University, Langelandsgade 140, 8000 Aarhus, Denmark
cHaldor Topsøe A/S, New Business R&D, Nymøllevej 55, 2800 Kgs, Lyngby, Denmark
dCarbon Dioxide Activation Center (CADIAC), The Interdisciplinary Center (iNANO), Biological and Chemical Engineering, Department of Engineering, Aarhus University, Finlandsgade 22, 8200 Aarhus N, Denmark
First published on 6th October 2017
Abstract
A protocol for the Pd-catalysed cyanation of aryl bromides using near stoichiometric and gaseous hydrogen cyanide is reported for the first time. A two-chamber reactor was adopted for the safe liberation of ex situ generated HCN in a closed environment, which proved highly efficient in the Ni-catalysed hydrocyanation as the test reaction. Subsequently, this setup was exploited for converting a range of aryl and heteroaryl bromides (28 examples) directly into the corresponding benzonitriles in high yields, without the need for cyanide salts. Cyanation was achieved employing the Pd(0) precatalyst, P(tBu)3-Pd-G3 and a weak base, potassium acetate, in a dioxane-water solvent mixture. The methodology was also suitable for the synthesis of 13C-labelled benzonitriles with ex situ generated 13C-hydrogen cyanide. Stoichiometric studies with the metal complexes were undertaken to delineate the mechanism for this catalytic transformation. Treatment of Pd(P(tBu)3)2 with H13CN in THF provided two Pd-hydride complexes, (P(tBu)3)2Pd(H)(13CN), and [(P(tBu)3)Pd(H)]2Pd(13CN)4, both of which were isolated and characterised by NMR spectroscopy and X-ray crystal structure analysis. When the same reaction was performed in a THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture in the presence of KOAc, only (P(tBu)3)2Pd(H)(13CN) was formed. Subjection of this cyano hydride metal complex with the oxidative addition complex (P(tBu)3)Pd(Ph)(Br) in a 1:1 ratio in THF led to a transmetallation step with the formation of (P(tBu)3)2Pd(H)(Br) and 13C-benzonitrile from a reductive elimination step. These experiments suggest the possibility of a catalytic cycle involving initially the formation of two Pd(II)-species from the oxidative addition of LnPd(0) into HCN and an aryl bromide followed by a transmetallation step to LnPd(Ar)(CN) and LnPd(H)(Br), which both reductively eliminate, the latter in the presence of KOAc, to generate the benzonitrile and LnPd(0).
:water mixture in the presence of KOAc, only (P(tBu)3)2Pd(H)(13CN) was formed. Subjection of this cyano hydride metal complex with the oxidative addition complex (P(tBu)3)Pd(Ph)(Br) in a 1:1 ratio in THF led to a transmetallation step with the formation of (P(tBu)3)2Pd(H)(Br) and 13C-benzonitrile from a reductive elimination step. These experiments suggest the possibility of a catalytic cycle involving initially the formation of two Pd(II)-species from the oxidative addition of LnPd(0) into HCN and an aryl bromide followed by a transmetallation step to LnPd(Ar)(CN) and LnPd(H)(Br), which both reductively eliminate, the latter in the presence of KOAc, to generate the benzonitrile and LnPd(0).
Introduction
Benzonitriles are a valuable class of compounds with a broad range of applications serving as important substructures in many agrochemicals, pharmaceuticals, dyes, polymers and other organic materials.1 The nitrile group is also a precursor to a variety of important functionalities such as aldehydes, ketones, carboxamides and carboxylates.2 Conventional means for the instalment of a cyanide group onto an aromatic ring include the Rosenmund-von Braun or Sandmeyer reactions applying stoichiometric CuCN with aryl halides or diazonium salts.3 Yet, in the last decade, transition metal catalysed cyanations of aromatic halides with metal complexes based on copper,4 nickel5 and palladium6,7 have held a key position. In particular, the protocols based on Pd-catalysis through the extensive work by Beller,7 Grushin8 and Buchwald9 are characterised by relatively mild reaction conditions and good functional group tolerance. A variety of different cyanide sources have been applied including salts or derivatives such as NaCN,8,10 KCN,6a,c,d,11 (Me)3SiCN,12 acetone cyanohydrin,13 K4Fe(CN)6,9a,14 Zn(CN)2,9b,15 CuCN,16etc. (Scheme 1). However, many of these sources also have their disadvantages. For example, metal contaminants can become an issue with the use of potassium ferrocyanide, whereas with the cyanohydrin of acetone, this chemical must be stored carefully and is approx. 20 times more costly than potassium cyanide.‡ Furthermore, considering the fact that all of these cyanide precursors originate from hydrogen cyanide (HCN), which is produced on a million-ton scale size via the Andrussow17 and BMA-Degussa processes18 from natural gas and ammonia or as a byproduct in acrylonitrile production,18,19 there could be an interest in the development of a cyanation process of aryl halides exploiting this original source of cyanide, namely gaseous HCN.20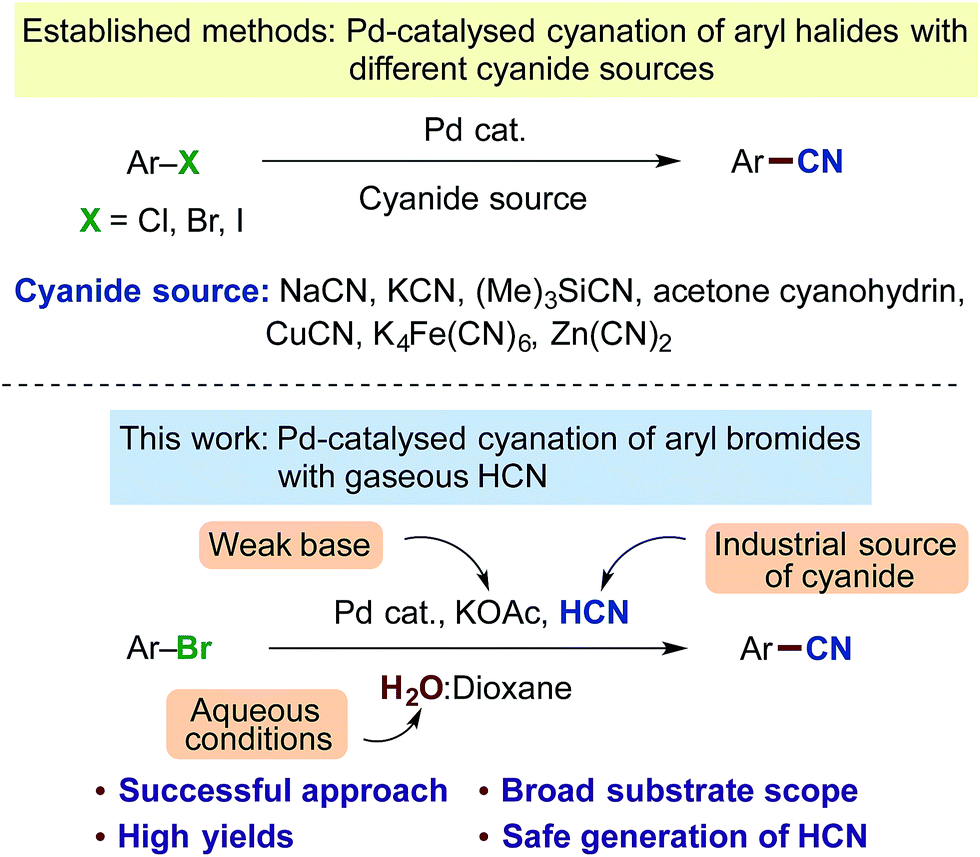 | ||
| Scheme 1 Previous developments in Pd-catalysed cyanation protocols and our new approach applying gaseous HCN. | ||
Only three reports are found in the literature applying HCN directly or by its in situ formation. In 2015, Buchwald and Hooker reported a 11C-cyanation study for the synthesis of aromatic cyanides with applications for positron emission tomography (PET).21 Here, submicromolar amounts of gaseous H11CN dissolved in THF were introduced to a 1000-fold excess of preformed oxidative addition palladium(II) complex, affording the desired 11C-benzonitriles nearly instantaneously. A follow-up paper by the same two groups demonstrated the possibilities of this approach for the 11C-labelling of unprotected peptides.22 Alternatively, Beller and co-workers reported the in situ release of HCN from acetone cyanohydrin although this was carried out in the presence of sodium carbonate, which undoubtedly leads to the direct formation of the reactive cyanide anion species.13
Nevertheless, there is a reluctance to apply HCN (b.p. 27 °C) as a cyanation reagent in an academic setting or an R&D laboratory, which is undoubtedly linked to the high toxicity and explosive nature of this gaseous one-carbon reagent, thereby complicating its handling, storage and transportation.23 Another challenge in the use of hydrogen cyanide as a cyanation reagent is how to control its dosage in stoichiometric amounts as Pd-catalysed cyanations of aryl halides are particularly sensitive to the cyanide concentrations. The strong binding of cyanide anion to transition metals can lead to catalyst deactivation if present in elevated concentrations.7,24 With other cyanide reagents, this complication is normally addressed by their slow addition to the reaction mixture, slow mass transport of the cyanide anion by specific water/organic solvents mixtures or by use of cyanide complexes with low solubilities.8,9b,13,14 Grushin and co-workers have published impressive and detailed studies on the mechanism of cyanide induced catalyst deactivation in the Pd-catalysed cyanation of aromatic halides. In their work, they report that addition of an excess of 13C-labelled potassium cyanide (K13CN) to different intermediates of the catalytic cycle in the Pd-catalysed cyanation of iodobenzene led to coordinatively saturated and catalytically inactive palladium complexes such as [Pd(CN)4]2−, [HPd(CN)3]2− or [ArPd(CN)3]2−.25 More interestingly, the same group revealed that traces of water in the reaction mixture combined with K13CN forms H13CN that immediately undergoes oxidative addition with Pd(0) leading to the same coordinatively saturated and off-cycle palladium complexes with concurrent formation of hydrogen gas.26
In this paper, we report on the direct use of industrially important hydrogen cyanide in the palladium-catalysed cyanation of (hetero)aromatic bromides (Scheme 1). These results deviate from conventional wisdom, since Grushin's earlier work demonstrated the propensity of palladium(0) to undergo fast oxidative addition into the H–CN bond shutting down the catalytic cycle. And yet, our results demonstrate that conditions can be identified whereby the presence of HCN does not terminate the cyanation reaction, but enhances the reactivity vis-à-vis cyanide salts. Secondly and most important, we describe a simple and safe setup whereby HCN is delivered in stoichiometric amounts by ex situ generation in a two-chamber reactor, thereby providing a simple and safe setting for handling gaseous HCN in small-scale reactions. We demonstrate the usefulness of this setup not only for the Pd-catalysed cyanation of aryl bromides, but also for the Ni-mediated hydrocyanation of styrenes as a test reaction. With respect to the cyanation reactions good functional group tolerance was obtained, and the method proved amenable to scale-up, but also to carbon-13 isotope labelling applying H13CN. Surprisingly, the developed conditions proved to be dependent on water as the co-solvent and the presence of a weak base, KOAc. In their absence, catalytic shutdown was observed, thereby indicating the operation of an alternative mechanism to Pd-catalysed cyanations applying cyanide salts. Studies and isolation of the oxidative addition complexes suggest a mechanism involving the transmetallation between two palladium(II) species, both formed by the oxidative addition of palladium(0) into either HCN or the aryl bromide electrophile.
Results and discussion
Hydrogen cyanide releasing studies
In order to provide a system for the controlled dosage of hydrogen cyanide in stoichiometric quantities, we envisioned that the two-chamber system previously utilised for the ex situ liberation of carbon monoxide,27 hydrogen28 and ethylene29 from specific nongaseous precursors could be exploited. Such an approach would provide a simple and safe setup without the direct handling of HCN gas. Hence, our focus was first directed to identifying a suitable system for the generation of HCN gas under a closed environment, and the well-established nickel-catalysed hydrocyanation of olefins was exploited as a test system for the optimisation.30–32 After considerable experimentation on the hydrocyanation of styrene (see ESI†), we finally adopted the following conditions as illustrated in Scheme 2. A combination of KCN, ethylene glycol and AcOH provided the desired release of gaseous HCN. Ethylene glycol serves a dual purpose acting both as solvent while simultaneously ensuring separation of the AcOH from KCN until stirring is initiated. By utilising this setup for the HCN producing chamber in combination with the Ni-catalysed hydrocyanation conditions in the second chamber (Ni(COD)2, XantPhos), a range of different styrenes bearing both electron donating and withdrawing groups could successfully be hydrocyanated to products 1–7 in near quantitative yields. Full conversion of all styrene derivatives was attained applying only 1.5 equivalents of KCN for the production of HCN, indicating the effectiveness of this gas generator.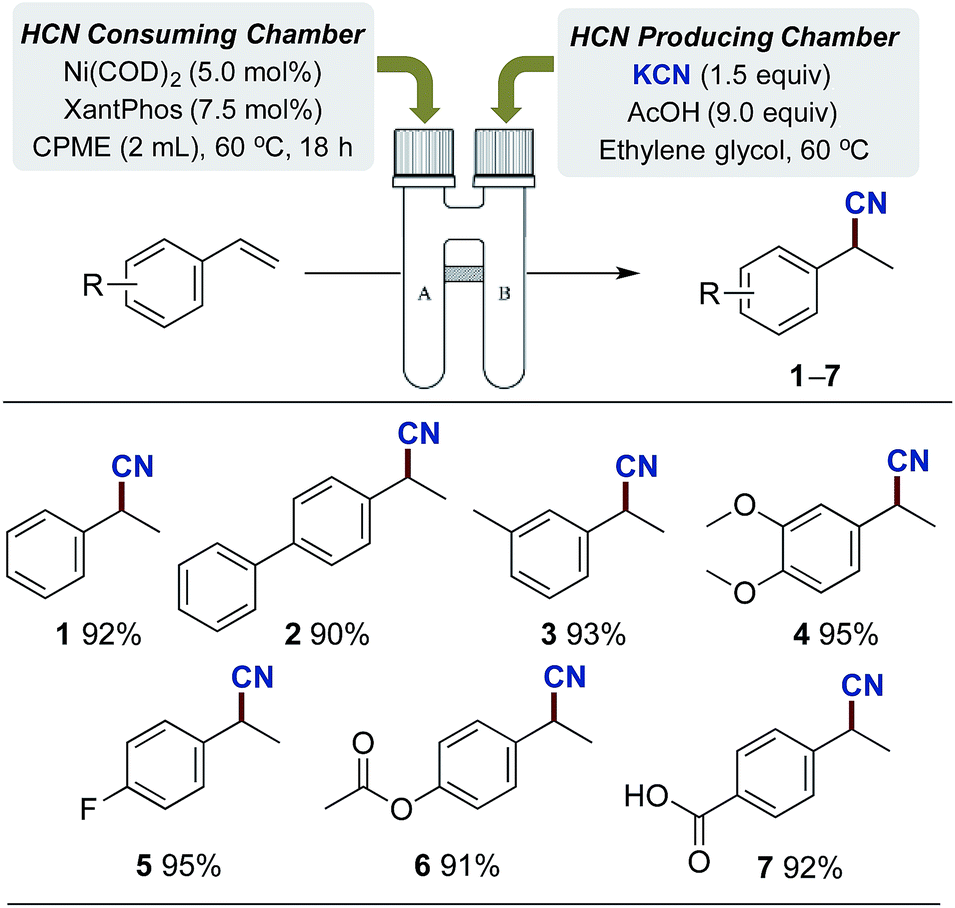 | ||
| Scheme 2 Ni-catalysed hydrocyanation of styrenes employing a two-chamber system with ex situ generated HCN. aChamber A: styrene (1.0 mmol), Ni(COD)2 (5.0 mol%), XantPhos (7.5 mol%) and CPME (2 mL). Chamber B: KCN (1.5 mmol), ethylene glycol (1 mL), AcOH (9.0 mmol). Isolated yields are given as an average of 2 runs. | ||
Studies on the cyanation of (hetero)aryl bromides
With a simple and convenient system for the release of gaseous HCN in hand, we next turned our attention towards the Pd-catalysed cyanation of (hetero)aryl bromides. As depicted in the scheme of Table 1, initial optimisation results were carried out with 4-bromoanisole. This study revealed that reaction conditions consisting of a catalyst formed from Pd(dba)2 with P(tBu)3, in the presence of hydrogen cyanide (1.5 equiv.), a weak base such as KOAc, and a solvent mixture of water and dioxane could generate 4-methoxybenzonitrile (8) with a conversion of 78% according to the 1H NMR analysis of the crude reaction mixture (Table 1, entry 1). P(tBu)3 as a ligand proved to be superior to other monodentate and bidentate ligands (entries 2–6; see ESI† section for full optimisation studies), which is in accord with earlier results reported by Grushin and co-workers.8,26 In general, weak bases such as KOAc and NaOAc provided better conversions to the desired 4-methoxybenzonitrile, whereas stronger bases including Cy2NMe, DBU and Et3N all proved inferior (entries 7−9). Increasing to three equivalents of KOAc provided a slight increase in the conversion to 82% (entry 10). The efficiency of the reaction could also be increased further exploiting the Buchwald pre-catalyst, P(tBu)3-Pd-G3, providing a 90% isolated yield of benzonitrile 8 after column chromatography (entry 11). Control experiments with omission of either base or palladium precursors resulted in low or no conversion, resp.|
|
||
|---|---|---|
| Entry | Deviation from lead conditions | Conversionb (%) |
| a Chamber A: 4-bromoanisole (1.0 mmol), Pd(dba)2 (2.5 mol%), P(tBu)3 (5.0 mol%), KOAc (2.0 mmol) in dioxane (1 mL) and H2O (1 mL). Chamber B: KCN (1.5 mmol), ethylene glycol (1 mL) and AcOH (9.0 mmol). b Determined by 1H NMR using mesitylene as an internal standard. P(tBu)3-Pd-G3 = third generation Buchwald precatalyst with the tri-tert-butylphosphine ligand. c Isolated yield. | ||
| 1 | None | 78 |
| 2 | Ligand: PCy3 | 0 |
| 3 | Ligand: XPhos | 60 |
| 4 | Ligand: tBu-XPhos | 74 |
| 5 | Ligand: DPPF | 0 |
| 6 | Ligand: XantPhos (2.5 mol%) | 0 |
| 7 | Base: Cy2NMe | 38 |
| 8 | Base: DBU | 5 |
| 9 | Base: Et3N | 67 |
| 10 | Base: KOAc (3.0 equiv.) | 82 |
| 11 | Pd/ligand: P(tBu) 3 -Pd-G3 KOAc (3.0 equiv.) | 90 (90) |
| 12 | Pd/ligand: P(tBu)3-Pd-G3 solvent: THF | 77 |
| 13 | Pd/ligand: P(tBu)3-Pd-G3 solvent: Toluene | 61 |

Different water:solvent combinations were tested and dioxane proved to be the solvent of choice (entries 12 and 13, see also ESI†). Notably, the presence of water as the co-solvent is of key importance as similar experiments applying only dioxane as the solvent resulted in complete catalytic shutdown. In our scope studies, we later discovered that a 2:1 mixture of water and dioxane provided higher yields for certain substrates, and hence this ratio was used for all subsequent cyanations. Not surprisingly and in accord with Grushin's previous results, carrying out the Pd-catalysed cyanation in a single chamber reactor applying KCN directly with or without the addition of acetic acid under the conditions developed led to no formation of 8 (result not shown).
With the optimised reaction conditions in hand, we set out to explore the scope of the Pd-catalysed cyanation using gaseous HCN. All yields are reported as an average of two runs and the results are depicted in Scheme 3. Aryl bromides carrying electron donating substituents were initially examined. Methoxy-, hydroxy-, alkyl- and aryl-substituted aryl bromides provided the desired compounds in high yields ranging from 90% to 97% (compounds 8–11). Electron withdrawing substituents such as cyano, acyl and carboxylate afforded compounds 12, 13 and 14 in yields attaining quantitative. Even p-bromobenzoic acid underwent successful coupling affording p-cyanobenzoic acid (15) in an 86% yield, using 4 equiv. of KOAc combined with 5 mol% of Buchwald's precatalyst. The use of phenol and benzoic acid derivatives in the Pd-catalysed cyanation with NaCN were previously shown by Ushkov and Grushin to lead to catalyst deactivation due to the formation of HCN.8p-Bromobenzaldehyde also proved reactive with the isolation of 16 in a somewhat lower yield. This slight reduction in isolated yield can possibly be explained by competing benzoin condensation of both starting material and product. The substitution pattern of bromoanilines interfered significantly with the developed conditions and installment of the cyano-functionality onto 2-, 3-, and 4-bromoaniline was achieved in yields of 95%, 77% and 47% respectively (compounds 17–19). Aryl bromides displaying ortho-substitutions were also effective for these substitution reactions as illustrated with compounds 17 and 20–22, all isolated in yields between 92% and 96%. However, 2-bromo-1,3-dimethylbenzene did require an increase of the catalyst loading to achieve the high 96% isolated yield of 21. Lowering the reaction temperature of the HCN consuming chamber, from 60 °C to 45 °C, ensured a chemoselective activation of the aromatic bromide in the presence of the p-chloride affording 23 in an 88% isolated yield. Subsequently, an aryl bromide possessing a benzylic alcohol, and five heteroaromatic bromides were tested under the developed conditions. All underwent successful cyanation to afford the desired target molecules in yields ranging from 75% to 95% (compounds 22, 24–28). It should be mentioned though that despite the successful coupling of these handful of heteroaryl bromides, two other heterocycles tested such as 2-acetyl-5-bromothiophene and 3-bromobenzothiophene failed to provide the corresponding nitrile product. The reasons for this catalytic shutdown are not completely understood (see Discussion in next section). Finally, attempts to perform catalytic cyanation on 4-biphenyl triflate, applying the developed protocol, were non-rewarding with no conversion observed for this electrophile.
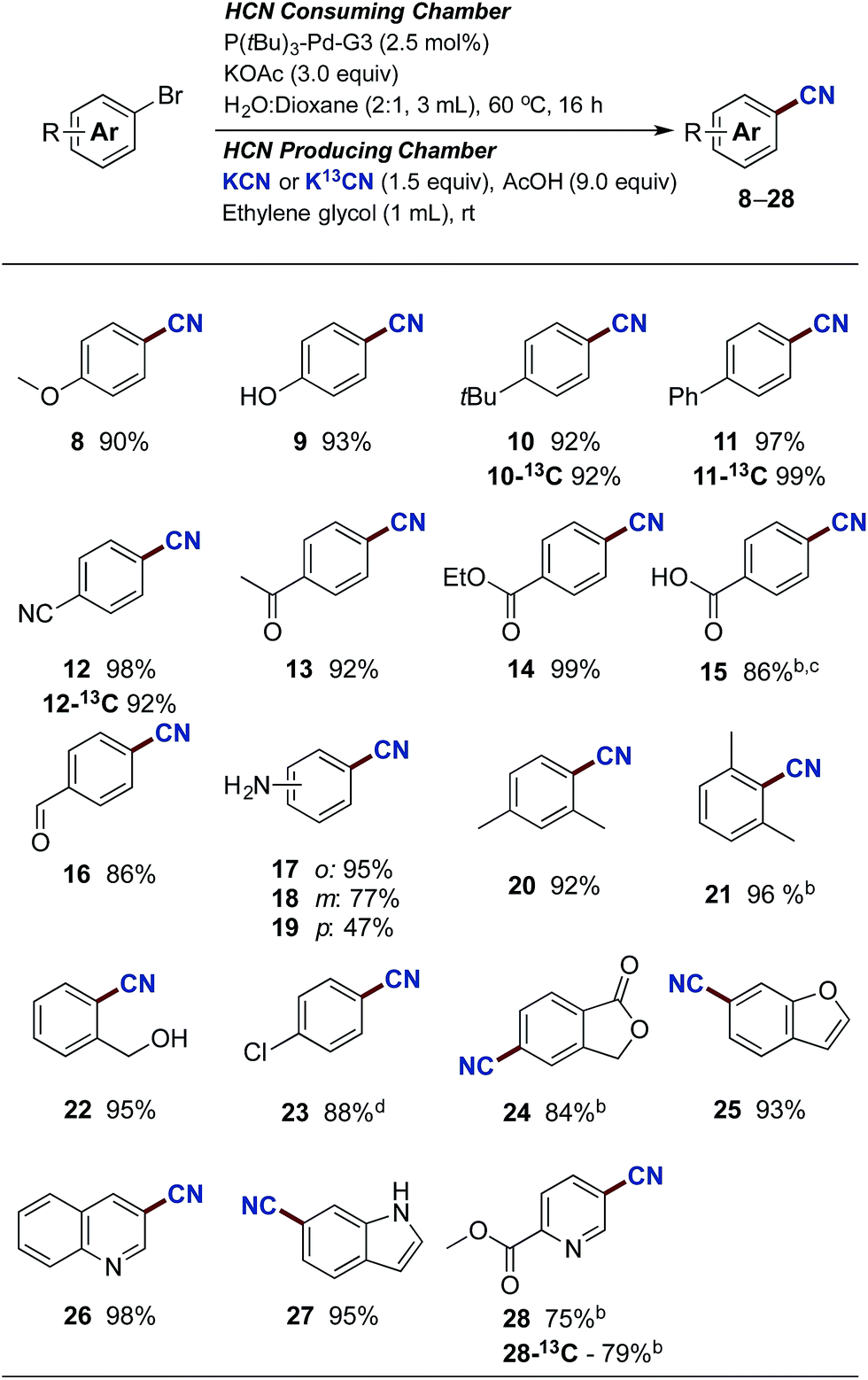 | ||
| Scheme 3 Pd-catalysed cyanation of aryl bromides. aChamber A: aryl bromide (1.0 mmol), P(tBu)3-Pd-G3 (2.5 mol%) and KOAc (3.0 mmol) in dioxane (1 mL) and H2O (2 mL). Chamber B: KCN (1.5 mmol) or K13CN (1.5 mmol) ethylene glycol (1 mL) and AcOH (9 mmol). Isolated yields are given as an average of 2 runs. b5.0 mol% catalyst used. c4.0 equiv. of KOAc used. dHCN consuming chamber only heated to 45 °C. | ||
Next, attention was directed to implementing this developed protocol for 13C-labelling. By simply substituting KCN with its 13C-labelled counterpart under otherwise unchanged conditions, direct access to isotopically labeled benzonitrile derivatives were achieved. Four entries from Scheme 3 were selected for labelling, and all compounds were isolated in near identical yields to the unlabeled compounds (compounds 10-13C, 11-13C, 12-13C and 28-13C).
Finally, the developed protocol was tested for the synthesis of three active pharmaceutical ingredients, namely dapivirine (reverse transcriptase inhibitor), citalopram (serotonin reuptake inhibitor) and letrozole (non-steroidal aromatase inhibitor), the results of which are depicted in Scheme 4.33 Dapivirine (29) and citalopram (30) were obtained in satisfactory 96% and 86% isolated yields, respectively, from the corresponding bromide precursors applying 5.0 mol% of the Buchwald precatalyst. For the synthesis of letrozole (31) a double cyanation is required with the dibromide 31a. Doubling the loading of KCN for HCN release to 3 equivalents combined with an increase in KOAc to 4 equivalents, gratifyingly afforded the dicyanide 31 in an excellent 97% isolated yield again with 5.0 mol% precatalyst. The synthesis of letrozole was then attempted on a fivefold scale combined with a 100 mL two-chamber reactor to afford a near identical isolated yield of 95%. Finally, scaling up to 10 mmol in a 200 mL two-chamber reactor, with an HCN release from 30 mmol of KCN, corresponding to more than 700 mL of gaseous HCN, afforded the desired pharmaceutical in a good 85% isolated yield.
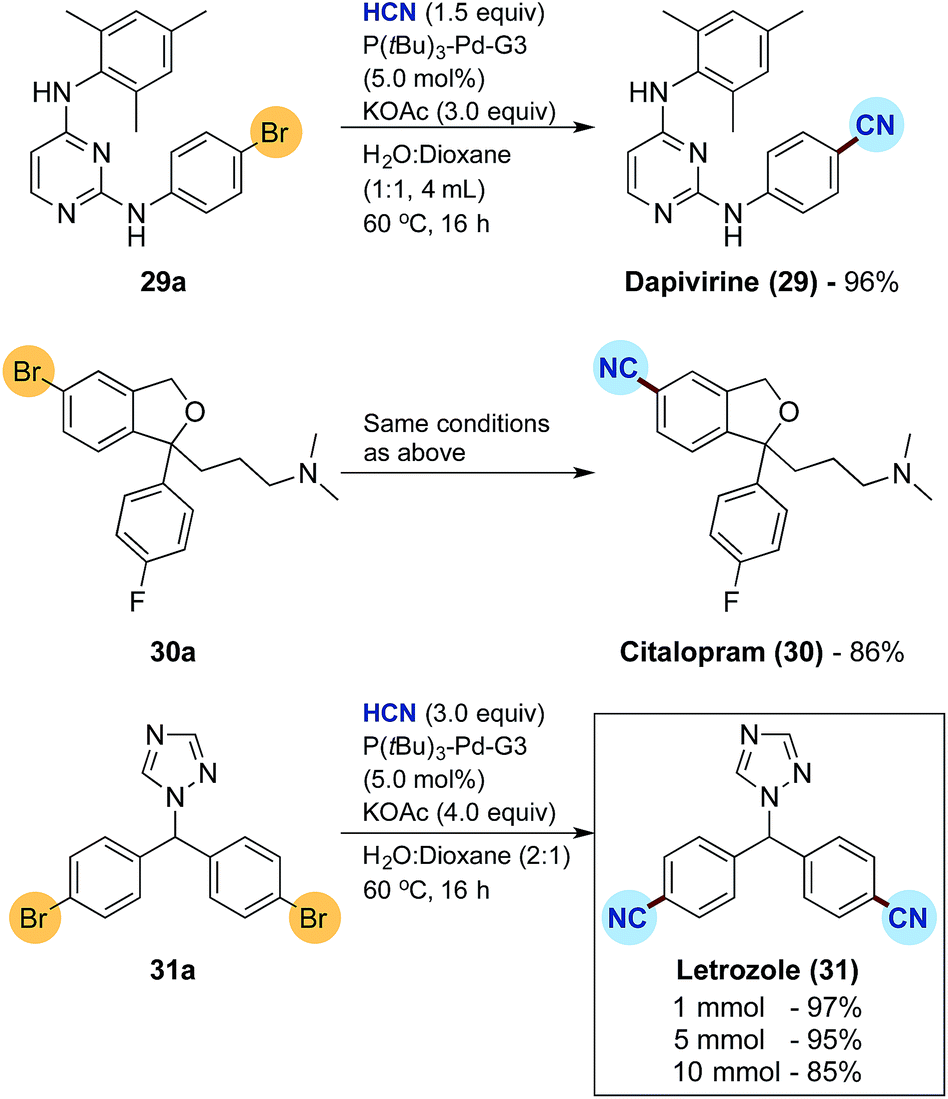 | ||
| Scheme 4 Synthesis of pharmaceuticals by Pd-catalysed cyanation and scale up studies. aFor reactions on 1.0 mmol scale, yields are an average of two runs. | ||
Mechanistic investigations
Having established the scope of the Pd-catalysed cyanation of (hetero)aryl bromides with hydrogen cyanide, some questions still remained. Given a pKa difference in water for acetic acid (4.76) and HCN (9.21) of almost 4.5 units, indicates that HCN is potentially the reactive species in solution and not the free cyanide anion. However, previous reports by Grushin and co-workers clearly demonstrated that the presence of free HCN formed from the protonation of KCN with trace amounts of water, quickly leads to shutdown of the catalytic activity.8,26 Still this deactivation is a combined result of Pd(0) undergoing oxidative addition into the H–CN bond followed by trapping of this species with excess cyanide anion to generate off-cycle palladium(II) cyanide complexes. Furthermore, there was not a clear indication why certain heteroaryl bromides were successful coupling partners with HCN, whereas others were not. With the exception of the protocol developed by Buchwald and Hooker, in which H11CN is used to form PET tracers, no reports are found on palladium-catalysed cyanation of aryl halides using gaseous HCN as the reactant.21 Although it should be noted under the conditions used for 11C-isotope-labelling, the Pd(II)-aryl complex is in an approx. 1000-fold excess compared to the H11CN generated. With this in mind it was decided to take a closer look at the possible mechanism for the developed protocol.The overall accepted mechanism for the Pd-catalysed cyanation using sodium or potassium cyanide is believed to go through an initial oxidative addition of the Pd(0) complex into the aryl halide, nucleophilic displacement by cyanide on the formed palladium(II) centre, and lastly a reductive elimination furnishing the desired benzonitrile (Scheme 5).6h,7,8,26 When metal complexes such as Zn(CN)2 are utilised, a change in mechanism occurs, whereby nucleophilic substitution is replaced with a transmetallation step.9b,34
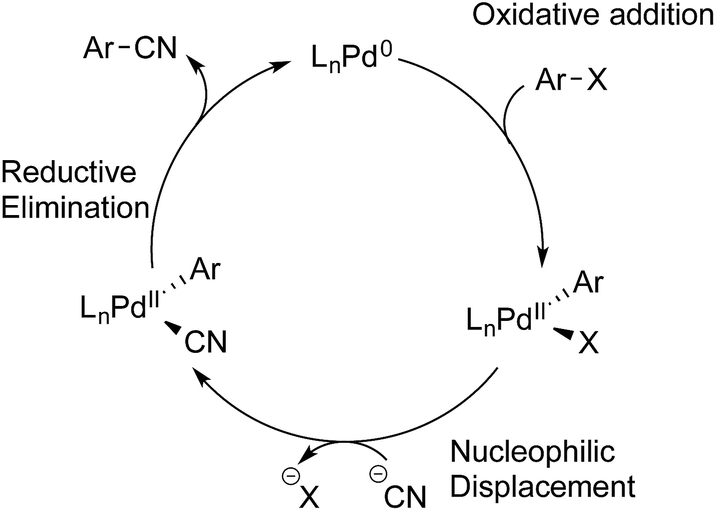 | ||
| Scheme 5 Accepted mechanism for the cyanation of aryl halides with cyanide salts. | ||
During the optimisation of the catalytic system, different bases were tested (Table 1, entries 7–10). A clear trend can be abstracted from this base screening, where an increase in base strength leads to a decrease in conversion. This can be explained by the higher degree of deprotonation of HCN with strong bases leading to catalytic shutdown. The effect is highest for the strong base DBU affording a mere 5% conversion to product 8. The optimum conditions found in this work applies KOAc as base, suggesting that HCN is the reactive species and not cyanide itself.
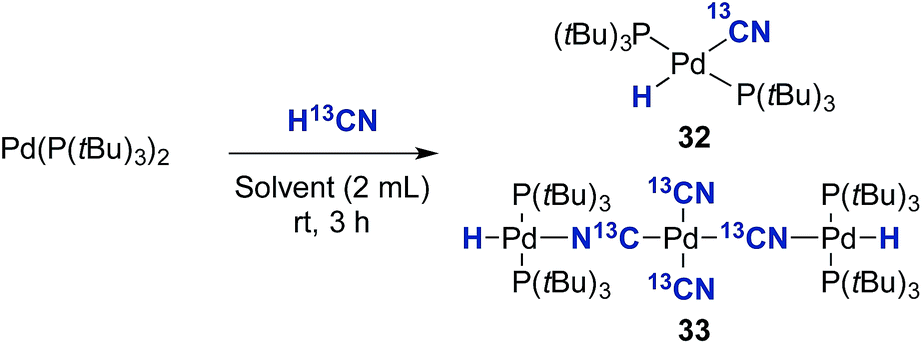
Initially, the oxidative addition of Pd(P(tBu)3)2 to H13CN in THF as the solvent was investigated using the two-chamber reactor (see ESI† for reaction details).§ This afforded two palladium-hydride species in roughly a 6:1 ratio, as observed by 1H NMR analysis of the reaction mixture, with the hydride signals residing at −11.42 and 16.79 ppm (Fig. 1 and 3).35 Careful manipulation of the reaction mixture allowed for the isolation and crystallisation of both hydride species 32 and 33, the structures of which were confirmed by X-ray crystal structural analysis (Fig. 2 and 4).
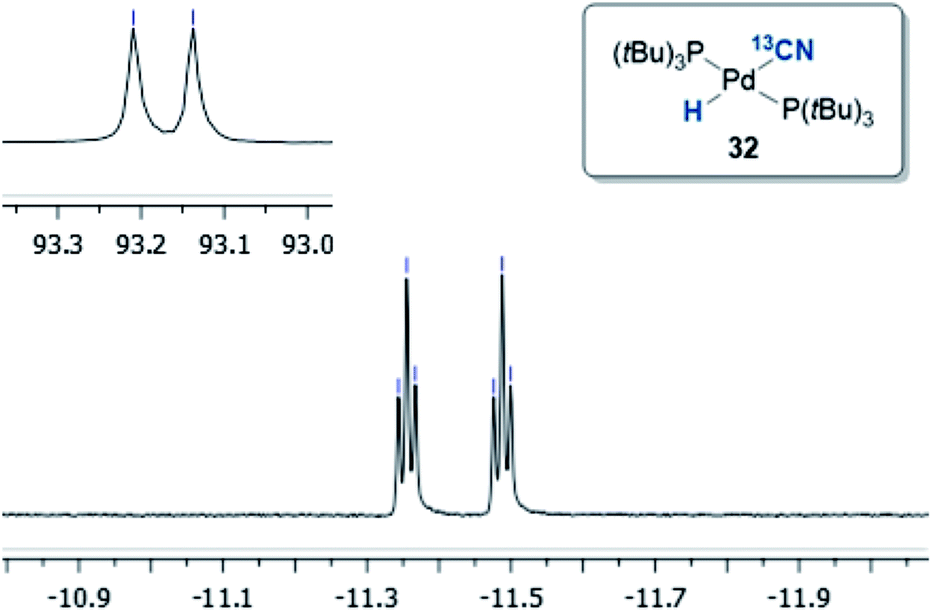 | ||
| Fig. 1 1H NMR (JH–C = 53.0 Hz, JH–P = 4.7 Hz) and 31P NMR (JP–C = 11.7 Hz) of compound 32 in THF-d8. | ||
 | ||
| Fig. 2 ORTEP representation of complex 32. | ||
 | ||
| Fig. 3 1H NMR and 31P NMR of compound 33 in CDCl3. | ||
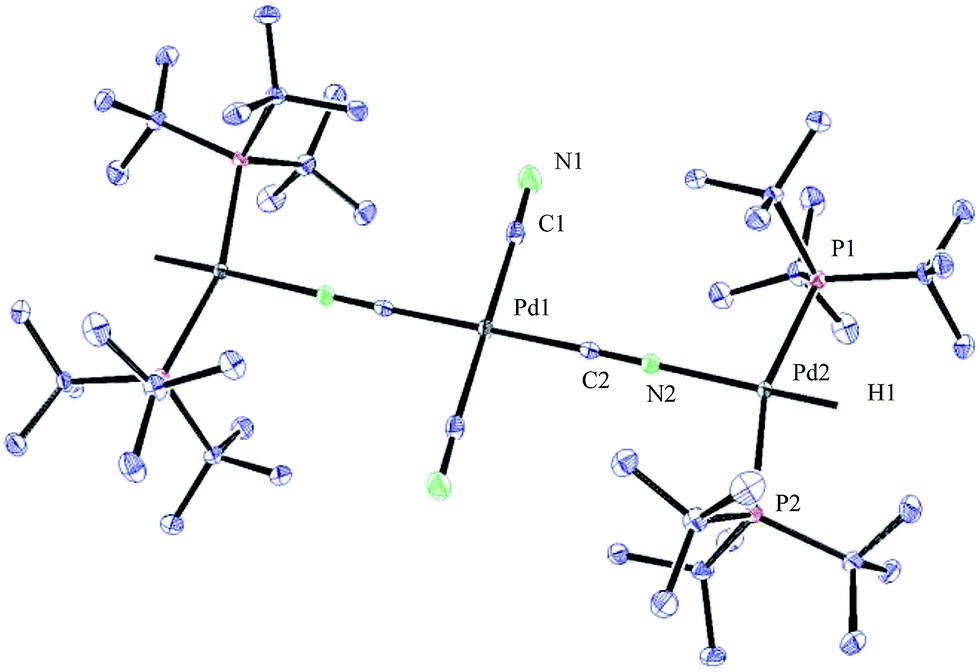 | ||
| Fig. 4 ORTEP representation of complex 33. | ||
Compound 32 was identified as the oxidative addition complex with H13CN, (P(tBu)3)2Pd(H)(13CN). While others have reported the corresponding HBr and HCl adducts,36,37 it is to the best of our knowledge the first time compound 32 has been isolated. The hydride signal is located at −11.42 ppm as a double triplet in the 1H NMR spectrum (Fig. 1), whereby the multiplicity originates from a large trans-coupling with the 13-carbon of the cyanide, and small cis-coupling with the two equivalent phosphine ligands.¶ The 31P NMR analysis of 32 reveals a doublet at 93.1 ppm arising from P–C coupling with the 13C-labelled cyanide. Due to disturbance in the crystal structure, the hydride could not be located. However, in view of the combined data obtained from the NMR and the X-ray crystal structural analysis, we are confident that the structure of 32 is as indicated in Fig. 2.
To our surprise, the X-ray crystal structure analysis of the palladium-hydride complex 33 revealed it to be a tri-metallic species as illustrated in the ORTEP representation of Fig. 4. In the 1H NMR spectrum a hydride signal is located at −16.79 ppm as a multiplet (Fig. 3), whereas for the 31P NMR spectrum, a singlet at 84.7 ppm can be found. A closer look at this Pd3-complex, shows its resemblance to K2[Pd(13CN)4], which can also be produced from the reaction of Pd(0) with H13CN in turn formed from the hydrolysis of K13CN with water.26 In structure 33, the anionic core, [Pd(13CN)4]2−, carries two cationic counter ions in the form of [(P(tBu)3)Pd(H)]+.
To investigate the importance of the reaction conditions and influence on the formation of 32 and 33, a series of experiments were performed the results of which are shown in Table 2. As can be seen from entries 1–3, neither the amount of H13CN nor the presence of water appears to influence the distribution between 32 and 33. However, the addition of KOAc and water resulted in the exclusive formation of the Pd-hydride complex 32 (entry 4).|| Increasing the H13CN loading from 1.0 to 3.0 equiv. provided a 92% conversion to 32. Full conversion to 32 was obtained with 3 equiv. of H13CN in combination with 6 equiv. of KOAc (entry 6). Finally, direct formation of 33 from 32 can also be achieved by heating 32 in THF at 60 °C, and after 3 h, a 3:1 ratio between 32 and 33 is achieved. This result could be explained by the slow transmetallation between two Pd-hydride species 32 leading to the formation of (P(tBu)3)2Pd(H)2 and (P(tBu)3)2Pd(13CN)2.35,38 Whereas, the former can reductively eliminate generating Pd(0) and dihydrogen, we speculate that (P(tBu)3)2Pd(13CN)2 could abstract cyanide from two Pd-hydride complexes 32 ultimately leading to the formation of the Pd3-complex 33.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | H13CN (equiv.) | KOAc (equiv.) | Ratio 32:33b |
| a Reactions performed on a 0.1 mmol scale. Reactions stopped after 3 h. b Values in brackets are given as conversions. | ||||
| 1 | THF | 1.0 | — | 85:15 (100%) |
| 2 | THF | 3.0 | — | 84:16 (100%) |
| 3 | THF:H2O |
1.0 | — | 86:14 (100%) |
| 4 | THF:H2O |
1.0 | 2.0 | >95:5 (29%) |
| 5 | THF:H2O |
3.0 | 2.0 | >95:5 (92%) |
| 6 | THF:H2O |
3.0 | 6.0 | >95:5 (97%) |
To further probe the necessity of the bulky and electron rich P(tBu)3 ligand, Pd(PPh3)4 was dissolved in THF and treated with H13CN (Scheme 6). This resulted in the sole formation of (PPh3)2Pd(13CN)2 (34), the structure of which was confirmed by both NMR and X-ray analysis (see ESI†).** Possibly, the transmetallation event involving two (PPh3)2Pd(H)(13CN) species is significantly faster than that for complex 32, and therefore cyanide abstraction from (PPh3)2Pd(H)(13CN) with complex 34 cannot compete as with similar palladium species bearing the P(tBu)3 ligand.
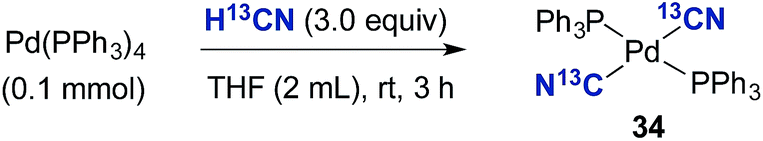 | ||
| Scheme 6 Reaction between Pd(PPh3)4 and H13CN. aReaction performed on a 0.1 mmol scale with a two-chamber system. The reaction was stopped after 3 h. | ||
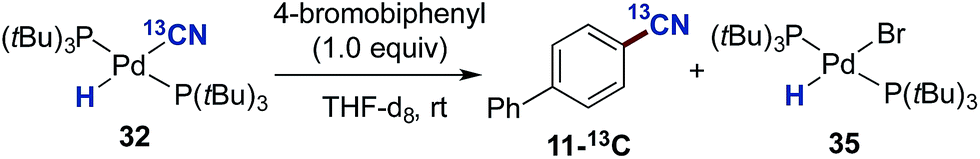
Next, we addressed the question whether complex 32 represents an active participant in the catalytic cycle. To this end, we examined the reaction between 4-bromobiphenyl and 32 in THF-d8 at room temperature with mesitylene as an internal standard (Scheme 7). The reaction progress was followed by both 1H- and 31P-NMR. From the 1H NMR spectrum it was clear that as the reaction progressed a build-up of the palladium-hydride species 35 in addition to the formation of the aromatic nitrile 11-13C was observed. The reaction turned out to be relatively slow at 25 °C despite a 1:1 relationship between complex 32 and 4-bromobiphenyl. Nevertheless, after 12 h, the conversion into 11-13C had reached 55%.
 | ||
| Scheme 7 Reactivity studies between complex 32 with 4-bromo-biphenyl. a0.02 mmol of both 32 and 4-bromobiphenyl were added to a NMR-tube. Mesitylene was used as an internal standard. | ||
To verify that the structure of the new Pd-hydride species formed indeed is as proposed for compound 35, a sample of this complex was prepared according to a known literature procedure involving the treatment of Pd(P(tBu)3)2 with pyridinium bromide in toluene.39 As can be seen in Fig. 5, complex 35 produced a triplet at −15.63 ppm in the 1H NMR spectrum arising from the coupling to the two equivalent phosphorus atoms. In the 31P NMR spectrum, 35 gives rise to a doublet located at 83.5 ppm. These spectroscopic data were in accord with those observed from the reaction of 32 with 4-bromobiphenyl. Finally, by dissolving 35 in a small amount of CH2Cl2 layered with heptane resulted in crystals that were suitable for X-ray analysis (Fig. 6).
 | ||
| Fig. 5 1H NMR (JH–P = 7.2 Hz) and 31P NMR (J = 1.9 Hz) of compound 35 in THF-d8. | ||
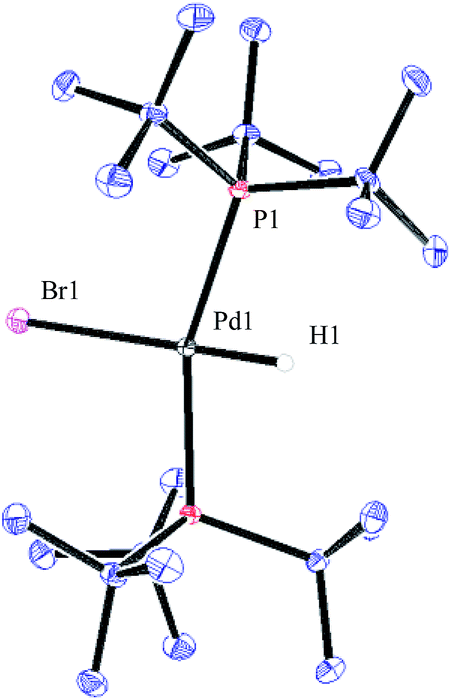 | ||
| Fig. 6 ORTEP representation of complex 35. | ||
The formation of aryl nitrile 11-13C is only possible if complex 32 can reductively eliminate generating Pd(P(tBu)3)2 and H13CN, thereby implying that the addition of HCN to Pd(0) is a reversible process.40 The Pd(0) species produced from this reductive elimination step can then either undergo an oxidative addition to H13CN again or alternatively to the aryl bromide generating (P(tBu)3)Pd(Ar)(Br).41 With the presence in solution of both this complex and 32, a transmetallation event could take place between these two species leading to the formation of the Pd-hydride 35 and (P(tBu)3)Pd(Ar)(13CN), which subsequently undergoes reductive elimination to the desired benzonitrile 11-13C and Pd(0).††
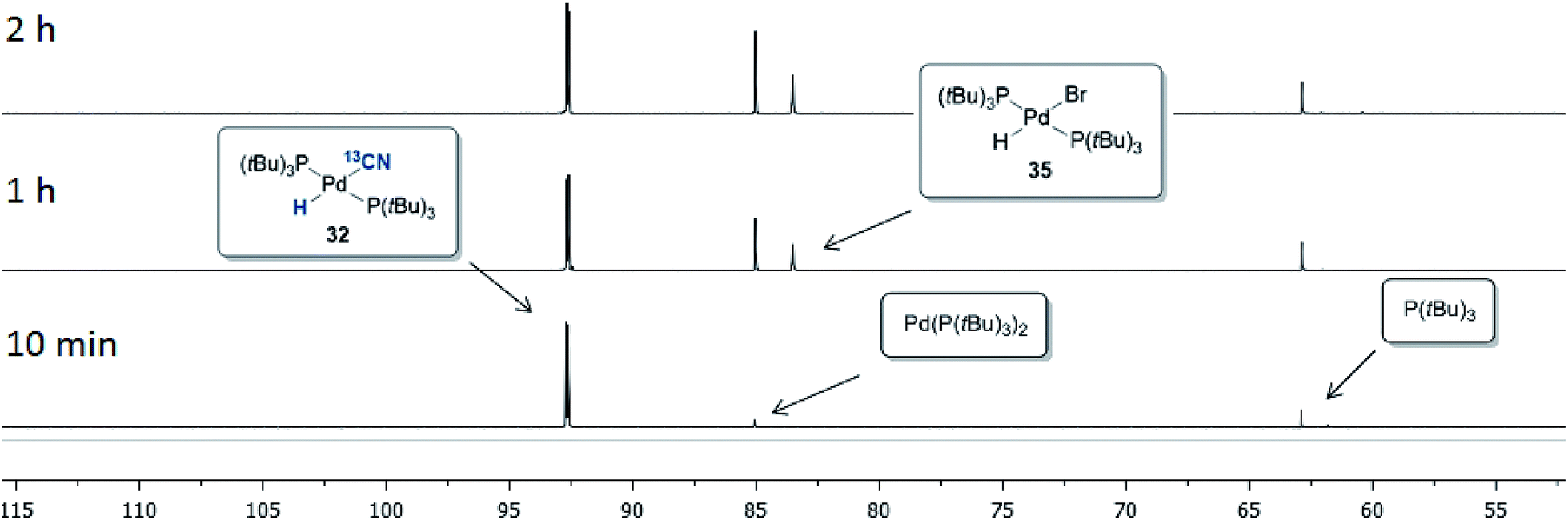
The progress of the reaction between 32 and p-bromobiphenyl was monitored by 31P NMR spectroscopy, the results of which are shown in Fig. 7. Within the first 10 minutes Pd(P(tBu)3)2 (85.5 ppm) and free P(tBu)3 (63.4 ppm) were formed, which is consistent with the observations by Fu and coworkers when a P(tBu)3/Pd(0) ration of 2:1 is used.42 The depletion of complex 32 then occurred with concurrent build-up of the Pd-hydride 35 residing at 83.5 ppm. No signal from the oxidative addition complex (P(tBu)3)Pd(Ar)(Br) was observed, suggesting that the oxidative addition of Pd(P(tBu)3) to 4-bromobiphenyl is the rate-determining step in this setup.
 | ||
| Fig. 7 31P NMR spectra of the reaction between complex 32 and p-bromobiphenyl. | ||
From these initial experiments, a possible mechanistic scenario for the Pd-catalysed cyanation of aryl bromides with gaseous HCN is depicted in Scheme 8, which accounts for the above observations. After formation of the catalytically active palladium(0) species, two reversible oxidative addition events take place forming LnPd(H)(CN) and LnPd(Ar)(Br). The necessity for the oxidative addition steps to be reversible at least for the latter complex may explain why aryl triflates are not competent electrophiles for this catalytic protocol. The two complexes can then undergo transmetallation providing LnPd(Ar)(CN) and LnPd(H)(Br). Similar transmetallative events have previously been reported for two Pd(II)-aryl species.43 Finally, the benzonitrile and the active LnPd(0) species are reformed through two reductive elimination events involving both LnPd(Ar)(CN) and LnPd(H)(Br). In the latter case, base is required to initiate the regeneration of LnPd(0), and the presence of aqueous KOAc should be sufficient for promoting this reduction step at the metal centre. Whereas stronger bases would be able to carry out this step as well, they would also lead to the deprotonation of HCN resulting in increased concentrations of cyanide anion in solution. Subsequently, this cyanide would trap the active species in the catalytic cycle as multi-cyano palladium complexes and as such lead to catalytic shutdown.
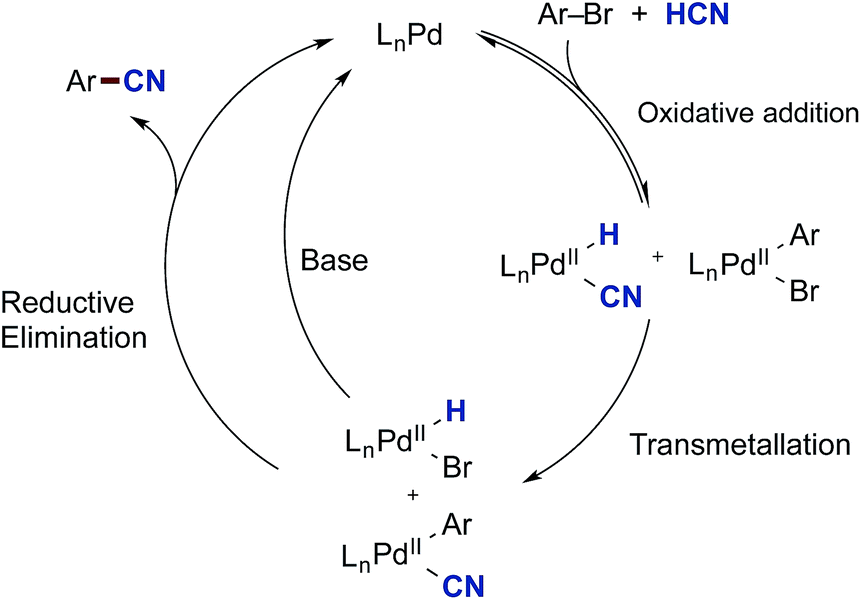 | ||
| Scheme 8 A proposed mechanism for the Pd-catalysed cyanation of aryl bromides with HCN. | ||
The mechanism depicted in Scheme 8 also supports the findings made by Buchwald and Hooker.21,22 In their setup, H11CN is introduced to a large excess of the oxidative addition complex of the same type as 36. Only a small portion of this Ar–Pd(II) complex would have to undergo reductive elimination to afford a Pd(0) species that could trap the added H11CN leading to product formation.
In order to investigate the viability of such a transmetallation step between two Pd(II) species of the proposed catalytic cycle in Scheme 8, we investigated the reaction between Pd-hydride 32 and the preformed oxidative addition complex 36 (Scheme 9a). The aryl–Pd complex 36 was prepared in a 79% isolated yield via the oxidative addition of Pd(P(tBu)3)2 to bromobenzene according to a literature procedure.44 Complexes 32 and 36 were dissolved in THF-d8, and using mesitylene as the internal standard, their transformation was followed by 31P NMR spectroscopy. From this experiment, three species were formed as the reaction progressed, being 13C-benzonitrile (37), (P(tBu)3)2Pd(H)(Br) (35) and the Pd3-complex 33. The transformation was completed in less than 30 min, and applying the internal standard, 37 was formed in a 53% yield while most of the remaining 13C-labelled cyanide could be accounted for from the formation of 33 (11% yield).
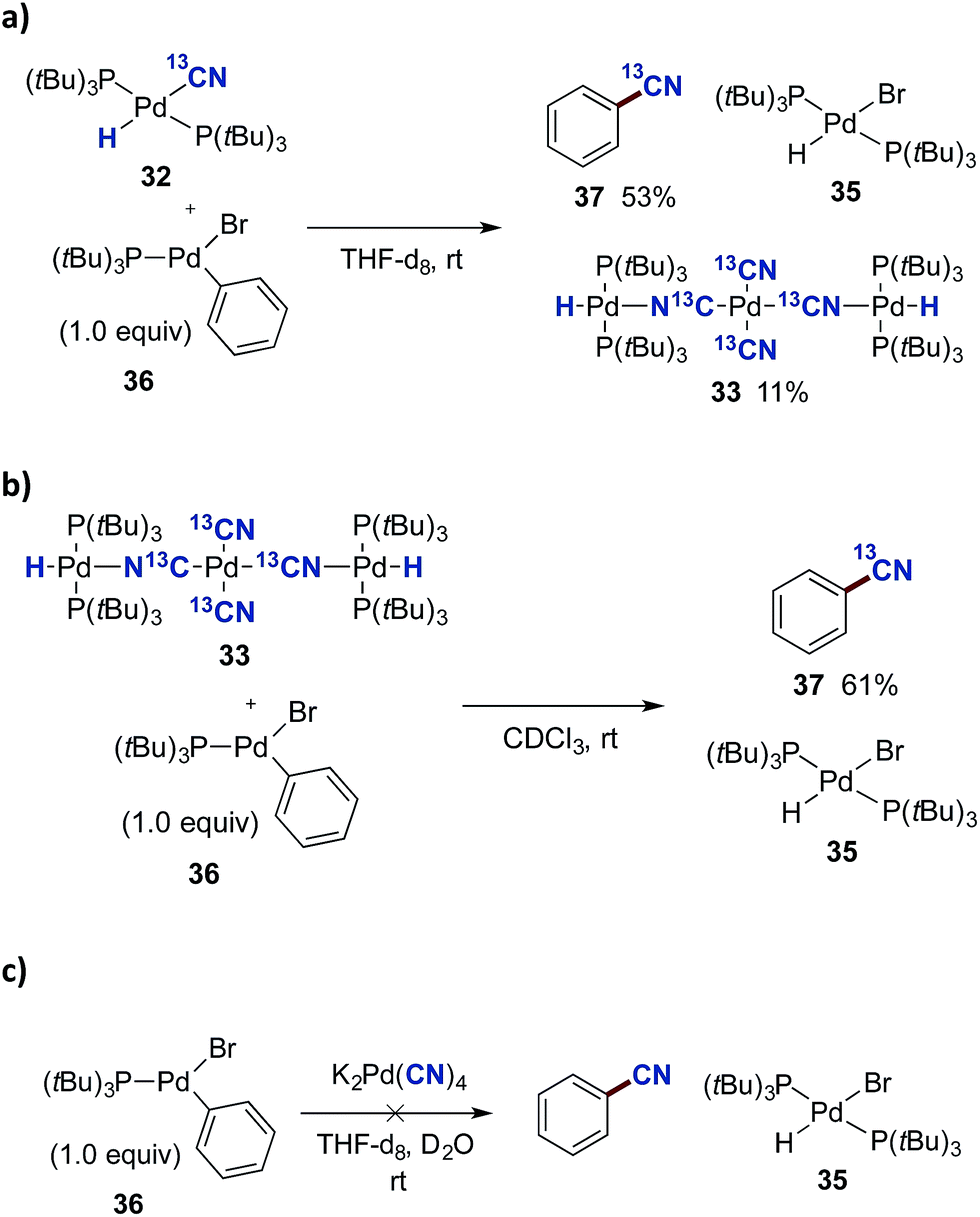 | ||
| Scheme 9 Transmetallation studies with complex 36. a0.02 mmol of either 32, 33 or K2[Pd(CN)4] with 36 were added to a NMR-tube. Mesitylene was used as an internal standard. | ||
Given the undoubtedly strong binding mode of cyanide to palladium(II), the possibility of free cyanide in solution is most likely absent, thereby eliminating the possibility for the formation of 35 through a nucleophilic displacement pathway. This in turn indicates that 35 forms as a result of a transmetallation step between 36 and one or both of complexes 32 and 33.
As mentioned above, the Pd3-complex 33 is presumably not formed under the optimised conditions. Nevertheless, we investigated whether 33 could also promote benzonitrile formation alone. The trinuclear complex 33 was mixed with 36 in a 1:1 relationship in CDCl3, and followed by 1H NMR spectroscopy. Surprisingly, complete formation of 13C-benzonitrile (37) and Pd-hydride 35 was achieved after only 2 h (Scheme 9b). To evaluate the importance of the [(P(tBu)3)2Pd(H)]+ cation in 33, the direct reaction between K2[Pd(CN)4] and 36 was tested (Scheme 9c). However, following the reaction by 1H NMR spectroscopy, no conversion was observed between these two palladium complexes as seen from the absence of signals for benzonitrile and Pd-hydride 35.
Next, we examined whether the different Pd-cyanide species are catalytically competent species and can promote the catalytic conversion of aryl bromides to aryl nitriles with HCN. By using 32 as the palladium source we could isolate biphenyl nitrile 11 in quantitative yield (Scheme 10). On the other hand, with 33 as the palladium source no conversion to product was observed. This is in perfect accordance with the observations shown in Table 2, demonstrating that the presence of aqueous KOAc prevents formation of Pd3-complex 33 and that K2[Pd(CN)4], which is most likely produced from 33 under these conditions, is inactive as a catalyst precursor.‡‡
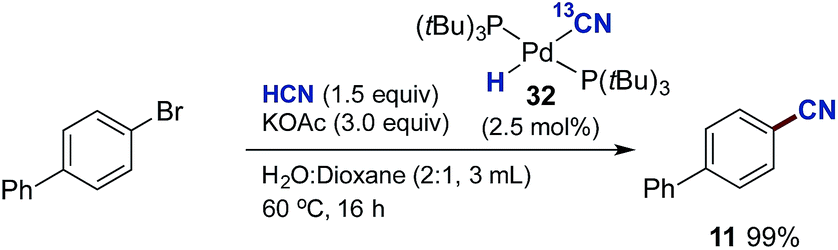 | ||
| Scheme 10 Pd-Catalysed cyanation of aryl bromides using 32 instead of P(tBu3)-Pd-G3. aChamber A: 4-bromobiphenyl (1.0 mmol), 33 (2.5 mol%) and KOAc (3.0 mmol) in dioxane (1.0 mL) and H2O (2.0 mL). Chamber B: KCN (1.5 mmol), ethylene glycol (1.0 mL) and AcOH (9.0 mmol). | ||
Finally, while the substrate scope of the Pd-catalysed cyanation of aryl bromides using HCN proved broad, some heteroaryl bromides provided low or even no conversion to product. One of these inactive bromides, 2-acetyl-5-bromothiophenyl bromide, was added to the cyanation reaction of 4-bromophenol, which under our standard condition with HCN and in the absence of the heteroaryl bromide provided the corresponding 4-cyanophenol in high yield as depicted in Scheme 3.45§§ However, under the exact same conditions but with the addition of this inactive heteroaryl bromide, the formation of benzonitrile 9 was completely inhibited (Scheme 11). Next, we attempted to isolate the oxidative addition complex of Pd(P(tBu)3)2 and 2-acetyl-5-bromothiophene in order to test the stoichiometric reaction with Pd-hydride 32. Despite extensive experimentation, i.e. different solvents and temperatures, we only observed the formation of Pd2(μ-Br)2(P(tBu)3)2 (38) and 5,5′-diacetyl-2,2′-bithiophene (39) in all cases (Scheme 12).
 | ||
| Scheme 11 Robustness screening on the formation of benzonitrile 9. aChamber A: 4-bromophenol (1.0 mmol), 2-acetyl-5-bromothiophene (1.0 mmol) P(tBu)3-Pd-G3 (2.5 mol%) and KOAc (3.0 mmol) in dioxane (1.0 mL) and H2O (2.0 mL). Chamber B: KCN (1.5 mmol), ethylene glycol (1.0 mL) and AcOH (9.0 mmol). | ||
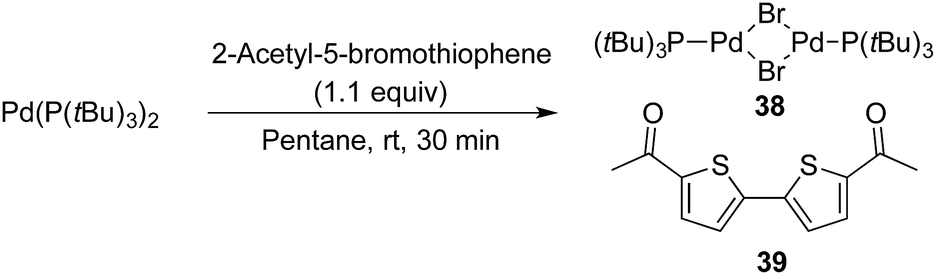 | ||
| Scheme 12 Formation of Pd2(μ-Br)2(P(tBu)3)2 and 5,5′-diacetyl-2,2′-bithiophene from Pd(P(tBu)3)2. aReaction performed on a 0.1 mmol scale. | ||
The Pd-complex 38 has previously been synthesised by Hartwig and co-workers by a similar protocol using Pd(dba)2, P(tBu)3 and a tenfold excess of 2-bromothiophene.46 The formation of 38 is fast at 25 °C when using either THF or pentane. Given that the developed conditions for the cyanation of aryl bromides operate at 60 °C, production of 38 is fast leading to apparent shutdown of the catalytic system. The formation of complex 38 offers an explanation to why some heteroaromatic bromides fail under the developed cyanation protocol, however further investigations are needed to understand the reasons for this divergence.47,48
Conclusions
In summary, a new protocol for the direct use of stoichiometric gaseous hydrogen cyanide in the Pd-catalysed cyanation of aryl bromides has been developed. Contrary to previous studies, the use of HCN did not lead to catalytic shutdown, but instead provided a robust and reproducible method. A broad range of aryl bromides and a few heteroaromatic bromides afforded the desired benzonitrile derivatives and given the simple setup, utilising ex situ generation of HCN, labelling with H13CN was also straightforward. The presence of water as co-solvent and the use of the mild base KOAc proved imperative for catalytic efficiency. In particular, the suitability of this weak base indicated that possibly the mechanism in operation deviates from previous catalytic cyanation studies as the concentration of free cyanide would be virtually non-existent. This led to the proposal of a mechanism based on a transmetallation between two Pd-complexes produced from the oxidative addition of Pd(0) into hydrogen cyanide and an aryl bromide. This proposal was based on mechanistic indications that co-aligns with observations reported by Grushin, Beller and others. Further work is now on-going to examine a similar protocol under nickel catalysis, as well as examining other electrophiles than aryl halides. The results of this work will be reported in due course.Conflicts of interest
Troels Skrydstrup and Anders T. Lindhardt are co-owners of SyTracks a/s, which commercialises the two-chamber technology.Acknowledgements
This study was funded by the BioValue SPIR, Strategic Platform for Innovation and Research on value added products from biomass, which is co-funded by The Innovation Fund Denmark, case no: 0603-00522B. We are also deeply appreciative of generous financial support from Haldor Topsøe A/S, the Danish National Research Foundation (grant no. DNRF118 and DNRF93) and Aarhus University.Notes and references
- (a) P. Pollak, G. Romeder, F. Hagedorn and H. P. Gelbke, Nitriles, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, June 15, 2000, http://onlinelibrary.wiley.com/doi/10.1002/14356007.a17_363/full Search PubMed; (b) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS PubMed; (c) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical substances: Syntheses, Patents, Applications of the most relevant APIs, Georg Thieme Verlag, Stuttgart, New York, 2001 Search PubMed; (d) L. J. Goujon, A. Khaldi, A. Maziz, C. Plesse, G. T. M. Nguyen, P.-H. Aubert, F. Vidal, C. Chevrot and D. Teyssié, Macromolecules, 2011, 44, 9683 CrossRef CAS; (e) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597 RSC; (f) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027 CrossRef CAS.
- Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publisher, New York, 1970 Search PubMed.
- (a) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 52, 1749 CrossRef; (b) J. Lindley, Tetrahedron, 1984, 40, 1433 CrossRef CAS; (c) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633 CrossRef; (d) H. H. Hodgson, Chem. Rev., 1947, 40, 251 CrossRef CAS PubMed; (e) J. K. Kochi, J. Am. Chem. Soc., 1957, 79, 2942 CrossRef CAS.
- For a recent review see: (a) Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and P. Wang, Tetrahedron Lett., 2014, 55, 1271 CrossRef CAS; for selected examples, see: (b) J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS PubMed; (c) H.-J. Cristau, A. Ouali, J.-F. Spindler and M. Taillefer, Chem.–Eur. J., 2005, 11, 2483 CrossRef CAS PubMed; (d) T. Schareina, A. Zapf, W. Mägerlein, N. Müller and M. Beller, Chem.–Eur. J., 2007, 13, 6249 CrossRef CAS PubMed; (e) T. Schareina, A. Zapf, A. Cotté, M. Gotta and M. Beller, Adv. Synth. Catal., 2011, 353, 777 CrossRef CAS.
- (a) L. Cassar, J. Organomet. Chem., 1973, 54, C57 CrossRef CAS; (b) L. Cassar, S. Ferrara and M. Foa, Adv. Chem. Ser., 1974, 132, 252 CrossRef CAS; (c) L. Cassar, M. Foa, F. Montanari and G. P. Marinelli, J. Organomet. Chem., 1979, 173, 335 CrossRef CAS; (d) Y. Sakakibara, F. Okuda, A. Shimobayashi, K. Kirino, M. Sakai, N. Uchino and K. Takagi, Bull. Chem. Soc. Jpn., 1988, 61, 1985 CrossRef CAS; (e) K. Takagi and Y. Sakakibara, Chem. Lett., 1989, 18, 1957 CrossRef; (f) K. Sasaki, M. Sakai, Y. Sakakibara and K. Takagi, Chem. Lett., 1991, 20, 2017 CrossRef; (g) Y. Sakakibara, Y. Ido, K. Sasaki, M. Saki and N. Uchino, Bull. Chem. Soc. Jpn., 1993, 66, 2776 CrossRef CAS; (h) Y. Sakakibara, K. Sasaki, F. Okuda, A. Hokimoto, T. Ueda, M. Sakai and K. Takagi, Bull. Chem. Soc. Jpn., 2004, 77, 1013 CrossRef CAS; (i) R. K. Arvela and N. E. Leadbeater, J. Org. Chem., 2003, 68, 9122 CrossRef CAS PubMed; (j) X. Zhang, A. Xia, H. Chen and Y. Liu, Org. Lett., 2017, 19, 2118 CrossRef CAS PubMed.
- (a) K. Takagi, T. Okamoto, Y. Sakakibara and S. Oka, Chem. Lett., 1973, 2, 471 CrossRef; (b) A. Sekiya and N. Ishikawa, Chem. Lett., 1975, 4, 277 CrossRef; (c) K. Takagi, T. Okamoto, Y. Sakakibara, A. Ohno, S. Oka and N. Hayama, Bull. Chem. Soc. Jpn., 1975, 48, 3298 CrossRef CAS; (d) K. Takagi, T. Okamoto, Y. Sakakibara, A. Ohno, S. Oka and N. Hayama, Bull. Chem. Soc. Jpn., 1976, 49, 3177 CrossRef CAS; (e) Y. Akita, M. Shimazaki and A. Ohta, Synthesis, 1981, 974 CrossRef CAS; (f) K. Takagi, K. Sasaki and Y. Sakakibara, Bull. Chem. Soc. Jpn., 1991, 64, 1118 CrossRef CAS; (g) B. A. Andersen, E. C. Bell, F. O. Ginah, N. K. Harn, L. M. Pagh and J. P. Wepsiec, J. Org. Chem., 1998, 63, 8224 CrossRef; (h) M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem.–Eur. J., 2003, 9, 1828 CrossRef CAS PubMed.
- For a recent review on Pd-catalysed cyanation of aryl halides covering the extensive work by Beller, see: P. Anbarasan, T. Scareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC ; see also references listed herein.
- A. V. Ushkov and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 10999 CrossRef CAS PubMed.
- (a) T. D. Senecal, W. Shu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 10053 CrossRef PubMed; (b) D. T. Cohen and S. L. Buchwald, Org. Lett., 2015, 17, 202 CrossRef CAS PubMed.
- T. Okano, M. Iwahara and J. Kiji, Synlett, 1998, 243 CrossRef CAS.
- M. Sundermeier, M. Zapf, M. Beller and J. Sans, Tetrahedron Lett., 2001, 42, 6707 CrossRef CAS.
- (a) N. Chatani and T. Hanafusa, J. Org. Chem., 1986, 51, 4714 CrossRef CAS; (b) M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 CrossRef CAS.
- M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 1661 CrossRef CAS PubMed.
- (a) T. Schareina, A. Zapf and M. Beller, Chem. Commun., 2004, 1388 RSC; (b) D. Zhang, H. Sun, L. Zhang, Y. Zhou, C. Li, H. Jiang, K. Chen and H. Liu, Chem. Commun., 2012, 48, 2909 RSC; (c) S. A. Weissman, D. Zewge and C. Chen, J. Org. Chem., 2005, 70, 1508 CrossRef CAS PubMed; (d) O. Grossman and D. Gelman, Org. Lett., 2006, 8, 1189 CrossRef CAS PubMed; (e) T. Schareina, A. Zapf, W. Mägerlein, N. Müller and M. Beller, Tetrahedron Lett., 2007, 48, 1087 CrossRef CAS; (f) T. Schareina, R. Jackstell, T. Schulz, A. Zapf, A. Cotté, M. Gotta and M. Beller, Adv. Synth. Catal., 2009, 351, 643 CrossRef CAS.
- (a) D. M. Tschaen, R. Desmond, A. O. King, M. C. Fortin, B. Pipik, S. King and T. R. Verhoeven, Synth. Commun., 1994, 24, 887 CrossRef CAS; (b) P. E. Maligres, M. S. Waters, F. Fleitz and D. Askin, Tetrahedron Lett., 1999, 40, 8193 CrossRef CAS; (c) M. Alterman and A. Hallberg, J. Org. Chem., 2000, 65, 7984 CrossRef CAS PubMed; (d) R. Chidambaram, Tetrahedron Lett., 2004, 45, 1441 CrossRef CAS; (e) R. S. Jensen, A. S. Gajare, K. Toyota, M. Yoshifuji and F. Ozawa, Tetrahedron Lett., 2005, 46, 8645 CrossRef CAS; (f) M. T. Martin, B. Liu, B. E. Cooley Jr and J. F. Eaddy, Tetrahedron Lett., 2007, 48, 2555 CrossRef CAS.
- T. Sakamoto and K. Ohsawa, J. Chem. Soc., Perkin Trans. 1, 1999, 2323 RSC.
- D. Andrussow, Angew. Chem., 1935, 48, 593 CrossRef.
- H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Industrial Organic Chemicals, John Wiley And Sons Ltd, 2013 Search PubMed.
- Areas of large scale-application for HCN includes the DuPont Adiponitrile process (monomer in the synthesis of Nylon 6,6), which annually produces around 1 million tons of adiponitrile, see: L. Bini, C. Müller and D. Vogt, ChemCatChem, 2010, 2, 590 CrossRef CAS.
- For excellent examples on the development and application of transfer hydrocyanations, see: (a) X. Fang, P. Yu and B. Morandi, Science, 2016, 351, 832 CrossRef CAS PubMed; (b) X. Fang, P. Yu, G. Prina Cerai and B. Morandi, Chem.–Eur. J., 2016, 22, 15629 CrossRef CAS PubMed; (c) B. N. Bhawal and B. Morandi, ACS Catal., 2016, 6, 7528 CrossRef CAS.
- H. G. Lee, J. P. Milner, M. S. Placzek, S. L. Buchwald and J. M. Hooker, J. Am. Chem. Soc., 2015, 137, 648 CrossRef CAS PubMed.
- W. Zhao, H. G. Lee, S. L. Buchwald and J. M. Hooker, J. Am. Chem. Soc., 2017, 139, 7152 CrossRef CAS PubMed.
- G. Romeder, Hydrogen Cyanide, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001 Search PubMed.
- For a recent review on deactivation of homogeneous transition metal catalysis see: R. H. Crabtree, Chem. Rev., 2015, 115, 127 CrossRef CAS PubMed.
- K. D. Dobbs, W. J. Marshall and V. V. Grushin, J. Am. Chem. Soc., 2007, 129, 30 CrossRef CAS PubMed.
- S. Erhardt, V. V. Grushin, A. H. Kilpatrick, S. A. Macgregor, W. J. Marshall and C. R. Roe, J. Am. Chem. Soc., 2008, 130, 4828 CrossRef CAS PubMed.
- (a) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061 CrossRef CAS PubMed; (b) S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594 CrossRef CAS PubMed.
- (a) A. Modvig, T. L. Andersen, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Org. Chem., 2014, 79, 5861 CrossRef CAS PubMed; (b) K. T. Neumann, S. Klimczyk, M. N. Burhardt, B. Bang-Andersen, T. Skrydstrup and A. T. Lindhardt, ACS Catal., 2016, 6, 4710 CrossRef CAS.
- G. K. Min, K. Bjerglund, S. Kramer, T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, Chem.–Eur. J., 2013, 19, 17603 CrossRef CAS PubMed.
- Hydrocyanations using HCN: (a) A. L. Casalnuovo, T. V. RajanBabu, T. A. Ayers and T. H. Warren, J. Am. Chem. Soc., 1994, 116, 9869 CrossRef CAS; (b) L. Bini, C. Müller, J. Wilting, L. von Chrzanowski, A. L. Spek and D. Vogt, J. Am. Chem. Soc., 2007, 129, 12622 CrossRef CAS PubMed; (c) J. Wilting, M. Janssen, C. Müller, M. Lutz, A. L. Spek and D. Vogt, Adv. Synth. Catal., 2007, 349, 350 CrossRef CAS; (d) L. Bini, E. A. Pidko, C. Müller, R. A. van Santen and D. Vogt, Chem.–Eur. J., 2009, 15, 8768 CrossRef CAS PubMed; (e) A. Falk, A. Cavalieri, G. S. Nichol, D. Vogt and H.-G. Schmalz, Adv. Synth. Catal., 2015, 357, 3317 CrossRef CAS.
- Hydrocyanations using HCN-surrogates: (a) M. de Greef and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 551 CrossRef CAS PubMed; (b) A. Falk, A.-L. Göderz and H.-G. Schmalz, Angew. Chem., Int. Ed., 2013, 52, 1576 CrossRef CAS PubMed.
- For recent reviews on Ni-catalysed hydrocyanations: (a) L. Bini, C. Müller and D. Vogt, Chem. Commun., 2010, 46, 8325 RSC; (b) T. V. RajanBabu, Org. React., 2011, 75, 1 Search PubMed.
- F. F. Flemming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shock, J. Med. Chem., 2010, 53, 7902 CrossRef PubMed.
- F. G. Buono, R. Chidambaram, R. H. Mueller and R. E. Waltermire, Org. Lett., 2008, 10, 5325 CrossRef CAS PubMed.
- For a review on palladium hydrides, see: V. V. Grushin, Chem. Rev., 1996, 96, 2011 CrossRef CAS PubMed.
- (a) R. Vilar, D. M. P. Mingos and C. J. Cardin, J. Chem. Soc., Dalton Trans., 1996, 4313 RSC; (b) V. Durà-Vilà, D. M. P. Mingos, R. Vilar, A. J. P. White and D. J. Williams, J. Organomet. Chem., 2000, 600, 198 CrossRef.
- (a) A. B. Goel and S. Goel, Inorg. Chim. Acta, 1980, 45, L85 CrossRef; (b) R. G. Goel and W. O. Ogini, Organometallics, 1982, 1, 654 CrossRef CAS; (c) I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 13178 CrossRef CAS PubMed.
- E. H. Brooks and F. Glockling, J. Chem. Soc. A, 1967, 1030 RSC.
- A. G. Sergeev, A. Spanneberg and M. Beller, J. Am. Chem. Soc., 2008, 130, 15549 CrossRef CAS PubMed.
- It has been earlier shown that the oxidative addition to AcOH is a reversible process: C. Amatore, A. Jutland, G. Meyer, I. Carelli and I. Chiarotto, Eur. J. Inorg. Chem., 2000, 1855 CrossRef CAS.
- The oxidative addition of Pd(P(tBu)3) into aryl bromides has previously been shown to be a reversible process: (a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 1232 CrossRef CAS PubMed; (b) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 13944 CrossRef CAS PubMed; (c) A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 1533 CrossRef CAS.
- A. D. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- (a) F. Ozawa, M. Fujimori, T. Yamamoto and A. Yamamoto, Organometallics, 1986, 5, 2144 CrossRef CAS; (b) A. L. Casado, J. A. Casares and P. Espinet, Organometallics, 1997, 16, 5730 CrossRef CAS; (c) Y. Tan, F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 3683 CrossRef CAS PubMed; (d) D. Wang, Y. Izawa and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 9914 CrossRef CAS PubMed; (e) D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 5704 CrossRef CAS PubMed.
- J. P. Stambuli, C. D. Incarvito, M. Bühl and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 1184 CrossRef CAS PubMed.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed.
- M. W. Hooper, M. Utsunomiya and J. F. Hartwig, J. Org. Chem., 2003, 68, 2861 CrossRef CAS PubMed.
- The formation and role of 38 has recently been elucidated by Schoenebeck and Colacot. See: C. C. C. Johansson Seechurn, T. Sperger, T. G. Scrase, F. Schoenebeck and T. J. Colacot, J. Am. Chem. Soc., 2017, 139, 5194 CrossRef CAS PubMed.
- For other examples from the Schoenebeck group exploiting 38 and its iodo-analogue as a Pd(0) precatalyst, see: (a) F. Proutiere, M. Aufiero and F. Schoenebeck, J. Am. Chem. Soc., 2012, 134, 606 CrossRef CAS PubMed; (b) K. J. Bonney, F. Proutiere and F. Schoenebeck, Chem. Sci., 2013, 4, 4434 RSC; (c) G. Yin, I. Kalvet and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 6809 CrossRef CAS PubMed; (d) M. Aufiero, T. Sperger, A. S.-K. Tsang and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 10322 CrossRef CAS PubMed; (e) T. Sperger, C. K. Stirner and F. Schoenebeck, Synthesis, 2017, 49, 115 CAS; (f) I. Kalvet, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 1581 CrossRef CAS PubMed; (g) I. Kalvet, T. Sperger, T. Scattolin, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 7078 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1504904–1504908. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03912c |
| ‡ According to the Sigma-Aldrich catalogue, 5 g of acetone cyanohydrin costs the same as approximately 100 g of potassium cyanide. |
| § H13CN was used because this isotope eased the spectroscopic analysis of the complexes formed. |
| ¶ In the non-isotopically labelled version, the multiplicity is seen as a triplet. |
| || Complex 33 or the corresponding potassium salt (K2[Pd(13CN)4]) could not be detected by either 31P NMR or 13C NMR spectroscopic analysis. |
| ** Compound 34 has previously been synthesised by Grushin and co-workers by the reaction of Pd(PPh3)4 with K13CN in the presence of water. See ref. 26. |
| †† During the reaction, no clear phosphine signals are observed from either the oxidative addition complex (i.e. (P(tBu)3)Pd(Ar)(Br)) or the complex after transmetallation (i.e. (P(tBu)3)Pd(Ar)(CN)). This further indicates that the rate determining step is the oxidative addition into the aryl bromide, since the generated P(tBu)3)Pd(Ar)(Br) is consumed almost instantaneously (see J. L. Klinkenberg and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5748, which studies the reductive elimination of LnPd(Ar)(CN) complexes. |
| ‡‡ K2[Pd(CN)4] was also tested. This resulted in no conversion to benzonitrile 11. |
| §§ Lowering the amount of 2-acetyl-5-bromothiophenyl bromide to 0.5 equivalents or using other heteroaromatic bromides, such as 5-bromopyridine, gave the same outcome. |
| This journal is © The Royal Society of Chemistry 2017 |