
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-linking Zr-based metal–organic polyhedra via postsynthetic polymerization†
Dongsik
Nam
a,
Jihyun
Huh
a,
Jiyoung
Lee
a,
Ja Hun
Kwak
b,
Hu Young
Jeong
c,
Kyungmin
Choi
d and
Wonyoung
Choe
 *a
*a
aDepartment of Chemistry, Ulsan National Institute of Science and Technology, 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: choe@unist.ac.kr
bDepartment of Chemical Engineering, Ulsan National Institute of Science and Technology, 50 UNIST-gil, Ulsan 44919, Republic of Korea
cUNIST Central Research Facilities, Ulsan National Institute of Science and Technology, 50 UNIST-gil, Ulsan 44919, Republic of Korea
dDepartment of Chemical and Biological Engineering, Sookmyung Women’s University, 100 Cheongpa-ro 47-gil, Seoul 04310, Korea
First published on 25th September 2017
Abstract
Metal organic polyhedra (MOPs) have potential as supramolecular building blocks, but utilizing MOPs for postsynthetic polymerization has not been explored. Although MOPs with flexible organic moieties have been recently reported to target enhanced processability, permanent porosity has not been demonstrated. Here, a novel synthetic strategy involving the cross-linking of MOPs via a covalent bond is demonstrated by exploiting a condensation reaction between the MOP and flexible organic linkers. An amine-functionalized Zr-based MOP is cross-linked with acyl chloride linkers in the crystalline state to form cross-linked MOPs. The condensation reaction results in a cross-linked system without significant changes to the structure of the Zr-based MOP. Such cross-linked MOPs provide a microporous tetrahedral cage based on gas sorption analysis. This cross-linking strategy highlights the potential of MOPs as building blocks and provides access to a new class of porous material.
Introduction
The phenomenal success of metal–organic frameworks (MOFs) achieved over the past two decades stems from several factors, one of which is certainly the rich choice of building blocks, namely metal nodes and organic linkers, leading to the generation of over 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOFs with a vast array of advanced functions.1–3 The underlying synthetic protocol for these MOFs, however, is rather simple, involving self-assembly of the metal nodes and organic building blocks.4–6 Instead of this simple protocol, sequential self-assembly utilizes nano-sized building blocks such as metal–organic polyhedra (MOPs) to build higher-dimensional metal–organic materials.7–9 Some MOPs are intrinsically porous and are therefore attractive building blocks for the cage-based construction of porous materials.10–13 The distinct advantage of such a modular, multi-step synthesis protocol is a delicate control of the topology and functions by utilizing pre-designed building blocks with high connectivity and symmetry.13,14
000 MOFs with a vast array of advanced functions.1–3 The underlying synthetic protocol for these MOFs, however, is rather simple, involving self-assembly of the metal nodes and organic building blocks.4–6 Instead of this simple protocol, sequential self-assembly utilizes nano-sized building blocks such as metal–organic polyhedra (MOPs) to build higher-dimensional metal–organic materials.7–9 Some MOPs are intrinsically porous and are therefore attractive building blocks for the cage-based construction of porous materials.10–13 The distinct advantage of such a modular, multi-step synthesis protocol is a delicate control of the topology and functions by utilizing pre-designed building blocks with high connectivity and symmetry.13,14
Such elaborate conceptual design attracted synthetic chemists to utilize MOPs and MOFs as building blocks for porous materials.8–11 A few examples include the formation of 3D MOFs, by connecting MOPs with pyridyl pillars to build 3D MOFs, as reported by Zhou, Su, Wang, and others.15–22 In these cases, rigid organic linkers connect discrete MOP cages via coordination-driven self-assembly. However, connecting MOPs with flexible organic linkers is highly desirable because both moieties can bring the features of polymers and MOFs, thus combining the advantages of two different classes of material to enhance chemical stability and processability.23,24
A similar synthetic approach, cross-linking with flexible organic moieties in MOFs, has been well-established, as pioneered by Lee25 in 2000, and recently demonstrated by Sada26,27 and Wang.28 MOF–polymer hybrid materials often lack molecular-level homogeneity due to the size of MOF particles being a few hundred nanometers (Fig. 1a).28 On the other hand, MOP counterparts, built from metal–organic nanosized cages (1–3 nm) with intrinsic pores, can be potentially useful in energy applications such as gas separation.29 Cohen,30 Nitschke,31 and Johnson32,33 have used polymeric ligands together with cage moieties for the synthesis of 3D polymeric frameworks (Fig. 1b). Recently, Kitagawa and Furukawa developed MOPs attached with flexible molecules.34,35 None of these MOP–polymer hybrid materials, however, have demonstrated intrinsic porosity originating from the MOP cages, possibly due to the fragile nature of the MOPs.36,37 Furthermore, cross-linking MOPs with covalent bonds, i.e. postsynthetic polymerization28,38 of MOPs, has not been demonstrated to our knowledge.
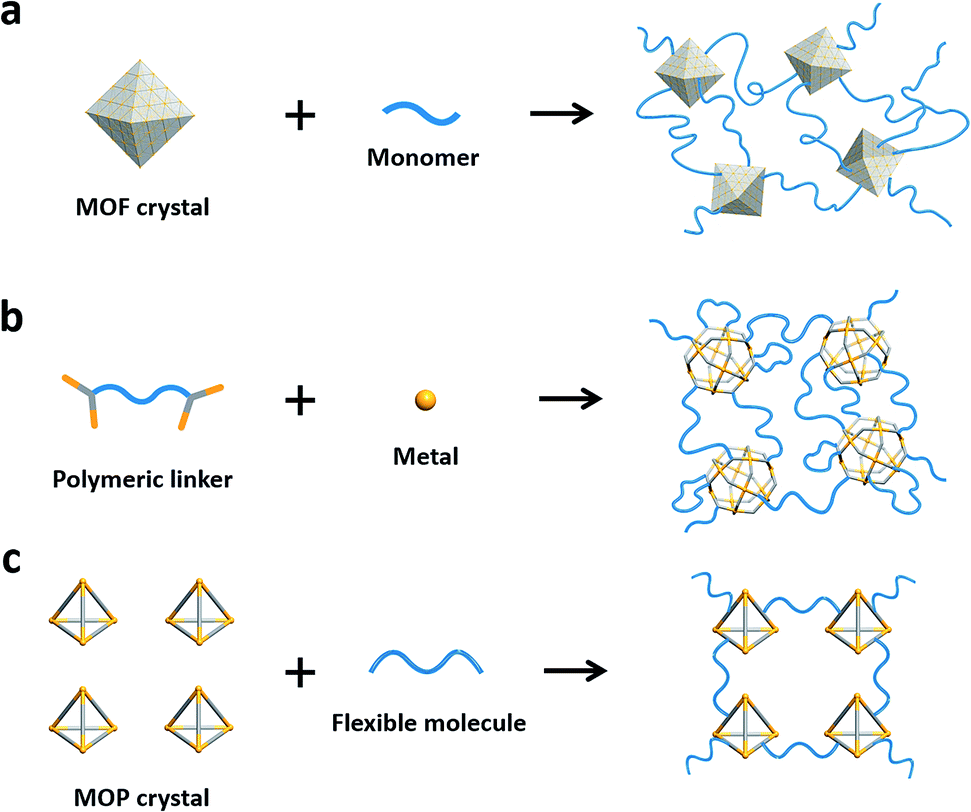 | ||
| Fig. 1 (a) Postsynthetic polymerization of MOF nanoparticles. (b) Synthesis of MOP–polymer hybrid materials using polymeric linkers. (c) Cross-linking of MOPs with flexible organic molecules. | ||
Herein, we demonstrate cross-linking MOPs via a condensation reaction between MOP cages and flexible organic molecules (Fig. 1c and 2). First, we synthesized a robust amine-functionalized Zr-based MOP (UMOP-1-NH2, UMOP for UNIST metal–organic polyhedra), [Cp3Zr3O(OH)3]4[BDC-NH2]6[(C2H5)2NH2]2Cl6, with a tetrahedral cage and connectivity similar to that found in the iconic MOF, UiO-66.39 Utilizing this robust MOP as a starting material, we carefully selected a simple and effective condensation reaction between the amine group of the Zr-MOP cage and the acyl chloride group of the linker to avoid the use of catalysts or high temperature. A series of highly crystalline and porous cross-linked MOPs (hereafter CLMOPs) was thus synthesized. These results demonstrate that MOPs can be used in the organic cross-linking reaction, providing easy access to a new class of crystalline porous material for advanced applications.
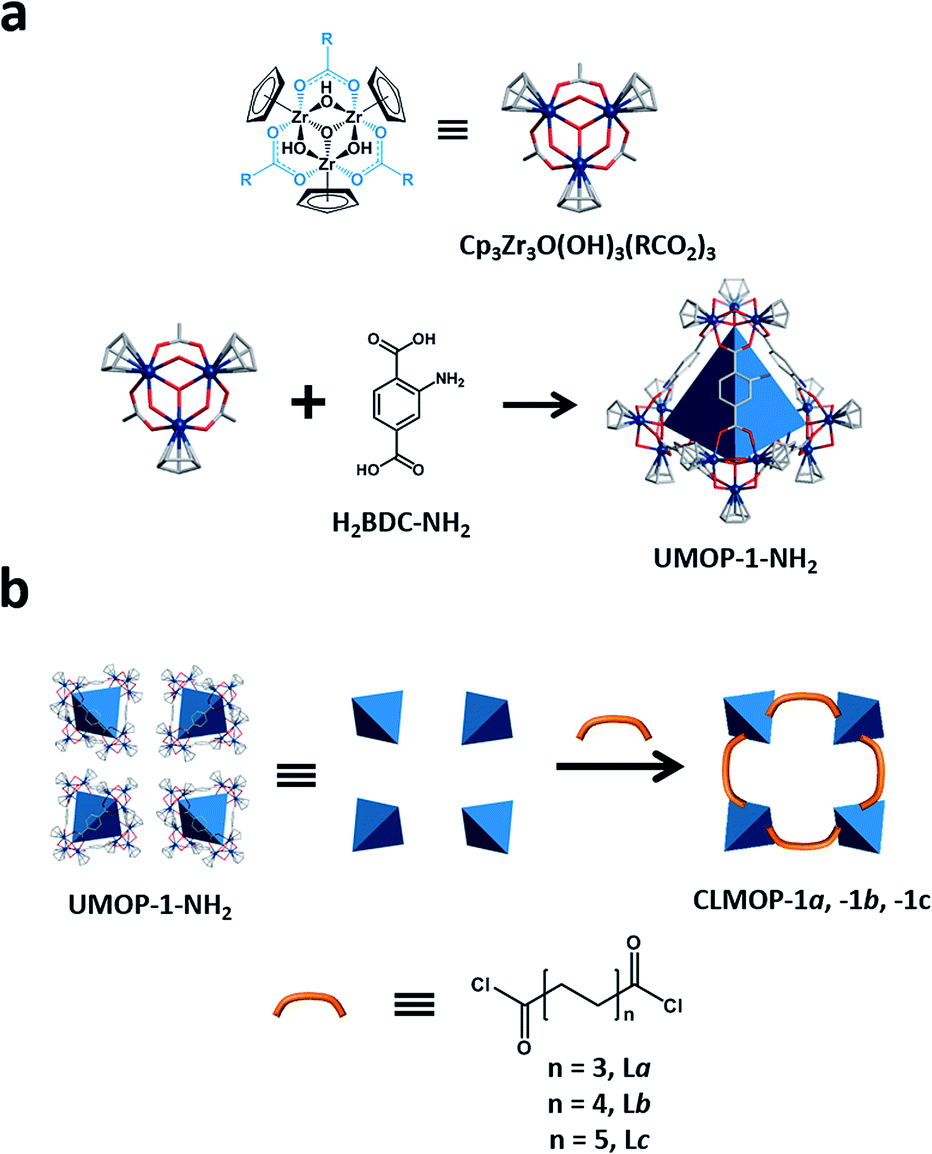 | ||
| Fig. 2 Scheme for the synthesis of CLMOPs. (a) Zr clusters [Cp3Zr3O(OH)3(RCO2)3] and organic linkers (H2BDC-NH2) form UMOP-1-NH2 with a tetrahedral cage (blue). (b) UMOP-1-NH2 reacts with acyl chloride linkers (La, Lb, and Lc) to form CLMOPs. Cross-linking is achieved by condensation between the amine groups and acyl chloride linkers. | ||
Results and discussion
A Zr-based MOP (UMOP-1-NH2) was synthesized from Cp2ZrCl2 (Cp = η5-C5H5) and 2-aminoterephthalic acid (H2BDC-NH2). Single crystal X-ray diffraction (SCXRD) showed that UMOP-1-NH2 crystallized in the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group with a cell parameter of 36.75 Å (Table S1†). The structure consists of tetrahedral cages with trinuclear Zr clusters ([Cp3Zr3O(OH)3(RCO2)3]+) at the vertexes. The Zr clusters are formed by hydrolysis of Cp2ZrCl2.40,41 The BDC-NH2 ligands are coordinated to the Zr clusters, forming the edge of the cage. Surprisingly, the crystal structure of UMOP-1-NH2 is quite similar to that of UiO-66 (Fig. 3). Both systems have structurally analogous tetrahedral cages. The cages in UMOP-1-NH2 are packed with rhombicuboctahedral and cubic voids (Fig. S1†), interacting via hydrogen bonding with Cl− ions. In the cubic voids, eight [Cp3Zr3O(OH)3(RCO2)3]+ clusters are interconnected via twelve Cl− ions forming hydrogen bonds with μ2-OH in the Zr clusters (Fig. S2 and S3†). A neutral charge is satisfied by four (C2H5)2NH2+ cations, produced from decomposition of N,N-diethylformamide (DEF), as evidenced by the 1H-NMR spectra of UMOP-1-NH2 (Fig. S4†). Although a similar tetrahedral MOP (ZrT-1) was reported by Yuan and co-workers, the packing pattern of the tetrahedral MOP cage is quite different.40
m space group with a cell parameter of 36.75 Å (Table S1†). The structure consists of tetrahedral cages with trinuclear Zr clusters ([Cp3Zr3O(OH)3(RCO2)3]+) at the vertexes. The Zr clusters are formed by hydrolysis of Cp2ZrCl2.40,41 The BDC-NH2 ligands are coordinated to the Zr clusters, forming the edge of the cage. Surprisingly, the crystal structure of UMOP-1-NH2 is quite similar to that of UiO-66 (Fig. 3). Both systems have structurally analogous tetrahedral cages. The cages in UMOP-1-NH2 are packed with rhombicuboctahedral and cubic voids (Fig. S1†), interacting via hydrogen bonding with Cl− ions. In the cubic voids, eight [Cp3Zr3O(OH)3(RCO2)3]+ clusters are interconnected via twelve Cl− ions forming hydrogen bonds with μ2-OH in the Zr clusters (Fig. S2 and S3†). A neutral charge is satisfied by four (C2H5)2NH2+ cations, produced from decomposition of N,N-diethylformamide (DEF), as evidenced by the 1H-NMR spectra of UMOP-1-NH2 (Fig. S4†). Although a similar tetrahedral MOP (ZrT-1) was reported by Yuan and co-workers, the packing pattern of the tetrahedral MOP cage is quite different.40
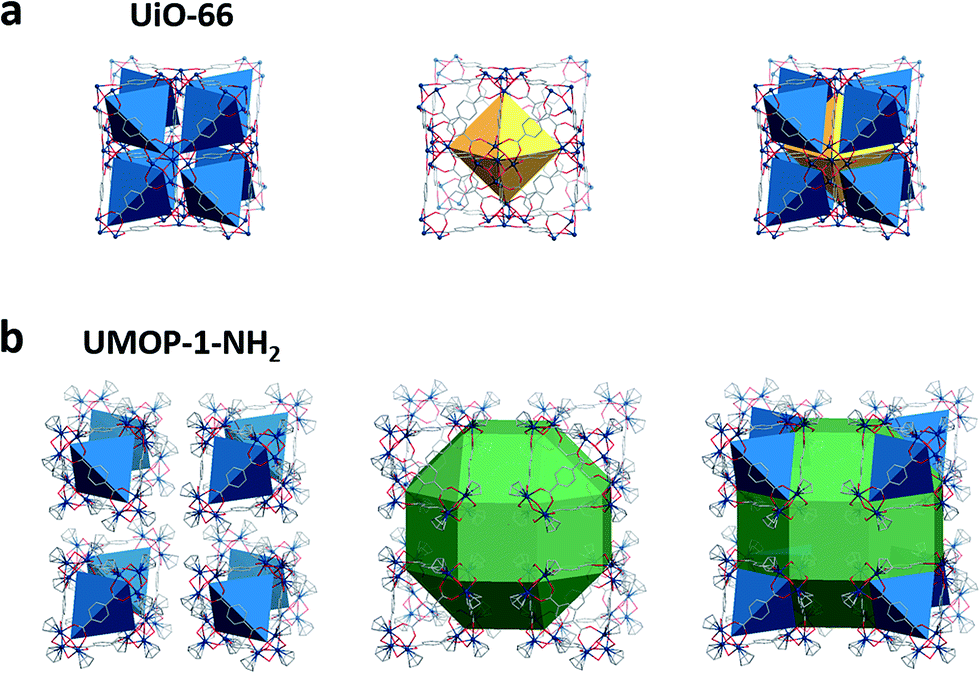 | ||
| Fig. 3 Packing patterns of UiO-66 and UMOP-1-NH2. The structure of (a) UiO-66 and (b) UMOP-1-NH2 with structurally similar tetrahedral cages (blue). In UiO-66, the cages are adjacent to each other with a central octahedron (yellow). In UMOP-1-NH2, the cages are separated around a rhombicuboctahedron (green). Chloride ions are omitted in UMOP-1-NH2. | ||
The permanent porosity of UMOP-1-NH2 was confirmed by N2 sorption analysis at 77 K (Fig. 4a) with notable BET and Langmuir surface areas of 782 m2 g−1 and 900 m2 g−1, respectively. Although UMOP-1-NH2 lost its crystallinity in activation (Fig. S5†), the pore size distribution (PSD) data (Fig. 4b and S6†) obtained from the N2 adsorption isotherm clearly indicate a pore width of 6 Å, corresponding to the pore of the tetrahedral cage. The pore sizes of the cube and rhombicuboctahedron (10 Å and 18 Å, respectively) were also observed in the PSD data, supporting that the pore structure is partly maintained despite amorphization in the activation. The structural similarity between UMOP-1-NH2 and UiO-66-NH2 is further supported by the PSD data, representing a tetrahedral pore of 6 Å in both structures (Fig. 4b). The CO2 isotherms of UMOP-1-NH2 acquired at 273 K and 293 K indicated a moderate CO2 uptake of 2.3 mmol g−1 and 1.4 mmol g−1 at 1 bar, respectively (Fig. S7†). The isosteric heat of adsorption (Qst) calculated by fitting the CO2 isotherms to the dual-site Langmuir–Freundlich equation indicated an initial Qst value of 26.5 kJ mol−1 (Fig. S8 and S9†), comparable to the values for UiO-66 (26.5 kJ mol−1)42 and UiO-66-NH2 (28.6 kJ mol−1).43 Interestingly, the powder X-ray diffraction (PXRD) pattern of activated UMOP-1-NH2 was restored to the original when the sample was soaked in N,N-diethylformamide (DEF) (Fig. S12†), suggesting that the arrangement of tetrahedral cages was not totally disordered. We further studied the stability of the discrete UMOP-1-NH2 cage by dissolving it in water. An electrospray ionization (ESI) mass spectrum of the UMOP-1-NH2 solution in water was obtained after 1 week and revealed a mass to charge ratio (m/z) consistent with a fully intact cage, [U]4+ (U: a cage without Cl− ions), with additional peaks for [U–H]3+ and [U–2H]2+ (Fig. 4c). The observed m/z values of the intact cage represent the remarkable stability of the UMOP-1-NH2 cage in water, while typical MOPs are not stable in water.44 The stability in MeOH was also identified with an ESI mass spectrum of a UMOP-1-NH2 solution in MeOH (Fig. S13†). The discrete UMOP-1-NH2 cage with a size of 1.5 nm was confirmed with a high-angle annular dark-field (HAADF) scanning transmission electron microscopy image (Fig. 4d). Elemental mapping images showing the distribution of Zr supported that the observed particle was UMOP-1-NH2 (Fig. S14†).
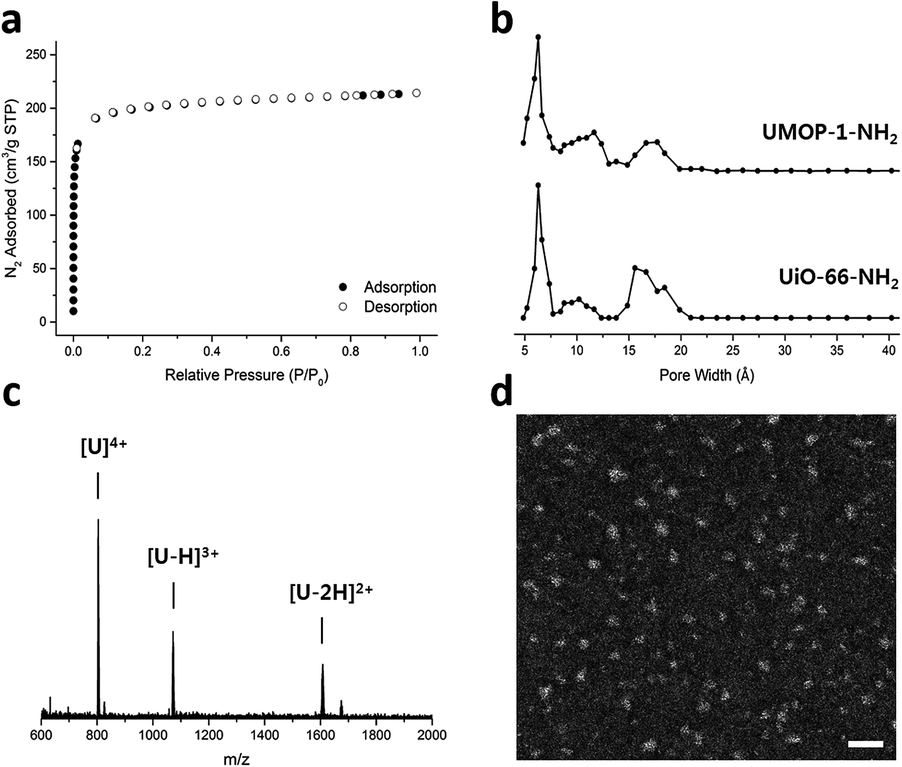 | ||
| Fig. 4 (a) N2 isotherm of UMOP-1-NH2 at 77 K. (b) Pore size distribution data of UMOP-1-NH2 and UiO-66-NH2. A tetrahedral pore with a size of 6 Å was commonly observed. (c) Electrospray ionization mass spectrum of the UMOP-1-NH2 solution in water. Mass to charge ratio values of [U]4+, [U–H]3+, and [U–3H]2+ were observed at 804.4, 1072.2, and 1608.2, respectively (U: one UMOP-1-NH2 cage without Cl− ions). The theoretical values are 804.7, 1072.6, and 1608.3, respectively. (d) HAADF image of the UMOP-1-NH2 solution in methanol obtained with a scanning transmission electron microscope. The scale bar indicates 5 nm. | ||
The CLMOPs were designed to exploit the condensation reaction between UMOP-1-NH2 and the acyl chloride linkers. The target reaction is the condensation between the amine and acyl chloride groups, resulting in the formation of a secondary amide and HCl. Three acyl chloride linkers with different lengths were used for cross-linking, i.e., suberoyl chloride (La) for CLMOP-1a, sebacoyl chloride (Lb) for CLMOP-1b, and dodecanedioyl dichloride (Lc) for CLMOP-1c, as shown in Fig. 2b.
To directly confirm successful condensation between the amine and acyl chloride groups forming the secondary amide, FT-IR spectra of UMOP-1-NH2 and the CLMOPs were obtained. The blue region in Fig. 5a shows ν(C–H) bands, derived from the alkyl chain in the CLMOPs. The existence of the secondary amide can be confirmed by the appearance of the ν(C–N) + δ(CNH) and δ(NH) + δ(OCN) bands, distinguished from the peaks of the aromatic amine.45 In the spectra of the CLMOPs, the peaks in the orange region in Fig. 5a (observed with shoulders) correspond to ν(C–N) + δ(CNH). In the green region in Fig. 5a, peaks for δ(NH) + δ(OCN) were observed around 1305 cm−1 for the CLMOPs. It is notable that a peak around 1340 cm−1 for the primary amine in UMOP-1-NH2 was not observed in the spectra of the CLMOPs. This result indicates that the secondary amide is a major group in the CLMOPs. The peak changes observed in the transformation of UMOP-1-NH2 to the CLMOPs were similar to those observed in postsynthetic modification of UiO-66-NH2.45,46
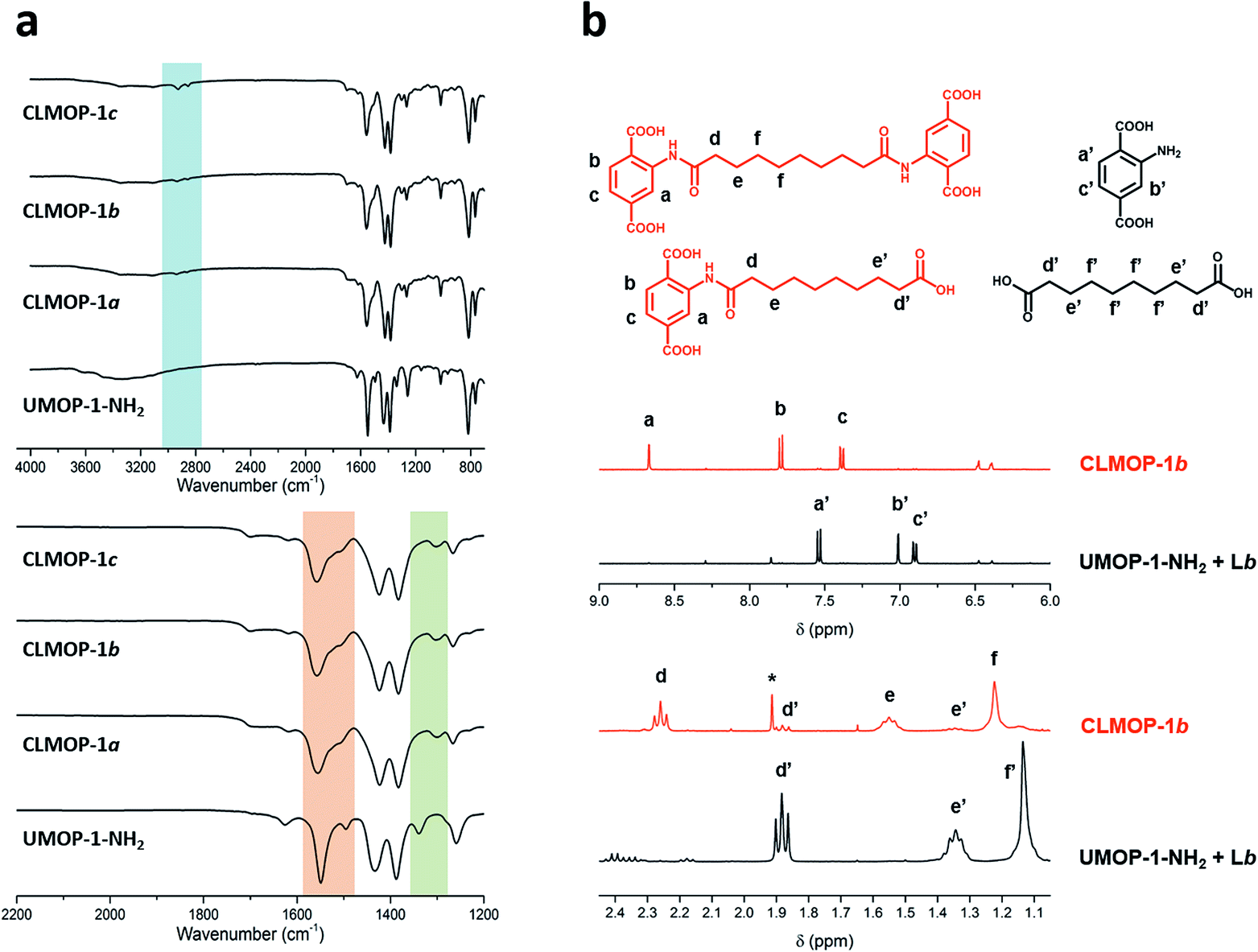 | ||
| Fig. 5 Evidence of cross-linking. (a) FTIR spectra of UMOP-1-NH2 and the CLMOPs. The region for ν(C–H) bands (blue). The regions for secondary amide bands (orange and green). The CLMOPs showed secondary amide bands, distinct from the amine bands observed in UMOP-1-NH2. (b) 1H-NMR spectra of digested CLMOP-1b (orange) and a mixture of digested UMOP-1-NH2 and Lb (black). The amide dimer and monomer produced from the digestion of CLMOP-1b (top left, orange). H2BDC-NH2 produced from the digestion of UMOP-1-NH2, and Lb (top right, black) (*: residual dimethylacetamide). | ||
As another convincing point of evidence supporting successful condensation, the amide dimer was directly detected through 1H-NMR measurement after digestion of the CLMOPs (Fig. 5b and S15–S17†). CLMOP-1b was analyzed as a representative of the CLMOPs (orange graph; Fig. 5b). A mixture of UMOP-1-NH2 and Lb (black graph) was also analyzed to prove that condensation between UMOP-1-NH2 and Lb did not occur under the digestion conditions. Peaks of the aromatic protons of the amide dimer were detected around 8.7 ppm, 7.8 ppm, and 7.4 ppm (a, b and c in Fig. 5b, respectively), whereas peaks of the protons of H2BDC-NH2 were observed around 7.5 ppm, 7 ppm, and 6.9 ppm (a′, b′ and c′ in Fig. 5b). This proves that almost all the amines reacted with the acyl chlorides during cross-linking, while UMOP-1-NH2 did not undergo condensation with Lb under the digestion conditions. Peaks of the alkyl chain in the amide dimer were observed at three different chemical shifts in the region of 1.1–2.4 ppm (d, e and f in Fig. 4b). We also detected the amide monomer, indicating that cross-linking was not perfectly achieved. The peaks for the protons labeled d′ and e′ in the amide monomer (in Fig. 5b) were observed upfield, as distinguished from the d and e peaks, due to their proximity to the carboxyl group produced by the reaction of acyl chloride with water. The proportion of interconnected ligands was calculated by comparing the integrated value of the b, d, and d′ peaks. 85% of the total amount of BDC-NH2 was interconnected by Lb. 13% of the total amount reacted with Lb, but terminated with a carboxyl group. The remaining 2% was from H2BDC-NH2, produced by hydrolysis of the amide group during digestion. The data show that a meaningful portion of the cages were cross-linked in the CLMOPs. We also confirmed that 88% of the total amount of the ligand was interconnected in CLMOP-1c (Table S3†), but CLMOP-1a was unable to be analyzed due to peak overlap.
After cross-linking, a drastic solubility change was observed (Fig. S18†). UMOP-1-NH2 dissolved rapidly in MeOH, but the CLMOPs did not dissolve in MeOH and maintained a crystal shape even after being shaken for 2 days in MeOH. Considering that the UMOP-1-NH2 cages remained fully intact in MeOH, the observed solubility change suggests successful cross-linking of the cages, thereby preventing the crystals from dissolving in MeOH.
The CLMOPs were further characterized via PXRD, thermogravimetric analysis (TGA), and gas sorption analysis. The PXRD patterns of the as-synthesized CLMOPs were identical to those of UMOP-1-NH2 (Fig. 6a). The data show the robustness of the tetrahedral cages and their packing even under HCl produced from the condensation. In the TGA analysis, the CLMOPs indicated a larger weight loss at high temperature than UMOP-1-NH2 (Fig. S19†). The greater weight loss in the CLMOPs corresponds to decomposition of a larger amount of the organic fragment, caused by the addition of the alkyl chains. The permanent porosity of the CLMOPs was analyzed by acquisition of N2 isotherms (Fig. 6b). CLMOP-1a, CLMOP-1b, and CLMOP-1c had respective total N2 uptakes of 140 cm3 g−1, 130 cm3 g−1, and 75 cm3 g−1. The BET surface areas were 502 m2 g−1, 469 m2 g−1, and 277 m2 g−1, respectively. The decreased N2 uptake and BET surface area of the CLMOPs, compared to UMOP-1-NH2, can be ascribed to the reduced space in the pores due to incorporation of the alkyl chains. The pore size distribution of the CLMOPs indicated a pore width of 6 Å, consistent with the pore width of the tetrahedral cage (Fig. S20†). The excellent agreement between the pore size of UMOP-1-NH2 and the CLMOPs suggests that the tetrahedral cages remain fully intact even after condensation. CO2 sorption studies of CLMOP-1a, -1b, and -1c showed respective CO2 uptakes of 1.9 mmol g−1, 1.8 mmol g−1, and 1.6 mmol g−1 at 1 bar at 273 K (Fig. S21†). The initial Qst values for the CLMOPs were compared with the values for UMOP-1-NH2 (Fig. 6c). The values for the CLMOPs were significantly increased (32.2 kJ mol−1 for CLMOP-1a, 30.7 kJ mol−1 for CLMOP-1b, and 36.2 kJ mol−1 for CLMOP-1c). These values were even higher than the values for UiO-66 (26.5 kJ mol−1)42 and UiO-66-NH2 (28.6 kJ mol−1).43 The increased enthalpy is ascribed to the van der Waals interaction between the alkyl groups and CO2 molecules.47 As observed in UMOP-1-NH2, the PXRD pattern of activated CLMOP-1c revealed an amorphous structure, but the crystallinity was recovered when the sample was soaked in DEF (Fig. S25†), implying that the cage structure was stable during activation.
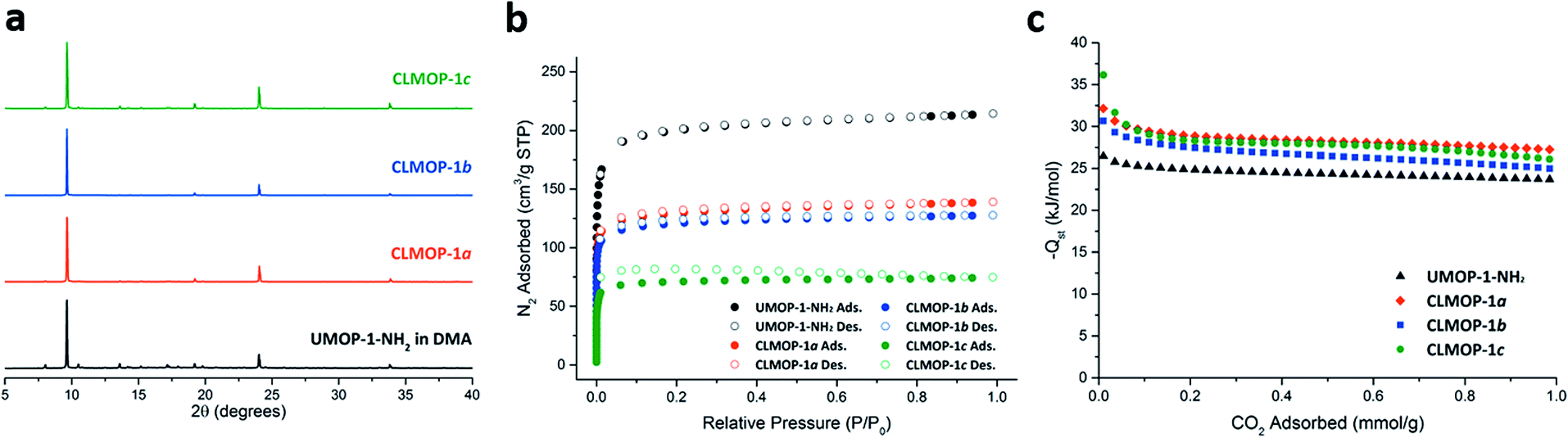 | ||
| Fig. 6 (a) PXRD patterns of UMOP-1-NH2 soaked in DMA for 3 days and the CLMOPs. (b) N2 isotherms (77 K) of UMOP-1-NH2 and the CLMOPs. (c) Isosteric heats of adsorption (Qst) calculated from the CO2 isotherms of UMOP-1-NH2 and the CLMOPs. | ||
Structural models of the CLMOPs are suggested by considering the connection by the acyl chloride linkers between the tetrahedral cages, based on the structure of UMOP-1-NH2. Three possible connections are shown in Fig. 7a with distances of 8.1 Å, 14.2 Å, and 18.4 Å in UMOP-1-NH2. When the amide dimers are fully stretched, the distances between the centers of the phenyl rings are approximately 17 Å, 19 Å, and 22 Å for the linkers La, Lb, and Lc, respectively (Fig. 7b). La and Lb are possibly long enough to connect units with the distance of 8.1 Å, but not long enough to create links over the distances of 14.2 Å and 18.4 Å (Fig. S26†). In contrast, Lc is sufficiently long to connect even the longest distances. In Fig. 7c, we show a structure model for CLMOP-1c with only one of three connections over the distance of 18.4 Å (orange), representatively.
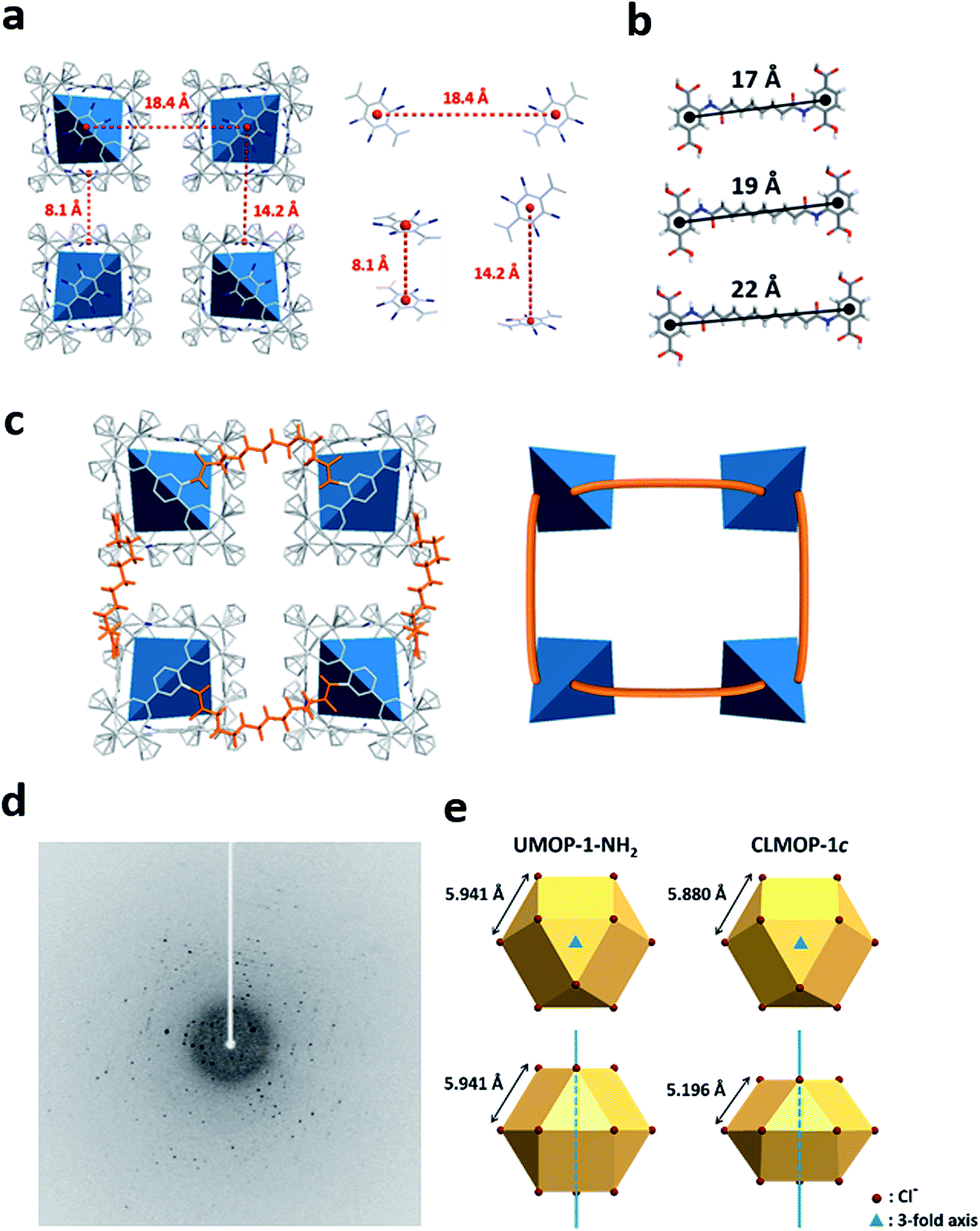 | ||
| Fig. 7 Single crystal X-ray diffraction and structural model of CLMOP-1c. (a) Three possible sites that can be connected by acyl chloride linkers in UMOP-1-NH2. (b) Fully stretched amide dimers. (c) A structure model for CLMOP-1c (left) with a connection over a distance of 18.4 Å and a simplified illustration (right). (d) Single crystal X-ray diffraction image of CLMOP-1c. (e) A comparison of cuboctahedral Cl− ions in UMOP-1-NH2 and CLMOP-1c. The geometry of the Cl− ions in CLMOP-1c reveals compression along the 3-fold axis. | ||
Surprisingly, we obtained SCXRD data for CLMOP-1c with acceptable quality (R = 7.9%; Table S6†), while the CLMOP-1a and CLMOP-1b crystals used for the SCXRD analysis represented poor single crystallinity based on the diffraction images (Fig. S27†). The SCXRD image of CLMOP-1c showed the single crystal nature (Fig. 7d). The longest connection by Lc possibly keeps the tetrahedral cages intact, otherwise rotational rearrangement of the cages might be involved. The structure of CLMOP-1c was solved in the Rm space group, while UMOP-1-NH2 crystallized in the Fmm space group. Although the positions of the tetrahedral cages were clearly assignable for CLMOP-1c, we were unable to model the alkyl chains. This is a common phenomenon for MOFs with alkyl chains, as observed for modified IRMOF-3.48 The packing pattern of CLMOP-1c is essentially the same as that of UMOP-1-NH2, except for distortion corresponding to compression along the 3-fold axis of rotational symmetry (Fig. 7e). Such structural difference is also observed in the PXRD pattern of CLMOP-1c, supporting the Rm space group (Fig. S28†).
Conclusions
In this work, we synthesized a Zr-based MOP, UMOP-1-NH2, with a structure similar to an iconic MOF, UiO-66. UMOP-1-NH2 showed permanent porosity and stability in water unlike typical fragile MOPs. Exploiting such a robust MOP, we demonstrated cross-linking with flexible organic molecules. Reactive condensation between the amines in UMOP-1-NH2 and acyl chlorides was employed. Even after cross-linking accompanied by the production of HCl, the porosity and crystallinity of the CLMOPs were maintained, providing evidence that the tetrahedral cages remained fully intact. It is remarkable that the CLMOPs are microporous while permanent porosity of other MOPs combined with flexible organic moieties has not been demonstrated. We confirmed that the Zr-based MOP, which was sufficiently stable, can be utilized in the condensation reaction while preserving the pore structure. To our knowledge, the CLMOPs are the first examples of the covalent cross-linking of MOPs, highlighting the potential of MOPs as building blocks in polymerization. Using this approach, a combination of MOPs with various organic linkers is now possible. For example, amine-rich organic linkers will provide a strong interaction with CO2 molecules and multitopic linkers will form denser cross-linked systems with highly tunable physical properties. We expect that this cross-linking strategy can be applied to advanced applications, contributing to the bridging of two classes of material, namely metal–organic materials and polymers, both of which are extensively studied to target high selectivity and processability in molecular separation. Specifically, we expect that such materials might provide an important clue to the synthesis of polymeric MOP membranes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research Foundation (NRF) of Korea (NRF-2015R1D1A1A02061738) and the Ulsan National Institute of Science and Technology (UNIST). The authors acknowledge the Pohang Accelerator Laboratory (PAL) for 2D beamline use (2014-3rd-2D-024) and 6D beamline use (2016-2nd-6D-A011).Notes and references
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and F.-J. David, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS PubMed.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- B. J. Burnett and W. Choe, Dalton Trans., 2012, 41, 3889–3894 RSC.
- B. J. Burnett, P. M. Barron, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 9984–9987 CrossRef CAS PubMed.
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- D. J. Tranchemontagne, Z. Ni, M. O’Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS.
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1–27 CrossRef CAS.
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- J. Park, S. Hong, D. Moon, M. Park, K. Lee, S. Kang, Y. Zou, R. P. John, G. H. Kim and M. S. Lah, Inorg. Chem., 2007, 46, 10208–10213 CrossRef CAS PubMed.
- J.-R. Li, D. J. Timmons and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368–6369 CrossRef CAS PubMed.
- H.-N. Wang, X. Meng, G.-S. Yang, X.-L. Wang, K.-Z. Shao, Z.-M. Su and C.-G. Wang, Chem. Commun., 2011, 47, 7128–7130 RSC.
- H.-N. Wang, F.-H. Liu, X.-L. Wang, K.-Z. Shao and Z.-M. Su, J. Mater. Chem. A, 2013, 1, 13060–13063 CAS.
- A.-L. Cheng, N. Liu, J.-Y. Zhang and E.-Q. Gao, Inorg. Chem., 2007, 46, 1034–1035 CrossRef CAS PubMed.
- L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201–210 CrossRef CAS PubMed.
- V. Brega, M. Zeller, Y. He, H. P. Lu and J. K. Klosterman, Chem. Commun., 2015, 51, 5077–5080 RSC.
- H. Jung, D. Moon and H. Chun, Bull. Korean Chem. Soc., 2011, 32, 2489–2492 CrossRef CAS.
- J. Lee, J. H. Kwak and W. Choe, Nat. Commun., 2017, 8, 14070 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 6238 CrossRef PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920–925 CrossRef CAS PubMed.
- Y.-H. Kiang, G. B. Gardner, S. Lee and Z. Xu, J. Am. Chem. Soc., 2000, 122, 6871–6883 CrossRef CAS.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427–5432 CrossRef CAS PubMed.
- Y. Furukawa, T. Ishiwata, K. Sugikawa, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2012, 51, 10566–10569 CrossRef CAS PubMed.
- Y. Zhang, X. Feng, H. Li, Y. Chen, J. Zhao, S. Wang, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2015, 54, 4259–4263 CrossRef CAS PubMed.
- M. Kitchin, J. Teo, K. Konstas, C. H. Lau, C. J. Sumby, A. W. Thornton, C. J. Doonan and M. R. Hill, J. Mater. Chem. A, 2015, 3, 15241–15247 CAS.
- T.-H. Chen, L. Wang, J. V. Trueblood, V. H. Grassian and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 9646–9654 CrossRef CAS PubMed.
- J. A. Foster, R. M. Parker, A. M. Belenguer, N. Kishi, S. Sutton, C. Abell and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 9722–9729 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. Zhong, E. G. Keeler, V. K. Michaelis, J. E. P. Sun, M. J. A. Hore, D. J. Pochan, R. G. Griffin, A. P. Willard and J. A. Johnson, Nat. Chem., 2016, 8, 33–41 CrossRef CAS PubMed.
- Y. Wang, M. Zhong, J. V. Park, A. V. Zhukhovitskiy, W. Shi and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 10708–10715 CrossRef CAS PubMed.
- N. Hosono, M. Gochomori, R. Matsuda, H. Sato and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 6525–6531 CrossRef CAS PubMed.
- R. Kawano, N. Horike, Y. Hijikata, M. Kondo, A. Carné-Sánchez, P. Larpent, S. Ikemura, T. Osaki, K. Kamiya, S. Kitagawa, S. Takeuchi and S. Furukawa, Chem, 2017, 2, 393–403 CAS.
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
- W. Lu, D. Yuan, A. Yakovenko and H.-C. Zhou, Chem. Commun., 2011, 47, 4968–4970 RSC.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- G. Liu, Z. Ju, D. Yuan and M. Hong, Inorg. Chem., 2013, 52, 13815–13817 CrossRef CAS PubMed.
- F. Boutonnet, M. Zablocka, A. Igau, J. Jaud, J.-P. Majoral, J. Schamberger, G. Erker, S. Werner and C. Krüger, J. Chem. Soc., Chem. Commun., 1995, 823–824 RSC.
- A. D. Wiersum, E. Soubeyrand-Lenoir, Q. Yang, B. Moulin, V. Guillerm, M. B. Yahia, S. Bourrelly, A. Vimont, S. Miller, C. Vagner, M. Datrui, G. Clet, C. Serre, G. Maurin and P. L. Llewellyn, Chem.–Asian J., 2011, 6, 3270–3280 CrossRef CAS PubMed.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- Y.-H. Kang, X.-D. Liu, N. Yan, Y. Jiang, X.-Q. Liu, L.-B. Sun and J.-R. Li, J. Am. Chem. Soc., 2016, 138, 6099–6102 CrossRef CAS PubMed.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848–9851 RSC.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- S. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920–1925 CrossRef CAS PubMed.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, instrumentation, and details for the characterization of UMOP-1-NH2 and CLMOPs. CCDC 1520037 and 1520038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03847j |
| This journal is © The Royal Society of Chemistry 2017 |