
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTertiary amine synthesis via reductive coupling of amides with Grignard reagents†
Lan-Gui
Xie
 and
Darren J.
Dixon
*
and
Darren J.
Dixon
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 11th September 2017
Abstract
A new iridium catalyzed reductive coupling reaction of Grignard reagents and tertiary amides affording functionalised tertiary amine products via an efficient and technically-simple one-pot, two-stage experimental protocol, is reported. The reaction – which can be carried out on gram-scale using as little as 1 mol% Vaska's complex [IrCl(CO)(PPh3)2] and TMDS as the terminal reductant for the initial reductive activation step – tolerates a broad range of tertiary amides from (hetero)aromatic to aliphatic (branched, unbranched and formyl) and a wide variety of alkyl (linear, branched), vinyl, alkynyl and (hetero)aryl Grignard reagents. The new methodology has been applied directly to bioactive molecule synthesis and the high chemoselectivity of the reductive coupling of amide has been exploited in late stage functionalization of drug molecules. This reductive functionalisation of tertiary amides provides a new and practical solution to tertiary amine synthesis.
Introduction
Tertiary amines are chemical motifs commonly found in molecules of importance to chemistry, biology and medicine. Their Lewis basic properties arising from the lone pair of electrons make them ideal as ligands for transition metal catalysts and organometallic complexes. Moreover, the Brønsted basicity of the tertiary amine moiety has been exploited extensively for sequestering acidic side products in coupling reactions, and, in metal-free catalysts exploiting acid/base equilibria for substrate activation in enantioselective carbon–carbon bond formation.1 In nature they are often found contained within alkaloid secondary metabolites produced by organisms such as bacteria, fungi, and plants. Classical synthetic routes typically rely on C–N bond forming reactions such as amine-carbonyl reductive amination, amine alkylation and C–N cross-coupling.1,2 However, amine synthesis through carbon–carbon bond formation alpha to nitrogen offers complementarity especially for accessing α-branched products, and, as such, the synthetic utility of the Petasis–Mannich reaction3 as well as the directed amine lithiation/functionalisation approach of Beak, Hoppe, and others, are well-documented.4 Furthermore, α-functionalisation of amines via radical intermediates produced through photoredox activation have recently been achieved.5Reductive functionalisation of amide functionality to generate carbon–carbon bonds alpha to nitrogen has also been demonstrated as a viable approach for amine synthesis.6–8 In this context, approaches using stoichiometric amounts of strongly electrophilic reagents, such as Tf2O and POCl3, for amide pre-activation prior to reductive functionalisation, have been described9 (Scheme 1A). Weinreb amides have also been demonstrated as alternative substrates for reductive pre-activation, using stoichiometric DIBAL-H or Schwartz's reagent (Cp2ZrHCl), affording alkoxy amine products after, for example, allylation and cyanation reactions in the presence of Lewis acids for reactive iminium ion formation10 (Scheme 1B). Although the above stoichiometric approaches are demonstrably successful, catalysed variants of these reductive functionalization processes offer a number of potential benefits, especially in terms of chemoselectivity and functional group tolerance. For example, in presence of Vaska's complex and (Me2HSi)2O (TMDS), Chida/Sato11 demonstrated the formation of hemiaminal intermediates from Weinreb amide substrates, which with the assistance of stoichiometric BF3·Et2O, undergo substitution by preformed nucleophiles such as silyl ketene acetals and allyltin reagents. Our group, and others, have been exploring amine synthesis through exploitation of enamine and iminium ion intermediates formed by catalytic partial reduction of tertiary amides.12 In this respect, we have reported the iridium catalysed reductive intramolecular nitro-Mannich reaction of lactams for the synthesis of bicyclic nitroamines, and the broad-scope iridium catalysed reductive Strecker reaction of tertiary amides for the efficient synthesis of α-amino nitrile products.
 | ||
| Scheme 1 Reductive functionalisation of amides with carbon-centered nucleophiles. | ||
In a continuation of our program, and with the ultimate aim of expanding the range of amine products accessible using the two stage reductive activation approach, we considered a reductive coupling of tertiary amides and Grignard reagents (Scheme 1C). Grignard reagents are widely available commercial organometallic reagents and are simply and reliably prepared in the laboratory. Similarly, tertiary amides are commonly found in biologically active natural products and drug compounds, and are widespread throughout pharmaceutical and agrochemical companies. When combined with the ease at which they can be prepared through standard amide coupling reactions,13 a mild reductive coupling protocol that could efficiently and chemoselectively target this functional group to generate tertiary amine products from Grignard reagents would likely find numerous applications in preparative scale synthesis, library generation, late-stage functionalisation, and total synthesis alike. Herein we wish to report our findings.
Results and discussion
We chose the reductive benzylation of dimethyl benzoylamide as a model reaction and selected Vaska's catalyst and tetramethyldisiloxane (TMDS) for the key reductive activation step (Table 1). Initial investigations employing THF or toluene as the reaction solvent were encouraging; in both cases a two-stage protocol – where the reductive activation step (using 1 mol% Vaska's catalyst and 2 eq. of TMDS) was allowed to run to completion prior to addition of benzylmagnesium chloride (1 M in THF) – afforded moderate yet encouraging yields of the desired tertiary amine 1 (entry 1 and 2). Further studies identified dichloromethane as the solvent of choice and the ideal concentration of amide starting material to be 0.1 M. Following optimisation, our two-stage one-pot protocol enabled the synthesis of 1 in 89% isolated yield after flash column chromatography (entry 6).|
|
||||
|---|---|---|---|---|
| Entry | Cat. loading (mol%) | Solvent | Concentration (M) | Yieldb (%) |
| a Reactions were performed on 0.3 mmol of amide. b NMR yields with 3,5-dibromoanisole as internal standard. c Isolated yield. | ||||
| 1 | 1 | Tetrahydrofuran | 0.05 | 54 |
| 2 | 1 | Toluene | 0.05 | 64 |
| 3 | 1 | CH2Cl2 | 0.05 | 82 |
| 4 | 3.3 | CH2Cl2 | 0.05 | 85 |
| 5 | 0.5 | CH2Cl2 | 0.05 | 53 |
| 6 | 1 | CH2Cl2 | 0.1 | 95(89)c |
| 7 | 1 | CH2Cl2 | 0.025 | 35 |
| 8 | 1 | CH2Cl2 | 0.5 | 80 |

With optimised conditions in hand the scope of the reductive benzylation reaction with respect to the tertiary amide was assessed (Scheme 2). Benzoylamides derived from cyclic amines pyrrolidine, azepine, azocane, morpholine and 6,7-dimethoxytetrahydroisoquinoline respectively, all furnished the target tertiary amines (2–6) in good to excellent yields (70–90%). Methylbenzylbenzoylamide was also a good substrate yielding benzylamine (7) in excellent yield and thus expanding the potential application of the protocol to secondary amine synthesis via subsequent hydrogenolysis. The Weinreb amide of benzoic acid was an excellent substrate and afforded α-benzylated methoxyamine product 8 in 87% yield. Methylaniline derived benzoyl amide was also amenable to reductive benzylation, and furnished a substituted aniline product 9. The acceptable yield of 57% in this case required the use of an increased loading of Vaska's complex (5 mol%) in the initial reductive activation step to overcome diminished substrate reactivity. Great tolerance with respect to the arene moiety of the dimethylamide was witnessed under the optimised reaction conditions. Thus electron deficient, electron-rich and halogen-substituted benzoic acid derivatives gave the respective tertiary amine products (14–18) in yields ranging from 84 to 97%. Similarly a 2-furanyl substrate gave the desired amine product (19) in 94% yield. Aliphatic dimethyl amides, derived from n-decanoic acid, cyclohexane carboxylic acid and pivalic acid were all good substrates for the reductive alkylation protocol and yielded tertiary amine products 10–12 in respectable yields. N-Formyl piperidine was also an excellent substrate and gave product 13 in 82% yield. The ability to dialkylaminomethylate organomagnesium reagents using tertiary formamides should find many synthetic applications within target synthesis, methodology development and drug discovery programs. Unfortunately, under the current reaction conditions, reductive coupling of lactams with Grignard reagents were not successful.14
 | ||
| Scheme 2 Scope with respect to the tertiary amide. Standard condition: amide 0.3 mmol, IrCl(CO)(PPh3)2 1 mol%, TMDS 0.6 mmol, CH2Cl2 3 mL, BnMgCl 0.6 mmol; isolated yields are given; Bpin = pinacol boronate; Boc = t-butyloxy carbonyl. a5 mol% of IrCl(CO)(PPh3)2 was used; bperformed at 0 °C for 6 h after the addition of BnMgCl (1.2 eq.); cPd/C catalysed hydrogenolysis of the N-benzyl group was performed on the tertiary amine product and overall yield is given. | ||
Despite the use of a relatively reactive organometallic coupling partner (BnMgCl) the functional group tolerance of the reductive functionalisation reaction was found to be high. Nitrile, aryl ketone, pinacol boronate ester, tert-butyl carbamate and ester functionality all survived when the amount of Grignard reagent in the second stage was lowered to 1.2 equivalents and the reaction temperature kept below 0 °C. The ability to tolerate halide functionality and pinacol boronate esters establishes this transformation as fully compatible with other cross-coupling reactions, indicating its potential for the synthesis of amine derived building blocks.
To demonstrate the synthetic utility of this newly developed reductive carbon–carbon bond forming methodology, we selected several amine-containing drug molecules as targets. NMDA receptor antagonists, diphenidine (25), methoxphenidine (26) and fluorolintane (27), which are generally used as dissociative anesthetics,15 were obtained directly and efficiently by submitting the corresponding and readily prepared parent amides to the standard protocol. Likewise ephenidine (28), a secondary amine drug molecule,16 was synthesised by partial hydrogenolysis of the initially generated tertiary amine from its benzylethyl benzoylamide precursor, in 67% yield over the two steps. Furthermore, piperazine-derived opioid analgesic drug MT-45(29)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 was prepared in near quantitative yield.
17 was prepared in near quantitative yield.
The scope with respect to the Grignard reagent was also found to be broad. As depicted in Scheme 3, methyl, linear-alkyl and branched-alkyl magnesium reagents were compatible with this protocol and yielded amine products (30–32) in good to excellent yields. (Trimethylsilyl)methylmagnesium chloride was also an excellent reagent for new carbon–carbon bond formation under the standard conditions. Importantly, a reductive methylation of N,N-dimethyl-p-toluamide was carried out on gram scale following the standard protocol and afforded methylated product 34 in 85% yield. Reductive vinylation using vinylmagnesium bromide and reductive alkynylation using hexynyl magnesium bromide yielded allylamine 35 and propargylamine 36 in 74% and 86% yield, respectively. Phenyl magnesium bromide was also found to be a competent nucleophilic coupling partner and afforded benzylic tertiary amine product 37 in 66% respectively. Reductive phenylation [or (hetero)arylation] of amides will likely find many applications within the pharmaceutical sector and here we have exemplified it through the synthesis of the antiemetic piperazine-containing drug cinnarizine (38).18 Using the two-step protocol 38 was successfully synthesised in 67% yield using phenylmagnesium bromide in the carbon–carbon bond-forming step.
 | ||
| Scheme 3 Scope with respect to the Grignard reagent. Standard condition: amide 0.3 mmol, IrCl(CO)(PPh3)2 1 mol%, TMDS 0.6 mmol, CH2Cl2 3 mL, RMgX 0.6 mmol; isolated yields are given. aPerformed on 1.01 g of amide with no additional optimisation; bGrignard reagent prepared by Mg/I exchange with i-PrMgBr; cwith 1.5 eq. Grignard reagent; dperformed at 0 °C for 6 h after Grignard reagent addition; eyield based on Grignard reagent (0.67 eq.). | ||
Encouraged by the high reactivity of the silylated hemiaminal intermediates towards organomagnesium reagents, we then chose to apply Knochel-type Grignard reagents to the new reductive carbon–carbon bond forming methodology. The diversity of organomagnesium reagents accessible by the Knochel procedure is impressive and would therefore boost the nature and type of amine products accessible using this approach.19 To this end, para-trifluoromethylphenyl organomagnesium reagent, prepared by magnesium/iodide exchange with isopropylmagnesium bromide, was investigated in the reductive arylation of the pyrrolidine amide of benzoic acid and indeed was found to be compatible with our protocol, yielding 39 in 65% yield. Similarly a range of aryl and pyrimidine-based heteroaryl Knochel-type Grignard reagents were successfully generated, then reacted with N-formyl piperidine following our standard protocol, furnishing homologated amine products (40–42) in satisfactory yields. Furthermore, organomagnesium reagents derived from α-D-glucofuranose and cholesterol derivatives using the Knochel procedure were also compatible.
The synthetic utility of this chemistry was further demonstrated through the late stage reductive functionalisation of drug molecules. Thus, piperine20 (an extract of black pepper used as traditional medicine and insecticide) was successfully modified by benzylation (45, 93%) and butenylation (46, 83%). Also, the psychoactive drug fipexide21 underwent a reductive coupling reaction with benzylmagnesium chloride affording 47 in 73% yield. While by magnesium/proton exchange of the acetylenic proton of pargyline22 (an irreversible selective monoamine oxidase MAO-B inhibitor) and then homologation with N-formyl piperidine, amine derivative 48 was obtained in 54% yield.
Conclusions
A new two-stage, iridium catalysed reductive coupling of tertiary amides and Grignard reagents has been developed. Due to its good functional group tolerance, high chemoselectivity and broad scope with respect to both the amide and Grignard coupling partners, this new and practical methodology should find many synthetic applications in research laboratories within both industry and academia.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Marie Curie Actions for a fellowship to L.-G. X. (H2020-MSCA-IF-2015, 707559). We also thank Heyao Shi, Dr Angel L. Fuentes de Arriba, Dr Amber L. Thompson and Dr Kirsten E. Christensen (Oxford Chemical Crystallography Service) for X-ray structure determination.Notes and references
- Amino group chemistry: from synthesis to the life sciences, ed. A. Ricci, Wiley VCH, Weinheim, 2008 Search PubMed.
- (a) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed; (b) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed.
- (a) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS PubMed; (b) T. R. Ramadhar and R. A. Batey, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, 2nd edn, 2011, vol. 2, pp. 427–477 Search PubMed.
- (a) P. Beak, A. Basu, D. J. Gallagher, Y. S. Park and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552 CrossRef CAS; (b) D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282 CrossRef CAS; (c) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092 CrossRef CAS PubMed.
- (a) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef CAS PubMed; (b) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304 CrossRef CAS PubMed; (c) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173 RSC; (d) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed; (e) J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 7266 CrossRef CAS PubMed; (f) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002 RSC.
- For reviews of amide alkylation and reduction, see: (a) D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421 CrossRef CAS PubMed; (b) A. Volkov, F. Tinnis, T. Slagbrand, P. Trilloa and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685 RSC; (c) S. A. Ruider and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 13856 CrossRef CAS PubMed; (d) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697 CrossRef CAS; (e) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS PubMed.
- For selected examples of (thio)amide dialkylation and related, see: (a) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228 CrossRef CAS PubMed; (b) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037 CrossRef CAS PubMed; (c) T. Murai and F. Asai, J. Am. Chem. Soc., 2007, 129, 780 CrossRef CAS PubMed; (d) T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968 CrossRef CAS PubMed; (e) Y. Tominaga, S. Kohra and A. Hosomi, Tetrahedron Lett., 1987, 28, 1529 CrossRef CAS; (f) Y. Tamaru, T. Harada, S. Nishi and Z. Yoshida, Tetrahedron Lett., 1982, 23, 2383 CrossRef CAS.
- For selected examples of tertiary amide reduction, see: (a) F. Tinnis, A. Volkov, T. Slagbrand and H. Adolfsson, Angew. Chem., Int. Ed., 2016, 55, 4562 CrossRef CAS PubMed; (b) D. Mukherjee, S. Shirase, K. Mashima and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 13326 CrossRef CAS PubMed; (c) A. Augurusa, M. Mehta, M. Perez, J. Zhu and D. W. Stephan, Chem. Commun., 2016, 52, 12195 RSC; (d) S. Das, Y. Li, C. Bornschein, S. Pisiewicz, K. Kiersch, D. Michalik, F. Gallou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 12389 CrossRef CAS PubMed; (e) A. Tahara, Y. Miyamoto, R. Aoto, K. Shigeta, Y. Une, Y. Sunada, Y. Motoyama and H. Nagashima, Organometallics, 2015, 34, 4895 CrossRef CAS; (f) Y. Li, J. A. Molina de La Torre, K. Grabow, U. Bentrup, K. Junge, S. Zhou, A. Bruckner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 11577 CrossRef CAS PubMed; (g) S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS PubMed; (h) S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032 CrossRef CAS PubMed; (i) Y. Motoyama, M. Aoki, N. Takaoka, R. Aoto and H. Nagashima, Chem. Commun., 2009, 1574 RSC; (j) J. T. Spletstoser, J. M. White, A. R. Tunoori and G. I. Georg, J. Am. Chem. Soc., 2007, 129, 3408 CrossRef CAS PubMed.
- (a) M. M. Heravi and N. Nazari, Curr. Org. Chem., 2015, 19, 2358 CrossRef CAS; (b) P.-Q. Huang, Y.-H. Huang, K.-J. Xiao, Y. Wang and X.-E. Xia, J. Org. Chem., 2015, 80, 2861 CrossRef CAS PubMed; (c) M. Mewald, J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2014, 53, 11634 CrossRef CAS PubMed; (d) J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572 CrossRef CAS PubMed; (e) K.-J. Xiao, A.-E. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314 CrossRef CAS PubMed; (f) R. Larouche-Gauthier and G. Belanger, Org. Lett., 2008, 10, 4501 CrossRef CAS PubMed.
- (a) S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato and N. Chida, J. Am. Chem. Soc., 2016, 138, 5246 CrossRef CAS PubMed; (b) K. Shirokane, Y. Tanaka, M. Yoritate, N. Takayama, T. Sato and N. Chida, Bull. Chem. Soc. Jpn., 2015, 88, 522 CrossRef CAS; (c) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512 CrossRef CAS PubMed; (d) M. Nakajima, Y. Oda, T. Wada, R. Minamikawa, K. Shirokane, T. Sato and N. Chida, Chem.–Eur. J., 2014, 20, 17565 CrossRef CAS PubMed; (e) G. Vincent, R. Guillot and C. Kouklovsky, Angew. Chem., Int. Ed., 2011, 50, 1350 CrossRef CAS PubMed; (f) K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2010, 49, 6369 CrossRef CAS PubMed.
- M. Nakajima, T. Sato and N. Chida, Org. Lett., 2015, 17, 1696 CrossRef CAS PubMed.
- (a) A. W. Gregory, A. Chambers, A. Hawkins, P. Jakubec and D. J. Dixon, Chem.–Eur. J., 2015, 21, 111 CrossRef CAS PubMed; (b) P. W. Tan, J. Seayad and D. J. Dixon, Angew. Chem., Int. Ed., 2016, 55, 13436 CrossRef CAS PubMed; (c) P.-Q. Huang, W. Ou and F. Han, Chem. Commun., 2016, 52, 11967 RSC; (d) A. L. Fuentes de Arriba, E. Lenci, M. Sonawane, O. Formery and D. J. Dixon, Angew. Chem., Int. Ed., 2017, 56, 3655 CrossRef CAS PubMed.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
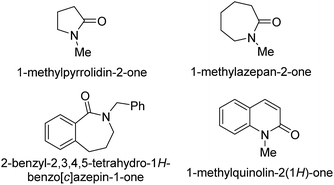
- Submission of lactams 1-methylpyrrolidin-2-one, 1-methylazepan-2-one, 2-benzyl-2,3,4,5-tetrahydro-1H-benzo[c]azepin-1-one and 1-methylquinolin-2(1H)-one to the standard procedure, led mainly to the corresponding over-reduction products without C–C bond formation
 .
. - J. Wallach, H. Kang, T. Colestock, H. Morris, Z. A. Bortolotto, G. L. Collingridge, D. Lodge, A. L. Halberstadt, S. D. Brandt and A. Adejare, PLoS One, 2016, 11, e0157021 Search PubMed.
- H. Morris and J. Wallach, Drug Test. Anal., 2014, 6, 614 CrossRef CAS PubMed.
- K. Natsuka, H. Nakamura, H. Uno and S. Umemoto, J. Med. Chem., 1975, 18, 1240 CrossRef CAS PubMed.
- M. Lucertini, N. Mirante, M. Casagrande, P. Trivelloni and V. Lugli, Physiol. Behav., 2007, 91, 180 CrossRef CAS PubMed.
- (a) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed. Engl., 2003, 42, 4302 CrossRef CAS PubMed; (b) P. Knochel, M. A. Schade, S. Bernhardt, G. Manolikakes, A. Metzger, F. M. Piller, C. J. Rohbogner and M. Mosrin, Beilstein J. Org. Chem., 2011, 7, 1261 CrossRef CAS PubMed.
- K. Srinivasan, Crit. Rev. Food Sci. Nutr., 2007, 47, 735 CrossRef CAS PubMed.
- C. Missale, G. Pasinetti, S. Govoni, P. F. Spano and M. Trabucchi, Boll. Chim. Farm., 1983, 122, 79 CAS.
- Z. Fisar, J. Hroudova and J. Raboch, Neuroendocrinol. Lett., 2010, 31, 645 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, copies of 1H and 13C NMR spectra and crystallographic data. CCDC 1552384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03613b |
| This journal is © The Royal Society of Chemistry 2017 |