
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly of strongly phosphorescent hetero-bimetallic and -trimetallic [2]catenane structures based on a coinage metal alkynyl system†
Xiao-Yong
Chang
 a,
Guang-Tao
Xu
a,
Bei
Cao
a,
Juan-Yu
Wang
a,
Jie-Sheng
Huang
*a and
Chi-Ming
Che
*ab
a,
Guang-Tao
Xu
a,
Bei
Cao
a,
Juan-Yu
Wang
a,
Jie-Sheng
Huang
*a and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Institute of Molecular Functional Materials, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: jshuang@hku.hk; cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen 518053, China
First published on 4th October 2017
Abstract
Homo-metallic metal alkynyl complexes exhibit interesting catenane structures, but their hetero-metallic catenane counterparts are under-developed. In this work, we report rare examples of bimetallic Au–Cu (DtbpC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C− ligand; Dtbp = 3,5-di-tert-butylphenyl), Au–Ag (tBuCC− ligand), and Au–Cu, Au–Ag (C6-FluoCC− ligand; C6-Fluo = 9,9-dihexyl-9H-fluoren-2-yl) complexes as well as a trimetallic Au–Ag–Cu (C6-FluoCC− ligand) complex, which feature [2]catenane structures. The formation of the [2]catenane structure is significantly affected by the coinage metal ion(s) and change of the structure of the alkynyl ligand. These hetero-metallic [2]catenane structures are strongly luminescent with tunable emission λmax from 503 to 595 nm and Φ values up to 0.83.
C− ligand; Dtbp = 3,5-di-tert-butylphenyl), Au–Ag (tBuCC− ligand), and Au–Cu, Au–Ag (C6-FluoCC− ligand; C6-Fluo = 9,9-dihexyl-9H-fluoren-2-yl) complexes as well as a trimetallic Au–Ag–Cu (C6-FluoCC− ligand) complex, which feature [2]catenane structures. The formation of the [2]catenane structure is significantly affected by the coinage metal ion(s) and change of the structure of the alkynyl ligand. These hetero-metallic [2]catenane structures are strongly luminescent with tunable emission λmax from 503 to 595 nm and Φ values up to 0.83.
Introduction
The construction of intriguing types of interpenetrated structures, including catenanes, continues to be an active area in supramolecular science. Various strategies, including π–π stacking, hydrogen bonding, metal templating, and hydrophobic interactions, have been developed to direct the assembly of interpenetrated structures.1 Closed-shell metallophilic interactions could also be appealing driving forces for the formation of catenanes, as demonstrated by the [2]catenane structures of [(tBuCCAu)6]2,2 (RCCAu)10,3 and Aun(SR)n (n = 10,4 11,5 and 12 (ref. 4)) which feature AuI–AuI interactions (Au–Au 2.88–3.30 Å, e.g.Fig. 1, upper). We previously reported a [3]catenane structure of (tBuCCCu)20.6 This type of catenane, first reported by Mingos and co-workers,2 is based on homoleptic homo-metallic alkynyl or thiolate complexes. Puddephatt and co-workers reported heteroleptic AuI-alkynyl/phosphine complexes adopting [2]catenane structures, which also feature AuI–AuI interactions.7 The quest remains for hetero-metallic catenanes based on a homoleptic metal alkynyl system.
 | ||
| Fig. 1 Upper: Literature examples of homo-metallic [2]catenanes. Lower: Hetero-metallic [2]catenane 1, an example of five hetero-metallic [2]catenanes reported in this work. The peripheral groups (tBu, R or Dtbp) are not shown. The metallophilic interactions are depicted as dashed lines. | ||
An important feature of AuI-alkynyl catenanes is the presence of two linear RCC–Au–CCR units in the locking center.2,3 These units function as a template to facilitate the formation of the first ring and as a building block for the second ring. In view of the RCC–M–CCR species commonly seen in the literature,8 homoleptic hetero-metallic coinage metal alkynyl complexes might also be suitable candidates for the construction of catenanes. A key issue is the control of the complex size, which is tunable by adjusting the bulkiness and/or substitution pattern of the alkynyl ligand.9 However, the design of new structures with specific configurations is hampered by the complexity and limited understanding of the structures of such complexes,8a,10,11 particularly for trimetallic ones.10c,d Also, in view of their intriguing phosphorescence and potential materials application,12,13 the exploration of new structures of hetero-metallic alkynyl complexes with high stability could be rewarding. Based on our previous work on a CuI–alkynyl system,9 we employed bulky alkynyl ligands RCC− (R = 3,5-di-tert-butylphenyl (Dtbp), 9,9-dihexyl-9H-fluoren-2-yl (C6-Fluo), or tBu) to construct novel assemblies of hetero-metallic alkynyl complexes. Herein, we described the formation of five hetero-metallic alkynyl [2]catenanes (including bimetallic 1, 4, 5 and 7, and trimetallic 6, Fig. 1 and 2) by the self-assembly of homoleptic coinage metal alkynyl systems. As revealed by the structures of these complexes and two other hetero-metallic complexes 2 and 3 (Fig. 2), the proper combination of coinage metal ions and alkynyl ligands is crucial to the formation of the catenane structure.
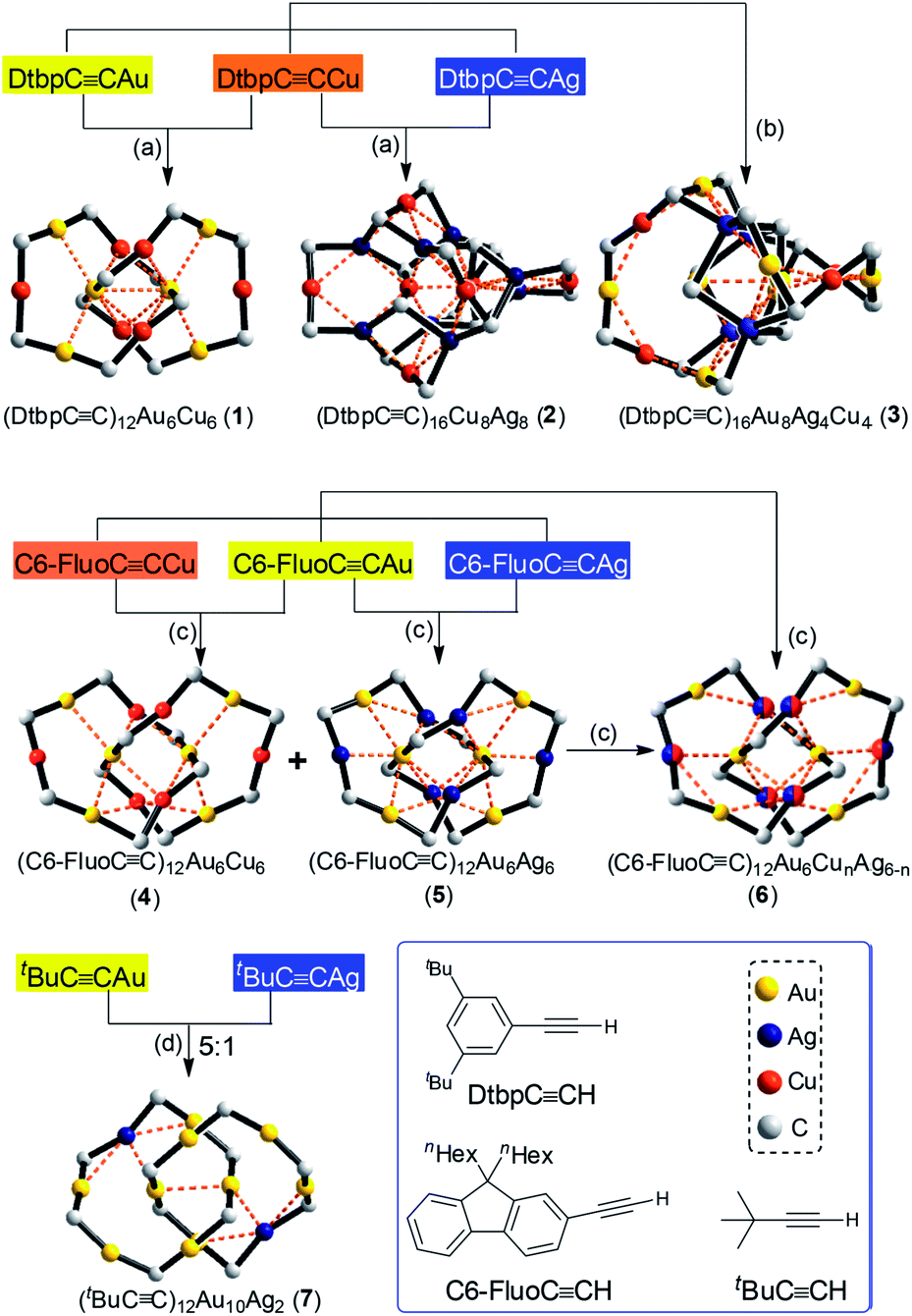 | ||
| Fig. 2 The synthetic scheme and coordination cores of hetero-metallic coinage metal complexes 1–7. The metallophilic interactions are depicted as dashed lines; for each alkynyl ligand, only the α carbon atom is shown. Reagents and conditions: (a) CH2Cl2/MeCN; (b) chlorobenzene; (c) toluene/MeCN; (d) CH2Cl2/Et3N. | ||
Results and discussion
The bimetallic complexes 1, 2, 4, 5 and 7 were prepared by mixing two homoleptic metal complexes in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 5:1 (for 7) molar ratio, and the trimetallic complexes 3 and 6 were prepared by mixing the homoleptic gold, silver and copper alkynyl complexes in a 2:1:1 molar ratio or by mixing the hetero-metallic Au–Cu and Au–Ag alkynyl complexes in a 1:1 molar ratio. Complexes 1–3 were also accessible from the reactions of alkynes with Au(SMe2)Cl, AgOTf and/or [Cu(MeCN)4]PF6 (2:1:1 for 1 and 2 and 3:1:1:1 for 3) in the presence of Et3N (yields: 27–78%). X-ray diffraction-quality crystals of 1–7 were obtained by the slow evaporation of the CH2Cl2/MeCN, chlorobenzene or toluene/MeCN solutions and their structures were determined by X-ray crystallography (Tables S1 and S2 in the ESI†).
:1 or 5:1 (for 7) molar ratio, and the trimetallic complexes 3 and 6 were prepared by mixing the homoleptic gold, silver and copper alkynyl complexes in a 2:1:1 molar ratio or by mixing the hetero-metallic Au–Cu and Au–Ag alkynyl complexes in a 1:1 molar ratio. Complexes 1–3 were also accessible from the reactions of alkynes with Au(SMe2)Cl, AgOTf and/or [Cu(MeCN)4]PF6 (2:1:1 for 1 and 2 and 3:1:1:1 for 3) in the presence of Et3N (yields: 27–78%). X-ray diffraction-quality crystals of 1–7 were obtained by the slow evaporation of the CH2Cl2/MeCN, chlorobenzene or toluene/MeCN solutions and their structures were determined by X-ray crystallography (Tables S1 and S2 in the ESI†).
The Au–Cu complex 1, with a formula of (DtbpCC)12Au6Cu6, has a crystallographic D2 symmetry and features two twisted (DtbpCC)6Au3Cu3 rings that are interlocked to form a [2]catenane structure (Fig. 3). Each ring is composed of three linear DtbpCC–Au–CCDtbp units and three Cu ions with relatively weak Au–Cu (2.9632(10) Å) and Au–Au (2.9625(3)–3.1497(8) Å) interactions (cf. the sums of the metallic radii: Au–Cu 2.72 Å and Au–Au 2.88 Å (ref. 14)), which are comparable to the metallophilic interactions in the well documented AuI-alkynyl and -thiolate [2]catenanes (Au–Au 2.88–3.30 Å).2–5 The bridging alkynyl groups each coordinate to the Au and Cu ions in the η1- and η2-modes, respectively. The M–C distances (Au–C 1.979(7)–2.009(6) Å and Cu–C 2.018(7)–2.285(14) Å) are comparable to those in the homo-metallic [2]catenane [(tBuCCAu)6]2 (Au–C 1.85(4)–2.26(3) Å)2 (Fig. 4). These observations make it reasonable to describe 1 as a hetero-metallic [2]catenane. In contrast, the PhCC− counterpart of 1, (PhCC)12Au6Cu6, adopts a non-catenane structure in which the six AuI ions are co-planar and the six CuI ions form a trigonal prism with the PhCC− ligands situated on the two opposite sides, rendering the Ph groups (for each side) rather close to each other10i and reflecting the unique role of the bulky Dtbp groups of 1 in the assembly/stabilization of the [2]catenane structure of the Au6Cu6 alkynyl complex. It is probable that the non-catenane structure of (PhCC)12Au6Cu6, upon changing its alkynyl ligands to the bulkier and more basic DtbpCC− ligands, is destabilized owing to the increased steric hindrance [resulting from the bulkiness of the electron-donating tBu substituents and stronger metal-alkynyl binding (cf. Au–C 2.004(6)–2.045(5) Å in (PhCC)12Au6Cu6 (ref. 10i) vs. 1.979(7)–2.009(6) Å in 1)], and as such the steric hindrance is minimized in the [2]catenane structure of the Au–Cu complex 1.
 | ||
| Fig. 3 The [2]catenane structure of 1. Upper left: the coordination core. Upper right: one of the two interlocked (DtbpCC)6Au3Cu3 rings. The tBu groups and hydrogen atoms are omitted for clarity, and the metallophilic interactions are depicted as dashed lines. Lower: a wire frame representation with the omission of the metallophilic interactions. | ||
 | ||
| Fig. 4 The comparison of the M–C distances in the [2]catenanes 1 (left) and [(tBuCCAu)6]2 (right). Only one ring is depicted in each case; the metallophilic interactions are not shown. | ||
By changing Au–Cu to Cu–Ag, but with the same DtbpCC− ligand unchanged, a non-catenane complex (DtbpCC)16Cu8Ag8 (2) was obtained. Complex 2 has a structure with an approximate S4 symmetry (Fig. 5a, Cu–Ag 2.6824(3)–3.0416(4) Å) and contains a rather complicated metallacycle core (Fig. 6a, Cu–C 1.855(3)–1.889(3) Å, Ag–C 2.236(3)–2.643(2) Å); the topology of its metallophilic interactions is similar to that of the recently reported Au–Ag counterpart (DtbpCC)16Au8Ag8, also with a non-catenane structure.11b In the case of 3, its structure (Fig. 5b) features a metallacycle core (Au–C 1.950(7)–2.025(9) Å, Cu–C 1.919(8)–2.038(6) Å, Ag–C 2.364(6)–2.618(7) Å) which is markedly different from that formed by simply replacing the four Ag ions of the (DtbpCC)16Au8Ag8 molecule11b with four Cu ions. As shown in Fig. 6b, compared with (DtbpCC)16Au8Ag8, 3 features central Au–Au distance (Au2–Au5 2.9591(9)11b ∼0.22 Å longer vs. Au3–Au7 3.1825(4) Å) and substantially larger C–M–C (M = Cu3, Cu4) angles (139.4(5)–142.6(4)° (M = Ag1, Ag2)11bvs. 172.4(3)–177.0(4)°).
 | ||
| Fig. 5 A perspective view of 2 (a) and 3 (b). The Dtbp groups of the alkynyl ligands are omitted for clarity. The metallophilic interactions are depicted as dashed lines. | ||
 | ||
| Fig. 6 The metallacycle cores in 2 (a) and 3 (b) as compared with that in (DtbpCC)16Au8Ag8 (ref. 11b) (inset) with the omission of all the metallophilic interactions (for each alkynyl ligand, only the α carbon atom is shown). | ||
Changing the R group of RCC− from Dtbp to the bulkier C6-Fluo resulted in the formation of bimetallic (C6-FluoCC)12Au6Cu6 (4) and (C6-FluoCC)12Au6Ag6 (5) and trimetallic (C6-FluoCC)12Au6CunAg6−n (6), and all of the three complexes adopt a [2]catenane structure (Fig. 7). The arrangement of the six RCC–Au–CCCR units in 4–6 is similar to that in 1; the connection of the C6-FluoCC–Au–CCC6-Fluo units by π-CC–Cu/Ag coordination forms the [2]catenane structures. The Cu and Ag atoms in 6 are in substitutional disorder (Fig. 7c): each π-CC–M is partially occupied by Cu and Ag atoms, the occupancy of the Ag atom of the outlier positions 1 and 2 (0.77 and 0.62, respectively) is slightly higher (Fig. 7c), and the overall Cu/Ag ratio (3.1:2.9) is close to the molar ratio (1:1) of 4 and 5 used in the preparation of 6. The average Cu/Ag–C(α) distance in 6 is 0.14 Å longer than the average Cu–C(α) distance in 4 and 0.12 Å shorter than the average Ag–C(α) distance in 5, while the difference of the average Au–C(α) distances between 4–6 is <0.03 Å.
 | ||
| Fig. 7 A perspective view of the C6-FluoCC− ligand based complexes 4 (a), 5 (b) and 6 (c). All of the hydrogen atoms and parts of the C6-FluoCC− ligands are omitted for clarity. The metallophilic interactions are depicted as dashed lines. | ||
As [2]catenane 6 can be formed by mixing the [2]catenanes 4 and 5 in solution, we also mixed the previously reported [2]catenane [(tBuCCAu)6]2 (ref. 2) with [tBuCCAg]n (molar ratio 5:1), and obtained 7, adopting a [2]catenane structure similar to that of [(tBuCCAu)6]2 except for the replacement of one bis(η2-CC) coordinated Au ion in each ring by one Ag ion (Fig. 8).
 | ||
| Fig. 8 A perspective view of the [2]catenane 7. Inset: a perspective view of [(tBuCCAu)6]2 (ref. 2) for comparison. The tBu groups are omitted for clarity. The metallophilic interactions are depicted as dashed lines. | ||
We examined the solution behavior of the hetero-metallic [2]catenanes 1 and 4–7, which are stable in solution at a concentration >10−4 M, using ESI-MS and 1H NMR measurements (see the ESI†); the results for 1 are discussed here as examples. The ESI mass spectrum of 1 (∼10−4 M) in CH2Cl2 features a prominent cluster peak at m/z 4146.4 attributed to [1 + Na]+ (Fig. S1, ESI†), like the observation of cluster peaks at m/z 5876.4, 6142.2, and 6009.3, which are attributed to [4 + Na]+, [5 + Na]+, and [6 + Na]+ for 4, 5, and 6, respectively. In the 1H NMR spectrum of 1 in CD2Cl2 and 1,2-dichlorobenzene-d4 (∼10−2 M, Fig. 9), three sets of coordinated DtbpCC− signals were observed at room temperature (consistent with the D2 symmetry in the crystal structure of 1), which were broadened into one set upon increasing the temperature to 353 K and were then recovered by cooling back to room temperature (Fig. 9, upper right). These spectral changes could be associated with the dependence of the metallophilic interactions in 1 on temperature. We further examined the solution of 1 in CD2Cl2 at room temperature by 1H DOSY NMR measurements; the spectrum obtained (Fig. S4, ESI†) reveals that the observed signals of DtbpCC− belong to a single complex (diffusion constant D = 8.32 × 10−10 m2 s−1), thus providing additional evidence for the purity of 1 in solution.
 | ||
| Fig. 9 The variable-temperature 1H NMR spectra (in the aromatic region) of 1 in CD2Cl2 (left, 298 K → 213 K) and in 1,2-dichlorobenzene-d4 (right, 298 K → 353 K → 298 K). | ||
DFT calculations were performed to examine the electronic details of the hetero-metallic [2]catenanes using 1 as an example. The DFT-optimized geometry of 1 is comparable to that determined by X-ray crystal analysis. For example, the computed structure of 1 features average values of Au–C 1.997 Å, C–Au–C 176.2°, and Cu–C 2.053 Å; these values compare well with the corresponding ones in the crystal structure of 1 (average values: Au–C 1.995(7) Å, C–Au–C 176.9(8)° and Cu–C 2.097(7) Å). To gain insight into why the [2]catenane of (DtbpCC)12Au6M6 was obtained for M = Cu (1) but not for M = Ag, we attempted to perform DFT optimization of (DtbpCC)12Au6Ag6 with a hypothetical similar [2]catenane structure, which did not converge. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of 1 are depicted in Fig. 10. The HOMO is mainly localized on the 5dz2 orbitals of the two Au atoms in the locking center of the [2]catenane structure, while the LUMO is distributed on the empty 6p orbitals of the same two Au atoms.
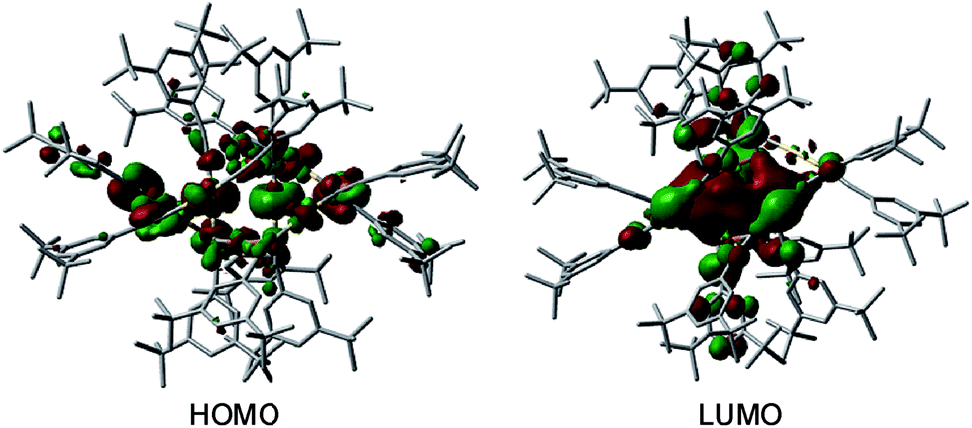 | ||
| Fig. 10 The HOMO and LUMO of 1. The hydrogen atoms are omitted for clarity. Color code: carbon (grey), gold (gold), and copper (orange). | ||
Complexes 1–7 are emissive in the solid state (Fig. 11). In view of their structural fluxional behavior in solution, their photo-physical properties in solution were not included in this study. The [2]catenanes 1 and 4–6 exhibit moderate yellow to strong orange emissions in the solid state (Φ = 0.37–0.83). Changing the ligand from DtbpCC− to C6-FluoCC− resulted in a bathochromic shift in emission energy and a significant improvement in the quantum yield. Notably, the emission energy and efficiency show only minor variation with the metal compositions of 4–6 (λmax = 588–595 nm, Φ = 0.71–0.83, and τ = 0.7–1.0 μs). The comparison of the emission spectra of (DtbpCC)12Au8Ag8 (λmax = 489 nm (ref. 11b)) and 3 reveals that the replacement of the four Ag ions by four Cu ions resulted in a broader and red-shifted emission band (λmax = 542 nm). The excitation of 7 in the solid state gave a strong green emission at λmax 503 nm with a tail up to 710 nm (Φ = 0.82). The wide span in emission energy (λmax from 503 to 595 nm) and high solid state emission quantum yields highlight the prospect of hetero-metallic [2]catenanes based on a coinage metal alkynyl system as useful photo-functional molecular materials.
 | ||
| Fig. 11 The emission spectra of 1–7 in the solid state. | ||
The use of the C6-FluoCC− ligand to result in the assembly of hetero-bimetallic and hetero-trimetallic [2]catenanes (RCC)12Au6M6 (M = Cu 4 and Ag 5) and (RCC)12Au6CunAg6−n (6) is remarkable. As 4–6 are nearly isostructural, it appears that their C6-FluoCC− ligands dominate the intermolecular interactions, with the effect of the Ag and Cu ions being minor in these cases. As revealed by the crystal structures of 4 and 5, replacing the Cu ions with Ag ions slightly expands the metallacycle core owing to the longer Ag–C than Cu–C distances (Fig. S5, ESI†). The expansion of the metallacycle core would reduce the repulsion between the peripheral alkynyl ligands and increase the tendency of the complex to re-assemble to higher nuclearity species. The bulky C6-FluoCC− ligand with flexible C6-alkyl chains is likely to restrict such tendency. For the complexes of the DtbpCC− ligand, which is sterically less demanding and relatively rigid, the replacement of the Cu ions by Ag ions leads to a core enlargement from M12 (1) to M16 (3). On the other hand, an Ag ion, compared with a Cu ion, is a stronger Lewis acid and is inclined to form weak interactions with more alkynyl ligands (cf.2 and 3 in Fig. 5); the extra π-CC–Ag interactions may distort the ring unit and then break the [2]catenane structure. Moreover, the preference of AuI for a linear two-coordinate configuration should also play an important role in the assembly of the hetero-metallic [2]catenanes in view of the core enlargement from M12 (1) to M16 (2), upon replacing the Au ions with Ag ions, and the presence of linear RCC–AuI–CCR units in all of the [2]catenanes 1, 4–6, and 7.
Conclusions
We have prepared and structurally characterized five hetero-metallic [2]catenanes based on coinage metal alkynyl complexes, including bimetallic Au–Cu and Au–Ag complexes and a trimetallic Au–Cu–Ag complex, by employing bulky DtbpCC−, C6-FluoCC−, and tBuCC− ligands. The structure of the trimetallic [2]catenane 6 is analogous to its corresponding Au–Ag bimetallic [2]catenanes with some of the Ag atoms replaced by Cu atoms; mixing the Au–Cu and Au–Ag complexes is a feasible and efficient method to prepare the trimetallic complex. The formation of [2]catenanes relies upon a delicate balance between various intermolecular interactions. The structural characterization of 1–7 provides useful insight into a better understanding of hetero-metallic coinage metal alkynyl complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the National Key Basic Research Program of China (No. 2013CB834802), the University Grants Committee of Hong Kong (Area of Excellence Program AoE/P-03/08), and the Strategic Research Theme of HKU on New Materials.Notes and references
- (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (b) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed.
- D. M. P. Mingos, J. Yau, S. Menzer and D. J. Williams, Angew. Chem., Int. Ed., 1995, 34, 1894–1895 CrossRef CAS.
- I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, S. I. Selivanov, J. Jänis, M. Haukka, T. Pakkanen, S. P. Tunik and P.-T. Chou, Inorg. Chem., 2012, 51, 7392–7403 CrossRef CAS PubMed.
- M. R. Wiseman, P. A. Marsh, P. T. Bishop, B. J. Brisdon and M. F. Mahon, J. Am. Chem. Soc., 2000, 122, 12598–12599 CrossRef CAS.
- (a) S. S.-Y. Chui, R. Chen and C.-M. Che, Angew. Chem., Int. Ed., 2006, 45, 1621–1624 CrossRef CAS PubMed; (b) F. Bertorelle, I. Russier-Antoine, N. Calin, C. Comby-Zerbino, A. Bensalah-Ledoux, S. Guy, P. Dugourd, P.-F. Brevet, Ž. Sanader, M. Krstić, V. Bonačić-Koutecký and R. Antoine, J. Phys. Chem. Lett., 2017, 8, 1979–1985 CrossRef CAS PubMed.
- S. S.-Y. Chui, M. F. Y. Ng and C.-M. Che, Chem.–Eur. J., 2005, 11, 1739–1749 CrossRef CAS PubMed.
- (a) C. P. McArdle, S. Van, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2002, 124, 3959–3965 CrossRef CAS PubMed; (b) R. J. Puddephatt, J. Organomet. Chem., 2015, 792, 13–24 CrossRef CAS.
- (a) S.-K. Yip, C.-L. Chan, W. H. Lam, K.-K. Cheung and V. W.-W. Yam, Photochem. Photobiol. Sci., 2007, 6, 365–371 RSC; (b) I. O. Koshevoy, Y.-C. Lin, A. J. Karttunen, M. Haukka, P.-T. Chou, S. P. Tunik and T. A. Pakkanen, Chem. Commun., 2009, 2860–2862 RSC; (c) I. O. Koshevoy, A. J. Karttunen, S. P. Tunik, M. Haukka, S. I. Selivanov, A. S. Melnikov, P. Y. Serdobintsev and T. A. Pakkanen, Organometallics, 2009, 28, 1369–1376 CrossRef CAS; (d) I. O. Koshevoy, C.-L. Lin, A. J. Karttunen, J. Jänis, M. Haukka, S. P. Tunik, P.-T. Chou and T. A. Pakkanen, Inorg. Chem., 2011, 50, 2395–2403 CrossRef CAS PubMed; (e) Y. Wang, H. Su, C. Xu, G. Li, L. Gell, S. Lin, Z. Tang, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 4324–4327 CrossRef CAS PubMed.
- X.-Y. Chang, K.-H. Low, J.-Y. Wang, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 10312–10316 CrossRef CAS PubMed.
- (a) O. M. Abu-Salah, A.-R. A. Al-Ohaly and C. B. Knobler, J. Chem. Soc., Chem. Commun., 1985, 1502–1503 RSC; (b) O. M. Abu-Salah, M. S. Hussain and E. O. Schlemper, J. Chem. Soc., Chem. Commun., 1988, 212–213 RSC; (c) O. M. Abu-Salah, A. R. A. Al-Ohaly and Z. F. Mutter, J. Organomet. Chem., 1990, 391, 267–273 CrossRef CAS; (d) O. M. Abu-Salah, Polyhedron, 1992, 11, 951–955 CrossRef CAS; (e) M. Ul-Haque, W. Horne and O. M. Abu-Salah, J. Crystallogr. Spectrosc. Res., 1992, 22, 421–425 CrossRef; (f) M. S. Hussain and O. M. Abu-Salah, J. Organomet. Chem., 1993, 445, 295–300 CrossRef CAS; (g) M. S. Hussain, M. Ul-Haque and O. M. Abu-Salah, J. Cluster Sci., 1996, 7, 167–177 CrossRef CAS; (h) O. Schuster, U. Monkowius, H. Schmidbaur, R. S. Ray, S. Krüger and N. Rösch, Organometallics, 2006, 25, 1004–1011 CrossRef CAS; (i) I. O. Koshevoy, L. Koskinen, M. Haukka, S. P. Tunik, P. Y. Serdobintsev, A. S. Melnikov and T. A. Pakkanen, Angew. Chem., Int. Ed., 2008, 47, 3942–3945 CrossRef CAS PubMed; (j) G. F. Manbeck, W. W. Brennessel, R. A. Stockland Jr and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 12307–12318 CrossRef CAS PubMed; (k) I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, M. Haukka, T. Pakkanen and P.-T. Chou, J. Am. Chem. Soc., 2012, 134, 6564–6567 CrossRef CAS PubMed; (l) I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, J. R. Shakirova, J. Jänis, M. Haukka, T. Pakkanen and P.-T. Chou, Chem.–Eur. J., 2013, 19, 5104–5112 CrossRef CAS PubMed.
- (a) W.-J. Guo, Y.-T. Wang, D.-X. Kong, J.-Y. Wang, Q.-H. Wei and G.-N. Chen, Chem.–Eur. J., 2015, 21, 4205–4208 CrossRef CAS PubMed; (b) R. Zhang, C. Zhao, X. Li, Z. Zhang, X. Ai, H. Chen and R. Cao, Dalton Trans., 2016, 45, 12772–12778 RSC.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- (a) Y. Ma, C.-M. Che, H.-Y. Chao, X. Zhou, W.-H. Chan and J. Shen, Adv. Mater., 1999, 11, 852–857 CrossRef CAS; (b) L.-J. Xu, J.-Y. Wang, X.-F. Zhu, X.-C. Zeng and Z.-N. Chen, Adv. Funct. Mater., 2015, 25, 3033–3042 CrossRef CAS.
- A. F. Wells, Structural Inorganic Chemistry, Clarendon Press, Oxford, 5th edn, 1984 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. CCDC 1499658 (1), 1499630 (2), 1499654 (3), 1554172 (4), 1554173 (5), 1554171 (6), and 1554174 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03529b |
| This journal is © The Royal Society of Chemistry 2017 |