
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystalline boron-linked tetraaminoethylene radical cations†
Yuanting
Su
,
Yongxin
Li
,
Rakesh
Ganguly
 and
Rei
Kinjo
*
and
Rei
Kinjo
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Nanyang Link 21, Singapore 637371. E-mail: rkinjo@ntu.edu.sg
First published on 12th September 2017
Abstract
Single-electron oxidation of neutral boryl-linked tetraaminoethylene derivatives 4 led to the formation of radical cations 4˙+, which have been isolated and fully characterized. X-ray diffraction analysis, EPR spectroscopy, and computational studies revealed that the unpaired electron is delocalized over the B2N4C2 skeleton and the spin density mainly resides on the carbon and boron atoms.
Introduction
Boron-containing radicals have been extensively studied due to their importance in fundamental chemistry and promising applications in various organic syntheses involving radical reactions.1 Thus far, a number of stable anionic2 and neutral3 boron radicals have been reported. By stark contrast, isolable boron radical cations are extremely rare probably due to the intrinsic electron deficient nature of boron. Indeed, only a handful of boron radical cations supported by the strong electron donors L have been isolated and structurally characterized by the Bertrand (I),4 Braunschweig (II),5 Xie (III),6 Himmel (IV),7 and Harman (V)8 groups (Fig. 1a). While two boron-containing radical cations (VI9 and VII10) without Lewis base stabilization have also been reported, the unpaired electron was most likely localized in the substituents rather than the boron atoms.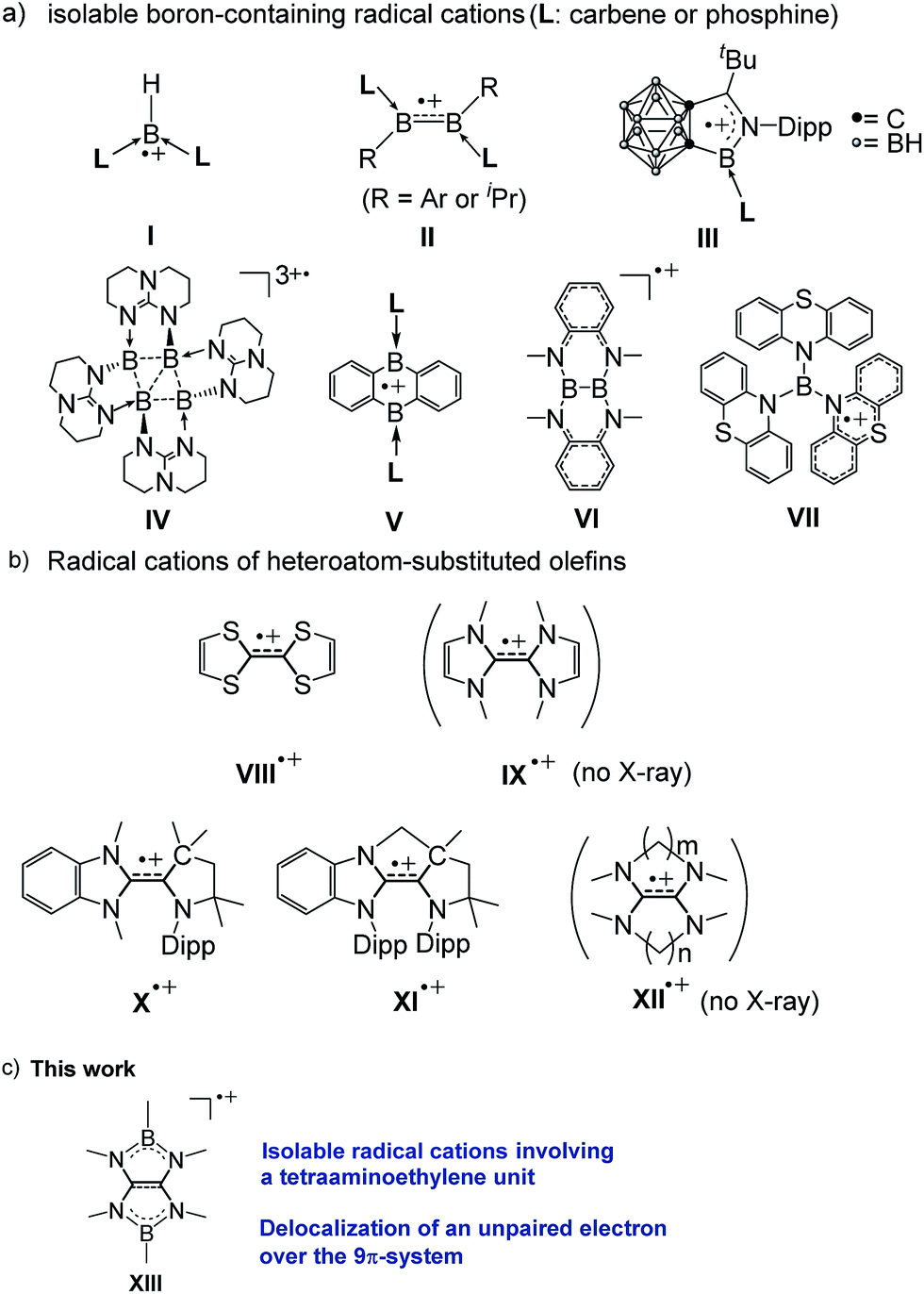 | ||
| Fig. 1 (a) Structurally characterized boron-containing radical cations. (b) Examples of radical cations of electron-rich olefins (Dipp = 2,6-diisopropylphenyl). (c) Present work. | ||
Owing to their peculiar ability to serve as powerful organic reducing reagents, electron-rich olefins (EROs) have received a surging interest more than two decades.11,12 During the redox reactions with EROs, the corresponding oxidized forms of EROs, especially the radical cations are concomitantly generated and therefore have been considered as key species (Fig. 1b). Among them, the radical cation of tetrathiafulvalene (TTF) VIII13 has been extensively studied owing to its diverse utilities in various applications.14–17 Compared to TTF, tetraazafulvalenes (TAFs) IX feature more negative potentials18 which makes them attractive organic super-electron donors11d–f,19 and organocatalysts.12 However, their corresponding radical cations IX˙+ have never been isolated thus far, mainly due to the facility of two-electron oxidation process attributed to the small electronic coupling. Recently, the Bertrand group reported two isolable triazaolefin radical cations (X˙+ and XI˙+) and demonstrated the methylene-tethered XI˙+ exhibits a larger electronic coupling.20 Similarly, it has been reported that aliphatic tetraaminoethylene (TAEs) XII linked by (poly)methylene chains increases the electronic coupling, which is, however, still not large enough to isolate the cation radical species XII˙+, and indeed no structural authentication of such species has been done to date.11d–f,19 Because of the isoelectronic and isosteric relationships between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and B–N units, boron atoms binding to nitrogen atoms have readily been incorporated into π-conjugation system.21,22 Accordingly, we envisaged that linking the nitrogen atoms of tetraaminoethylene by two boryl groups may effectively increase the electron coupling, and the corresponding radical cation could be stabilized through delocalization of both the positive charge and the unpaired electron over the B2C2N4 framework as it should mitigate the electron deficiency of the boron centers. Consequently, synthetically challenging Lewis bases-free boron radical cations would be accessible. Herein, we show that in fact radical cations XIII involving a tetraaminoalkene unit (Fig. 1c) can be isolated. Their spectroscopic properties and structures are also presented.
C and B–N units, boron atoms binding to nitrogen atoms have readily been incorporated into π-conjugation system.21,22 Accordingly, we envisaged that linking the nitrogen atoms of tetraaminoethylene by two boryl groups may effectively increase the electron coupling, and the corresponding radical cation could be stabilized through delocalization of both the positive charge and the unpaired electron over the B2C2N4 framework as it should mitigate the electron deficiency of the boron centers. Consequently, synthetically challenging Lewis bases-free boron radical cations would be accessible. Herein, we show that in fact radical cations XIII involving a tetraaminoalkene unit (Fig. 1c) can be isolated. Their spectroscopic properties and structures are also presented.
Results and discussion
Treatment of tetraaryloxalamidine 1 (ref. 23) with one equivalent of dibromophenylborane in the presence of two equivalents of diisopropylethylamine in toluene afforded 2a as a yellow powder in 78% yield. A subsequent reaction between 2a and a stoichiometric amount of dibromophenylborane in toluene immediately afforded a brown precipitate, which was collected by filtration and then washed with hexane to give 3a as a white solid in 75% yield. A toluene solution of 3a with two equivalents of potassium graphite (KC8) was stirred overnight under ambient condition, and after work up, boryl-linked tetraazaolefin derivative 4a was obtained as a yellowish green powder in 54% yield (Scheme 1). The 11B NMR spectrum of 4a displays a broad singlet at 25.4 ppm, which is shifted downfield with respect to that (9.8 ppm) of 3a. Compound 4a is thermally stable both in the solid state and in solutions, and it melts at 339 °C without decomposition. When para-substituted dibromophenylboranes with F and tBu groups were employed under the same reaction procedures, the corresponding derivatives 4b and 4c were obtained in moderate yields. In the 11B NMR spectra, a broad singlet appears at δ = 26.4 ppm (4b) and δ = 26.1 ppm (4c), respectively. Compounds 4a–c can be deemed inorganic/organic hybrid versions of pentalene dianion,24 which has been widely utilized as an ancillary ligand of metal complexes.25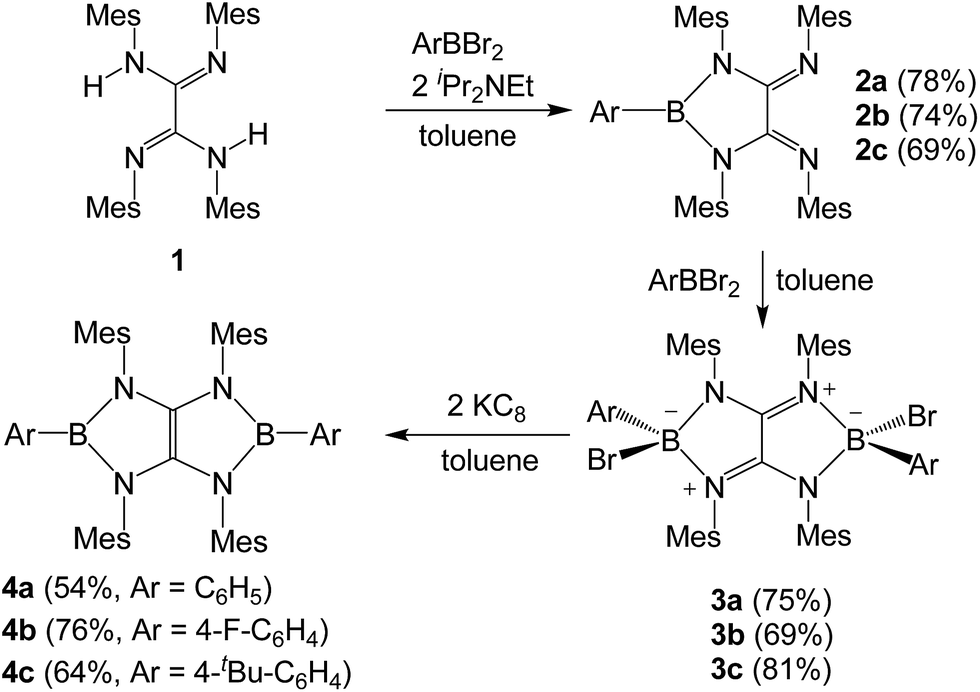 | ||
| Scheme 1 Synthesis of compound 4 (Mes = 2,4,6-trimethylphenyl). | ||
X-ray diffraction analysis of 4a confirmed the essentially planar B2N4C2 framework with a propeller-like orientation of the six aryl groups (Fig. 2a). Two boron atoms adopt perfectly trigonal-planar geometry (ΣB = 360°) with the N1–B1–N2 bond angle of 105.8(4)°. The equal B–N (1.460(5) Å) bond distances are significantly shorter than the corresponding bonds (1.581(4) Å and 1.585(4) Å) in 3a (Fig. S71†) and in the range between typical B–N single bond (1.50 Å) and BN double bond (1.31 Å), indicating the partial multiple bond property. Compared to those of 3a, there are lengthening of the C16–N1 bond (1.396(3) Å) and the C17–N2 bond (1.397(4) Å) and a markedly shortening of the C16–C17 bond (1.323(7) Å), which is close to those found in tetrakis(dimethylamino)ethylene (TDAE) (1.350(2) Å),26X (1.3459(16) Å) and XI (1.344(2) Å).20 Compound 4b (Fig. S72†) exhibits metric parameters similar to those of 4a. DFT calculation performed on 4a at the M062X/6-31G(d,p) level of theory shows that the HOMO of 4a corresponds to the π-system over the B2C2N4 skeleton featuring a node between each NBN and the central CC π-unit (Fig. 2b). Natural bond orbital (NBO) analysis gave Wiberg bond index values of the C–C bond (1.51), the B–N bonds (0.94 and 0.93) and the C–N bonds (1.05 and 1.05) of the B2C2N4 skeleton.
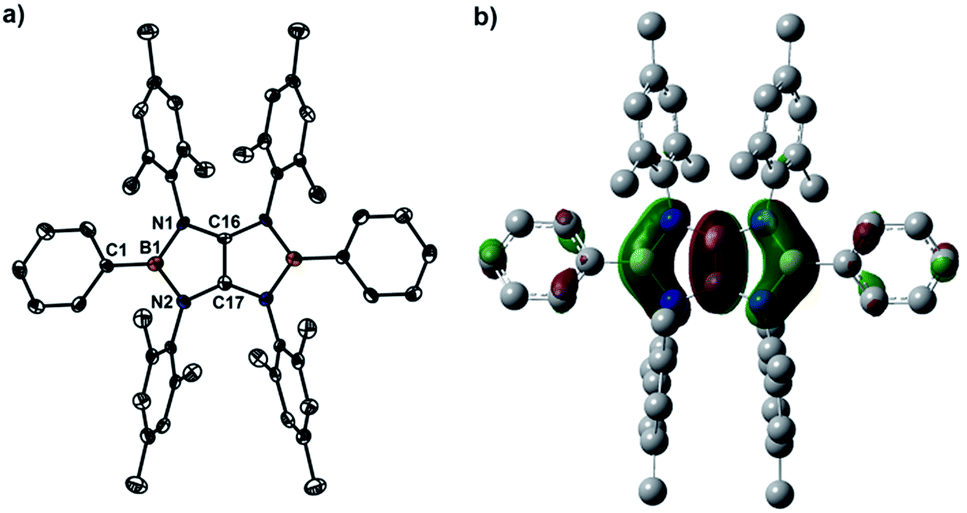 | ||
| Fig. 2 (a) Solid-state structures of 4a (hydrogen atoms are omitted for clarity). Thermal ellipsoids are set at the 50% probability level. Selected bond lengths [Å] and angles [°]: B1–C1 1.551(6), B1–N1 1.460(5), B1–N2 1.460(5), C16–N1 1.396(3), C17–N2 1.397(4), C16–C17 1.323(7), C1–B1–N1 127.1(4), C1–B1–N2 127.1(4), N1–B1–N2 105.8(4). (b) The plot of the HOMO of 4a. | ||
Cyclic voltammetry of 4a in CH2Cl2 (0.1 M nBu4NPF6 as the electrolyte) showed two well-separated oxidation waves (Fig. S79†), which is in sharp contrast to TAFs IX exhibiting very close redox waves (ΔE1/2 < 0.3 V).11d–f,18 The first oxidation of 4a at E1/2 = −0.44 V (referenced against the ferrocene/ferrocenium (Fc/Fc+) couple) was found to be a reversible process associated with the formation of the radical cation species whereas the second oxidation centered at E1/2 = 0.43 V was irreversible. Cyclic voltammograms of 4b and 4c show similar oxidation events with the first reversible (−0.44 V for 4b and −0.56 V for 4c) and second irreversible (0.41 V for 4b and 0.33 V for 4c) waves, respectively (Fig. S80 and 81†).
The first oxidation potentials of 4a–c are less negative than those of tetraazafuvalenes IX (around −1.00 V vs. SCE, Fc/Fc+: E1/2 = 0.46 V vs. SCE). Thus, upon incorporation of the boryl groups, the reducing power of compounds 4a–c becomes weaker than that of tetraazafuvalenes IX.
It has been reported that tetraaminoethylene derivative typically undergoes the addition reaction with an electrophile at the central CC moiety.27 To investigate the chemical behavior of 4, we performed their reactions with electrophiles. Treatment of a toluene solution of 4a with an equimolar amount of trifluoromethane sulfonic acid (HOTf) at room temperature afforded the corresponding conjugated acid 5a as a white powder in 85% yield (Scheme 2). The 11B NMR spectrum displays a broad singlet at 38.1 ppm which is shifted downfield with respect to that (25.4 ppm) of 4a. In the 1H and 13C NMR spectra, a singlet at 7.71 ppm for NCHN proton and 82.2 ppm and 187.4 ppm for the corresponding NCHN and NCN carbons were observed, respectively. Under the same conditions, 4b and 4c underwent protonation with HOTf to afford the respective products 5b and 5c in good yields. An X-ray diffraction studies of 5a revealed that both boron centers feature the trigonal-planar geometry. One of the central carbon atoms in the B2C2N4 ring is in the tetragonal configuration with a hydrogen atom whereas its adjacent cationic carbon exhibits the trigonal-planar geometry with a formal charge of +1 (Fig. 3a). The distance of the C25–C26 (1.490(5) Å) bond is significantly longer than the corresponding bond (1.323(7) Å) in 4a, and comparable to typical single C–C bond length. These results demonstrate the tetraaminoethylene-type nature of 4.
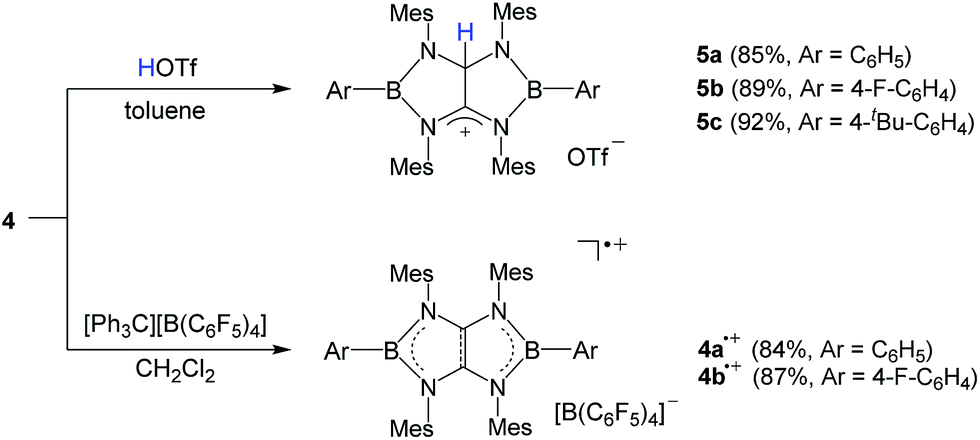 | ||
| Scheme 2 Synthesis of 4˙+ and 5. | ||
 | ||
| Fig. 3 (a) Solid-state structures of 5a (hydrogen atoms are omitted for clarity). Thermal ellipsoids are set at the 50% probability level. Selected bond lengths [Å] and angles [°]: B1–N1 1.514(6), B1–N2 1.422(6), B1–C1 1.546(7), B2–N3 1.430(6), B2–N4 1.489(6), B2–C36 1.542(7), N1–C26 1.330(5), N2–C25 1.469(5), N3–C25 1.484(5), N4–C26 1.365(5), C25–C26 1.490(5), N1–B1–N2 107.3(4), N1–B1–C1 123.3(4), N2–B1–C1 129.2(4), N3–B2–N4 107.0(4), N3–B2–C36 126.7(4), N4–B2–C36 1276.3(4). (b) Solid-state structures of 4a˙+ (hydrogen atoms are omitted for clarity). Thermal ellipsoids are set at the 50% probability level. Selected bond lengths [Å] and angles [°]: B1–C3 1.545(3), B1–N1 1.458(3), B1–N2 1.474(3), B2–N3 1.464(2), B2–N4 1.472(2), C1–N1 1.356(3), C1–N3 1.365(2), C2–N2 1.356(2), C2–N4 1.352(2), C1–C2 1.414(3), C3–B1–N1 126.64(17), C3–B1–N2 126.69 (17), N1–B1–N2 106.66(16), C27–B2–N3 125.82(16), C27–B2–N4 127.73(17), N3–B2–N4 106.40(15). | ||
In line with the oxidation potentials, chemical single-electron oxidations of 4a and 4b were readily accomplished by using of [Ph3C][B(C6F5)4] as an oxidizing agent. Reactions of 4a and 4b with [Ph3C][B(C6F5)4] in CH2Cl2 at ambient temperature smoothly generated 4a˙+ and 4b˙+ as NMR-silent red (4a˙+: 84%) and reddish brown powder (4b˙+: 87%), respectively (Scheme 2). Crystals suitable for X-ray crystallographic studies were obtained by recrystallization from a mixture of CH2Cl2 and toluene solution. Like the neutral 4a and 4b, 4a˙+ and 4b˙+ bearing the 9π-system retain the planar B2N4C2 skeletons with trigonal planar sp2 boron atoms (Fig. 3b and S73†). Upon oxidation of 4a to 4a˙+, the B–N bonds (1.458(3)–1.474(3) Å) and the CC (1.414(3) Å) bond are very slightly lengthened, while the endocyclic C–N bonds (1.352(2)–1.365(2) Å) are shortened, which is in good agreement with the electronic structure of 4. Thus, upon removal of one electron from the HOMO of 4 (Fig. 2b), the electron density in the CC and N–B–N π-bonding orbitals decreases, leading to the elongation of the CC and B–N distances, while the repulsion between the bonding (N–B–N and CC) electron pairs decreases resulting in the shortening of the C–N bond distances. Note that 4˙+ represent the first example of radical cations derived from BN heterocycles featuring some aromatic nature (Fig. S84, Table S2†).21,28 Moreover, 4˙+ correspond to the anion radical of organic pentalene, among which only the derivatives featuring thermodynamically highly stabilizing benzo substituents have been structurally characterized.29
The EPR spectrum of 4a˙+ measured in fluorobenzene at room temperature displays a complex system (g = 2.0050) arising from couplings with two boron (a(11B) = 1.16 G, a(10B) = 0.63 G), four nitrogen (a(14N) = 1.53 G), and four hydrogen atoms (a(1H) = 1.32 G) at the ortho-positions of phenyl groups (Fig. 4a). The 11B hyperfine coupling constant is smaller than those of I (6.43 G)4 and isolable boron radical anions,2 but comparable to those of II (≤1.18 G)5 and reported stable neutral boron radicals (0.96–8.5 G).3 The moderate a(11B) coupling constant together with relative small a(14N) values suggests delocalization of the unpaired electron over the B2N4C2 core. To gain more insight into the electronic structure of 4a˙+, unrestricted DFT calculations were performed at the UM062X/6-31G(d,p) level. NBO method confirmed that the spin density is entirely delocalized over the B2N4C2 framework with some extension to the two Ph groups on the B atoms (Fig. 4b). The spin density is estimated to be present mainly on the carbon (0.26e × 2) and the boron (0.19e × 2) atoms with a relatively small contribution of the nitrogen atoms (0.02e × 4). While the several resonance forms including 4x˙+, 4y˙+, 4z˙+ can be drawn for 4˙+ (Fig. 4c), the X-ray diffraction analysis, ESR spectra in combination with DFT results indicate that compared with the N-centered radical form 4z˙+, contribution of others including the tetraaminoalkene radical cation 4x˙+ and boron radical cation 4y˙+ is more significant to the actual electronic structure of 4˙+.
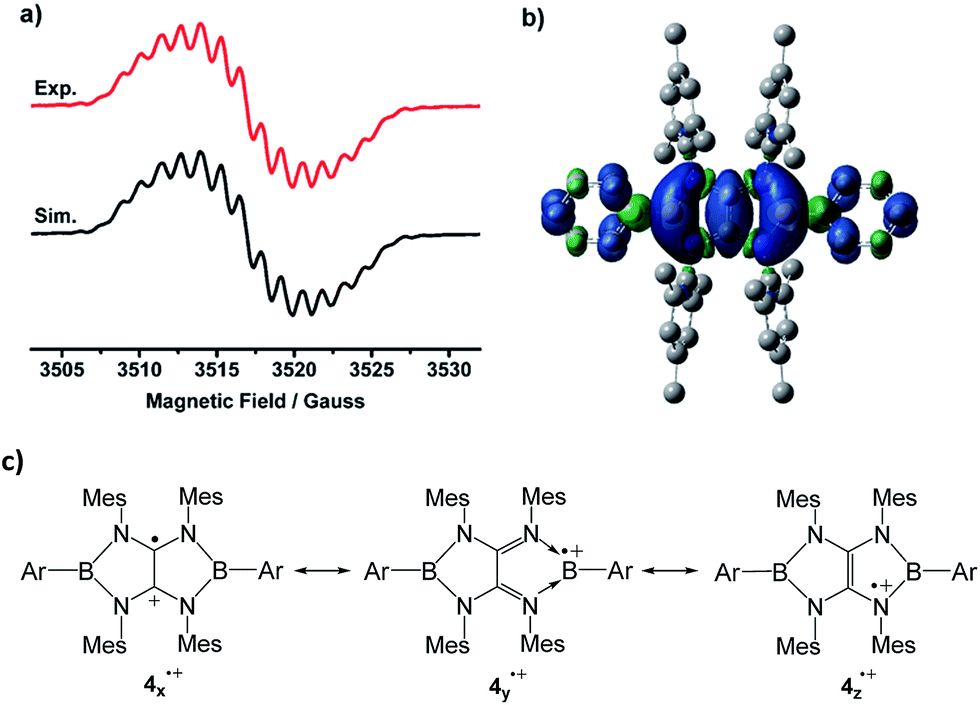 | ||
| Fig. 4 (a) Experimental (red) and simulated (black) EPR spectra of 4a˙+ in fluorobenzene at room temperature. (b) The plot of the spin density of 4a˙+. (c) The selected resonance forms of 4˙+. | ||
Conclusions
In summary, we have shown that boryl-tethered tetraaminoalkene derivatives 4 can be readily synthesized and they undergo protonation reaction and one-electron oxidation reaction. The former afforded the corresponding salts 5 whereas the latter led to the formation of isolable radical cations 4˙+. X-ray diffraction analysis, EPR spectra as well as computational studies of 4˙+ revealed the unpaired electron delocalized over the B2N4C2 framework mainly resides on the carbon and boron atoms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to to Nanyang Technological University (NTU) and the Singapore Ministry of Education (MOE2015-T2-2-032) for financial support. We also sincerely appreciate Prof. Richard Webster for the EPR characterizations.Notes and references
- (a) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC; (b) J. C. Walton, Angew. Chem., Int. Ed., 2009, 48, 1726–1728 CrossRef CAS PubMed; (c) W. Kaim, N. S. Hosmane, S. Záliš, J. A. Maguire and W. N. Lipscomb, Angew. Chem., Int. Ed., 2009, 48, 5082–5091 CrossRef CAS PubMed; (d) D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed; (e) X. Pan, E. Lacôte, J. Lalevée and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 5669–5674 CrossRef CAS PubMed; (f) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (g) T. Chivers and J. Konu, in Comprehensive Inorganic Chemistry II: From Elements to Applications, Vol. 1: Main-Group Elements, Including Noble Gases, ed. T. Chivers, Elsevier, Amsterdam, 2013, pp. 349–373 Search PubMed.
- (a) M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1986, 108, 4235–4236 CrossRef CAS; (b) W. J. Grigsby and P. P. Power, Chem.–Eur. J., 1997, 3, 368–375 CrossRef CAS; (c) H. Braunschweig, V. Dyakonov, J. O. C. Jimenez-Halla, K. Kraft, I. Krummenacher, K. Radacki, A. Sperlich and J. Wahler, Angew. Chem., Int. Ed., 2012, 51, 2977–2980 CrossRef CAS PubMed; (d) T. Kushida and S. Yamaguchi, Organometallics, 2013, 32, 6654–6657 CrossRef CAS; (e) A. Hübner, A. M. Diehl, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2014, 53, 4832–4835 CrossRef PubMed; (f) Y. Zheng, J. Xiong, Y. Sun, X. Pan and J. Wu, Angew. Chem., Int. Ed., 2015, 54, 12933–12936 CrossRef CAS PubMed; (g) A. Hübner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2015, 137, 3705–3714 CrossRef PubMed; (h) L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS PubMed; (i) H. Asakawa, K.-H. Lee, K. Furukawa, Z. Lin and M. Yamashita, Chem.–Eur. J., 2015, 21, 4267–4271 CrossRef CAS PubMed; (j) N. Yuan, W. Wang, Z. Wu, S. Chen, G. Tan, Y. Sui, X. Wang, J. Jiang and P. P. Power, Chem. Commun., 2016, 52, 12714–12716 RSC; (k) L. Wang, Y. Fang, H. Mao, Y. Qu, J. Zuo, Z. Zhang, G. Tan and X. Wang, Chem.–Eur. J., 2017, 23, 6930–6936 CrossRef CAS PubMed; (l) R. Mondol, D. A. Snoeken, M.-C. Chang and E. Otten, Chem. Commun., 2017, 53, 513–516 RSC; (m) N. Yuan, W. Wang, Y. Fang, J. Zuo, Y. Zhao, G. Tan and X. Wang, Organometallics, 2017, 36, 2498–2501 CrossRef CAS.
- (a) C.-W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 1723–1725 CrossRef CAS PubMed; (b) Y. Aramaki, H. Omiya, M. Yamashita, K. Nakabayashi, S. Ohkoshi and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 19989–19992 CrossRef CAS PubMed; (c) R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed; (d) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed; (e) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed; (f) A. D. Ledet and T. W. Hudnall, Dalton Trans., 2016, 45, 9820–9826 RSC; (g) L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2016, 138, 2500–2503 CrossRef CAS PubMed; (h) L. E. Longobardi, P. Zatsepin, R. Korol, L. Liu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2017, 139, 426–435 CrossRef CAS PubMed; (i) M. F. S. Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135–1140 CrossRef PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, T. Kupfer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 5689–5693 CrossRef CAS PubMed; (b) P. Bissinger, H. Braunschweig, A. Damme, C. Hörl, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2015, 54, 359–362 CrossRef CAS PubMed.
- H. Wang, J. Zhang, Z. Lin and Z. Xie, Organometallics, 2016, 35, 2579–2582 CrossRef CAS.
- S. Litters, E. Kaifer and H.-J. Himmel, Angew. Chem., Int. Ed., 2016, 55, 4345–4347 CrossRef CAS PubMed.
- J. W. Taylor, A. M. Skimming, C. F. Guzman and W. H. Harman, J. Am. Chem. Soc., 2017, 139, 11032–11035 CrossRef CAS PubMed.
- X. Xie, C. J. Adams, M. A. M. Al-Ibadi, J. E. McGrady, N. C. Norman and C. A. Russell, Chem. Commun., 2013, 49, 10364–10366 RSC.
- S. Suzuki, K. Yoshida, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2013, 52, 2499–2502 CrossRef CAS PubMed.
- (a) K. Deuchert and S. Hünig, Angew. Chem., Int. Ed. Engl., 1978, 17, 875–886 CrossRef; (b) S. Hünig and H. Berneth, Top. Curr. Chem., 1980, 92, 1–44 CrossRef; (c) J. A. Murphy, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley-VCH, Weinheim, 2012, p. 817 Search PubMed; (d) J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed; (e) E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073–6087 RSC; (f) J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384–413 CrossRef CAS PubMed.
- For recent examples, see: (a) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673–3676 CrossRef CAS PubMed; (b) E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 52, 2239–2242 CrossRef CAS PubMed; (c) E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934–10937 CrossRef CAS PubMed; (d) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS PubMed; (e) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., Int. Ed., 2016, 55, 4492–4496 CrossRef CAS PubMed; (f) J. Broggi, M. Rollet, J.-L. Clément, G. Canard, T. Terme, D. Gigmes and P. Vanelle, Angew. Chem., Int. Ed., 2016, 55, 5994–5999 CrossRef CAS PubMed.
- F. Wudl, D. Wobschall and E. J. Hufnagel, J. Am. Chem. Soc., 1972, 94, 670–672 CrossRef CAS.
- D. Lorcy, N. Bellec, M. Fourmigué and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS.
- M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153–164 RSC.
- (a) M. Bendikov and F. Wudl, Chem. Rev., 2004, 104, 4891–4946 CrossRef CAS PubMed; (b) U. Geiser and J. A. Schlueter, Chem. Rev., 2004, 104, 5203–5242 CrossRef CAS PubMed; (c) A. L. Kanibolotsky, N. J. Findlay and P. J. Skabar, Beilstein J. Org. Chem., 2015, 11, 1749–1766 CrossRef CAS PubMed; (d) R. Pfattner, S. T. Bromley, C. Rovira and M. Mas-Torrent, Adv. Funct. Mater., 2016, 26, 2256–2275 CrossRef CAS.
- D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- J. Garnier, D. W. Thomson, S. Zhou, P. I. Jolly, L. E. A. Berlouis and J. A. Murphy, Beilstein J. Org. Chem., 2012, 8, 994–1002 CrossRef CAS PubMed.
- N. Wiberg, Angew. Chem., Int. Ed. Engl., 1968, 7, 766–779 CrossRef CAS.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed.
- (a) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed; (b) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef; (c) Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed.
- See also: (a) B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS PubMed; (b) D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS PubMed; (c) B. Su, Y. Li, R. Ganguly, J. Lim and R. Kinjo, J. Am. Chem. Soc., 2015, 137, 11274–11277 CrossRef CAS PubMed.
- R. Bauer, Ber. Dtsch. Chem. Ges., 1907, 40, 2650–2662 CrossRef CAS.
- (a) J. J. Stezowski, H. Hoier, D. Wilhelm, T. Clark and P. v. R. Schleyer, J. Chem. Soc., Chem. Commun., 1985, 1263–1264 RSC; (b) T. J. Katz, M. Rosenberger and R. K. O'Hara, J. Am. Chem. Soc., 1964, 86, 249–252 CrossRef CAS; (c) T. J. Katz and M. Rosenberger, J. Am. Chem. Soc., 1962, 84, 865–866 CrossRef CAS.
- H. Butenschön, Angew. Chem., Int. Ed. Engl., 1997, 36, 1695–1697 CrossRef.
- H. Bock, H. Borrmann, Z. Havlas, H. Oberhammer, K. Ruppert and A. Simon, Angew. Chem., Int. Ed., 1991, 30, 1678–1681 CrossRef.
- (a) D. M. Lemal, R. A. Lovald and K. I. Kawano, J. Am. Chem. Soc., 1964, 86, 2518–2519 CrossRef CAS; (b) J. Hocker and R. Merten, Angew. Chem., Int. Ed., 1972, 11, 964–973 CrossRef CAS; (c) R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schütz, Angew. Chem., Int. Ed., 2004, 43, 5896–5911 CrossRef CAS PubMed; (d) M. F. Lappert, S. Álvarez, G. Aullón, R. Fandos, A. Otero, A. Rodríguez, S. Rojas and P. Terreros, Eur. J. Inorg. Chem., 2009, 1851–1860 CrossRef CAS.
- (a) L. Weber, Coord. Chem. Rev., 2001, 215, 39–77 CrossRef CAS; (b) E. R. Abbey and S.-Y. Liu, Org. Biomol. Chem., 2013, 11, 2060–2069 RSC; (c) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS.
- M. Saito, Y. Hashimoto, T. Tajima, K. Ishimura, S. Nagase and M. Minoura, Chem.–Asian J., 2012, 7, 480–483 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, crystallographic data, crystal structures, UV-vis spectra, cyclic voltammograms, EPR spectra, and theoretical calculation. CCDC 1548460–1548466. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03528d |
| This journal is © The Royal Society of Chemistry 2017 |