
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in the selective dispersion of single-walled carbon nanotubes using conjugated polymers
Darryl
Fong
and
Alex
Adronov
 *
*
Department of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, ON, Canada. E-mail: adronov@mcmaster.ca
First published on 7th August 2017
Abstract
A significant barrier that impedes the commercialization of single-walled carbon nanotube-related applications is that all known synthetic methods produce a complicated mixture of semiconducting and metallic species. For device applications, pure semiconducting or pure metallic samples are desirable. Thus far, the purification methods that have been identified are capable of separating individual carbon nanotube species on a microgram scale, but purification on a large scale has remained elusive. The use of conjugated polymers to selectively disperse specific nanotube species is a promising approach to resolve the scalability issue, but a comprehensive understanding of the selectivity mechanism has not yet been achieved. Here, several of the trends reported in the literature are outlined to further the rational design of conjugated polymers for nanotube sorting. Numerous variables influence dispersion selectivity, including polymer structure and molecular weight, nanotube type used, sonication temperature, amount of polymer relative to nanotube, and solvent. We have organized these seemingly disparate parameters into two simple categories: conjugated polymer structure, and dispersion preparation conditions. Most importantly, we consider the mechanistic arguments that have been proposed, and provide additional insights based on the observations in the literature.
1. Introduction
Within the realm of nano-scale materials, the recently discovered allotropes of carbon, including fullerenes, carbon nanotubes, and graphene, have attracted significant research attention. In particular, since the first deliberate preparation methods of single-walled carbon nanotubes (SWNTs) were reported,1,2 a substantial number of studies have focused on exploiting the unique properties of this nanomaterial. The high tensile strength,3 aspect ratio,4 thermal and electrical conductivity,5–8 and extraordinary optical characteristics9–11 of SWNTs find utility in numerous applications, including high-strength nanocomposites,12–14 field-effect transistors (FETs),15 sensors,16–19 photodetectors,20 organic photovoltaics (OPVs),21–23 flexible electronics,24,25 touch screens,26 microelectronic interconnects,27 and other devices.28 Indeed, semiconducting SWNTs (sc-SWNTs) have demonstrated performance superior to that of traditional silicon-based semiconductors,15 while metallic SWNTs (m-SWNTs) are superior conductors to traditional metals, such as copper.29 Though SWNT commercialization has begun,27 the promise of transformative technologies has not materialized, especially in cases where nanotube purity is critical to the application. Nanotube purity is a challenge because all known commercial synthetic methods, including high-pressure carbon monoxide disproportionation (HiPCO),30 chemical vapour deposition (CVD),31 arc-discharge,32 laser ablation,33 and plasma torch growth,34 produce a roughly 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of sc-SWNTs and m-SWNTs. In the pursuit of electronically-pure SWNTs, differences in the exploitable properties used to separate the electronic types are exceedingly small, and thus their separation is extremely difficult.
:1 mixture of sc-SWNTs and m-SWNTs. In the pursuit of electronically-pure SWNTs, differences in the exploitable properties used to separate the electronic types are exceedingly small, and thus their separation is extremely difficult.
Several methods for isolating electronically-enriched SWNTs have recently been developed. The first, and arguably most transformative, is density gradient ultracentrifugation (DGU), described in the seminal papers by Hersam and co-workers that appeared in 200535 and 2006.36 This technique allowed, for the first time, access to electronically-enriched samples of SWNTs, and enabled follow-up studies that developed a variety of SWNT-based devices.28 DGU is capable of separating individual SWNT species; also referred to as chiralities (for a thorough discussion on SWNT properties, including chirality, chiral vectors, chiral angles, and electronics, we refer the reader to the excellent review by Charlier37). Subsequently, agarose gel filtration,38 electrophoresis,39 two-phase extraction,40,41 and size-exclusion chromatography42,43 were developed as alternative separation methods for m- and sc-SWNTs. As well, single-stranded DNA, in combination with ion exchange chromatography, was found to be capable of separating individual SWNT species.44–46 However, with all the aforementioned purification methods, the yield of electronically-enriched SWNTs is a major limitation. These methods typically isolate microgram quantities of enriched material after extensive purification procedures, and the difficulty in obtaining large quantities of enriched material is reflected in the exorbitant cost of commercially-available enriched SWNTs. At the time of writing this Perspective, one milligram of >99% enriched sc- or m-SWNT samples could be purchased from Raymor Industries Inc. at a cost of $695 and $899 USD, respectively (prices quoted directly from Raymor in June, 2017).
An alternative strategy that has the potential to address the scalability challenge involves the selective dispersion of SWNTs using conjugated polymers (CPs). Since the initial reports that both aromatic small molecules (i.e., pyrene)47 and CPs [i.e., poly(phenylene vinylene) (PPV)]48,49 exhibit interactions with SWNTs, nanotube dispersions have been prepared using a variety of CP structures.50–52 The potential for selective CP–SWNT dispersions went practically unnoticed (except for a brief report in the patent literature)53 until the ground-breaking work of Nicholas and co-workers in 2007.54 This group was the first to demonstrate that simple commercially-available CPs, including poly(9,9-di-n-octylfluorene) (PFO), can selectively disperse a small subset of sc-SWNT chiralities. Since then, studies of CP–SWNT interactions have dramatically increased, with numerous reports of highly enriched sc-SWNT dispersions having appeared, some with purities in excess of 99.9%.55 Recently, polymer structures that utilize backbone conformational changes or depolymerization to desorb from the SWNT surface have been developed to allow for the isolation of pristine SWNTs,52 with some systems demonstrating both enrichment and release of sc-SWNTs.56–58 Overall, the selective dispersion of SWNTs using CPs is arguably the most viable method for time-efficient and scalable SWNT subtype enrichment.
Despite the examples of selective sc-SWNT dispersions using CPs reported thus far, a fundamental understanding of the underlying principles behind the observed selectivity is limited. Currently, the ability to selectively isolate substantial quantities of a desired SWNT subtype or chirality from its parent mixture using a rationally designed CP remains elusive. The difficulty in identifying polymer characteristics that dictate nanotube selectivity arises from the fact that systematic variation of CP structure is non-trivial. Structural changes to monomers result in differences in solubility, stability, and polymerization reactivity, the combination of which makes it difficult to control polymer molecular weight and dispersity (Đ). This is problematic because polymer molecular weight is known to impact dispersion selectivity, and so a rigorous analysis of structure–selectivity relationships necessitates that polymers within a series under investigation have comparable degrees of polymerization. Although this requirement is infrequently satisfied, the numerous published examples of selective dispersions allow consideration of the most important polymer characteristics that dictate dispersion selectivity. Analysis of these examples make it possible to glean insight into mechanistic considerations that can be used to improve our understanding of this observed selectivity in the future.
In this Perspective, we focus on recent advances in the selective dispersion of SWNTs using CPs, and attempt to illuminate the general features of polymer structure and preparation conditions that control dispersion selectivity. Although the number of potential variables is vast, we hope to provide researchers with a methodical framework to organize the observations on dispersion selectivity to date. As depicted in Fig. 1, the general process of dispersing SWNTs begins with the sonication of a mixture of SWNT powder and solubilized CP. Careful consideration must be made with respect to polymer structure and molecular weight, as well as the SWNT source, sonication temperature, CP:SWNT mass ratio, and identity of the solvent. Sonication is followed by mild centrifugation to remove SWNT bundles and isolate polymer-wrapped, individualized SWNTs. Within this protocol, two opportunities arise for control over the outcome of dispersion selectivity: CP structure and dispersion preparation conditions. We emphasize that these two categories can be used to organize the observations that have been made thus far, and to simplify the initially overwhelming number of possible variables. The intent of this Perspective is to help elucidate consistent trends in dispersion selectivity, and to guide future explorations aimed at unravelling the selectivity mechanism.
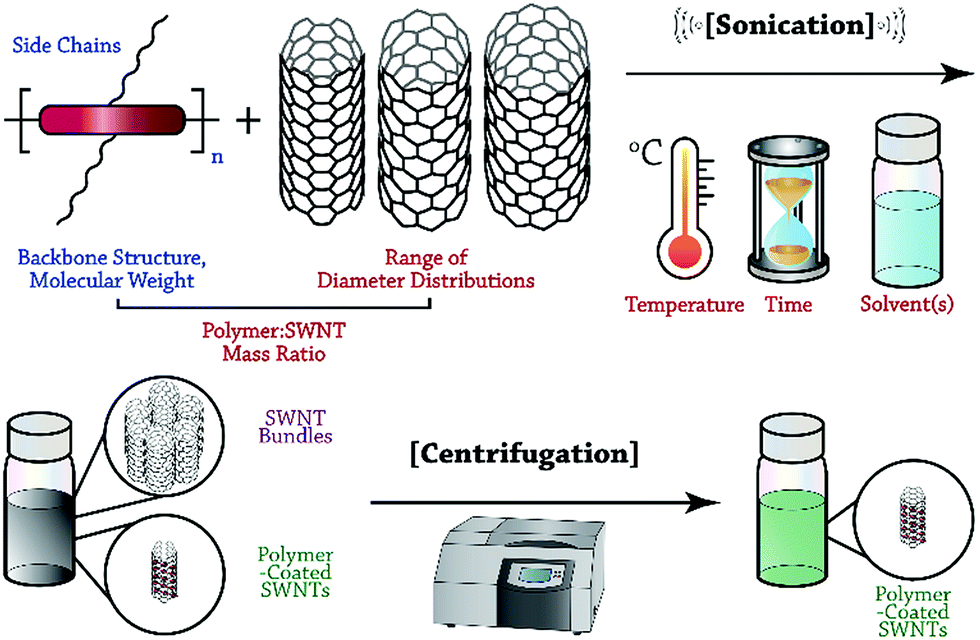 | ||
| Fig. 1 General overview of the SWNT dispersion protocol. Soluble CP is sonicated with insoluble SWNT powder, and then SWNT bundles are removed by mild centrifugation. The variables involved in dispersion selectivity are categorized, where polymer features are labelled in blue and preparation conditions are labelled in red. | ||
2. Conjugated polymer structure
Although numerous reports about the interactions of CPs with SWNTs have appeared in the literature, we consider here only reports explicitly discussing selectivity toward specific SWNT species. Though this work has led to the investigation of a wide array of CP–SWNT complexes, the results of these investigations are non-trivial to assemble into a cohesive discussion because of the significant differences in the way these studies were carried out. We have attempted to gather the most relevant experimental findings, and summarize the most important messages garnered. Before delving into the different polymer features that influence dispersion selectivity, it is instructive to point out that commercial syntheses of SWNTs lead to samples that differ in their diameter ranges, depending on the synthetic methodology used. In order from smallest to largest average diameters, the different SWNT types include: CoMoCAT (0.7–0.9 nm), HiPCO (0.8–1.2 nm), plasma torch (1.1–1.5 nm), arc-discharge (1.2–1.7 nm), and TUBALL (1.5–2.0 nm).52 In addition, batch-to-batch variability within any given SWNT synthesis is significant, and warrants that, if results are to be directly compared, experiments should be performed using SWNTs of the same type and from the same batch. Generally, researchers list the SWNT type used in their experimental work, and provide the batch number. Exact reproduction of results necessitates that the same SWNT type and, preferably, batch number be used, which may not always be feasible.Backbone structure
Amongst the different CP structures that have been found selective for specific SWNT species, the vast majority have focused on polymers based on the fluorene monomer unit, followed by thiophene- and carbazole-based polymers (Fig. 2). Copolymers of these core monomers with other monomer types have also been considered. At the end of this section, we provide a summary of the most salient information from the numerous structures used to prepare selective dispersions.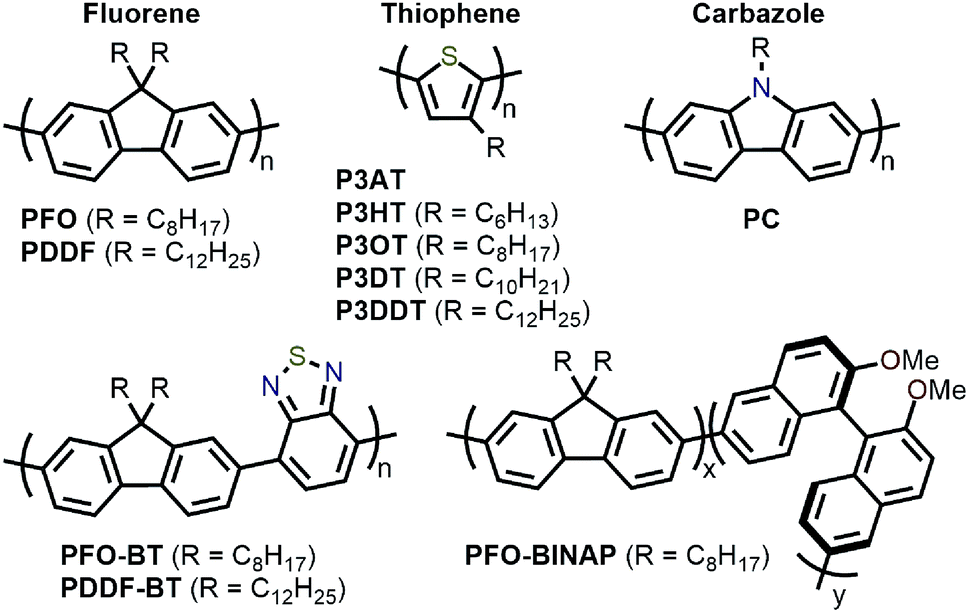 | ||
| Fig. 2 Chemical structures of homopolymers derived from fluorene, thiophene, and carbazole monomers, as well as copolymers of fluorene with benzothiadiazole (BT) or binaphthol (BINAP) units. | ||
Arguably, the investigation of selective CP–SWNT dispersions began with the seminal work by Nicholas and co-workers, in which the notable selectivity of PFO for sc-SWNTs was first reported.54 In this work, dispersions produced by sonicating SWNTs in the presence of PFO were compared to dispersions produced using the non-selective surfactant, sodium dodecylbenzenesulfonate (SDBS). It was found that the PFO–SWNT dispersion in toluene only contained a small subset of the original SWNT population (Fig. 3a), and had a substantial enrichment of sc-SWNTs compared to the non-selective SDBS surfactant (Fig. 3b). Interestingly, addition of benzothiadiazole (BT) as a co-monomer to produce poly[(9,9-di-n-octylfluorene)-alt-(benzo[2,1,3]thiadiazole)] (PFO–BT) biased the selectivity toward larger-diameter SWNTs (Fig. 3c), while still only dispersing a small subset of species (Fig. 3d). In a follow-up study, Nicholas and co-workers examined a range of dispersion solvents and (primarily) fluorene derivatives, and found that PFO and PFO–BT were still the most selective when dispersions were prepared in toluene or xylene.59 In THF, a significant decrease in selectivity was observed. Chloroform was also examined, but its density precluded sedimentation of bundles during centrifugation, and resulted in poor dispersion selectivity (for a discussion on solvent effects, vide infra). Based on this work, the authors postulated that increased rigidity of the polymer backbone enhanced interaction selectivity as a result of limited backbone conformational freedom, enabling more efficient π–π stacking with the SWNT surface.
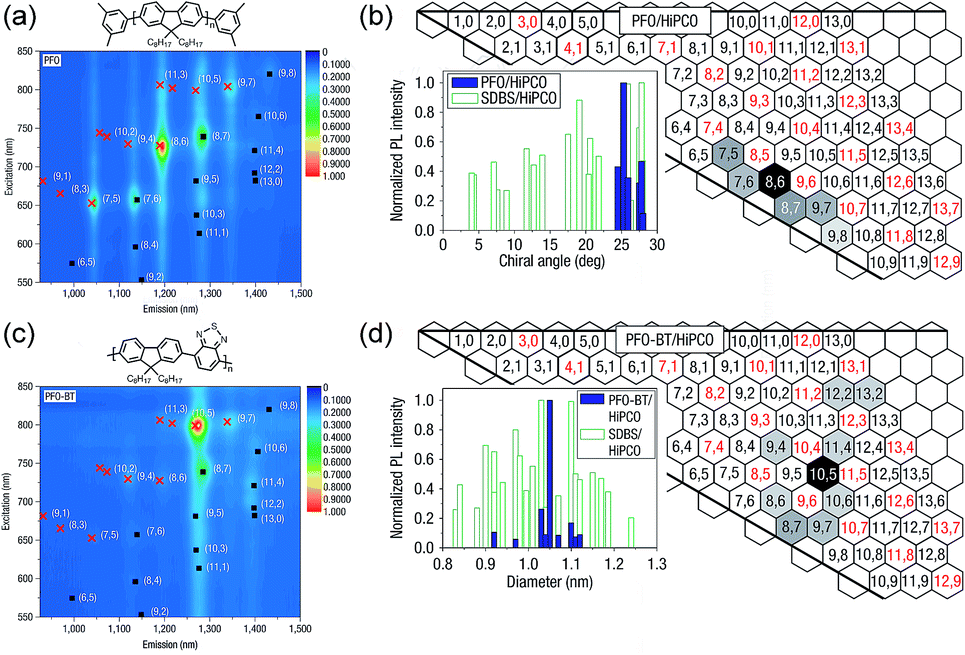 | ||
| Fig. 3 First example of the selective dispersion of SWNTs using CPs. (a) PL and (b) graphene sheet maps of PFO–SWNT dispersions in toluene. (c) PL and (d) graphene sheet maps of PFO–BT–SWNT dispersions in toluene. Graphene sheet maps use normalized PL map intensities and compare the relative fluorescence intensities of different SWNT species present in the SWNT dispersion. Reprinted with permission from ref. 54. Copyright 2007, Nature Publishing Group. | ||
In a separate study, Chen et al. used the same set of polymers as in the original Nicholas work to disperse SWNT samples prepared using the cobalt catalyst, Co-MCM-41, which produced a narrower SWNT diameter distribution than what is found in commercial samples.60 The same selectivity trends were observed, with PFO dispersing smaller-diameter sc-SWNTs (0.83–1.03 nm) than PFO–BT (1.03–1.07 nm). When dispersing HiPCO SWNTs with these polymers, species selectivity was identical to that described by Nicholas, providing a valuable reproduction of the results. In addition, Kappes and co-workers further optimized the polymer concentration and CP:SWNT mass ratio for dispersions prepared using PFO and poly(9,9-di-n-dodecylfluorene) (PDDF) to produce samples that were predominantly enriched with a single SWNT species, including (9,7), (7,5), (7,6), or (10,5), by using various combinations of CP and SWNT starting material.61 Limiting the amount of CP relative to SWNT was critical to enriched single-chirality dispersions, with excess polymer being shown to decrease dispersion selectivity. These experiments illuminate the impact that preparation conditions have on the eventual composition of dispersed SWNTs, even when the CP structure remains the same. As a further extension of the selectivity with PFO-based polymers, Nakashima and co-workers examined the effect of chirality in the polymer backbone on dispersion selectivity.62 It was found that, in addition to maintaining selectivity for sc-SWNTs in toluene, changing the handedness of the polymer through the incorporation of a chiral binapthol (BINAP) co-monomer within the polymer backbone resulted in isolation of the two enantiomeric forms of the (6,5) and (7,5) SWNT species.
With the established selectivity of fluorene-based CPs for sc-SWNTs, a number of research groups have attempted to modify this polymer backbone to alter selectivity toward specific SWNT diameters. Small-diameter sc-SWNTs are of interest for OPVs, as the larger bandgap allows for the formation of an improved type-II heterojunction with fullerene acceptors.63 Large-diameter sc-SWNTs (>1.3 nm), meanwhile, are ideal for FETs that require low-bandgap semiconductors, as SWNT diameter is inversely proportional to bandgap.64 It has been posited that diameter selectivity is correlated to the flexibility of the CP backbone, with more flexible structures being able to conform to the larger surface curvatures of small-diameter sc-SWNTs; the opposite being the case for more rigid polymer backbone structures.63 Iijima and co-workers carried out a study with PFO–BT and enriched the (15,4) sc-SWNT chirality, confirming previous results that showed PFO–BT was selective for larger-diameter SWNTs compared to fluorene homopolymer (i.e., PDDF).65 In work by Gerstel et al., a library of triazole-containing polymers with different backbone structures was prepared using Cu-catalyzed azide–alkyne click chemistry (Fig. 4).66 Combinations of different fluorene and non-fluorene monomers were used to produce a series of polymers that varied in backbone structure, side-chain identity, and molecular weight. It was found that the most selective dispersions were prepared when only fluorene-based monomers were copolymerized, and that selectivity for sc-SWNTs was akin to other polyfluorene CPs that were prepared by Pd cross-coupling. The same group also investigated variation in CP backbone structure by producing a library of 23 different polymers that included naphthalene, anthracene, or anthraquinone monomers copolymerized with 9,9-di-n-dodecylfluorene or N-decylcarbazole.67 Unfortunately, the number average molecular weights (Mn's) and Đ's of this series of CPs ranged from 1–60 kDa and 1.1–5.7, respectively, making a comparison between different CP backbones difficult. Nevertheless, it was generally found that 2,7-linked CPs produced more stable SWNT dispersions than the 3,6-linked analogs (Fig. 5). In addition, the authors found that the alternating copolymer composed of 9,9-di-n-dodecylfluorene-2,7-diyl and anthracene-1,5-diyl units (PDDF-A; Mn = 21 kDa; Đ = 2.4) enriched sc-SWNT species in the upper diameter range of the HiPCO starting material (>0.95 nm). In a later study, a similar anthracene-containing polymer was shown by Blackburn and co-workers to be selective for primarily (10,8) sc-SWNTs, which have a diameter of 1.24 nm (Fig. 5).68 Alongside this work, other co-monomers that modified the conjugated backbone surface area have been investigated. It was consistently found that, within a given polymer series, increasing the aromatic surface area of repeat units within the backbone was correlated with the dispersion of larger-diameter sc-SWNTs.69–71
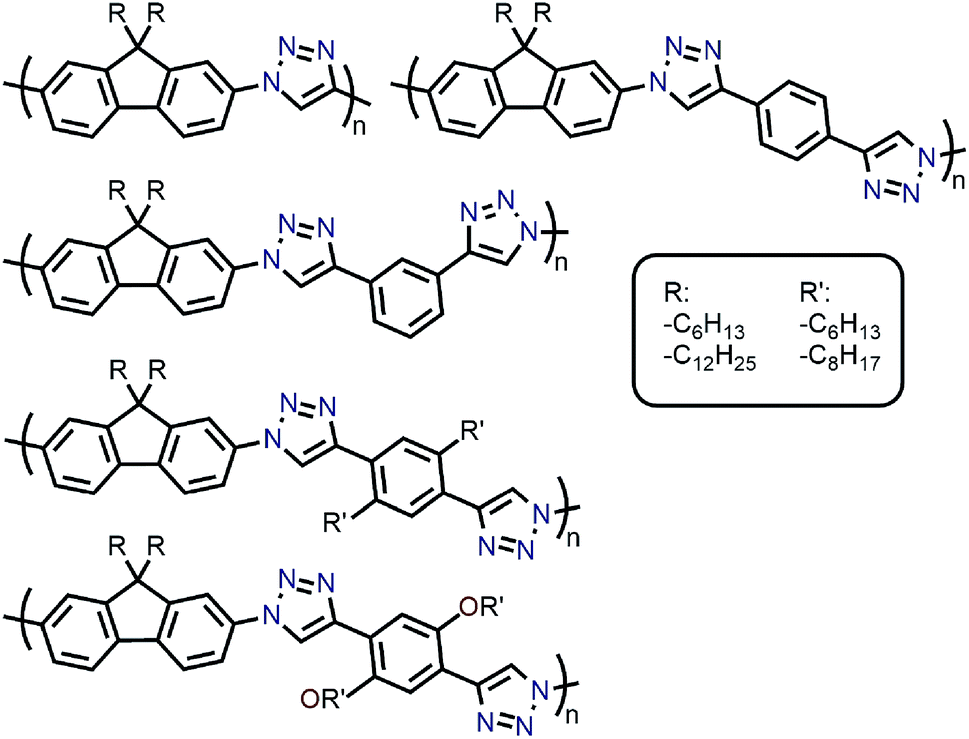 | ||
| Fig. 4 Chemical structures of triazole-containing CPs. | ||
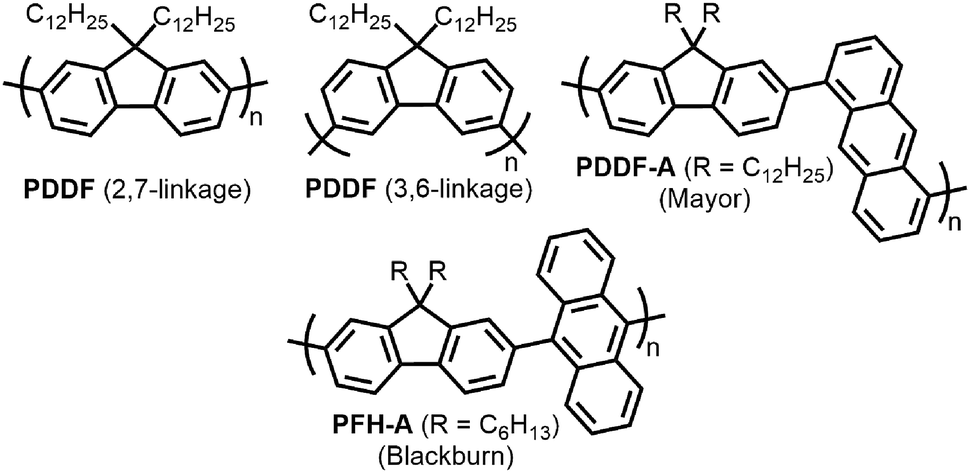 | ||
| Fig. 5 Chemical structures of PDDF with 2,7-linkages and 3,6-linkages, as well as copolymers containing fluorene and anthracene-based monomers. | ||
In the pursuit of large-diameter sc-SWNTs for FET applications, copolymers of fluorene with pyridine derivatives have been frequently utilized (Fig. 6). Tange et al. showed that the commercially-available copolymer of 9,9-di-n-octylfluorene and pyridine (PFO–Py; Mn = 7.1 kDa, Đ = 2.1) dispersed a narrow range of large-diameter sc-SWNTs (1.23–1.38 nm), which was narrower than what had been achieved previously using PFO–BT.72,73 These sc-SWNTs, mainly composed of the (13,5), (14,3), and (10,8) species, exhibited NIR emission bands in the 1500–1600 nm range, in line with telecommunication wavelengths. In work by Mayor and co-workers, it was also found that PFO–Py (Mn = 10.8 kDa, Đ = 2.28) was selective for a similar diameter range of sc-SWNTs (1.15–1.30 nm).74 Though the CP molecular weights and dispersities differ between the studies, these differences are within the errors of synthetic reproducibility and molecular weight determination. Despite the differences in the preparation conditions (CP:SWNT mass ratio and amount of solvent), the results are in reasonable agreement with Tange et al. Commercially-available polymers containing 9,9-di-n-octylfluorene copolymerized with bipyridine (PFO–BPy) were also shown to be selective for large-diameter sc-SWNTs.68 Blackburn and co-workers found that commercially-available PFO–BPy (American Dye Source, 10–100 kDa) could efficiently disperse large-diameter sc-SWNTs produced by laser vapourization (LV), resulting in concentrated inks. The authors demonstrated that this CP could disperse 33% of the sc-SWNTs originally present within the SWNT starting material. Interestingly, comparison to the analogous polymers containing pyridine and terpyridine co-monomers resulted in similar diameter selectivity, but did not produce the concentrated dispersions found when using PFO–BPy. In order to produce FETs, Arnold, Gopalan, and co-workers also exploited the ability of PFO–BPy to efficiently disperse large-diameter sc-SWNTs.75 The FETs produced from the PFO–BPy–SWNT dispersion exhibited a mobility of 46 cm2 V−1 s−1, an on-conductance of 7.3 μS μm−1, and an on/off ratio of 5 × 105. By preparing a large number of devices and measuring the frequency of device short-circuiting, the authors estimated the semiconducting purity of the dispersed SWNTs to be in excess of 99.9%. In subsequent work, the same collaborators were able to produce arrays of highly enriched and aligned sc-SWNTs on Si/SiO2 substrates using the Dose-controlled, Floating Evaporative Self-assembly (DFES) approach, relying on PFO–BPy as a selective CP dispersant.76 The combination of nanotube purity and alignment enabled FET optimization to achieve a mobility of 179 cm2 V−1 s−1, on conductance of 261 μS μm−1, while maintaining and on/off ratio of 2 × 105, which are amongst the best values reported for SWNT-based devices. It was subsequently shown, as seen in Fig. 7, that the bipyridyl units in PFO–BPy could be used to chelate Re metal centres, causing a conformational change in the polymer backbone and resulting in a structure that was no longer capable of dispersing SWNTs.77 Thus, the combination of selective sc-SWNT dispersion with PFO–BPy followed by polymer desorption upon metal complexation allowed for the isolation of electronically-enriched samples of pristine sc-SWNTs.
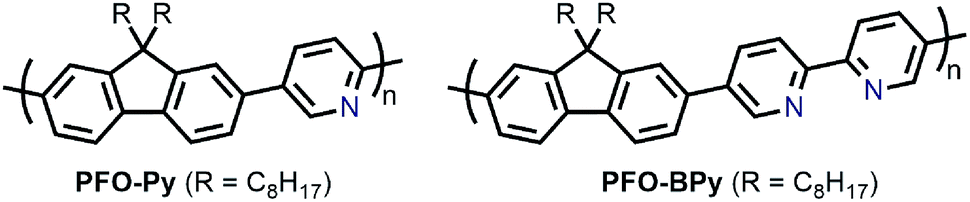 | ||
| Fig. 6 Chemical structures of PFO–Py and PFO–BPy. | ||
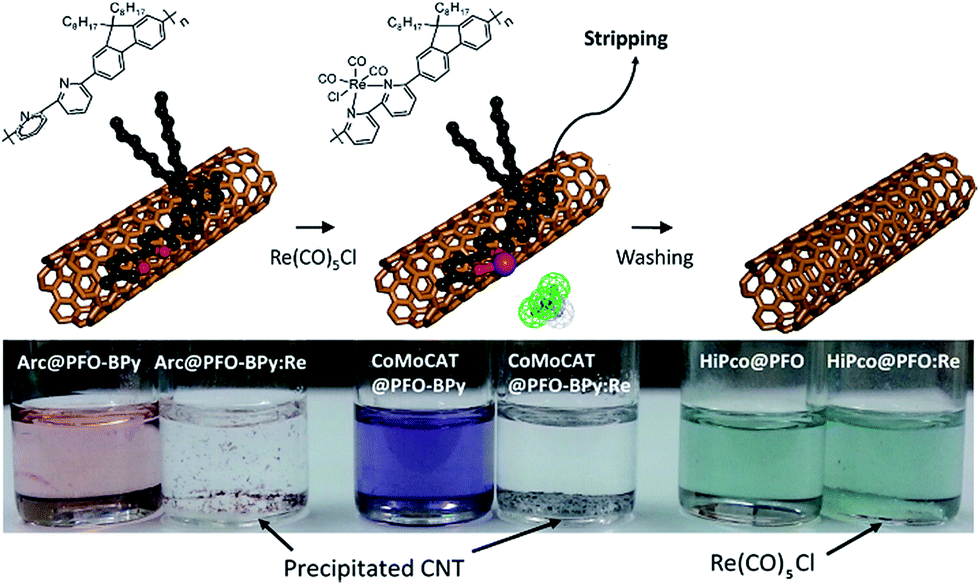 | ||
| Fig. 7 Selective dispersion and release of sc-SWNTs using PFO–BPy followed by metal complexation with a Re metal centre. Reprinted with permission from ref. 77. Copyright 2015, American Chemical Society. | ||
Recently, polymer backbones that are degradable, recyclable, and selectively disperse sc-SWNTs have been prepared, with polymer removal mechanisms including supramolecular metal coordination chemistry,56 hydrogen bond disruption,57 and imine hydrolysis (Fig. 8).58 In work by Toshimitsu and Nakashima, a fluorene unit appended to two phenanthroline moieties was used to form supramolecular metal-coordination polymers with perchlorate salts of Co(II), Ni(II), Cu(II), and Zn(II) in benzonitrile, which served as a “good” solvent.56 HiPCO SWNTs were then non-selectively dispersed in benzonitrile to prepare homogenous dispersions containing both sc-SWNTs and m-SWNTs. Addition of 50 vol% of toluene, a “poor” polymer solvent, resulted in the preferential flocculation of CP:m-SWNT complexes, with the Zn(II)-containing polymer affording the best removal of m-SWNTs. Subsequently, removal of the adsorbed polymer from the remaining dispersed sc-SWNT complexes was accomplished via depolymerization, which occurred upon addition of trifluoroacetic acid (TFA). To investigate the origin of selectivity, molecular mechanics simulations were performed, and it was found that stabilization energies were higher in toluene for the CP:sc-SWNT complexes relative to the CP:m-SWNT complexes. In an alternative approach to a similar goal, Bao and co-workers prepared a fluorene monomer flanked by self-complementary quadruply hydrogen-bonding 2-ureido-6[1H]-pyrimidinone (UPy) groups, which allowed the fluorene units to form a supramolecular polymer.57 It was shown that this polymer selectively dispersed sc-SWNTs, and could depolymerize on-demand to release the enriched sc-SWNT sample via addition of TFA, which acted as a hydrogen-bond disruptor. Subsequently, Bao and co-workers prepared imine-containing polyfluorenes, which were used to selectively disperse large-diameter sc-SWNTs.58 In this case, depolymerization and SWNT release were initiated via imine hydrolysis using catalytic TFA.
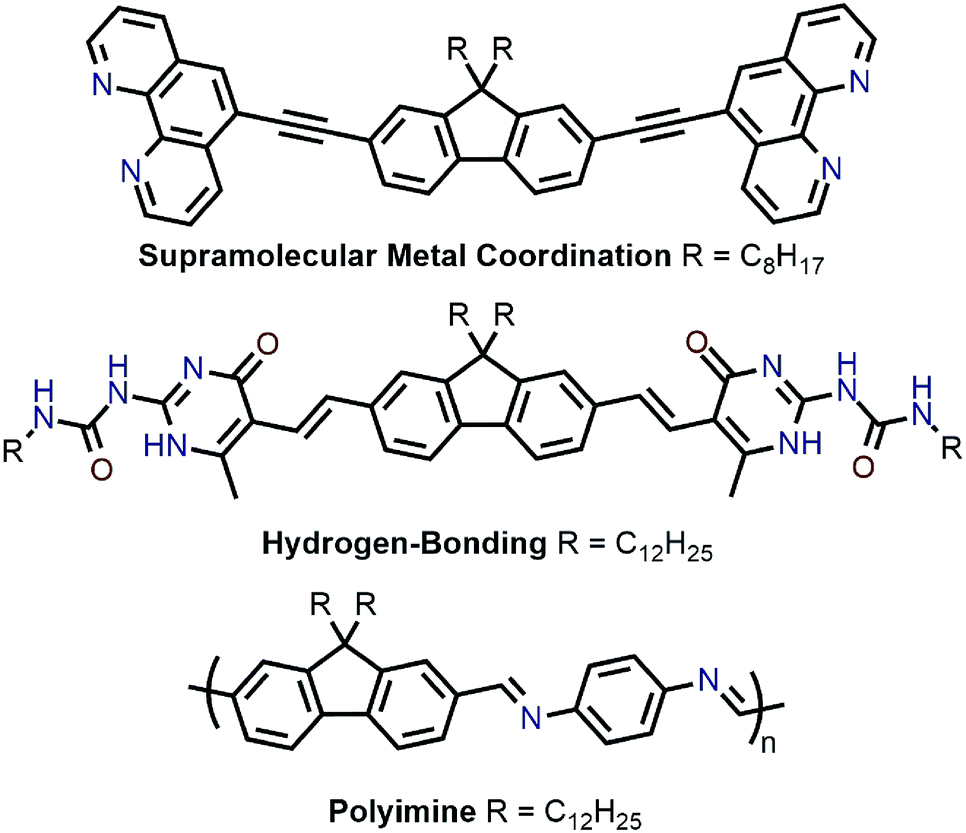 | ||
| Fig. 8 Chemical structures of monomers used for the enrichment and release of sc-SWNTs, as well as the PDDF-imine derivative. | ||
From the perspective of device fabrication, the use of CPs to selectively disperse sc-SWNTs is advantageous, with the caveat that the surface-bound polymer does not deleteriously affect device function. Regrettably, fluorene-containing polymers such as PFO, which exhibit good selectivity for sc-SWNTs, also have large bandgaps, and are not as suitable for device applications as small bandgap polymers such as poly(3-hexylthiophene) (P3HT). Nicholas and co-workers addressed this limitation by exploring a polymer exchange process after preparing an enriched sc-SWNT dispersion with PFO (Fig. 9).78,79 It was shown that surface-bound PFO could be quantitatively displaced with thiophene-containing polymers, such as P3HT, upon sonication. It was concluded that, although PFO was highly selective for sc-SWNTs, its SWNT binding energy was lower than that of the non-selective P3HT. Thus, it appeared that strongly-bound polymers were less selective for specific SWNT species. However, since thiophene-containing polymers are more suitable for device applications, this exchange process allowed for the initial dispersion of enriched sc-SWNTs with PFO, followed by its subsequent replacement with a more suitable surface-bound polymer.
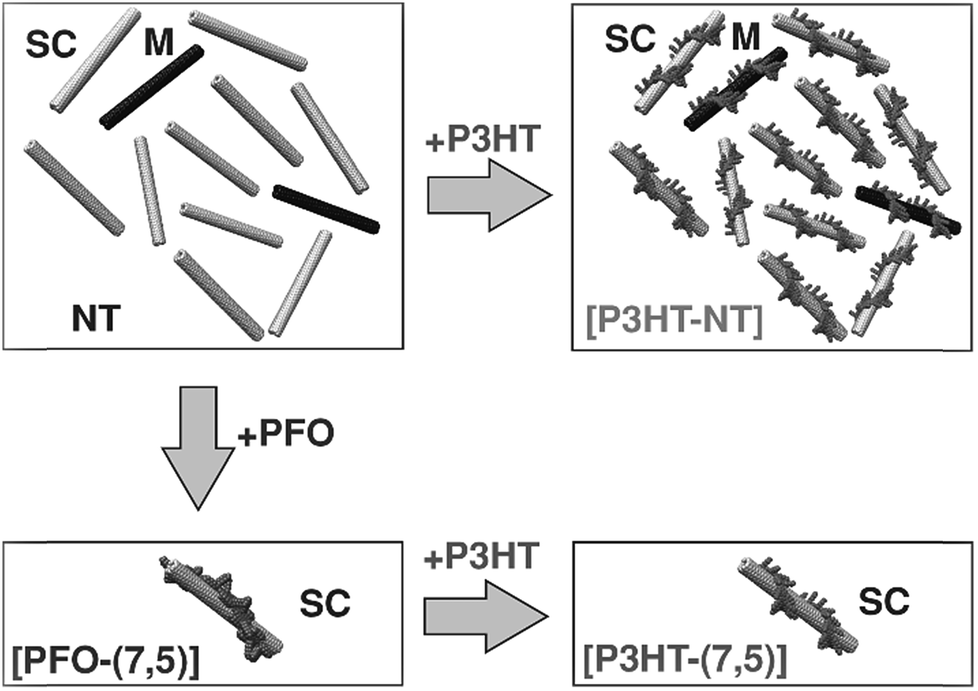 | ||
| Fig. 9 Schematic of the polymer exchange study between PFO and P3HT. PFO resulted in the selective dispersion of the (7,5) sc-SWNT species, while P3HT resulted in non-selective dispersions. However, P3HT could displace PFO on the SWNT surface. Reprinted with permission from ref. 78. Copyright 2013, John Wiley and Sons. | ||
Interestingly, although Nicholas and co-workers were unable to obtain selective dispersions with P3HT,78 regioregular poly(3-dodecylthiophene) (rr-P3DDT) containing longer side chains has been reported to selectively disperse sc-SWNTs by Bao and co-workers.80 It was shown that both the regioregularity of the CP side chains, as well as side-chain length and density, were important in controlling the dispersion selectivity of poly(3-alkylthiophene)s (P3ATs) (Fig. 10a). When significantly shorter side chains or insufficient amounts of C12 side chains were incorporated into the CP, dispersions obtained were either not achieved or non-selective. The authors conjectured that a tight polymer shell was produced on the nanotube surface, and that the polymer adopted a helical conformation with interdigitated side chains as a result of its regioregular structure. High quality FET devices were prepared and shown to be reproducible, with average hole mobilities of 8.5 cm2 V−1 s−1 and on/off ratios of 3 × 106 over 12 random devices, with no device shorting in more than 100 devices. The ability of rr-P3DDT to selectively disperse only sc-SWNTs was further verified in a subsequent study, in which short-channel SWNT-FETs were fabricated to probe 140 SWNTs, all of which were shown to be semiconducting.81 The comparison between the results by Nicholas and Bao illustrates that, for identical polymer backbone structures, side chain variation can consequentially impact dispersion selectivity. Following this work, several studies have appeared in which rr-P3DDT or other thiophene-containing polymers were used to selectively disperse sc-SWNTs, followed by utilization of the resulting “ink” to fabricate FET devices.82–84 In addition, selectivity for large-diameter SWNTs was achieved using copolymers of thiophene and dithiafulvalene-fluorene (DTFF)85 or diketopyrrolopyrrole (DPP) moieties (Fig. 11).64,86 In these studies, Bao and co-workers found that these polymers effectively dispersed sc-SWNTs with diameters in the 1.1–1.8 nm range. This is broadly consistent with the work on polyfluorenes, again showing that increased backbone rigidity and aromatic surface area is correlated with selectivity for larger diameter sc-SWNTs. Along similar lines, Loi and co-workers investigated a copolymer of bithiophene and a derivative of naphthalene dicarboximide (NDI), which also dispersed sc-SWNTs and allowed for the fabrication of FETs.87 Unfortunately, the diameters of the SWNTs dispersed by this polymer were not reported.
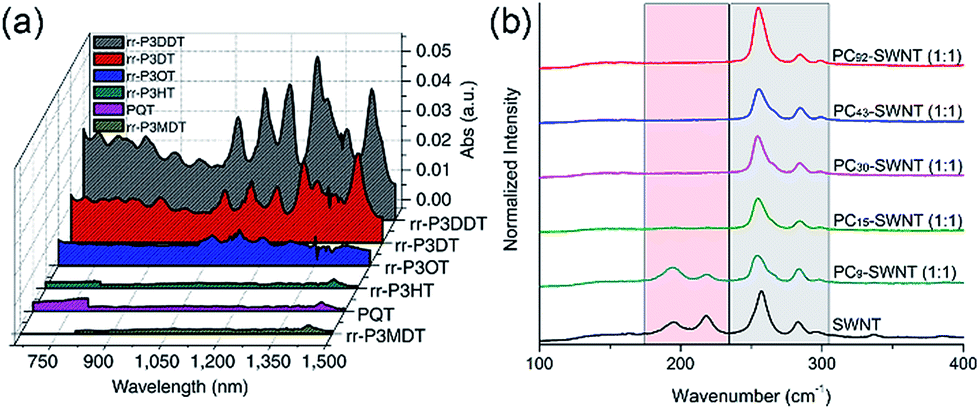 | ||
| Fig. 10 Spectroscopic data illustrating that CP backbone structure is insufficient for complete control over dispersion selectivity. (a) UV-Vis-NIR spectra of dispersions produced using P3ATs with different side-chain length, density, and regioregularity in toluene. (b) Raman spectra (using 633 nm excitation) of dispersions produced using different molecular weights of poly(2,7-N-alkylcarbazole), ranging from 9 to 92 kDa, with a 1:1 CP:SWNT mass ratio in THF. The gray box denotes the location of sc-SWNT peaks, while the pink box denotes the location of m-SWNT peaks. Reprinted with permission from ref. 80 and 91. Copyrights 2011, Macmillan Publishers Ltd., and 2015, John Wiley and Sons, respectively. | ||
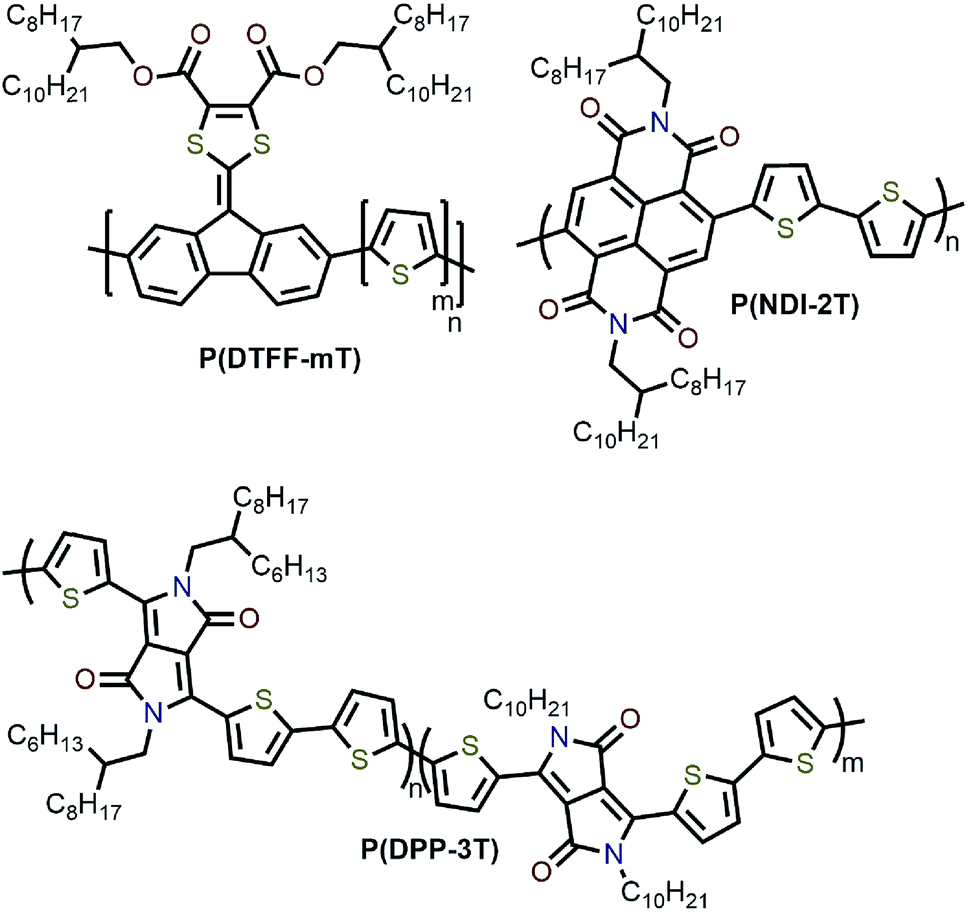 | ||
| Fig. 11 Chemical structures of several sc-SWNT-selective polythiophene derivatives. | ||
Aside from thiophenes, polycarbazoles are closely related to polyfluorenes, with the only difference being a bridging sp2-hybridized nitrogen atom in the central ring of the carbazole unit, instead of the sp3-hybridized carbon in fluorene. This difference is substantial, however, as carbazole exhibits increased π-conjugation and has only a single side chain attached, making the carbazole moiety less sterically encumbered than the fluorene moiety. Kappes and co-workers first reported the selective dispersion of sc-SWNTs with poly(N-decyl-2,7-carbazole) in toluene, and compared this to the dispersion produced by PFO.88 Due to the inherently poor solubility of polycarbazoles, the molecular weight of the polycarbazole structure used in this study was low (Mn = 3400, Đ = 1.6), constituting relatively short oligomers. Nevertheless, it was found that well-exfoliated, concentrated dispersions of sc-SWNTs could be achieved. Subsequently, high molecular weight polycarbazoles with large solubilizing groups were reported by Rice et al., and shown to be highly selective for small-diameter sc-SWNTs in both toluene and THF.89,90 In addition, it was found that selectivity for sc-SWNTs increased with polymer molecular weight (Fig. 10b), but this will be discussed separately below.91
In summary, a direct comparison of the numerous backbone structures examined is necessarily complicated due to the significant differences in molecular weight and side chain composition. Furthermore, selectivity is not solely a function of polymer structure, but is also influenced by the way in which the CP–SWNT complex is prepared. In spite of this, the following trends have been observed in multiple instances: (i) many polymers containing fluorene, thiophene, or carbazole derivatives are selective for sc-SWNTs, but there are no examples of polymer structures selective for only m-SWNTs; (ii) higher conformational rigidity of the polymer backbone is correlated to the dispersion of larger-diameter SWNTs, and vice versa; and (iii) increased surface area of the conjugated π-system is correlated to the dispersion of larger-diameter SWNTs. Beyond these trends, the thiophene and carbazole homopolymers are instructive examples demonstrating that, even for an identical polymer backbone, other polymer features can result in the transition from a non-selective to selective dispersion, whether it be side chains (polythiophene; Fig. 10a)80 or molecular weight (polycarbazole; Fig. 10b).91
Side chains
As mentioned above, the influence of side-chain length on dispersion selectivity was apparent when juxtaposing the results observed by Nicholas78,79 and Bao80 in the polythiophene homopolymer system. The effect of side-chain length and density on dispersion selectivity has subsequently been the subject of some investigation. Loi and co-workers examined polyfluorenes with varying side-chain length (6 to 18 carbon atoms).92 SWNT dispersions were prepared using HiPCO or arc plasma jet SWNTs in toluene. It was found that longer alkyl chain lengths resulted in higher SWNT concentrations, with the resulting dispersions showing a bias toward larger-diameter sc-SWNTs. The overall selectivity for semiconducting species versus metallic species, however, decreased with increasing side-chain length. MD simulations indicated that longer alkyl chains allowed for improved polymer coating of the SWNT surface, which prevented SWNT re-bundling. In similar work, Malenfant and co-workers showed that, for polyfluorenes, increasing side-chain length from 10 to 18 carbon atoms also increased the concentration of dispersed SWNTs, and biased the distribution toward larger diameters.93 Meanwhile, Bao and co-workers examined a series of rr-P3ATs with side chains of varying length (8 to 13 carbon atoms).63 Using CoMoCAT SWNTs, it was found that the nanotube dispersion yield increased from 4% to 31% as side-chain length was increased. In terms of diameter, the sc-SWNTs dispersed using polythiophenes with longer side chains were in the lower diameter range of CoMoCAT SWNTs (<0.76 nm). MD simulations indicated that the binding energy increased with alkyl chain length as a result of higher contact surface area (CSA) with longer alkyl chains. It was postulated that the higher binding energy for rr-P3ATs with longer alkyl chains was the origin of higher SWNT yield due to stronger CP–SWNT interactions.In addition to side-chain length, side-chain architecture and density have also been investigated. Bao and co-workers examined diketopyrrolopyrrole-co-trithiophene (PDPP-3T) random copolymers with varying compositions of branched and linear alkyl chains (0–20% linear alkyl chains) (Fig. 11).64,80 It was posited that increasing the proportion of linear alkyl side chains would decrease steric hindrance and allow for increased CP–SWNT interactions. Using arc-discharge SWNTs, it was found that increasing the proportion of linear alkyl chains increased the selectivity for sc-SWNTs. At 10% linear alkyl chains, an ideal balance between SWNT yield and sc-SWNT purity was found.64 DFT calculations were performed and suggested that the association energy between DPP with branched alkyl chains was 6.2 kcal mol−1 lower than DPP with linear alkyl chains. In terms of side-chain density, it was found that decreasing the number of side chains along the polymer backbone resulted in no observable SWNT dispersion.80 Meanwhile, in work by Ozawa et al., chiral and bulky side chains were introduced into a polyfluorene backbone and used to tune the selectivity toward specific sc-SWNT chiralities.94 Overall, these studies demonstrate that longer side chains increase the concentration of dispersed SWNTs, at the cost of selectivity for a small subset of SWNT species. Computational studies suggest that longer side chains increase CP–SWNT interaction strength (presumably via van der Waals interactions), which may account for these observations. Modulation of side-chain architecture and density may also play a role in dictating the CP–SWNT interaction strength for specific subsets of SWNTs. As for diameter selectivity, there appears to be no clear trend between side-chain length and the resulting sc-SWNT diameter distribution.
Molecular weight
The significance of molecular weight on dispersion concentration and selectivity was alluded to above, and several groups have conducted detailed studies in this area that warrant separate discussion. Imin et al. studied a series of poly[2,7-(9,9-di-n-octylfluorene)-alt-2,5-(3-dodecylthiophene)] (PFT) samples with Mn ranging from 5 to 85 kDa.95 It was found that molecular weights that were either too low or too high resulted in dilute SWNT dispersions, while intermediate molecular weights (10–30 kDa) produced concentrated dispersions. The decrease in dispersion concentration at higher molecular weights coincided with the decrease in CP solubility. In work by Mayor and co-workers, fluorene oligomers (n = 2–8) were investigated and compared to PDDF (Mn = 12.9 kDa).96 It was found that increasing the number of fluorene repeat units increased the stability of the resulting SWNT dispersion, and that dispersions could be further stabilized by exchange of the oligomer for a polymer such as PDDF–BT (Mn = 29.0 kDa). These results together indicate that low molecular weight oligomers are often insufficient in producing stable SWNT dispersions. With respect to selective dispersions, the effect of molecular weight was examined using poly(2,7-N-alkylcarbazole)s (Mn = 4.6–91.7 kDa) by Rice et al. (Fig. 10b).91 It was found that low molecular weight polymers (Mn = 4.6–15.2 kDa) dispersed both sc-SWNTs and m-SWNTs. Above a threshold molecular weight (Mn ≥ 27.4 kDa), sc-SWNTs were selectively dispersed. It should be noted that, although poly(2,7-N-alkylcarbazole)s tend to be insoluble, large solubilizing side chains were used in this study to produce highly soluble polymers, even at high molecular weights. Thus, in comparison to the work by Imin et al., a drastic decrease in dispersion concentration at high molecular weights was not observed. In work by Zaumseil and co-workers, PFO (Mn = 6.7–98.1 kDa) and PFO–BT (Mn = 7.4–62.8 kDa) were used to prepare SWNT dispersions in toluene or o-xylene.97 It was found that both molecular weight and solvent influenced dispersion selectivity, but that this was a complex process that possibly involved changes to viscosity. These studies, as a whole, indicate that molecular weight is an important factor to consider in the selective dispersion of SWNTs, and it should be kept in mind that polymer molecular weight can influence polymer solubility, which can impact the concentration of dispersed SWNTs.Backbone electronics
In the Backbone structure section, it was noted that many fluorene-, thiophene-, and carbazole-containing polymers were capable of selectively dispersing sc-SWNTs. Qualitatively, these polymers can be considered to possess electron-rich π-systems. These empirical observations lead to the hypothesis that electron-rich CPs may exhibit a preference for sc-SWNTs. If this is the case, then it may follow that, conversely, electron-poor CPs may preferentially disperse m-SWNTs. Interestingly, the electronic nature of small molecules has been demonstrated to influence the electronic structure of SWNTs.98,99 In a study by Lee and co-workers,98 arc-discharge SWNTs were sonicated in electron-rich and electron-poor aliphatic and aromatic solvents, and then the suspensions were filtered to obtain a SWNT thin film. Lee and co-workers analyzed the thin films using Raman spectroscopy, and found that electron-poor nitrobenzene and nitromethane suppressed the Breit–Wigner–Fano (BWF) line in the G-band of the Raman spectra excited at 633 nm (1.96 eV), which is a peak that corresponds to m-SWNTs. As well, the electron-poor solvent increased the intensity of the D-band, which corresponded with an increase in SWNT surface defects. These observations were attributed to the introduction of a bandgap in the m-SWNTs upon interaction with the electron-poor solvent molecule. In contrast, electron-rich aniline and butylamine did not suppress the BWF line at 633 nm. These results suggested that the electronic structure of m-SWNTs is more strongly influenced by electron-poor molecules than by electron-rich molecules. In a corroborating study by Rao and co-workers, the relative interaction strength of electron-rich and electron-poor molecules with SWNTs was examined using isothermal titration calorimetry.99 It was found that the interaction energy with 95% pure m-SWNTs increased with increasing electron affinity of the small molecule. In contrast to as-produced SWNTs containing a 2:1 mixture of sc-SWNTs:m-SWNTs, the interaction energy with electron-poor molecules was always larger with the pure m-SWNT sample. This suggested that electron-poor molecules interacted more strongly with m-SWNTs than sc-SWNTs. These reports, together with the empirical observation that many of the common electron-rich CP backbones selectively disperse sc-SWNTs (e.g. polyfluorene, polythiophene, polycarbazole, etc.) lead us to the operating hypothesis that an electron-poor conjugated π-system may interact preferentially with m-SWNTs over sc-SWNTs.
With this in mind, we examined the effect of the electronic nature of the CP π-system on dispersion selectivity.100 We prepared two fluorene copolymers with an electron-rich (p-dimethoxyphenyl) or electron-poor (p-dinitrophenyl) co-monomer. While maintaining identical side chains and similar DPs, we observed that the electron-rich copolymer selectively dispersed sc-SWNTs, while the electron-poor copolymer dispersed a mixture of sc-SWNTs and m-SWNTs (Fig. 12a). We found that decreasing the electron density in the polymer backbone increased the amount of m-SWNTs dispersed, which is consistent with our hypothesis. In a follow-up study, we investigated a fluorene-co-pyridine copolymer in which the π-system could be modified post-polymerization.101 By methylating the pyridine unit in the copolymer, a cationic charge is introduced into the polymer backbone to convert it from electron-rich to electron-poor in a single step (Fig. 12b). Using this synthetic methodology, the two polymer structures being compared were almost identical, with the only difference being a single methyl group repeated along the backbone. It was found that pre-methylation, sc-SWNTs were exclusively dispersed, while post-methylation, there was a large increase in the amount of dispersed m-SWNTs. Conductivity measurements on thin-films produced from SWNTs dispersed by the post-methylated polymer indicated an increase in bulk conductivity of four orders of magnitude compared to the dispersion prepared by the pre-methylated polymer, which is consistent with the presence of m-SWNTs. These studies agree with the hypothesis that decreasing the electron density in the CP backbone has a dramatic effect on the amount of m-SWNTs dispersed. This work warrants further investigation into the effect of polymer electronics on dispersion selectivity.
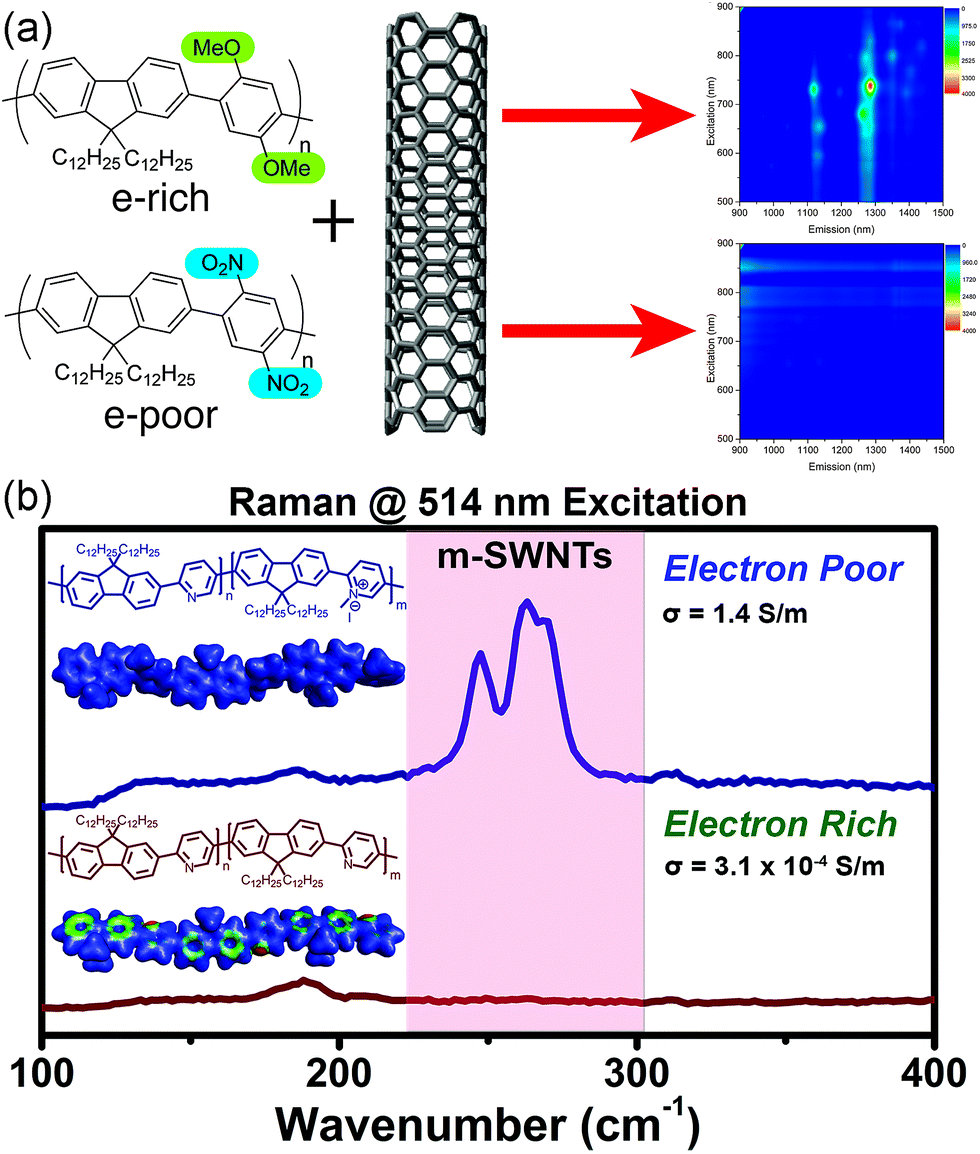 | ||
| Fig. 12 Schemes depicting the dispersion of SWNTs using electronically-distinct CPs. (a) PL map fluorescence quenching is observed in the dispersion prepared using the nitro group-containing polyfluorene (electron-withdrawing groups), in contrast to discernible sc-SWNT peaks in the dispersion prepared using the methoxy group-containing polyfluorene (electron-donating groups). (b) Raman spectra obtained (using 514 nm excitation) of SWNT dispersions prepared using the pre-methylated (neutral) and post-methylated (cationic) CPs. m-SWNTs are present in the sample prepared using the post-methylated CP. Reprinted with permission from ref. 100 and 101. Copyrights 2015, American Chemical Society, and 2016, John Wiley and Sons, respectively. | ||
3. Dispersion preparation conditions
General considerations
As mentioned above, several variables can be considered during the SWNT dispersion process to modulate dispersion selectivity: these include the SWNT type used, sonication temperature, CP:SWNT mass ratio, and choice of solvent. We reiterate that, for the same CP sample, different SWNT species may be dispersed depending on the starting SWNT material used, so investigation of different SWNT types is important. In addition, though not necessarily related to dispersion selectivity, there have been conflicting reports on the effect of sonication time on the length of SWNTs. Below, we compile the reports and establish that the impact of sonication on SWNT length is dependent on the sonication power used. Additionally, although centrifugation speed is not particularly relevant to dispersion selectivity, it should be noted that an adequate speed must be identified for each individual CP–SWNT system, such that bundles are efficiently removed. Overall, based on the numerous studies that have been done, we attempt to highlight the general trends with respect to each variable.
Sonication time
Sonication is used to temporarily exfoliate SWNT bundles, such that dispersant can access and coat the SWNT surface to prevent re-bundling. Bao and co-workers examined the effect of sonication time on SWNT concentration, length, and bundling, using the solvent NMP as the dispersant.102 SWNTs in NMP were sonicated at 225 W for up to 480 min, and aliquots were taken at regular intervals for analysis. It was found that increased sonication time decreased bundling, until sufficient diffusion of atmospheric water into NMP resulted in SWNT bundling (∼120 min); a known phenomenon.103 It was also found that increased sonication time decreased the average SWNT length and length polydispersity, and increased the dispersed SWNT concentration.102 The SWNT dispersions were used to prepare FETs, and it was determined that shorter sonication times (<30 min) were ideal to minimize sonication-induced SWNT defects. Contrary to these findings, Loi and co-workers found that sonication time up to 2 h had little effect on SWNT length.92 It is important to note that, in this study, sonication power was 90 W, compared to 225 W used by Bao and co-workers. Thus, we highlight that sonication-induced SWNT damage is dependent on the sonication apparatus used. It is imperative to optimize sonication time such that SWNTs can be sufficiently de-bundled to allow for polymer wrapping, while being aware that higher sonication power may potentially result in changes to the SWNT material.Sonication temperature
Bao and co-workers examined the effect of temperature on the selective dispersion of HiPCO SWNTs using rr-P3DDT.80 Temperature was varied from −40 to 90 °C, and it was found that the strongest SWNT absorption intensity at 1288 nm was obtained for the dispersion prepared at 50 °C, which coincided with the alkyl side chain melting temperature. The observations were modelled by the authors as follows: (i) at low temperatures, polymer aggregates cannot dissociate, even if the CP–SWNT complex is more thermodynamically stable; (ii) at high temperatures, entropic penalties may prohibit CP–SWNT complex formation; and (iii) at an intermediate temperature, enough energy is present to overcome polymer–polymer interactions and allow for equilibration to more stable CP–SWNT complexes, without excessive entropic penalties. In tandem with this study, Loi and co-workers also investigated rr-P3DDT dispersions with HiPCO SWNTs, varying sonication temperature from 0–80 °C.104 SWNT dispersions prepared with or without polymer aggregation resulted in no difference in the dispersion selectivity. It was found, in contrast to the aforementioned study, that sonication from 10–20 °C produced the most concentrated SWNT dispersions (Fig. 13a). It should be noted that preparation conditions were not identical. For instance, Bao and co-workers used a 2:1 CP:SWNT mass ratio, while Loi and co-workers used a 3:1 CP:SWNT mass ratio. The CP molecular weights and dispersities were also different (Mn = 39.1 kDa and Đ = 1.97, vs. Mn = 26.8 kDa and Đ = 1.08), as were the initial SWNT concentrations in toluene (0.20 mg mL−1vs. 0.33 mg mL−1). These differences in polymer features and dispersion preparation conditions highlight the difficulty in making generalizable statements about the effect of a single variable, even with extraneous variables held constant within a single study. Though it is clear that sonication temperature influences dispersion selectivity and cannot be ignored, the exact role it plays is unclear and warrants further examination.
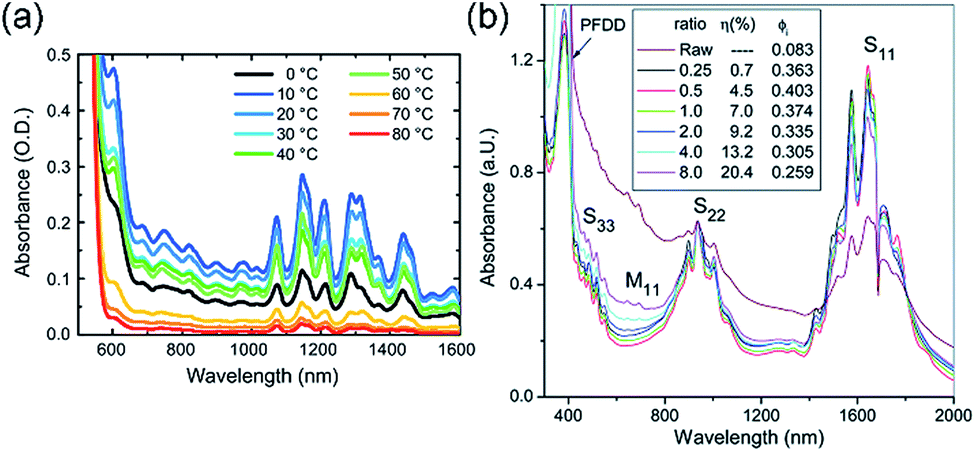 | ||
| Fig. 13 Spectroscopic data illustrating the effects of preparation conditions on dispersion selectivity. Absorption spectra of dispersions prepared in toluene (a) using rr-P3DDT with bath sonicator temperatures ranging from 0–80 °C, and (b) using PDDF with CP:SWNT mass ratios ranging from 0.25:1 to 8:1. Reprinted with permission from ref. 93 and 104. Copyrights 2015, Elsevier, and 2014, Royal Society of Chemistry, respectively. | ||
CP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :SWNT mass ratio
:SWNT mass ratio
Several groups have examined the CP:SWNT mass ratio and its effect on dispersion selectivity. In work by Malenfant and co-workers, it was shown that CP:SWNT mass ratios of 0.5:1 to 1:1 produced the highest sc-SWNT purity, while larger CP:SWNT mass ratios resulted in decreased sc-SWNT discrimination (Fig. 13b).93 Likewise, Rice et al.91 and Zaumseil and co-workers97 found similar results. The appropriate CP:SWNT mass ratio depends on the polymer system, but in general, there is an ideal mass ratio range where, (i) there is enough polymer to adequately coat SWNT surfaces and produce stable CP–SWNT dispersions, while (ii) there is not enough polymer present to saturate all SWNT surfaces and indiscriminately disperse all SWNTs. The CP:SWNT mass ratio should be optimized for each polymer system.64,91
Solvent
In general, solvent density must be lower than the buoyant density of SWNT bundles (∼1.3 g cm−3), otherwise bundles cannot sediment upon centrifugation. This necessarily excludes chlorinated solvents such as CH2Cl2 and CHCl3, as these solvents are too dense (densities of ∼1.3 and 1.5 g cm−3, respectively). Beyond this, several groups have examined different solvent properties. Cui and co-workers examined the effect of solvent dielectric constant on dispersion selectivity.105 Arc discharge SWNTs were dispersed using commercially available poly(9,9-di-n-octylfluorene-co-bithiophene) (PFO-T2) in various solvents (0–1.75 D). It was found that sc-SWNT selectivity only occurred for solvents with a dielectric constant between 0 and 0.5 D (toluene, m-xylene, and xylenes). It was argued that selectivity was driven by charge transfer that prevented the formation of the supramolecular CP–SWNT complexes required to disperse m-SWNTs, and that in solvents with a higher dielectric constant, this charge transfer could be inhibited. Within the solvent series, CH2Cl2 and CHCl3 were included as solvents with higher dielectric constants (1.60 and 1.04 D, respectively); yet the absence of selectivity can also be explained by the high densities of these solvents, a possibility that was left unaddressed. Meanwhile, Zaumseil and co-workers examined the relationship between solvent viscosity and dispersion selectivity.97 It was found that the fluorescence intensity for the (9,4) chirality was correlated with the magnitude of solvent kinematic viscosity for toluene, the individual xylene isomers, and mesitylene, while keeping the CP properties (molecular weight, backbone structure) constant. However, for the (10,5) chirality, fluorescence intensity was not linearly correlated with increasing solvent kinematic viscosity. The exact relationship between solvent viscosity and dispersion selectivity is therefore unclear. In work by Bao and co-workers, HiPCO SWNTs were dispersed using rr-P3DDT in THF, decalin, tetralin, m-xylene, and o-xylene.106 It was found that decalin and tetralin resulted in higher dispersion selectivity (fewer sc-SWNT chiralities). All the solvents, excluding THF, resulted in enriched sc-SWNT dispersions, which was attributed to solvent polarity differences between THF and the other solvents. Interestingly, the viscosities of the solvents that resulted in enriched sc-SWNT dispersions vary over a wide range (0.59–2.09 cP). It is evident from these studies that the effect of solvent on dispersion selectivity involves a complicated amalgamation of solvent density, polarity, and viscosity, and it is perhaps challenging or impossible to disentangle these individual effects. Overall, non-polar solvents such as toluene are commonly used for the preparation of selective dispersions, and, often, more polar solvents such as THF result in non-selective dispersions. We emphasize, however, that examples in the literature exist of selective sc-SWNT dispersions in THF,90,91,101 and that these trends are not absolute.4. Conjugated polymer sorting mechanism
To date, the mechanism behind dispersion selectivity is not completely understood. Bao and co-workers postulate that selectivity originates from the difference in SWNT polarizability, as m-SWNTs have been calculated to be ∼103 times more polarizable than sc-SWNTs.106 Consequently, it is postulated that the more polarizable CP:m-SWNT supramolecular complexes can aggregate in non-polar solvents due to dipole–dipole interactions, and sediment during centrifugation, since bundles are denser than polymer-wrapped, individualized SWNTs.50,52 This model is consistent with the observation that, in many cases, selective dispersions are achieved in non-polar solvents, such as toluene, xylene, and decalin, which are unable to adequately stabilize dipole moments.106 Conversely, dispersions prepared in THF tend to have poor selectivity, as this solvent is capable of stabilizing dipole moments, and would thus eliminate the driving force for m-SWNT aggregation. However, several examples in the literature exist where sc-SWNTs are selectively dispersed in THF;90,91,101 a fact that is inconsistent with this model. Malenfant and co-workers also invoke the differences in SWNT polarizability as the underpinning of selective dispersion. They propose that selectivity is strongly influenced by oxygen-driven p-doping in a pH-dependent manner.107 In experiments carried out using PDDF, the authors concluded that the polymer was not itself selective for sc-SWNTs over m-SWNTs, but instead responsible only for the stabilization of SWNTs in solution. It was proposed that, under ambient conditions, latent atmospheric O2 and H2O could oxidize sc-SWNTs to produce positively charged SWNTs, resulting in an induced dipole–dipole interaction with the highly polarizable m-SWNTs. The concomitant aggregate then precipitates, effectively removing m-SWNTs from solution and enriching the supernatant in sc-SWNTs.In our own work, we hypothesize that the electronic nature of the CP backbone may influence selectivity of the CP–SWNT interaction. Building on small molecule observations, we have found that, when variables such as molecular weight and side-chain length are kept relatively constant, electron-rich CPs selectively disperse sc-SWNTs, while electron-poor CPs disperse a mixture of sc- and m-SWNTs. Thus, changing the electronic nature of a CP appears to influence dispersion selectivity. The notion that electron-rich CPs exhibit selectivity for sc-SWNTs has been supported by numerous reports with a variety of different polymer structures. Conversely, only two examples of the interactions of electron-poor CPs with SWNTs have been examined,90,101 and the hypothesis that electron-poor CPs exhibit selectivity for m-SWNTs must therefore be tested more extensively.
We propose that the concept of selective SWNT dispersion can be considered in terms of the classic chemistry principles of kinetics and thermodynamics. Recalling the general SWNT dispersion protocol in Fig. 1, it is apparent that dispersion selectivity arises during the sonication step. From this perspective, CP–SWNT interactions result in either the selective exfoliation of a small subset of SWNTs, or, alternatively, a specific subset of SWNTs may selectively aggregate to form bundles followed by subsequent removal in the centrifugation step. The mechanisms proposed separately by Bao and Malenfant focus on the latter possibility involving SWNT aggregation, while our hypothesis focuses on the concept of selective exfoliation. These events are not necessarily mutually-exclusive, and may be partial components of a more complex mechanism. If the system is governed primarily by kinetics, then selectivity is driven by either the activation energy barrier (Ea) to CP–SWNT complex formation, or the Ea to aggregation (and flocculation) of specific SWNT species. In this case, if the exfoliation is carried out under conditions favouring kinetic control, the process with the lowest Ea will occur and dictate selectivity. Conversely, if the system is controlled primarily by thermodynamics, where enough energy is available to surmount every Ea and allow microscopic reversibility, then the stability of CP–SWNT complexes and/or the stability of SWNT bundles becomes the governing factor. Considering that sonication, a highly energetic process, is nearly always used to disperse SWNTs and promote the formation of CP–SWNT complexes, we postulate that thermodynamic control may be the dominant factor that dictates selectivity. This is consistent with the fact that CP exchange can occur when PFO–SWNT complexes are sonicated in the presence of P3HT, as shown by Nicholas and co-workers,78 or when fluorene oligomers are displaced by PDDF–BT, as shown by Mayor and co-workers.96 These exchange processes are unlikely if the system was under kinetic control. In addition, dispersion selectivity is often observed only when a limited amount of polymer is used (i.e., at low CP:SWNT mass ratios), which may indicate that only the most thermodynamically favourable complexes are dispersed under CP-limited conditions. When excess CP is used, it may be that less energetically favourable complexes can be formed under these conditions. Ultimately, the proposed mechanisms by Bao and Malenfant can be viewed thermodynamically as the energetically-favourable preference for SWNT bundle formation with m-SWNTs, while our hypothesis can be viewed thermodynamically as the potential for selective CP–SWNT interactions, depending on the electronic nature of the CP backbone. By considering the selectivity problem from the vantage point of kinetics versus thermodynamics, the logical consequences of these frameworks can be considered, and this may better enable the rational design of CP structures to selectively disperse specific SWNT species. Nevertheless, it is evident that the origin of dispersion selectivity is multifaceted and requires further investigation to identify a clear and predictive mechanism.
5. Concluding remarks
The fundamental process that results in the selective dispersion of SWNTs using CPs is a complex combination of CP structure and dispersion preparation conditions. The examples highlighted in this Perspective illustrate that backbone structure alone is insufficient in controlling dispersion selectivity, as exemplified in the case studies where polythiophene side chain or polycarbazole molecular weight dramatically influence dispersion selectivity. Beyond CP structure, the reports examined in this Perspective also indicate that dispersion preparation conditions have an essential role in dispersion selectivity. Maintaining consistency between these parameters within a single study is essential to allow for analysis of structure–selectivity relationships. Differences in the preparation conditions between studies, however, amplify the non-trivial nature of comparison between reports, and illustrate the difficulty in proposing generalizable CP design rules. It has been observed by many groups that using non-polar solvents, such as toluene, often results in the selective dispersion of sc-SWNTs. From this observation, selectivity mechanisms have been proposed based on the differences in polarizability between m-SWNTs and sc-SWNTs. However, recent reports that demonstrate the selective dispersion of sc-SWNTs in polar solvents, such as THF, highlight that these mechanistic proposals require further consideration. We have proposed that viewing the selectivity problem through the classic chemistry framework of kinetics versus thermodynamics can expand upon these existing mechanistic proposals, and may provide additional avenues to explore the rational design of CPs. In particular, we hypothesize that polymer backbone electronics may play a significant role in dispersion selectivity, and our findings from preliminary studies warrant further investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Natural Science and Engineering Research Council of Canada (NSERC) is gratefully acknowledged. D. F. is grateful for financial support from an NSERC PGS-D scholarship.References
- S. Iijima, Mater. Sci. Eng., B, 1993, 19, 172–180 CrossRef.
- D. S. Bethune, C. H. Kiang, M. S. de Vries, G. Gorman, R. Savoy, J. Vazquez and R. Beyers, Nature, 1993, 363, 605–607 CrossRef CAS.
- M.-F. Yu, B. S. Files, S. Arepalli and R. S. Ruoff, Phys. Rev. Lett., 2000, 84, 5552–5555 CrossRef CAS PubMed.
- M. Terrones, Annu. Rev. Mater. Res., 2003, 33, 419–501 CrossRef CAS.
- Z. Han and A. Fina, Prog. Polym. Sci., 2011, 36, 914–944 CrossRef CAS.
- P. Avouris, Z. Chen and V. Perebeinos, Nat. Nanotechnol., 2007, 2, 605–615 CrossRef CAS PubMed.
- P. Avouris, Acc. Chem. Res., 2002, 35, 1026–1034 CrossRef CAS PubMed.
- P. G. Collins and P. Avouris, Sci. Am., 2000, 283, 62–69 CrossRef CAS PubMed.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593–596 CrossRef PubMed.
- R. B. Weisman and S. M. Bachilo, Nano Lett., 2003, 3, 1235–1238 CrossRef CAS.
- H. Kataura, Y. Kumazawa, Y. Maniwa, I. Umezu, S. Suzuki, Y. Ohtsuka and Y. Achiba, Synth. Met., 1999, 103, 2555–2558 CrossRef CAS.
- G.-D. Zhan, J. D. Kuntz, J. Wan and A. K. Mukherjee, Nat. Mater., 2003, 2, 38–42 CrossRef CAS PubMed.
- L. Liu, A. H. Barber, S. Nuriel and H. D. Wagner, Adv. Funct. Mater., 2005, 15, 975–980 CrossRef CAS.
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- T. Dürkop, S. A. Getty, E. Cobas and M. S. Fuhrer, Nano Lett., 2003, 4, 35–39 CrossRef.
- P. Qi, O. Vermesh, M. Grecu, A. Javey, Q. Wang, H. Dai, S. Peng and K. J. Cho, Nano Lett., 2003, 3, 347–351 CrossRef CAS.
- M. Dionisio, J. M. Schnorr, V. K. Michaelis, R. G. Griffin, T. M. Swager and E. Dalcanale, J. Am. Chem. Soc., 2012, 134, 6540–6543 CrossRef CAS PubMed.
- G. A. Rivas, M. D. Rubianes, M. C. Rodríguez, N. F. Ferreyra, G. L. Luque, M. L. Pedano, S. A. Miscoria and C. Parrado, Talanta, 2007, 74, 291–307 CrossRef CAS PubMed.
- X. Pang, P. Imin, I. Zhitomirsky and A. Adronov, Macromolecules, 2010, 43, 10376–10381 CrossRef CAS.
- J.-H. Kim, K.-J. Yee, Y.-S. Lim, L. G. Booshehri, E. H. Haroz and J. Kono, Adv. Mater., 2011, 24, 4977–4994 Search PubMed.
- M. W. Rowell, M. A. Topinka, M. D. McGehee, H.-J. Prall, G. Dennler, N. S. Sariciftci, L. Hu and G. Gruner, Appl. Phys. Lett., 2006, 88, 233506 CrossRef.
- D. J. Bindl, N. S. Safron and M. S. Arnold, ACS Nano, 2010, 4, 5657–5664 CrossRef CAS PubMed.
- G. Keru, P. G. Ndungu and V. O. Nyamori, Int. J. Energy Res., 2014, 38, 1635–1653 CrossRef CAS.
- D.-M. Sun, C. Liu, W.-C. Ren and H.-M. Cheng, Small, 2013, 9, 1188–1205 CrossRef CAS PubMed.
- A. A. Green and M. C. Hersam, Nano Lett., 2008, 8, 1417–1422 CrossRef CAS PubMed.
- D. S. Hecht, D. Thomas, L. Hu, C. Ladous, T. Lam, Y. Park, G. Irvin and P. Drzaic, J. Soc. Inf. Disp., 2009, 17, 941 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- S. Hong and S. Myung, Nat. Nanotechnol., 2007, 2, 207–208 CrossRef CAS PubMed.
- P. Nikolaev, M. J. Bronikowski, R. K. Bradley, F. Rohmund, D. T. Colbert, K. Smith and R. E. Smalley, Chem. Phys. Lett., 1999, 313, 91–97 CrossRef CAS.
- J. Kong, A. M. Cassell and H. Dai, Chem. Phys. Lett., 1998, 292, 567–574 CrossRef CAS.
- P. Bernier, C. Journet, W. K. Maser, A. Loiseau, M. L. de la Chapelle, S. Lefrant, P. Deniard, R. Lee and J. E. Fischer, Nature, 1997, 388, 756–758 CrossRef.
- T. Guo, P. Nikolaev, A. Thess, D. T. Colbert and R. E. Smalley, Chem. Phys. Lett., 1995, 243, 49–54 CrossRef CAS.
- K. S. Kim, G. Cota-Sanchez, C. T. Kingston, M. Imris, B. Simard and G. Soucy, J. Phys. D: Appl. Phys., 2007, 40, 2375–2387 CrossRef CAS.
- M. S. Arnold, S. I. Stupp and M. C. Hersam, Nano Lett., 2005, 5, 713–718 CrossRef CAS PubMed.
- M. S. Arnold, A. A. Green, J. F. Hulvat, S. I. Stupp and M. C. Hersam, Nat. Nanotechnol., 2006, 1, 60–65 CrossRef CAS PubMed.
- J. C. Charlier, Acc. Chem. Res., 2002, 35, 1063–1069 CrossRef CAS PubMed.
- H. Liu, D. Nishide, T. Tanaka, H. Kataura and C. Mioskowski, Nat. Commun., 2011, 2, 309 CrossRef PubMed.
- T. Tanaka, H. Jin, Y. Miyata and H. Kataura, Appl. Phys. Express, 2008, 1, 114001 CrossRef.
- C. Y. Khripin, J. A. Fagan and M. Zheng, J. Am. Chem. Soc., 2013, 135, 6822–6825 CrossRef CAS PubMed.
- J. A. Fagan, C. Y. Khripin, C. A. Silvera Batista, J. R. Simpson, E. H. Hároz, A. R. Hight Walker and M. Zheng, Adv. Mater., 2014, 26, 2800–2804 CrossRef CAS PubMed.
- B. S. Flavel, M. M. Kappes, R. Krupke and F. Hennrich, ACS Nano, 2013, 7, 3557–3564 CrossRef CAS PubMed.
- B. S. Flavel, K. E. Moore, M. Pfohl, M. M. Kappes and F. Hennrich, ACS Nano, 2014, 8, 1817–1826 CrossRef CAS PubMed.
- M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. Mclean, S. R. Lustig, R. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338–342 CrossRef CAS PubMed.
- X. Tu, S. Manohar, A. Jagota and M. Zheng, Nature, 2009, 460, 250–253 CrossRef CAS PubMed.
- M. Zheng and E. D. Semke, J. Am. Chem. Soc., 2007, 129, 6084–6085 CrossRef CAS PubMed.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS PubMed.
- A. Star, J. F. Stoddart, D. Steuerman, M. Diehl, A. Boukai, E. W. Wong, X. Yang, S.-W. Chung, H. Choi and J. R. Heath, Angew. Chem., Int. Ed., 2001, 40, 1721–1725 CrossRef CAS PubMed.
- D. W. Steuerman, A. Star, R. Narizzano, H. Choi, R. S. Ries, C. Nicolini, J. F. Stoddart and J. R. Heath, J. Phys. Chem. B, 2002, 106, 3124–3130 CrossRef CAS.
- H. Wang and Z. Bao, Nano Today, 2015, 10, 737–758 CrossRef CAS.
- S. K. Samanta, M. Fritsch, U. Scherf, W. Gomulya, S. Z. Bisri and M. A. Loi, Acc. Chem. Res., 2014, 47, 2446–2456 CrossRef CAS PubMed.
- T. Lei, I. Pochorovski and Z. Bao, Acc. Chem. Res., 2017, 50, 1096–1104 CrossRef CAS PubMed.
- P. R. L. Malenfant, W. V. Cicha, P. A. Bui and D. L. Simone, US20060045838A1, 2006.
- A. Nish, J.-Y. Hwang, J. Doig and R. J. Nicholas, Nat. Nanotechnol., 2007, 2, 640–646 CrossRef CAS PubMed.
- G. J. Brady, Y. Joo, M.-Y. Wu, M. J. Shea, P. Gopalan and M. S. Arnold, ACS Nano, 2014, 8, 11614–11621 CrossRef CAS PubMed.
- F. Toshimitsu and N. Nakashima, Nat. Commun., 2014, 5, 5041 CrossRef CAS PubMed.
- I. Pochorovski, H. Wang, J. I. Feldblyum, X. Zhang, A. L. Antaris and Z. Bao, J. Am. Chem. Soc., 2015, 137, 4328–4331 CrossRef CAS PubMed.
- T. Lei, X. Chen, G. Pitner, H.-S. P. Wong and Z. Bao, J. Am. Chem. Soc., 2016, 138, 802–805 CrossRef CAS PubMed.
- J.-Y. Hwang, A. Nish, J. Doig, S. Douven, C.-W. Chen, L.-C. Chen and R. J. Nicholas, J. Am. Chem. Soc., 2008, 130, 3543–3553 CrossRef CAS PubMed.
- F. Chen, B. Wang, Y. Chen and L.-J. Li, Nano Lett., 2007, 7, 3013–3017 CrossRef CAS PubMed.
- N. Stürzl, F. Hennrich, S. Lebedkin and M. M. Kappes, J. Phys. Chem. C, 2009, 113, 14628–14632 CrossRef.
- K. Akazaki, F. Toshimitsu, H. Ozawa, T. Fujigaya and N. Nakashima, J. Am. Chem. Soc., 2012, 134, 12700–12707 CrossRef CAS PubMed.
- H. Wang, G. I. Koleilat, P. Liu, G. Jiménez-Osés, Y. C. Lai, M. Vosgueritchian, Y. Fang, S. Park, K. N. Houk and Z. Bao, ACS Nano, 2014, 8, 2609–2617 CrossRef CAS PubMed.
- T. Lei, Y. C. Lai, G. Hong, H. Wang, P. Hayoz, R. T. Weitz, C. Chen, H. Dai and Z. Bao, Small, 2015, 11, 2946–2954 CrossRef CAS PubMed.
- M. Tange, T. Okazaki and S. Iijima, J. Am. Chem. Soc., 2011, 133, 11908–11911 CrossRef CAS PubMed.
- P. Gerstel, S. Klumpp, F. Hennrich, O. Altintas, T. R. Eaton, M. Mayor, C. Barner-Kowollik and M. M. Kappes, Polym. Chem., 2012, 3, 1966 RSC.
- F. Lemasson, N. Berton, J. Tittmann, F. Hennrich, M. M. Kappes and M. Mayor, Macromolecules, 2012, 45, 713–722 CrossRef CAS.
- K. S. Mistry, B. A. Larsen and J. L. Blackburn, ACS Nano, 2013, 7, 2231–2239 CrossRef CAS PubMed.
- S. Liang, A. V Subrahmanyam, M. Khadem, Y. Zhao and A. Adronov, RSC Adv., 2016, 6, 25733–25740 RSC.
- X. Zhang, J. Zhao, M. Tange, W. Xu, W. Xu, K. Zhang, W. Guo, T. Okazaki and Z. Cui, Carbon, 2015, 94, 903–910 CrossRef CAS.
- H. Li, F. Zhang, S. Qiu, N. Lv, Z. Zhao, Q. Li and Z. Cui, Chem. Commun., 2013, 49, 10492 RSC.
- M. Tange, T. Okazaki and S. Iijima, ACS Appl. Mater. Interfaces, 2012, 4, 6458 Search PubMed.
- M. Tange, T. Okazaki and S. Iijima, Nanoscale, 2014, 6, 248–254 RSC.
- N. Berton, F. Lemasson, A. Poschlad, V. Meded, F. Tristram, W. Wenzel, F. Hennrich, M. M. Kappes and M. Mayor, Small, 2014, 10, 360–367 CrossRef CAS PubMed.
- G. J. Brady, Y. Joo, S. Singha Roy, P. Gopalan and M. S. Arnold, Appl. Phys. Lett., 2014, 104, 83107 CrossRef.
- Y. Joo, G. J. Brady, M. S. Arnold and P. Gopalan, Langmuir, 2014, 30, 3460–3466 CrossRef CAS PubMed.
- Y. Joo, G. J. Brady, M. J. Shea, M. B. Oviedo, C. Kanimozhi, S. K. Schmitt, B. M. Wong, M. S. Arnold and P. Gopalan, ACS Nano, 2015, 9, 10203–10213 CrossRef CAS PubMed.
- S. D. Stranks, A. M. R. Baker, J. A. Alexander-Webber, B. Dirks and R. J. Nicholas, Small, 2013, 9, 2245–2249 CrossRef CAS PubMed.
- S. D. Stranks, S. N. Habisreutinger, B. Dirks and R. J. Nicholas, Adv. Mater., 2013, 25, 4365–4371 CrossRef CAS PubMed.
- H. W. Lee, Y. Yoon, S. Park, J. H. Oh, S. Hong, L. S. Liyanage, H. Wang, S. Morishita, N. Patil, Y. J. Park, J. J. Park, A. Spakowitz, G. Galli, F. Gygi, P. H.-S. Wong, J. B.-H. Tok, J. M. Kim and Z. Bao, Nat. Commun., 2011, 2, 541 CrossRef PubMed.
- S. Park, H. W. Lee, H. Wang, S. Selvarasah, M. R. Dokmeci, Y. J. Park, S. N. Cha, J. M. Kim and Z. Bao, ACS Nano, 2012, 6, 2487–2496 CrossRef CAS PubMed.
- S. G. Bucella, J. M. Salazar-Rios, V. Derenskyi, M. Fritsch, U. Scherf, M. A. Loi and M. Caironi, Adv. Electron. Mater., 2016, 2, 1600094 CrossRef.
- W. Gomulya, V. Derenskyi, E. Kozma, M. Pasini and M. A. Loi, Adv. Funct. Mater., 2015, 25, 5858–5864 CrossRef CAS.
- P. Feng, W. Xu, Y. Yang, X. Wan, Y. Shi, Q. Wan, J. Zhao and Z. Cui, Adv. Funct. Mater., 2017, 27, 1604447 CrossRef.
- H. Wang, J. Mei, P. Liu, K. Schmidt, G. Jiménez-Osés, S. Osuna, L. Fang, C. J. Tassone, A. P. Zoombelt, A. N. Sokolov, K. N. Houk, M. F. Toney and Z. Bao, ACS Nano, 2013, 7, 2659–2668 CrossRef CAS PubMed.
- T. Lei, G. Pitner, X. Chen, G. Hong, S. Park, P. Hayoz, R. T. Weitz, H.-S. P. Wong and Z. Bao, Adv. Electron. Mater., 2016, 2, 1500299 CrossRef.
- J. M. Salazar-Rios, W. Gomulya, V. Derenskyi, J. Yang, S. Z. Bisri, Z. Chen, A. Facchetti and M. A. Loi, Adv. Electron. Mater., 2015, 1, 1500074 CrossRef.
- F. A. Lemasson, T. Strunk, P. Gerstel, F. Hennrich, S. Lebedkin, C. Barner-Kowollik, W. Wenzel, M. M. Kappes and M. Mayor, J. Am. Chem. Soc., 2011, 133, 652–655 CrossRef CAS PubMed.
- N. A. Rice and A. Adronov, Macromolecules, 2013, 46, 3850–3860 CrossRef CAS.
- N. A. Rice and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2738–2747 CrossRef CAS.
- N. A. Rice, A. V. Subrahmanyam, S. E. Laengert and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2510–2516 CrossRef CAS.
- W. Gomulya, G. D. Costanzo, E. J. F. De Carvalho, S. Z. Bisri, V. Derenskyi, M. Fritsch, N. Fröhlich, S. Allard, P. Gordiichuk, A. Herrmann, S. J. Marrink, M. C. Dos Santos, U. Scherf and M. A. Loi, Adv. Mater., 2013, 25, 2948–2956 CrossRef CAS PubMed.
- J. Ding, Z. Li, J. Lefebvre, F. Cheng, G. Dubey, S. Zou, P. Finnie, A. Hrdina, L. Scoles, G. P. Lopinski, C. T. Kingston, B. Simard and P. R. L. Malenfant, Nanoscale, 2014, 6, 2328–2339 RSC.
- H. Ozawa, T. Fujigaya, Y. Niidome, N. Hotta, M. Fujiki and N. Nakashima, J. Am. Chem. Soc., 2011, 133, 2651–2657 CrossRef CAS PubMed.
- P. Imin, F. Cheng and A. Adronov, Polym. Chem., 2011, 2, 1404–1408 RSC.
- N. Berton, F. Lemasson, F. Hennrich, M. M. Kappes and M. Mayor, Chem. Commun., 2012, 48, 2516–2518 RSC.
- F. Jakubka, S. P. Schießl, S. Martin, J. M. Englert, F. Hauke, A. Hirsch and J. Zaumseil, ACS Macro Lett., 2012, 1, 815–819 CrossRef CAS.
- H.-J. Shin, S. M. Kim, S.-M. Yoon, A. Benayad, K. K. Kim, S. J. Kim, H. K. Park, J.-Y. Choi and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 2062–2066 CrossRef CAS PubMed.
- N. Varghese, A. Ghosh, R. Voggu, S. Ghosh and C. N. R. Rao, J. Phys. Chem. C, 2009, 113, 16855–16859 CrossRef CAS.
- N. A. Rice, A. V. Subrahmanyam, B. R. Coleman and A. Adronov, Macromolecules, 2015, 48, 5155–5161 CrossRef CAS.
- D. Fong, W. J. Bodnaryk, N. A. Rice, S. Saem, J. M. Moran-Mirabal and A. Adronov, Chem.–Eur. J., 2016, 22, 14560–14566 CrossRef CAS PubMed.
- S. N. Barman, M. C. LeMieux, J. Baek, R. Rivera and Z. Bao, ACS Appl. Mater. Interfaces, 2010, 2, 2672–2678 Search PubMed.
- Z. Sun, I. O'Connor, S. D. Bergin and J. N. Coleman, J. Phys. Chem. C, 2009, 113, 1260–1266 CrossRef CAS.
- W. Gomulya, J. M. S. Rios, V. Derenskyi, S. Z. Bisri, S. Jung, M. Fritsch, S. Allard, U. Scherf, M. C. Dos Santos and M. A. Loi, Carbon, 2015, 84, 66–73 CrossRef CAS.
- L. Qian, W. Xu, X. Fan, C. Wang, J. Zhang, J. Zhao and Z. Cui, J. Phys. Chem. C, 2013, 117, 18243–18250 CrossRef CAS.
- H. Wang, B. Hsieh, G. Jiménez-Osés, P. Liu, C. J. Tassone, Y. Diao, T. Lei, K. N. Houk and Z. Bao, Small, 2014, 1–8 CrossRef.
- J. Ding, Z. Li, J. Lefebvre, X. Du and P. R. L. Malenfant, J. Phys. Chem. C, 2016, 120, 21946–21954 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |