
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A modular route to boron doped PAHs by combining borylative cyclisation and electrophilic C–H borylation†
D. L.
Crossley
,
R. J.
Kahan
 ,
S.
Endres
,
A. J.
Warner
,
R. A.
Smith
,
J.
Cid
,
J. J.
Dunsford
,
J. E.
Jones
,
I.
Vitorica-Yrezabal
and
M. J.
Ingleson
*
,
S.
Endres
,
A. J.
Warner
,
R. A.
Smith
,
J.
Cid
,
J. J.
Dunsford
,
J. E.
Jones
,
I.
Vitorica-Yrezabal
and
M. J.
Ingleson
*
School of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: Michael.ingleson@manchester.ac.uk
First published on 28th September 2017
Abstract
Heteroatom doping into polyaromatic hydrocarbons (PAHs) is a powerful approach for modifying key physical properties, however, there are extremely few modular routes that enable facile formation of B-, B2- and B,N-(specifically not containing direct B–N bonds) doped PAHs despite the growing importance of these materials. Sequential, one pot borylative cyclisation/intramolecular electrophilic C–H borylation of naphthyl-alkynes provides a simple new route to access novel B-, B,N- and B2-doped (PAHs). The initial products, dihydronaphthalene/dihydroquinoline B-mesityl PAHs, were reacted with [Ph3C][BF4]/pyridyl base to form the oxidised B-, and B,N-doped PAHs. However, for B-triisopropylphenyl (Trip) PAH congeners oxidation has to be performed prior to Trip installation due to preferential oxidation of an isopropylaryl moiety to the styrene. This alternative sequence enables access to Trip-B-PAHs and to structurally constrained B and B2-PAHs. Analysis of the solid state structures and optoelectronic properties of these PAHs confirm that frontier orbital energies, extended packing structures, Stokes shift and quantum yields all can be rationally modified using this methodology. The simplicity of this synthetic approach makes it a powerful tool for rapidly generating novel bench stable boron doped PAHs, which is important for facilitating further structure–property relationship studies and the wider utilisation of these materials in optoelectronic applications.
Introduction
Polyaromatic hydrocarbons (PAHs) are of significant topical interest as their attractive properties have resulted in a range of applications in the optoelectronic arena.1 Many key properties of PAHs are controlled by their frontier molecular orbitals, which in turn can be modulated by: (i) varying the size and shape of the PAH and, (ii) incorporating dopant (non-carbon) main group atoms.2 The introduction of electron deficient three coordinate boron atoms into PAHs is an attractive strategy to reduce the LUMO energy.3 Materials with low lying LUMO energy levels are highly desirable, for example as n-type or ambipolar semi-conductor materials.4 While there are many established routes to form all carbon PAHs, and PAHs doped with electron rich atoms,2 in contrast methods to form boron doped PAHs are relatively limited.4 While some notable progress has been made in this area this has principally generated mono-B and B–E (containing direct B–E bond(s), E = N or O) doped PAHs.3,5,6 This limitation significantly restricts the accessible compound space. Therefore the development of a simple and modular methodology that provides access to mono-B doped, and more importantly, to multiply B-doped and B, E doped (where there is no B–E bond) PAHs is highly desirable to facilitate fuller exploitation of these materials and enable greater understanding of structure property relationships. To the best of our knowledge there is only one modular route able to generate B, B2 and B, E (where there is no B–E bond) doped PAHs (Fig. 1, top left).7 This route requires the formation of 9-sila-anthracene (or 9-bora-anthracene) and then uses Peterson olefination, photocyclization, or ruthenium catalyzed cyclisation of aryl ene-ynes, and Si/B exchange reactions to form the B-PAHs.7 Therefore, new modular routes that are complementary to this work and ideally start from simpler precursors are required as a key enabler for this field.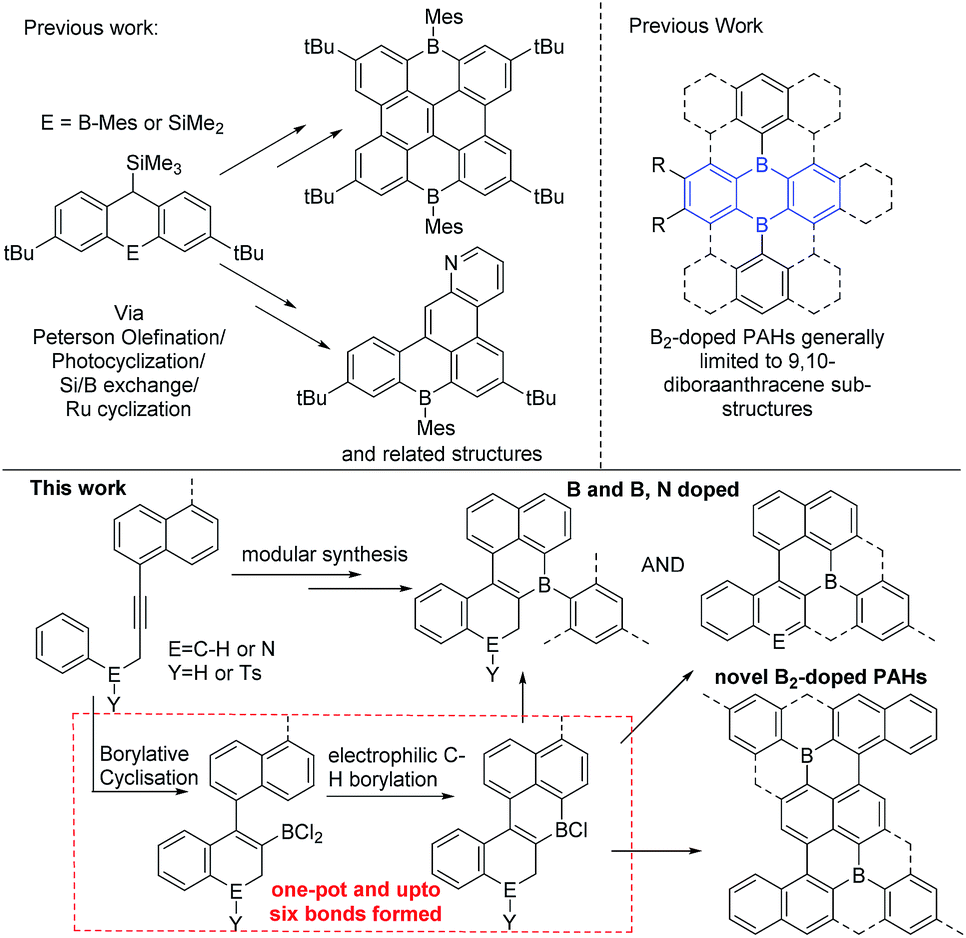 | ||
| Fig. 1 Top left, a current modular route to B doped PAHs. Top right, 9,10-dibora-anthracene based B2-PAHs. Bottom the modular route to novel B, B2 and B,N-doped PAHs reported herein. | ||
We envisaged combining alkyne borylative cyclisation8 and intramolecular electrophilic C–H borylation9 to generate a versatile method to form B, B2 and B,N doped PAHs in one pot (Fig. 1, bottom). Naphthalene functionalised alkynes were selected as: (i) they are available in one step from commercial materials (e.g. aryl bromides and terminal alkynes), (ii) post borylative cyclisation they are correctly arranged to form six membered boracycles by intramolecular electrophilic borylation,10 and (iii) non-planar PAHs will be formed (due to the presence of cove regions) enhancing solubility.11 Herein we report that this single methodology facilely leads to B-, B2- and B,N-doped PAHs all from simple alkyne precursors. This represents a facile modular route to generate PAH materials containing novel dopant atom distributions. This approach also increases the diversity of B2-doped PAHs by accessing structures that are not based on 9,10-dibora-anthracene units (Fig. 1, top right, blue substructure), with the latter ubiquitous in the B2-PAHs reported to date.3,12
Results and discussion
Synthesis of mono-boron-PAHs
1-(4-phenylbut-1-yn-1-yl)-naphthalene has been previously demonstrated to undergo borylative cyclisation using BCl3/2,4,6-tritertbutyl pyridine (TBP) to form a vinyl BCl2 species.8 In this work instead of pinacol protection, the vinyl-BCl2 species is converted by intramolecular electrophilic C–H borylation to 1-BCl. This is achieved by addition of stoichiometric quantities of AlCl3 and 2,6-dichloropyridine (Cl2-py).13 Compound 1-BCl was isolated in 72% yield (Scheme 1) with no five membered boracycle derived from substitution of H1 observed. As an alternative to isolating 1-BCl, the crude reaction mixture containing 1-BCl and protonated pyridine by-products from SEAr was reacted directly with MgBrMes to generate 1-BMes (76% yield) or with MgBrTrip (Trip = triisopropylbenzene) to produce 1-BTrip (71% yield). These are excellent yields for a three step one pot process forming four bonds direct from the alkyne. While 1-BCl proved sensitive to moisture both 1-BMes and 1-BTrip were stable to non-purified solvents and to silica gel column chromatography. The formulation as 1-BCl, 1-BMes and 1-BTrip was supported by multinuclear NMR spectroscopy (δ11B = 51, 58 and 60 ppm, respectively) and mass spectrometry for the latter two species. Furthermore, X-ray diffraction studies were performed on single crystals of 1-BCl and 1-BMes (Fig. 2). | ||
| Scheme 1 Sequential borylative cyclisation/C–H borylation to form boracycles. | ||
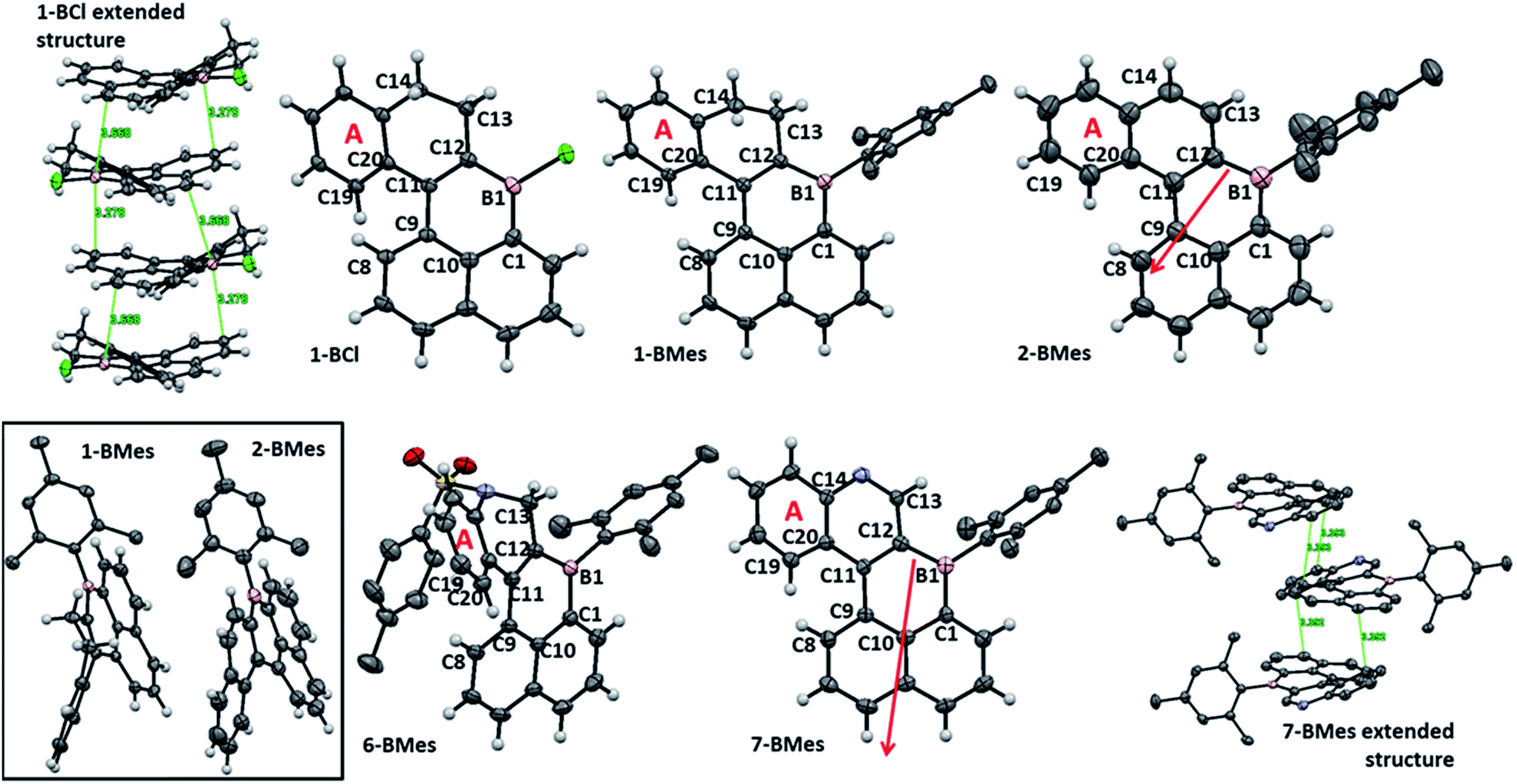 | ||
| Fig. 2 Solid state structures of B-PAHs. Ellipsoids at the 50% probability level, hydrogens on mesityl and tosyl groups are omitted for clarity. Top left and bottom right, extended structures for 1-BCl and 7-BMes. Red arrows indicate the dipole moment from the calculated structures of 2-BMes and 7-BMes at the M06-2X/6311G(d,p) level. | ||
The oxidation of BPin substituted dihydronaphthalenes to the naphthalene congeners using [Ph3C][BF4]/TBP in 1,2-dichloroethane (DCE) has been previously reported.8 This reagent combination was used to oxidise 1-BCl to 2-BCl and 1-BMes to 2-BMes (Scheme 2), with the latter amenable to recrystallization and X-ray diffraction studies. Notably, the oxidation procedure does not convert 1-BTrip to 2-BTrip and instead leads to a complex 1H NMR spectrum. This spectrum contained 1H integral diastereotopic resonances (at 3.66 and 3.20 ppm, 2JHH = 13.1 Hz) with the two triplets for the CH2 moieties of the dihydronaphthalene persisting suggesting that the locus of reactivity is at an isopropyl group. This was supported by reacting isopropylbenzene with [Ph3C][BF4]/TBP which led to similar diastereotopic resonances in the 1H NMR spectrum to that observed using 1-BTrip. [Ph3C][BF4] and α-methylstyrene are reported to undergo a formal [3 + 2] cyclo-addition reaction to form 1-methyl-1,3,3-triphenylindane,14 and this indane is the product formed from the reaction of isopropylbenzene with [Ph3C][BF4]/TBP. Thus on heating, [Ph3C][BF4]/TBP oxidises isopropylbenzenes to the respective styrene, which then undergoes a rapid reaction with a further equivalent of [Ph3C]+ to generate the indane. Therefore the product from the reaction of 1-BTrip with [Ph3C][BF4]/TBP is the indane 3 (Scheme 2, bottom right), fully consistent with multinuclear NMR spectroscopy and mass spectrometry (and 3 is isolated in 52% yield). To the best of our knowledge this frustrated Lewis pair type oxidation of isopropylbenzenes is unprecedented.15 Using an alternative sequence 2-BTrip was accessible in good yield (65%) from 1-BCl by first oxidising to form 2-BCl using [Ph3C][BF4]/TBP, and then in situ addition of MgBrTrip to form 2-BTrip. 2-BTrip was not amenable to crystallization in our hands, although the NMR data for 2-BMes and 2-BTrip are comparable (excluding the Mes/Trip resonances) and both have broad 11B resonances centered at 61 ppm.
 | ||
| Scheme 2 Oxidation of dihydronaphthalene B-PAHs. | ||
With kinetic stabilization post borylative cyclisation/SEAr using Mes and Trip groups demonstrated, the ability to kinetically protect the boron centre using the principle of structural constraint was explored.16 Structural constraint provides stability to ambient conditions by the rigid fused structure disfavouring pyramidalisation at boron. A previous example comparing the effects of protecting boron with mesityl groups versus structural constraint has indicated that there is minimal impact on frontier molecule orbital energies and character.17 However, there are dramatic effects on the extended solid state structures, thus both protection methods are important as they will lead to distinct bulk properties that are complementary for application purposes. Compound 1-BCl was oxidized to 2-BCl and then addition of (2,6-di(prop-1-en-2-yl)phenyl)lithium led to 4 (Scheme 3). In our hands 4 could not be isolated (and decomposed during column chromatography, consistent with previous reports on related compounds).17 Therefore, the crude reaction mixture derived from 1-BCl was reacted with Sc(OTf)3 at 75 °C for 16 h to effect double Friedel Crafts cyclisation. The resultant fully fused B-PAH 5 proved stable to ambient conditions and could be isolated by column chromatography in 26% yield directly from 1-BCl (the yield is only moderate but this is for 4 bond forming steps without any intermediate purification steps). While suitable single crystals were not obtained NMR and mass spectrometry were consistent with this formulation. Notably, the δ11B for 5 at 45 ppm is shifted upfield from that for 2-BMes/2-BTrip (δ11B 61 ppm) consistent with a planarised boracycle structure.17
 | ||
| Scheme 3 Synthesis of the structurally constrained B-PAH 5. | ||
With the methodology demonstrated for B-doped PAHs we next extended it to form B,N-doped PAHs. The borylative cyclisation/electrophilic borylation one pot methodology generates a B,N-doped PAH from 4-methyl-N-(3-(naphthalen-1-yl)prop-2-yn-1-yl)-N-phenylbenzene-sulfonamide (this alkyne is accessible in one step from commercial materials). Compound 6-BCl (Scheme 4) is generated in good conversion provided ≥ 2 eq. of AlCl3 are utilised, presumably the first equivalent of AlCl3 interacts preferentially with Lewis basic sites in the N-Ts moiety preventing activation of the vinylBCl2 until a second equivalent of AlCl3 is introduced. Post extraction of the crude mixture into toluene, compound 6-BCl can be protected with ZnMes2 to afford 6-BMes. MesMgBr was not effective in forming 6-BMes as this reagent led to competitive N-tosyl deprotection18 and complex intractable mixtures. The dihydroquinoline moiety in 6-BMes can be oxidised to furnish the quinoline congener, 7-BMes, using [Ph3C][BF4], with tosyl removal then achieved by addition of 4-DMAP. Compound 7-BMes was isolated in good yield (79%) with its formulation confirmed by multinuclear NMR spectroscopy (including a δ11B = 60 ppm), mass spectrometry and X-ray diffraction studies. Pyridyl containing B-doped PAHs (such as 7-BMes) are extremely rare, particularly those without B–N bonds.7b Indeed, the bicyclic B,N substructure in 7-BMes, (a borinino[2,3-c]pyridine, highlighted in blue, Scheme 4) is to the best of our knowledge the first reported example of this substructure.
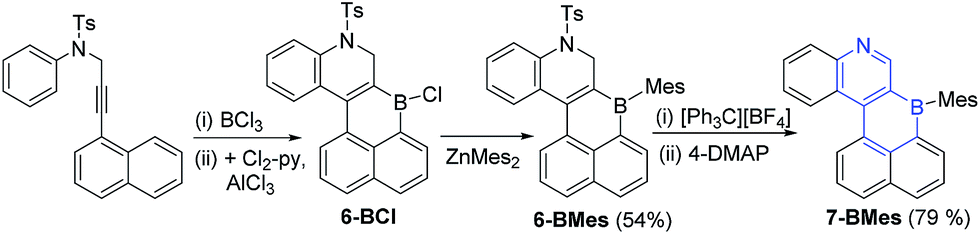 | ||
| Scheme 4 Borylative cyclisation/SEAr to form 6- and 7-BMes after oxidation. | ||
Solid state structures of mono-boron PAHs
The solid state structures (Fig. 2) of 1-BCl, 1-BMes, 2-BMes, 6-BMes and 7-BMes all contain trigonal planar boron centres and effectively planar boracycles. The mesityl substituents approach an orthogonal orientation to the polycyclic system in the latter four compounds. The short vinylic C11–C12 distances in 1-BCl, 1-BMes and 6-BMes (1.377(3), 1.374(3), and 1.366(2) Å, respectively) and long vinylC-B distances (shortest B1–C12 = 1.522(3) Å in 1-BCl which is comparable to the naphthylC-B distance in 1-BCl, B1–C1 = 1.525(3) Å) indicate the structure is dominated by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/vinylC-B resonance form. On oxidation to 2-BMes and 7-BMes the C11–C12 bond distances increase to 1.412(3) and 1.407(4) Å, respectively, as expected on transforming a vinyl into an aromatic system. Irrespective of oxidation level the C9–C11 bond distances are long (1.476–1.486 Å) indicating minimal aromaticity in the boracycle.
C/vinylC-B resonance form. On oxidation to 2-BMes and 7-BMes the C11–C12 bond distances increase to 1.412(3) and 1.407(4) Å, respectively, as expected on transforming a vinyl into an aromatic system. Irrespective of oxidation level the C9–C11 bond distances are long (1.476–1.486 Å) indicating minimal aromaticity in the boracycle.
Despite containing essentially planar boracycles there is considerable deviation from planarity in the fused pentacyclic structures due to steric crowding in the cove region. The dihedral angles C19–C20–C9–C8 are 45.8°, 48.0° and 44.2° for 1-BCl, 1-BMes and 6-BMes, respectively, which decrease to 36.9° in 2-BMes and 32.8° in 7-BMes. The greater deviation from planarity in 1-BCl, 1-BMes and 6-BMes partly arises from hinging of the partially saturated ring (Fig. 2, bottom left) which leads to angles of 33.4°, 36.3° and 35.4°, respectively, between the mean plane of the boracycle and the mean plane of the six atoms in ring A. For 2-BMes and 7-BMes the greater rigidity arising from oxidation leads to a smaller angle between the mean plane of the boracycle and the mean plane of the six atoms in ring A (21.6° in 2-BMes and 21.1° in 7-BMes) as expected. A comparison of structural constraint versus orthogonal B-aryl groups is provided for the B2-PAHs where both have been characterised by X-ray diffraction.17
Examination of the extended structures reveal that 1-BCl contains close packed 1D columnar formations (Fig. 2, top left) as expected as it does not contain any orthogonal aryl groups. While there is no significant differences in the structural metrics of the isoelectronic compounds 2-BMes and 7-BMes, comparison of the extended structures revealed notable differences. The former contains no close intermolecular contacts involving the pentacyclic core; however, 7-BMes forms 1D columns with short intermolecular C–C contacts of 3.39 Å between pentacyclic cores of adjacent molecules (Fig. 2, bottom right). This disparity can be explained by the permanent dipole moment which is greater and has a different orientation in 7-BMes relative to 2-BMes (calculated dipole for 2-BMes = 2.05 D and for 7-BMes = 5.13 D at the M06-2X/6311G(d,p) level, see red arrows Fig. 2). Thus inclusion of an additional heteroatom dramatically alters the physical properties as expected by increasing the polarization within the PAH. This polarization favours forming a head to tail stacking to correctly align adjacent dipoles. A related outcome has been recently observed on incorporation of a B–CC–N moiety to generate an indole derived-PAH.10 Comparison of 2-BMes and 7-BMes with dibenz[a,kl]anthracen-7-one (which is isoelectronic to 2-BCl, Fig. 3, left) reveals a similar distortion from planarity in the pentacyclic structure with a C19–C20–C9–C8 angle of 33.4° in this fused cyclic-ketone.19 Compound 2-BMes is an isomer of compound A and comparison reveals minimal differences in the B–C bond metrics with 2-BMes and A having similar C–B–C angles and B–C distances. However, the different benzannulation pattern in A leads to an effectively planar structure due to the absence of cove regions in A (and in the structurally constrained isomer B which can be viewed as an isomer of 5).17
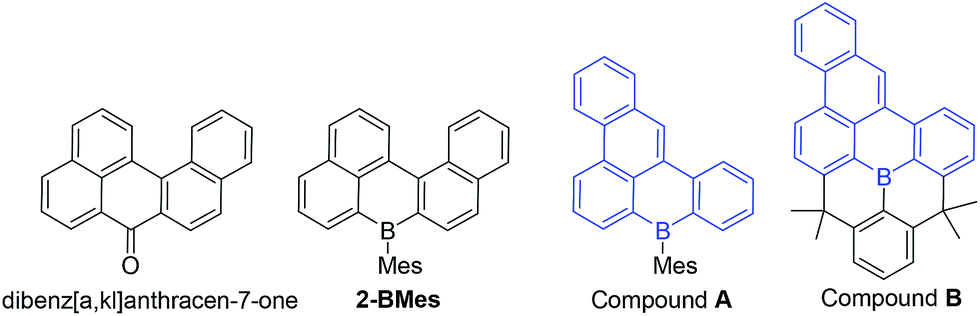 | ||
| Fig. 3 Isoelectronic (to 1-BCl) and isomeric structures of 2-BMes and 5. | ||
Synthesis of di-boron-PAHs
To further demonstrate the utility of sequential borylative-cyclisation/intramolecular C–H borylation an extended PAH containing two boron centres was targeted. Diyne 8 was synthesized in one step from 1,5-dibromonaphthalene and the commercially available terminal alkyne 3-butyn-1-yl-benzene. Notably, cyclisation of 8 will lead to a diboron-doped PAH containing a different arrangement of the two boron centres relative to that found in 9,10-dibora-anthracenes. Diyne 8 was converted to the double boracycle compound 9-BCl in one pot using identical conditions to that successful for the mono-B-PAHs. Specifically, BCl3/TBP was used to achieve borylative cyclisation which then was followed by addition of AlCl3/Cl2-py to effect intramolecular electrophilic C–H borylation. Compound 9-BCl is poorly soluble in chlorinated organic solvents and precipitated from solution on standing. This enabled its isolation in excellent yield (86% from 8) for a one pot reaction forming four B–C bonds and two C–C bonds. The identification of 9-BCl was based on 1H NMR spectroscopy, elemental analysis and an X-ray diffraction study (Fig. 4). Compound 9-BCl can be converted to 9-BMes by addition of MgBrMes. | ||
| Fig. 4 Solid state structures of B2-PAHs. Ellipsoids are drawn at the 50% probability level. | ||
Notably, 9-BMes has lower stability towards moisture than 1-BMes, although sufficient 9-BMes was obtained for characterisation by multinuclear NMR spectroscopy and for an X-ray diffraction study. In contrast, 9-BTrip was stable to ambient atmosphere and could be isolated post column chromatography as a red powder in 70% yield. Application of the oxidation conditions used for 1-BCl ([Ph3C][BF4]/TBP in DCE) resulted in conversion of 9-BCl to 10-BCl. Compound 10-BCl was protected in situ using MgBrTrip to produce 10-BTrip in 45% isolated yield post chromatography. The formation of 10-BTrip was confirmed by multinuclear NMR, mass spectrometry and an X-ray diffraction study. The 11B NMR chemical shifts for 9/10-BTrip are centred at 62 ppm as expected for boron containing PAHs. Compound 9-BCl also was converted through to the fully fused PAH 11 in one pot via10-BCl in 11% isolated yield (while low this is an overall yield for oxidation/arylation/two Friedel Crafts cyclisations for each boracycle with no intermediate purifications performed, so is for 8 bond forming steps in one pot at ca. 57% yield per bond forming step). Compound 11 is a large PAH containing fourteen annulated rings that is stable to column chromatography and ambient conditions with an X-ray diffraction study confirming its successful formation.
The combination of borylative cyclisation/electrophilic C–H borylation enables facile access to 9/10-BAryl which contain a previously unknown arrangement of two boron atoms for a B2-PAH, (highlighted in blue in Scheme 5, while aza-borapyrenes have been previously reported,20 no diborapyrene based PAHs have been previously reported to the best of our knowledge). Overall the 1,6-di-borapyrene unit is accessed rapidly via a short sequence of linear steps (only two isolation steps required to form 9-BCl starting from commercial materials). B2-PAHs that are not based on 9,10-dibora-anthracene units are extremely rare and those that have been reported to date are synthesised via relatively laborious methodologies (e.g. halogenation/lithiation/borylation) starting from complex precursors (e.g. 1,5-dihalo-2,6-ditriflato-naphthalenes),21 disfavouring the wider utilisation of these molecules.
 | ||
| Scheme 5 Synthesis of B2-doped PAHs, with the 1,6-diborapyrene core highlighted. | ||
Solid state structures of B2-PAHs
The solid state structures of 9-BCl, 9-BMes and 10-BTrip contain approximately planar B2C14 diborapyrene cores. Compounds 9-BCl, 9-BMes and 10-BTrip have geometric trends similar to that discussed for the 1/2-B series. For example, oxidation reduces the angle between the mean planes of ring A relative to that of the boracycle B1–C1–C10–C9–C11–C12 (for 9-BCl and 9-BMes = 39.2 and 34.8°, respectively, and for 10-BTrip = 23.1°). Displacement distances for the B2C14 atoms from the mean plane of the diborapyrene unit are all < 0.15 Å for 9-BCl and 9-BMes, but they are larger for 10-BTrip (all < 0.24 Å). The greater displacement distances of atoms from the diborapyrene mean plane in 10-BTrip (despite a lower angle between the planes of the proximal boracycle and A) is presumably due to structural distortions to reduce steric crowding in the cove region as oxidation prevents the terminal bicyclic unit hinging to alleviate sterics (as observed in 9-BCl and 9-BMes analogous to 1-BMes).Close comparison of 11 with 10-BTrip is noteworthy and revealed that 11 is less distorted from planarity (e.g. the angle between the mean plane of the boracycle B1–C1–C10–C9–C11–C12 and the mean plane of ring A = 20.8°). This is due to the more rigid structure formed on full fusion around boron which results in the linking of the naphthalene unit containing ring A to the diborapyrene core by three C–C bonds (in 11) instead of two (in 10-BTrip). The diborapyrene cores are generally comparable in metrics (excluding the shorter B–C distances in 11 analogous to that observed comparing A and B (Fig. 3))17 but in 11 it is more planar (deviations of atoms from the mean plane of the B2C14 core are all < 0.20 Å for 11) than 10-BTrip. Therefore in this case the protection at boron using the structural constraint approach increases the rigidity of the overall molecule and thus makes it a more planar PAH compared to protection using bulky aryl groups. The extended structures of 11 and 10-BTrip are also notably different with 10-BTrip containing no close intermolecular contacts involving the diborapyrene core and in fact only exhibiting one close face to face π–π contact involving the extended PAH system (through ring A, with one ring A approaching a ring A of an adjacent molecule above and below the plane with an intermolecular C–C distance of 3.348 Å, see Fig. S16†). In contrast, the extended structure of 11 contains multiple short intermolecular face to face π–π contacts, including involving the planarised diborapyrene cores of adjacent molecules (intermolecular C–C and B–C contacts = 3.351 and 3.395 Å see Fig. S17†). This results in a 1D columnar extended structure.
Notably, two symmetry related orthogonally orientated columnar structures are present in the extended structure which is attractive for providing charge mobility in two dimensions. This again highlights the ability of the structural constraint approach to engender more face to face π stacking interactions, as previously noted by Wagner and Yamaguchi.17
Computational studies
With eight B-doped PAHs in hand, structure–property relationships were investigated (10-BTrip was simplified to 10-BMes). The calculated structures have similar metrics and trends to those observed in the solid state. 1-BMes/2-BMes have similar energy and character LUMOs with significant boron character in both (Fig. 5). However, the HOMOs were more disparate with that of 2-BMes being 0.233 eV higher in energy than for 1-BMes. This is attributed to a greater degree of orbital delocalisation due to a more planar PAH structure post oxidation and a larger contribution to the HOMO from the additional CC bond. Comparison of the frontier orbitals of 9-BMes and 10-BMes also revealed closely comparable energy and character LUMOs which are delocalised throughout the central diborapyrene unit with significant contribution from boron in both cases. The HOMO of the oxidised congener 10-BMes, is higher in energy, which is attributed to the greater delocalisation of the HOMO on oxidation as discussed above for 2-BMes. On comparison of the B- and B2-PAHs (e.g.2-BMesvs.10-BMes) the most dramatic difference is in the energy of the LUMO which is calculated to be ∼0.7 eV lower in energy for the significantly more extended B2-PAH.
 | ||
| Fig. 5 Molecular orbital energy levels and molecular orbital contours (isovalue = 0.04) of the HOMO and LUMO of the B, B,N and B2-doped PAHs calculated at the M06-2X/6311G(d,p) level with PCM DCM. | ||
Comparison of 2-BMes, 5 and 7-BMes reveals minimal change in the distribution of the LUMO throughout the series. Whereas increasing the degree of planarization results in less boron character in the HOMO. The most notable change in this series is in the frontier orbital energies, with incorporation of the stronger acceptor quinoline moiety (relative to naphthalene) resulting in a significant reduction in the energy of the HOMO and LUMO relative to 2-BMes. In contrast, fully fused 5 has higher HOMO and LUMO energies relative to 2-BMes, which is consistent to that previously observed by Wagner and Yamaguchi for differentially protected 8H-8-borabenzo[gh]tetraphenes (compounds A and B, Fig. 3).17 This change in energy has been attributed previously to the inductive effect of the CMe2 substituents.
Analogous trends in frontier orbital energies are observed for 9-BMes, 10-BMes and 11. However, a notable disparity between compounds 11 and 10-BMes is that the HOMO is more delocalised in the former with significant character on the diborapyrene core. This is distinct to what was observed for compounds A and B where the frontier orbitals are effectively identical irrespective of the protecting strategy (mesityl vs. structural constraint). This disparity is presumably due to the fact that A and B both have fully planar PAH cores due to the absence of cove/bay regions, thus there are no geometric differences in the pentacycle units between compounds A and B. However, as discussed above, 11 is more planar than 10-BTrip which will favour greater delocalisation of molecular orbitals, as observed in the HOMO for 11 compared to 10-BMes. Therefore the different protecting methods available to stabilise boron doped PAHs, specifically structural constraint and orthogonal bulky aryls, can result in sufficient changes in geometry to impact the electronic structure of the boron doped PAHs.
Combined, the calculations/solid state structures indicate that this modular methodology to B-doped PAHs when coupled with the two different protecting strategies enables significant modification of (i) the frontier orbital energies (e.g.2-BMesvs.7-BMes), (ii) the frontier orbital distribution (1-BMesvs.5) and, (iii) the extended structures (e.g.10-BMesvs.11). These are all key variables that need to be controlled when optimising performance in organoelectronic devices.
Optoelectronic properties
Cyclic voltammetry confirmed the calculated trends in the LUMO energies with the diboron PAHs 9-BTrip and 10-BTrip being more readily reduced by ∼0.55 eV than 1-BMes and 2-BMes. It is also notable that 9-BTrip and 10-BTrip display two reversible reduction waves; in contrast, 1-BMes and 2-BMes exhibit a second reduction wave at a much more negative potential that is irreversible for a range of scan rates. For both the B- and B2-PAHs the fully fused congeners have slightly more negative reduction potentials than the non fused derivatives in line with calculations and previous observations.17 Furthermore, the reduction waves are non-reversible for 5 and 11. On replacing naphthalene for quinoline the reduction process occurs at a less negative potential (by 0.2 V for 7-BMesvs.2-BMes), confirming that the stronger acceptor moiety results in a notable reduction of the LUMO energy.Analysis of the photophysical data is also informative and supports the general trends derived from calculations (see ESI† for all the absorption and emission spectra). For example, the largest optical band gap is found for 7-BMes, while the smallest is found for 10-BTrip and 11 consistent with the calculated frontier orbital energies. The structurally constrained compounds 5 and 11 are significantly more emissive than their non-fused analogues 2-BMes and 10-BTrip, respectively (ΦPL values 24 vs. 42% and 23 vs. 66%, respectively) and that the emission of 5 and 11 is blue shifted relative to 2-BMes and 10-BTrip, respectively. Again this is in contrast to compounds A and B which have closely comparable photophysical properties (see Table 1), but a related increase in ΦPL has been observed on structurally constraining 9,10-dibora-anthracenes.16a,22 The smaller Stokes shift and higher quantum yields for 5 and 11 can be attributed to their more rigid structures (relative to 2-BMes and 10-BTrip), which can be expected to result in less structural reorganisation on excitation, and fewer rotational and vibrational degrees of freedom (reducing non-radiative decay pathways).23 Finally, compound 9-BTrip is effectively non emissive, and has a low energy emission band. This compound shows concentration dependence of emission intensity with the lowest energy emission bands centred at 628 and 650 nm increasing in intensity (relative to that at 524 nm) on increasing the concentration (see Fig. S4†). This is attributed to excimer formation, which despite the presence of the Trip substituents is possible through intermolecular interactions involving the terminal naphthalene units as observed in the solid state structure of 10-BTrip (which has face to face π stacking through ring A, Fig. S16†). In contrast, 10-BTrip shows no evidence for concentration dependence in relative emission intensity.
| Compound | 1st reduction wavea | 2nd reduction wavea | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E peak (V) | E 1/2 (V) | LUMO (eV) | E peak (V) | E 1/2 (V) | λmaxabs (nm)b | ε × 103 (M−1 cm−1) | λmaxem (nm)b | ΔEopt (eV)b | Φ PL (%)c | |
| a Measured in THF (1 mM) with [nBu4N][PF6] (0.1 M) as the supporting electrolyte at a scan rate of 50 mV s−1, potentials are given relative to Fc/Fc+ redox couple which is taken to be 4.80 eV below vacuum. LUMO energies calculated using the Eonset values (see Table S2). b Measured at 1 × 10−5 M in toluene, optical band gap from the onset of absorption. c absolute quantum yield values measured using an integrating sphere at 0.2 × 10−5 M in toluene (estimated error ± 10%), s = shoulder, λex (nm). d 351. e 336. f 348. g 359. h 338. i 396. j 487. k 483. l From ref. 17. | ||||||||||
| 1-BMes | −2.00 | −1.92 | −2.96 | −2.72 | — | 351, 367, 420 | 16.1, 12.9, 4.1 | 516d | 2.61 | 16d |
| 2-BMes | −2.00 | −1.92 | −2.95 | −2.65 | — | 336, 370, 428 | 15.8, 8.7, 7.2 | 512e | 2.59 | 24e |
| 5 | −2.10 | — | −2.87 | — | — | 310, 348, 431 | 11.6, 6.2, 3.3 | 486f | 2.62 | 42f |
| 6-BMes | −1.84 | −1.77 | −3.10 | −2.60 | — | 418s, 378, 359 | 5.1, 14.7, 17.1 | 411, 433, 536g | 2.54 | 7g |
| 7-BMes | −1.80 | −1.73 | −3.18 | −2.50 | — | 402, 374, 338 | 9.8, 10.2, 14.2 | 410, 478h | 2.76 | 19h |
| 9-BTrip | −1.40 | −1.33 | −3.53 | −2.00 | −1.93 | 320, 396, 413, 446, 476 | 51.0, 53.0, 49.4, 31.0, 28.5 | 524, 628, 650i | 2.49 | 1i |
| 10-BTrip | −1.46 | −1.39 | −3.48 | −1.93 | −1.86 | 318, 353, 487 | 46.5, 12.5, 21.5 | 592j | 2.31 | 23j |
| 11 | −1.56 | — | −3.38 | −1.89 | — | 326, 375, 411, 483, 511 | 28.4, 9.7, 4.9, 15.1, 17.2 | 553k | 2.30 | 66k |
| Compound Al | — | −2.05 | — | — | — | 311, 382, 400 | 22.4, 21.5, 30.9 | 411, 433, 460 | 3.02 | 85 |
| Compound Bl | — | −2.14 | — | — | — | 330, 379, 400 | 11.4, 22.3, 36.4 | 408, 430, 458 | 3.03 | 89 |
Conclusions
In summary, the combination of borylative cyclisation and intramolecular electrophilic C–H borylation represents a facile route to synthesize a range of boron doped PAHs. The modularity has been proven by forming mono-B doped, doubly B-doped and B,N doped PAHs; with a number of these PAHs containing arrangements of dopant atoms that have not been reported previously. Structure property relationship studies on this series have highlighted a number of approaches to control key properties, such as frontier orbital energies (readily modified for example, by exchanging naphthalene for quinoline). This methodology is particularly attractive as the alkyne precursors are available in one step from commercial materials and thus this approach will enable access to many other novel Bn-doped PAHs starting from suitably halogenated aromatic precursors (e.g., 1,4-dibromonaphthalene, dibromoanthracenes etc.). Therefore the facile, modular synthesis disclosed herein will facilitate the elucidation of structure property relationships and the fuller exploitation of these materials across a range of optoelectronic applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research leading to these results has received funding from the ERC under framework 7 (Grant number 305868), the University of Manchester, the Leverhulme Trust (RPG-2014-340) and the Royal Society (for a U. R. F. to M. J. I.). We also acknowledge the Erasmus + programme of the European Union and the EPSRC (grant number EP/K039547/1) for financial support. The authors would also like to acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London in carrying out elements of this work. Dr Louise Natrajan is thanked for assistance with photophysical measurements (Leverhulme grant: RL-2012-072). Additional research data supporting this publication are available as supplementary information accompanying this publication.Notes and references
- For recent reviews see: (a) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (b) Q. Ye and C. Chi, Chem. Mater., 2014, 26, 4046 CrossRef CAS; (c) L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J.-L. Brédas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500 CrossRef CAS PubMed.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef PubMed; (b) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (c) W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC.
- See: (a) F. Jäkle, Chem. Rev., 2010, 110, 3985 CrossRef PubMed; (b) A. Lorbach, A. Hübner and M. Wagner, Dalton Trans., 2012, 41, 6048 RSC; (c) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257 RSC; (d) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357 CrossRef CAS; (e) Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996 RSC; (f) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846 RSC.
- See: C. Y. Chiu, H. Wang, F. G. Brunetti, F. Wudl and C. J. Hawker, Angew. Chem., Int. Ed., 2014, 53, 3996 CrossRef CAS PubMed , and references therein.
- For B-N PAHs see: (a) M. M. Morgan and W. E. Piers, Dalton Trans., 2016, 45, 5920 RSC; (b) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074 CrossRef CAS PubMed; (c) Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242 CrossRef CAS PubMed; (d) X.-Y. Wang, J.-Y. Wang and J. Pei, Chem.–Eur. J., 2015, 21, 3528 CrossRef CAS PubMed.
- For select recent examples see: (a) X. Wang, F. Zhang, K. S. Schellhammer, P. Machata, F. Ortmann, G. Cuniberti, Y. Fu, J. Hunger, R. Tang, A. A. Popov, R. Berger, K. Müllen and X. Feng, J. Am. Chem. Soc., 2016, 138, 11606 CrossRef CAS PubMed; (b) D. Marinelli, F. Fasano, B. Najjari, N. Demitri and D. Bonifazi, J. Am. Chem. Soc., 2017, 139, 5503 CrossRef CAS PubMed; (c) A. John, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 5588 CrossRef CAS PubMed; (d) S. Natatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087 CrossRef PubMed; (e) K. Matsuo, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 11984 CrossRef CAS PubMedT. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210 CrossRef CAS PubMed; (f) H. Saito, S. Otsuka, K. Nogi and H. Yorimitsu, J. Am. Chem. Soc., 2016, 138, 15315 CrossRef CAS PubMed; (g) X.-Y. Wang, X.-C. Wang, A. Narita, M. Wagner, X. Y. Cao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 12783 CrossRef CAS PubMed; (h) S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Chem. Sci., 2016, 7, 219 RSC; (i) Y.-G. Shi, X. Wang, N. Wang, T. Peng and S. Wang, Organometallics, 2017, 36, 2677 CrossRef CAS; (j) J. S. A. Ishibashi, A. Dargelos, C. Darrigan, A. Chrostowska and S.-Y. Liu, Organometallics, 2017, 36, 2494 CrossRef CAS; (k) G. H. M. Davies, Z.-Z. Zhou, M. Jouffroy and G. A. Molander, J. Org. Chem., 2017, 82, 549 CrossRef CAS PubMed.
- (a) V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800 CrossRef CAS PubMed; (b) V. M. Hertz, J. G. Massoth, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J, 2016, 22, 13181 CrossRef CAS PubMed; (c) V. M. Hertz, H.-W. Lerner and M. Wagner, Org. Lett., 2015, 17, 5240 CrossRef CAS PubMed.
- A. J. Warner, J. R. Lawson, V. Fasano and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 11245 CrossRef CAS PubMed.
- V. Bagutski, A. Del Grosso, J. Ayuso Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed.
- A. Escande, D. L. Crossley, J. Cid, I. A. Cade, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 17160 RSC.
- (a) K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808 RSC; (b) K. Schickedanz, J. Radtke, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2017, 139, 2842 CrossRef CAS PubMed.
- For a representative early example of a B2-doped PAH based on the 9,10-diboraanthracene unit see: C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206 CrossRef CAS PubMed . For more recent examples see 6c and references therein.
- A. Del Grosso, J. Ayuso Carrillo and M. J. Ingleson, Chem. Commun., 2015, 51, 2878 RSC.
- N. Surapanich, P. Chalsulwan, C. Kihakarn and V. Reutrakul, Synlett, 2016, 27, 2689 CrossRef CAS.
- For the FLP based oxidation of N-heterocycles see: A. F. G. Maier, S. Tussing, T. Schneider, U. Flörke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219 CrossRef CAS PubMed.
- For pioneering studies on the structural constraint approach see: (a) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529 CrossRef CAS PubMed; (b) S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130 CrossRef CAS PubMed; (c) K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580 CAS.
- V. M. Hertz, N. Ando, M. Hirai, M. Bolte, H.-W. Lerner, S. Yamaguchi and M. Wagner, Organometallics, 2017, 36, 2512 CrossRef CAS.
- For the deprotection of N-Ts with Girgnard reagents see: A. Mustafa, W. Asker, O. H. Hoshmat, A. F. A. Shalaby and M. Kamel, J. Am. Chem. Soc., 1954, 76, 5447 CrossRef CAS.
- S. Fujisawa, I. Oonishi, J. Aoki, Y. Ohashi and Y. Sasada, Bull. Chem. Soc. Jpn., 1985, 58, 3356 CrossRef CAS.
- M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, Angew. Chem., Int. Ed., 2007, 46, 4940 CrossRef CAS PubMed.
- R. E. Messersmith, M. A. Siegler and J. D. Tovar, J. Org. Chem., 2016, 81, 5595 CrossRef CAS PubMed.
- T. Agou, M. Sekine and T. Kawashima, Tetrahedron Lett., 2010, 51, 5013 CrossRef CAS.
- M. Sauer, J. Hofkens and J. Enderlein, Handbook of Fluorescence Spectroscopy and Imaging, WILEY-VCH, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterisation data, copies of NMR spectra, CV, absorption and emission data and crystallographic data. CCDC 1537995, 1538224, 1538258, 1538386, 1546328, 1546329, 1553723, 1553725 and 1555383. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02793a |
| This journal is © The Royal Society of Chemistry 2017 |