
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A unified photoredox-catalysis strategy for C(sp3)–H hydroxylation and amidation using hypervalent iodine†
Guo-Xing
Li
a,
Cristian A.
Morales-Rivera
b,
Fang
Gao
a,
Yaxin
Wang
a,
Gang
He
a,
Peng
Liu
 *b and
Gong
Chen
*ac
*b and
Gong
Chen
*ac
aState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China. E-mail: gongchen@nankai.edu.cn
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA. E-mail: pengliu@pitt.edu
cDepartment of Chemistry, The Pennsylvania State University, 104 Chemistry Building, University Park, PA 16802, USA. E-mail: guc11@psu.edu
First published on 4th September 2017
Abstract
We report a unified photoredox-catalysis strategy for both hydroxylation and amidation of tertiary and benzylic C–H bonds. Use of hydroxyl perfluorobenziodoxole (PFBl–OH) oxidant is critical for efficient tertiary C–H functionalization, likely due to the enhanced electrophilicity of the benziodoxole radical. Benzylic methylene C–H bonds can be hydroxylated or amidated using unmodified hydroxyl benziodoxole oxidant Bl–OH under similar conditions. An ionic mechanism involving nucleophilic trapping of a carbocation intermediate by H2O or CH3CN cosolvent is presented.
Introduction
Methods for efficient and selective alkyl C–H oxidation could streamline the synthesis of fine chemicals, natural products, and drug metabolites.1,2 Despite rapid advances in the development of metal-catalyzed reactions3 and reagents,4 synthetically useful C(sp3)–H oxygenation chemistry is still in great demand.5,6 Recently, radical reactions mediated by hypervalent iodine(III) reagents have emerged as viable means to oxygenate C(sp3)–H bonds under mild conditions.7–11 Ochiai first reported the oxidation of activated C(sp3)–H bonds of benzyl and allyl ethers to the corresponding esters using t-butylperoxy benziodoxole (Bl–OOtBu, 1) via H-abstraction by benziodoxole radical Bl˙ 2 (Scheme 1A).9 Maruoka elegantly demonstrated the use of acyclic iodane reagents 3 and 5 in the selective oxidation of unactivated methylene C–H bonds of simple alkanes to the corresponding ketones, effected by more reactive iodanyl radical intermediates 4 and 6.10 Notably, Maruoka's oxygenation reactions proceed with a selectivity for secondary over tertiary C–H bonds. Herein, we report an efficient and broadly applicable photoredox-catalysis strategy for the selective hydroxylation of tertiary and benzylic C–H bonds using hydroxyl benziodoxoles as oxidant and H2O as cosolvent and hydroxylation reagent. This reaction system can be easily modulated to achieve tertiary and benzylic C–H amidation with high efficiency and selectivity using CH3CN as co-solvent and amidation reagent.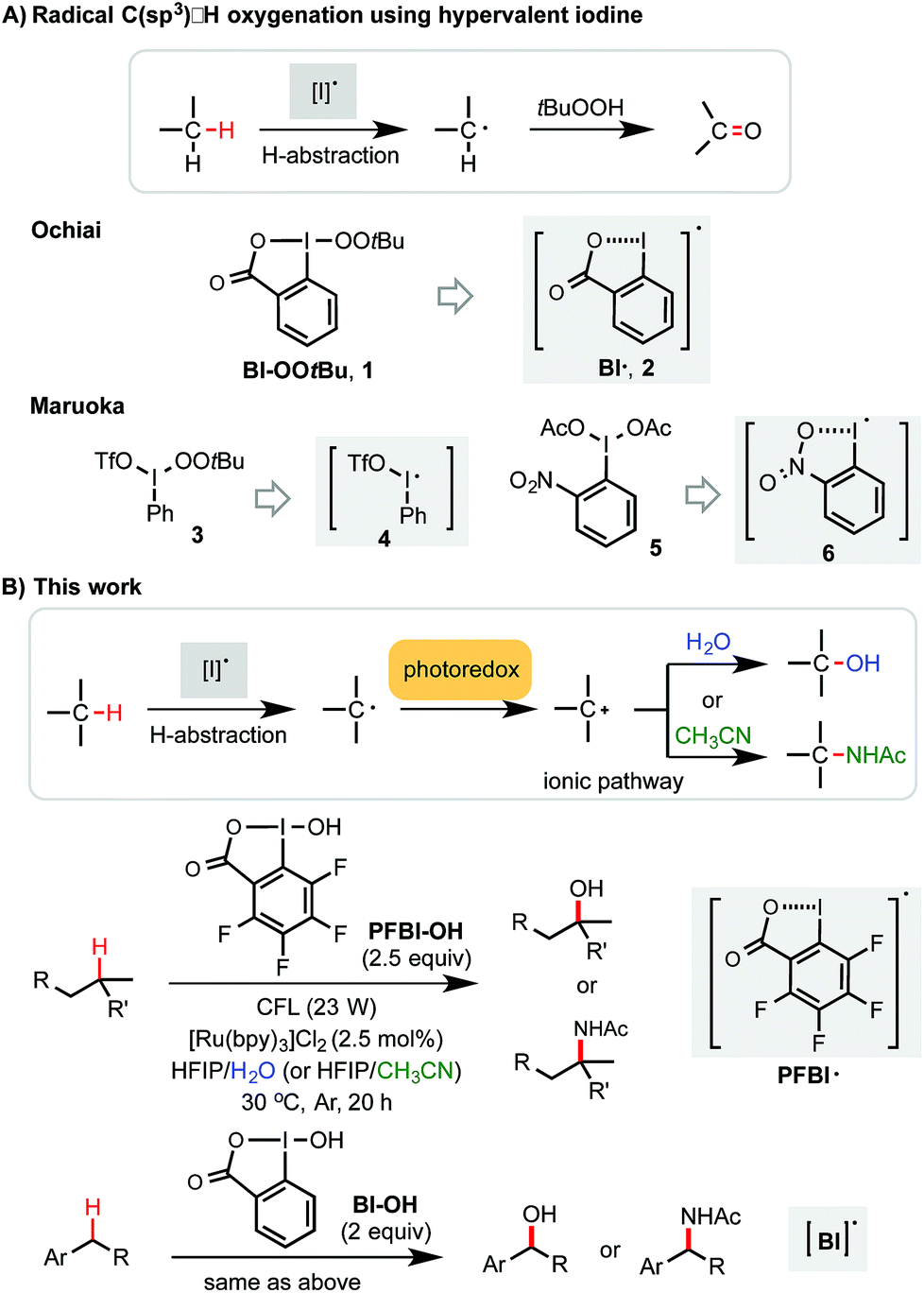 | ||
| Scheme 1 C(sp3)–H oxygenation and amination with hypervalent iodine(III). | ||
Results and discussion
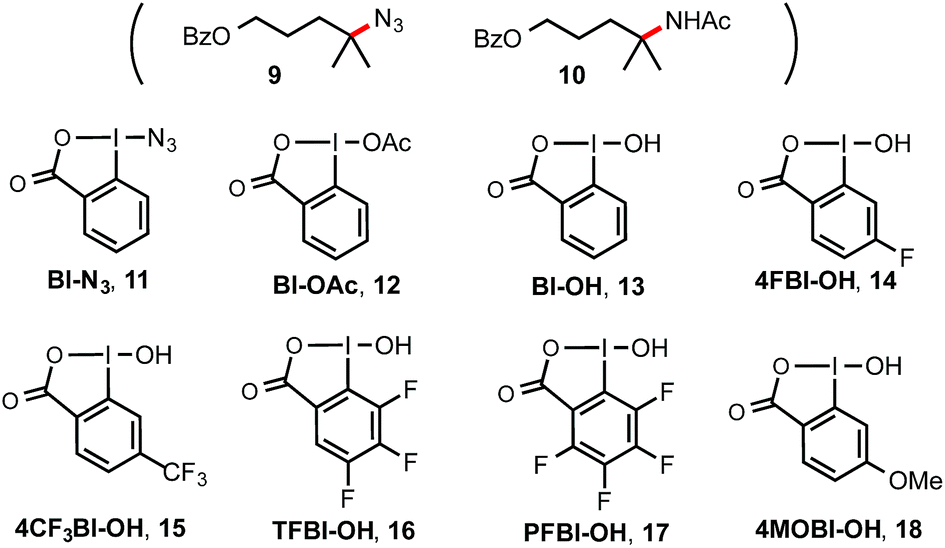
Previously, we discovered a visible light-promoted method for tertiary C–H azidation using Zhdankin reagent Bl–N311 (see entry 1, Table 1), [Ru(bpy)3]Cl2 photosensitizer, and household compact fluorescent lamp (CFL) irradiation.12–14 We proposed a radical chain mechanism for this azidation reaction, beginning with formation of Bl radical 2via single electron reduction of 11 by a photoexcited Ru(II)* species. Bl˙ 2 then selectively abstracts a H atom from the substrate (e.g. 4-methylpentyl benzoate 7), forming tertiary alkyl radical intermediate, which reacts with 11 to give C–H azidation product and regenerate radical 2, propagating a radical chain reaction. Encouraged by these results, we questioned whether the reaction with the corresponding hydroxyl benziodoxole could offer C–H hydroxylation product under similar conditions.|
|
|||
|---|---|---|---|
| Entry | Reagents (equiv.) | Solvents | Yielda (%) 8 |
| a Isolated yield on a 0.2 mmol scale, c ∼ 50 mM, ACS grade of HFIP was used. b 58% of 9 was obtained. c 8% of 7 was recovered. d Anhydrous HFIP and CH3CN dried over 4 Å molecular sieves were used. e c ∼ 30 mM, ∼10% of 7 was recovered. See ESI for more screening results. | |||
| 1 | Bl–N311 (2) | HFIP | <1b |
| 2 | BI–OH 13 (2) | HFIP | <2 |
| 3 | Bl–OH 13 (2) | HFIP/H2O (26/1) | 29 |
| 4 | 4FBI–OH 14 (2) | HFIP/H2O (26/1) | 32 |
| 5 | 4CF3BI–OH 15 (2) | HFIP/H2O (26/1) | 38 |
| 6 | TFBI–OH 16 (2) | HFIP/H2O (26/1) | 46 |
| 7 | PFBI–OH 17 (2) | HFIP/H2O (26/1) | 51 |
| 8 | 4MOBI–OH 18 (2) | HFIP/H2O (26/1) | 25 |
| 9 | BI–OAc 12 (2) | HFIP/H2O (26/1) | 18 |
| 10 | 17 (2.5) | HFIP/H 2 O (26/1) | 64 |
| 11 | 17 (2.5) | HFIP/H2O (10/1) | 55 |
| 12 | 17 (2.5), O2 (1 atm) | HFIP/H2O (26/1) | 29 |
| 13 | 17 (2.5), Ir(ppy)3 (2.5 mol%) | HFIP/H2O (26/1) | <2 |
| 14 | 17 (2.5), [Ru(bpz)3](PF6)2 (2.5 mol%) | HFIP/H2O (26/1) | <2 |
| 15 | 17 (2.5), in darkness | HFIP/H2O (26/1) | <2 |
| 16 | 17 (2.5), [Ru(bpy)3]Cl2 (1 mol%) | HFIP/H2O (26/1) | 40 |
| 17 | 17 (2.5) | HFIP | 4 |
| 18 | 17 (2.5) | DMSO/H2O (26/1) | <2 |
| 19 | 17 (2.5) | HFIP/CH3CN (26/1)d | <2 (+10% of 10) |
| 20 | 17 (2.5) | HFIP/CH 3 CN (4/3) | <2 (+56% of 10) |
| 21 | 13 (2.5) | HFIP/CH3CN (4/3)d | <2 (+8% of 10) |
|
|||
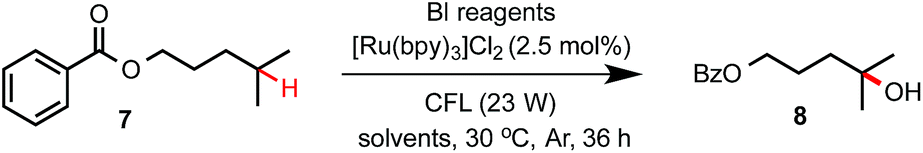
As shown in Table 1, we commenced the investigation of tertiary C–H hydroxylation of 7 with Bl–OH 13 under the irradiation of CFL (23 W) using [Ru(bpy)3]Cl2 as photocatalyst in hexafluoroisopropanol (HFIP) at 30 °C. Our previous work has shown that Bl–OH 13 can be used to generate Bl˙ 2 under similar conditions for a Minisci-type C–H alkylation reaction of N-heteroarenes with alkyl boronic acids.15,16 However, subjecting 7, 13, and [Ru(bpy)3]Cl2 to CFL irradiation produced only trace amount of the desired hydroxylation product 8, with 7 largely unconsumed (entry 2). However, adding H2O to the reaction increased the yield of 8 to 29% (entry 3). Our previous work has indicated that the spin density of Bl˙ is delocalized on both O and I atoms and that Bl˙ is more stable than benzoyloxy radical BzO˙.15 The stability of Bl˙ may explain the observed weak reactivity for H-abstraction and the low conversion of 7.17,18 We speculated that installation of electron-withdrawing groups on the aryl motif of Bl would increase its electrophilicity, and enhance its H-abstraction reactivity. As shown in entries 4–7, Bl–OH analogs 14–17 with different electron-withdrawing groups were prepared and evaluated (see ESI† for more details).19 We were pleased to find that these Bl–OH analogs provided improved results, and hydroxyl perfluorobenziodoxole (PFBl–OH, 17) gave the best yield.20,21 A 64% isolated yield of 8 was obtained when 2.5 equiv. of 17 was used (entry 10). Regarding the optimization of this hydroxylation reaction, we note: (1) addition of H2O is critical to obtain high yield (see entries 10 vs. 17). (2) 17 has high polarity and only dissolves well in polar solvents such as HFIP, DMSO, DMF; HFIP gives significantly better results than other solvent tested; (3) under O2 atmosphere, the reaction gave significantly diminished yield (entry 12); (4) in the dark, the reaction gave no product (entry 15); (5) only trace amount (<3% yield) of methylene C–H hydroxylation side product was detected. Interestingly, when the reaction was performed in mixed HFIP/CH3CN solvents (4/3) under similar conditions we obtained 56% yield of the C–H aminated product 10 with excellent selectivity (entry 20).
With optimized conditions in hand, we investigated the substrate scope of this C–H hydroxylation reaction (Scheme 2). In general, the reaction proceeds with excellent selectivity for tertiary C–H bonds and in good yield. Common functional groups including CN, iodo, esters, amide, imide and pyridine moiety are tolerated. When reaction of 7 was performed in a mixture of HIFP and H218O (97% of 18O), 18O-labelled product 19 was obtained. C–H hydroxylation of sulbactam and thalidomide derivatives (see 27 and 28) bearing β-lactam and imide groups proceeded in good yield. 28 was obtained in 60% yield on a gram scale. Both steric and electronic factors influence the reactivity of tertiary C–H bonds. For instance, tertiary C–H hydroxylation took place selectively at the more distal 3° carbon of 29. Hydroxylation of the sterically hindered and electron-poor tertiary C–H bond of phthaloyl valine methyl ester gave 32 in moderate yield. In comparison, C–H hydroxylation of leucine methyl ester 34 provided lactone product 33 in 52% yield. Moreover, short peptide substrates (see 35 and 36) can be C–H hydroxylated on the Leu residue with excellent selectivity under standard conditions.
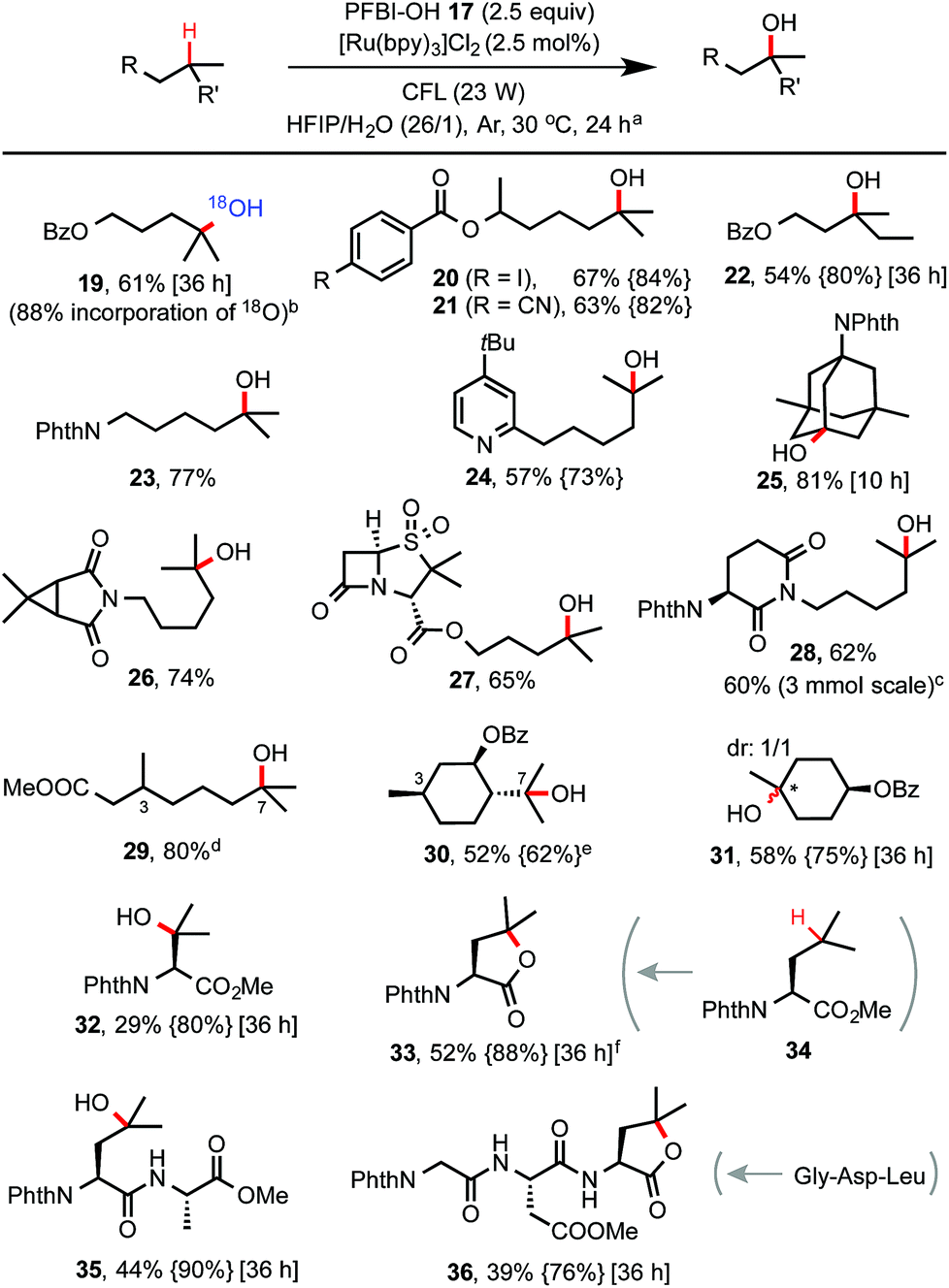 | ||
| Scheme 2 Substrate scope of tertiary C–H hydroxylation with PFBl–OH 17. (a) Isolated yield on 0.2 mmol scale under the standard conditions, c ∼ 50 mM. For reaction with <85% conversion of starting material, yields based on recovered SM (BRSM) were given in braces. (b) H218O (97% 18O) was used. (c) 3 mmol scale, 46 h. (d) C3 hydroxylation product <5%. (e) No C3 hydroxylation product was detected. (f) No free OH product was obtained. | ||
While a number of methods for oxidation of benzylic methylene groups to ketones have been developed,22 practical methods for C–H hydroxylation of these methylene groups to benzyl alcohols are sparse.23 As shown in Scheme 3, we subjected 4-ethylphenyliodide to our standard C–H hydroxylation conditions with PFBl-OH 17, and obtained the alcohol product 37 in 40% yield along with 22% of ketone 37′ and other unidentified by-products. We were delighted to find that use of 2 equiv. of Bl–OH 13 under the same conditions gave 37 in 71% yield along with 8% of ketone. More ketone 37′ (24%) was obtained when 4 equiv. of Bl–OH was used for extended reaction time (24 h). This Bl–OH mediated benzylic C–H hydroxylation exhibited excellent chemo-selectivity and broad substrate scope. The reaction tolerates functional groups such as iodo, ketone, amide, even pinacolyl boronate ester (see 42). Electron-deficient arenes are less reactive and require the use of 4 equiv. of Bl–OH 13 (see 41). Electron-rich substrates give good yield with 1.5 equiv. of Bl–OH (see 38). Reaction of ibuprofen methyl ester gave 43 in 64% yield without the formation of tertiary C–H hydroxylated product. The same reaction in H218O gave 18O-labelled product 44. Reaction of natural product celestolide gave product 45 in excellent yield.
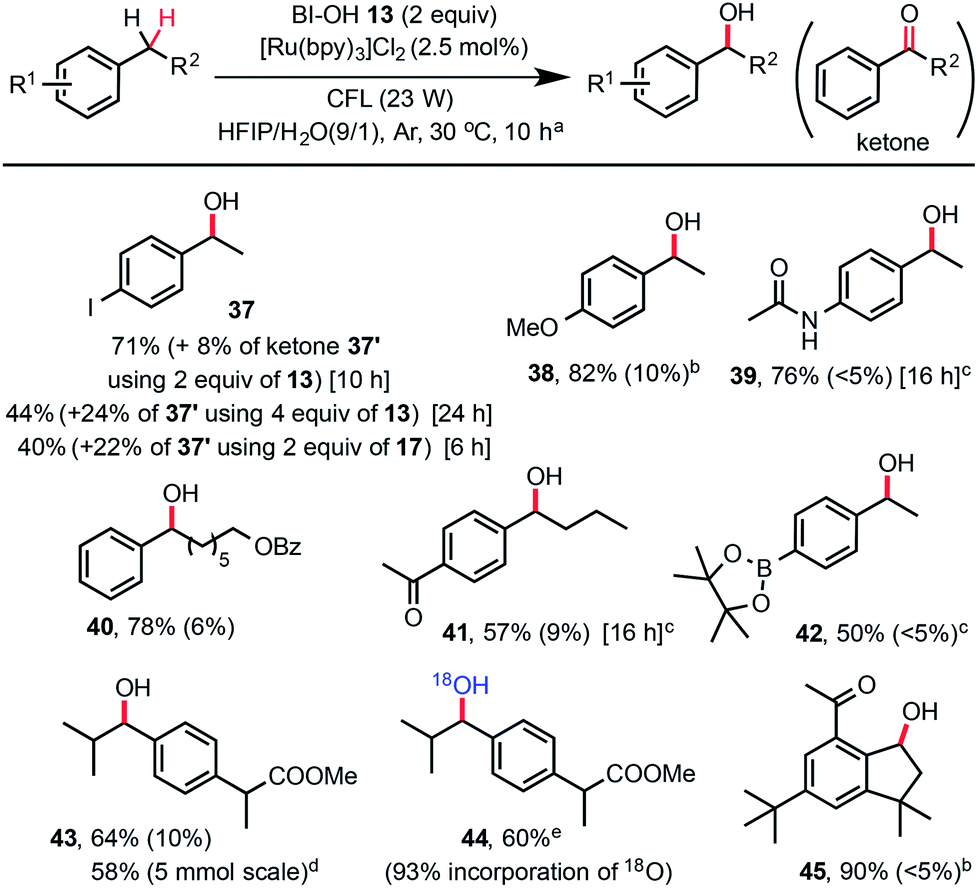 | ||
| Scheme 3 Substrate scope of benzylic C–H hydroxylation with Bl–OH 13. (a) Isolated yield on a 0.2 mmol scale under standard conditions, c ∼ 45 mM, yield of ketone by-product was given in parentheses. (b) 1.5 equiv. of 13 was used. (c) 4 equiv. of 13 was used. (d) 5 mmol scale, 20 h. (e) H218O (97% 18O) was used. | ||
As shown in Scheme 4, by simply switching to the HFIP/CH3CN solvents, this hydroxyl benziodoxole-mediated reaction system provides an excellent method for C(sp3)–H amidation, which remains a challenging transformation for C–H functionalization chemistry.24,25 Tertiary C–H amidation with PFBl–OH 17 and benzylic C–H amidation with Bl–OH 13 proceeded with yields and regio-selectivity similar to the corresponding C–H hydroxylations carried out in HFIP/H2O solvents. Unactivated methylene C–H bonds were generally unreactive with either 13 or 17. However, cycloalkanes such as cyclohexane were efficiently amidated with 17 (see 51), probably due to their slightly more activated C–H bonds and more favorable kinetics.25b Product 54 carrying a benzamide group was obtained in good yield using HFIP/PhCN solvent under similar conditions. Generally, the competing C–H hydroxylation reactions were well suppressed (<2% yield) in HFIP/nitrile solvents.
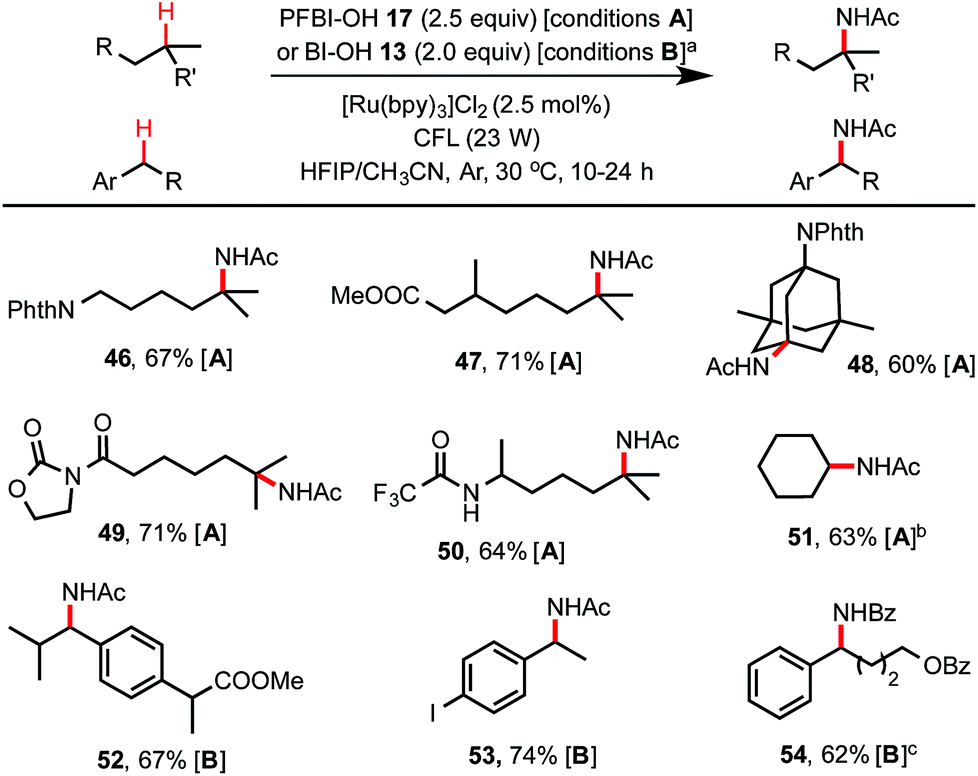 | ||
| Scheme 4 C(sp3)–H amidation with 13 or 17. (a) Conditions A for tertiary C–H amidation, HFIP/CH3CN (4/3), c ∼ 30 mM, 24 h; conditions B for benzylic C–H amidation, HFIP/CH3CN (8/3), c ∼ 35 mM, 10 h. Anhydrous HFIP and CH3CN dried over 4 Å molecular sieves were used. Isolated yield on a 0.2 mmol scale. (b) 1 equiv. of cyclohexane was used, 0.5 mmol scale. (c) PhCN was used as cosolvent, HFIP/PhCN (8/5), c ∼ 30 mM, 10 h. | ||
As shown in Scheme 5A, two C–O bond forming mechanisms were initially considered for this C–H hydroxylation reaction: nucleophilic trapping of a carbocation intermediate with H2O (pathway a) or a radical chain reaction with the hydroxyl benziodoxole reagents (pathway b).23c In contrast to the large quantum yield Φ observed in our previously reported visible light-promoted C–H azidation reaction with Bl–N311,12 a small Φ (0.85, measured by Yoon's method26) of the C–H hydroxylation reaction of 7 with PFBl–OH 17 suggested a non-radical chain mechanism (see ESI† for details). The dependence of the reactivity on the H2O co-solvent and the formation of amidation product in the presence of CH3CN product strongly support ionic pathway a. Stern–Volmer experiments confirmed that the excited state of photocatalyst [Ru(bpy)3]Cl2 can be quenched by the addition of PFBl–OH 17, while no obvious luminescence change of the photocatalyst was observed in the presence of substrate 7 (see ESI† for details).27
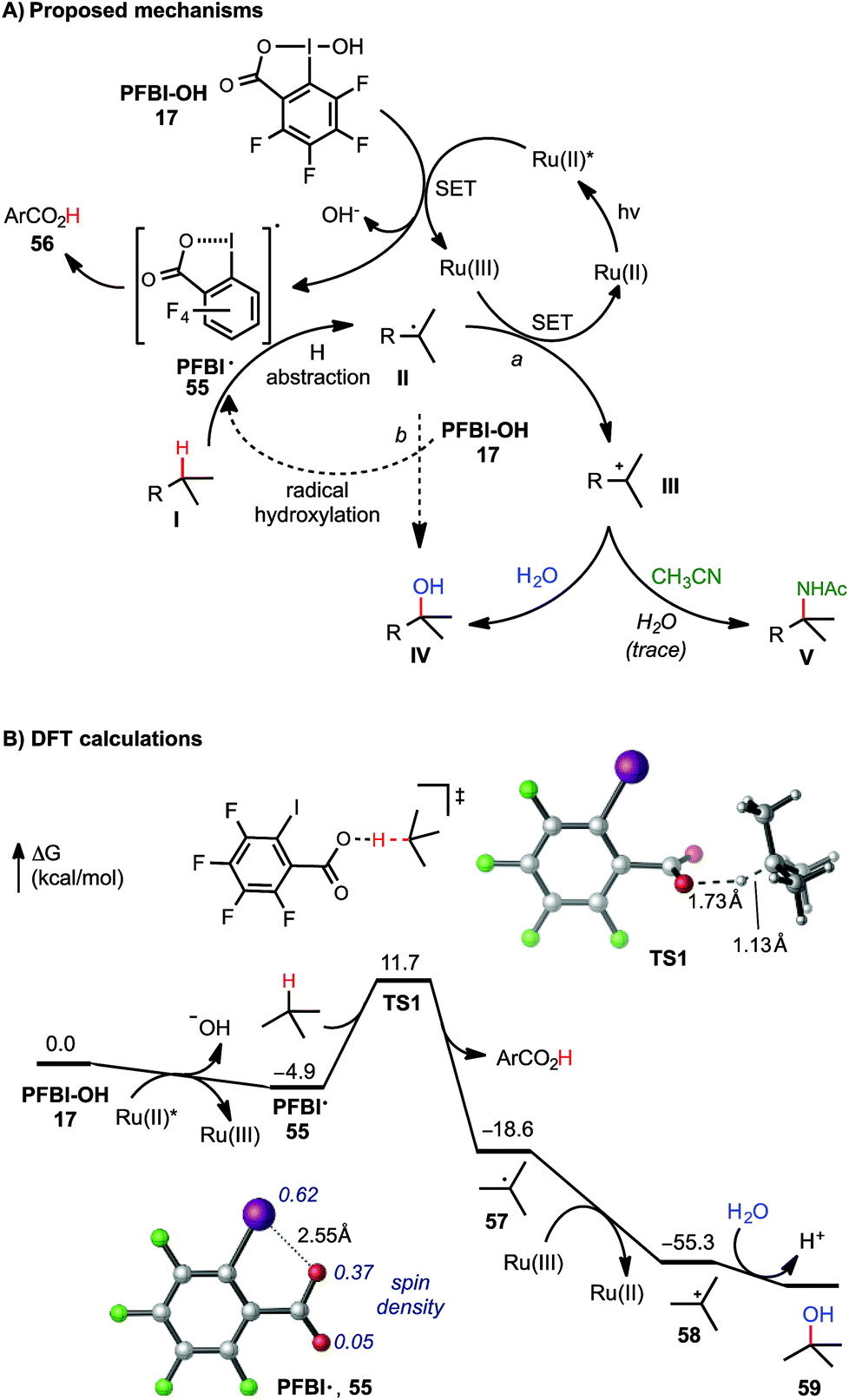 | ||
| Scheme 5 Mechanistic consideration of C(sp3)–H functionalization with PFBl–OH. DFT calculations were performed at the M06-2X/6-311++G(d,p)-SDD/SMD(HFIP)//M06-2X/6-31+G(d)-SDD level of theory. All energies are in kcal mol−1. See ESI† of DFT calculations with Bl–OH. | ||
The mechanism of tertiary C–H hydroxylation with PFBl–OH 17 likely begins with single electron transfer (SET) from photoexcited Ru(II)* to PFBl–OH 17, generating radical PFBl˙ 55. Radical 55 abstracts a H atom from alkane substrate I, forming tertiary carbon radical II. II can be oxidized by the Ru(III) species, forming tertiary carbocation intermediate III, and regenerating the photocatalyst. Finally, tertiary carbocation intermediate III is attacked by H2O to give hydroxylated product IV. Trapping of III by CH3CN can give the amidated product V following a Ritter reaction-type mechanism.25b We speculate that Bl–OH mediated benzylic C–H hydroxylation and amidation proceeds through a similar mechanism, involving cleavage of benzylic C–H bond with less electrophilic Bl˙ 2.
This mechanism is supported by density functional theory (DFT) calculations using t-butane as a model substrate (Scheme 5B). The initial SET reduction of PFBl–OH 17 to PFBl˙ 55 is significantly more exergonic than the SET with Bl–OH 13 to Bl˙ 2 (ΔG = −4.9 kcal mol−1 with PFBl–OH 15vs. −0.9 kcal mol−1 with Bl–OH 13).28,29 With its spin density delocalized over the O and I atoms, PFBl˙ 55 undergoes facile H-abstraction of t-butane through an O-centered transition state (TS1) with a ΔG‡ of 16.6 kcal mol−1 to give tBu˙.30 This H-abstraction process is promoted by the electron-deficient perfluoroaryl group. The corresponding H-abstraction with Bl˙ 2 requires a noticeably higher barrier of 18.2 kcal mol−1 (see ESI†). The subsequent oxidation of tBu˙ by Ru(III) to tbutyl cation is highly exothermic. Finally, the tbutyl cation is trapped with H2O, providing tBuOH. Taken together, the DFT calculations indicated the perfluorinated analogue PFBl–OH promotes both the initial SET reduction and the H-abstraction steps in the catalytic cycle of the tertiary C–H hydroxylation.
Conclusions
In summary, we have developed a unified photoredox-catalysis strategy for both C(sp3)–H hydroxylation and amidation using hydroxyl benziodoxole oxidant. This strategy allows the selective functionalization of tertiary and benzylic methylene C–H bonds under mild conditions. These reactions exhibit excellent substrate scope, and offer an efficient and convenient method for late-stage derivatization of complex substrates. Distinct from the radical chain mechanism invoked for our previous tertiary C–H azidation reaction with azido benziodoxole, we propose a new product-forming pathway: photoredox catalyzed formation of a carbocation intermediate, followed by nucleophilic trapping with H2O or nitrile cosolvent. Further expansion of the nucleophile scope and the functionalization of unactivated methylene C–H bonds using this reaction system are currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly thank the State Key Laboratory of Elemento-Organic Chemistry at Nankai University, and NSFC 21421062, 21672105 for financial support of this work. DFT calculations were performed using supercomputer resources at the Center for Simulation and Modeling at the University of Pittsburgh, and the Extreme Science and Engineering Discovery Environment supported by the National Science Foundation.Notes and references
- (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (b) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (c) J. Genovino, D. Sames, L. G. Hamann and B. B. Touré, Angew. Chem., Int. Ed., 2016, 55, 14218 CrossRef CAS PubMed.
- (a) P. A. Wender, M. K. Hilinski and A. V. W. Mayweg, Org. Lett., 2005, 7, 79 CrossRef CAS PubMed; (b) K. Chen and P. S. Baran, Nature, 2009, 459, 824 CrossRef CAS PubMed; (c) E. M. Stang and M. C. White, Nat. Chem., 2009, 1, 547 CrossRef CAS PubMed.
- For selected metal-catalyzed C(sp3)–H oxygenation reactions, see: (a) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS PubMed; (b) Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654 CrossRef CAS PubMed; (c) M. Zhou, N. D. Schley and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 12550 CrossRef CAS PubMed; (d) E. McNeill and J. Du Bois, J. Am. Chem. Soc., 2010, 132, 10202 CrossRef CAS PubMed; (e) M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 12796 CrossRef CAS PubMed.
- For selected C–H hydroxylation with dioxiranes and oxaziridines, see: (a) D. Yang, M.-K. Wong, X.-C. Wang and Y.-C. Tang, J. Am. Chem. Soc., 1998, 120, 6611 CrossRef CAS; (b) B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391 CrossRef CAS PubMed; (c) K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247 CrossRef CAS PubMed; (d) N. D. Litvinas, B. H. Brodsky and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 4513 CrossRef CAS PubMed; (e) S. Kasuya, S. Kamijo and M. Inoue, Org. Lett., 2009, 11, 3630 CrossRef CAS PubMed; (f) A. M. Adams and J. Du Bois, Chem. Sci., 2014, 5, 656 RSC.
- For other selected methods for C(sp3)–H oxygenation: (a) X. Li, X. Che, G.-H. Chen, J. Zhang, J.-L. Yan, Y.-F. Zhang, L.-S. Zhang, C.-P. Hsu, Y. Q. Gao and Z.-J. Shi, Org. Lett., 2016, 18, 1234 CrossRef CAS PubMed; (b) J. Ozawa, M. Tashiro, J. Ni, K. Oisaki and M. Kanai, Chem. Sci., 2016, 7, 1904 RSC.
- For a photoredox-catalyzed sulfonate-directed C(sp3)–H hydroxylation, see: A. Hollister, E. S. Conner, M. L. Spell, K. Deveaux, L. Maneval, M. W. Beal and J. R. Ragains, Angew. Chem., Int. Ed., 2015, 54, 7837 CrossRef PubMed.
- For selected reviews on hypervalent iodine, see: (a) Hypervalent Iodine Chemistry-Modern Developments in Organic Synthesis, Topics in Current Chemistry, ed. T. Wirth, M. Ochiai, V. V. Zhdankin, G. F. Koser, H. Tohma and Y. Kita, Springer, Berlin, 2003 Search PubMed; (b) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS PubMed; (c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (d) T. Dohi and Y. Kita, Chem. Commun., 2009, 16, 2073 RSC; (e) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS PubMed.
- (a) R. Samanta, K. Matcha and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769 CrossRef CAS; (b) T. Dohi and Y. Kita, ChemCatChem, 2014, 6, 76 CrossRef CAS; (c) L. Bering and A. P. Antonchick, Chem. Sci., 2017, 8, 452 RSC.
- M. Ochiai, T. Ito, H. Takahashi, A. Nakanishi, M. Toyonari, T. Sueda, S. Goto and M. Shiro, J. Am. Chem. Soc., 1996, 118, 7716 CrossRef CAS.
- (a) S. A. Moteki, A. Usui, T. Zhang, C. R. S. Alvarado and K. Marouka, Angew. Chem., Int. Ed., 2013, 52, 8657 CrossRef CAS PubMed; (b) S. A. Moteki, S. Selvakumar, T. Zhang, A. Usui and K. Marouka, Asian J. Org. Chem., 2014, 3, 932 CrossRef CAS.
- Y. Zhao and Y. Y. Yeung, Org. Lett., 2010, 12, 2128 CrossRef CAS PubMed . A carbonyl-directed methylene C–H oxygenation using PhI(OAc)2 and tBuOOH, yielding ketones, is described.
- Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679 RSC.
- (a) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192 CrossRef CAS; (b) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS PubMed.
- For selected reviews on photoredox catalysis reactions: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) T. P. Yoon, M. A. Ischay and J. N. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (d) J. Xie, H. Jin, P. Xu and C. Zhu, Tetrahedron Lett., 2014, 55, 36 CrossRef CAS.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407 RSC.
- For use of Bl–OH in photoredox-promoted reactions: (a) H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed; (b) H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374 CrossRef CAS PubMed; (c) K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514 CrossRef CAS PubMed.
- (a) J. Barluenga, E. Campos-Gomez, D. Rodriguez, F. Gonzalez-Bobes and J. M. Gonzalez, Angew. Chem., Int. Ed., 2005, 44, 5851 CrossRef CAS PubMed; (b) Ref. 9 and 13a .
- The relatively weak H-abstraction reactivity of Bl˙ is less consequential in the C–H azidation reaction because the reaction proceeds via a radical chain pathway.
- M. Linuma, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2014, 772 Search PubMed.
- For the original report of PFBl–OH 17, see: R. D. Richardson, J. M. Zayed, S. Altermann, D. Smith and T. Wirth, Angew. Chem., Int. Ed., 2007, 46, 6529 CrossRef CAS PubMed . PFBl–OH can be readily prepared on the gram scale in 2 steps from a cheap precursor (2,3,4,5-tetrafluorobenzoic acid, <$250 per kg) without column purification. It is a stable solid which can be stored in dark for 2 months and conveniently handled under air on the bench top.
- For a recent use of PFBl–OH 17 in photoredox-mediated reaction, see: K. Jia, Y. Pan and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 2478 CrossRef CAS PubMed.
- For recent examples of benzylic C–H oxygenation to form ketones: (a) T. Dohi, N. Takenaga, A. Goto, H. Fujioka and Y. Kita, J. Org. Chem., 2008, 73, 7365 CrossRef CAS PubMed; (b) J.-B. Xia, K. W. Cormier and C. Chen, Chem. Sci., 2012, 3, 2240 RSC.
- For recent examples of benzylic methylene C–H oxygenation to install NHPI, ONO2, or OTFA groups, see: (a) J. M. Lee, E. J. Park, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2008, 130, 7824 CrossRef CAS PubMed; (b) S. Kamijo, Y. Amaoka and M. Inoue, Tetrahedron Lett., 2011, 52, 4654 CrossRef CAS; (c) R. Sakamoto, T. Inada, S. Selvakumar, S. A. Moteki and K. Maruoka, Chem. Commun., 2016, 52, 3758 RSC.
- For selected reviews on C–H amination: (a) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926 RSC; (b) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed.
- For selected example of intermolecular C(sp3)–H amination: (a) X.-Q. Yu, J.-S. Huang, X.-G. Zhou and C.-M. Che, Org. Lett., 2000, 2, 2233 CrossRef CAS PubMed; (b) Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Soc. Chem., 2012, 134, 2547 CrossRef CAS PubMed; (c) J. L. Roizen, D. N. Zalatan and J. Du Bois, Angew. Chem., Int. Ed., 2013, 52, 11343 CrossRef CAS PubMed; (d) K. Kiyokawa, T. Kosaka, T. Kojima and S. Minakata, Angew. Chem., Int. Ed., 2015, 54, 13719 CrossRef CAS PubMed; (e) G. Pandey and R. Laha, Angew. Chem., Int. Ed., 2015, 54, 14875 CrossRef CAS PubMed; (f) K. Kiyokawa, K. Takemoto and S. Minakata, Chem. Commun., 2016, 52, 13082 RSC.
- For a leading reference characterizing chain processes in visible light photoredox catalysis based on the measurements of quantum yield Φ: M. A. Crismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC . A reaction with Φ ≫ 1 could only be consistent with a product-forming chain mechanism.
- N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, CA, 1978 Search PubMed.
- (a) M. J. Frisch, et al., DFT calculations were performed using Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMedGeometries were optimized at the M06-2X/6-31+G(d)-SDD level of theory in the gas phase. Single point energies were calculated at the M06-2X/6-311++G(d,p)-SDD level with the SMD solvation model in HFIP. See ESI† for computational details. For recent computational studies on photoredox mediated C–C bond formation reactions, see: (b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed; (c) T. B. Demissie, K. Ruud and J. H. Hansen, Organometallics, 2015, 34, 4218 CrossRef CAS.
- The experimental redox potentials of
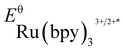 (−0.81 V vs. SCE in MeCN) and
(−0.81 V vs. SCE in MeCN) and 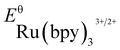 (+1.29 V vs. SCE in MeCN) were used in the computation of the SET reaction energies with Ru(ii)* and Ru(iii). See:
(a) C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 2909 CrossRef CAS. For DFT calculations of redox potentials, see: ref. 16 and
(b) P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217 CrossRef CAS;
(c) A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef CAS PubMed;
(d) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 CAS.
(+1.29 V vs. SCE in MeCN) were used in the computation of the SET reaction energies with Ru(ii)* and Ru(iii). See:
(a) C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 2909 CrossRef CAS. For DFT calculations of redox potentials, see: ref. 16 and
(b) P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217 CrossRef CAS;
(c) A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef CAS PubMed;
(d) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 CAS. - Our DFT calculation shows that the I-centered H-abstraction pathway from 55 yields a highly unstable hydridoiodane and is much less favourable (see ESI† for details).
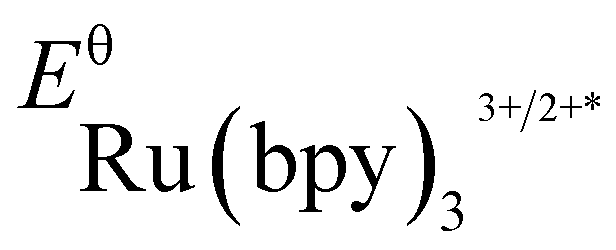
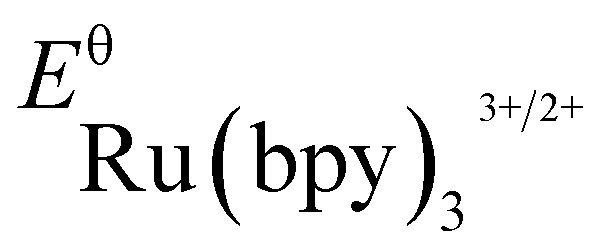
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02773g |
| This journal is © The Royal Society of Chemistry 2017 |