
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous visualization of the subfemtomolar expression of microRNA and microRNA target gene using HILO microscopy†
Yi-Zhen
Lin
a,
Da-Liang
Ou
b,
Hsin-Yuan
Chang
a,
Wei-Yu
Lin
a,
Chiun
Hsu
cd and
Po-Ling
Chang
 *a
*a
aDepartment of Chemistry, Tunghai University, Taichung 407, Taiwan. E-mail: poling@thu.edu.tw
bGraduate Institute of Oncology, College of Medicine, National Taiwan University, Taipei 100, Taiwan
cDepartment of Internal Medicine, National Taiwan University Hospital, Taipei 100, Taiwan
dDepartment of Oncology, National Taiwan University Hospital, Taipei 100, Taiwan
First published on 31st July 2017
Abstract
The family of microRNAs (miRNAs) not only plays an important role in gene regulation but is also useful for the diagnosis of diseases. A reliable method with high sensitivity may allow researchers to detect slight fluctuations in ultra-trace amounts of miRNA. In this study, we propose a sensitive imaging method for the direct probing of miR-10b (miR-10b-3p, also called miR-10b*) and its target (HOXD10 mRNA) in fixed cells based on the specific recognition of molecular beacons combined with highly inclined and laminated optical sheet (HILO) fluorescence microscopy. The designed dye-quencher-labelled molecular beacons offer excellent efficiencies of fluorescence resonance energy transfer that allow us to detect miRNA and the target mRNA simultaneously in hepatocellular carcinoma cells using HILO fluorescence microscopy. Not only can the basal trace amount of miRNA be observed in each individual cell, but the obtained images also indicate that this method is useful for monitoring the fluctuations in ultra-trace amounts of miRNA when the cells are transfected with a miRNA precursor or a miRNA inhibitor (anti-miR). Furthermore, a reasonable causal relation between the miR-10b and HOXD10 expression levels was observed in miR-10b* precursor-transfected cells and miR-10b* inhibitor-transfected cells. The trends of the miRNA alterations obtained using HILO microscopy completely matched the RT-qPCR data and showed remarkable reproducibility (the coefficient of variation [CV] = 0.86%) and sensitivity (<1.0 fM). This proposed imaging method appears to be useful for the simultaneous visualisation of ultra-trace amounts of miRNA and target mRNA and excludes the procedures for RNA extraction and amplification. Therefore, the visualisation of miRNA and the target mRNA should facilitate the exploration of the functions of ultra-trace amounts of miRNA in fixed cells in biological studies and may serve as a powerful tool for diagnoses based on circulating cancer cells.
Introduction
MicroRNAs (miRNAs) are a class of noncoding short oligoribonucleotides (∼22 nt) that have been demonstrated to downregulate specific genes, thereby affecting the function of a cell. The abnormal expression of miRNA has also been shown to be related to several severe diseases such as cancer and thus is called oncomiR.1,2 On the other hand, modulation of the miRNA levels has been shown to inhibit cancer metastasis and enhance anticancer therapy, and is already being tested in clinical trials.3–5 The modulation of the miRNA levels could be performed by means of miRNA replacement, a miRNA inhibitor or an oncolytic virus that is delivered into cells via a modifier of the miRNA, RNA duplex, liposomes, cationic lipid nanoparticles, or adenovirus.6,7 Therefore, a change in the quantity of the miRNAs may be considered as an important biomarker for the diagnosis and/or prognosis of diseases.Using extracted total RNA, the miRNA expression level can be evaluated more easily using stem-loop reverse transcription quantitative PCR (RT-qPCR)8 and droplet digital PCR (ddPCR).9 In addition to conventional assays, several published methods represent novel concepts regarding the evaluation of the expression levels of miRNA using different strategies. For instance, a series of sensitive miRNA assays based on the different modes of amplification have been proposed.10–16 In these studies, less than 100 copies of let-7a have been demonstrated to be detected in 10 pg of total RNA via exponential isothermal amplification and reached a dynamic range of ten orders of magnitude.13 Dong et al. proposed an ultra-sensitive assay for the determination of miR-21. Six order linear range with a detection limit of 52.5 zM was demonstrated using magnetic nanoprobe enrichment and enzyme-assisted strand cycle exponential amplification.15 In addition, Zhang et al. combined isothermal exponential amplification and single microbead based sensing for the determination of miRNA and reached the single molecule level.16 Recently, a 500 fM limit of detection has been shown for direct miRNA detection that utilizes single-molecule technology.17 Moreover, a multiplexed miRNA assay may be performed using capillary electrophoresis with laser-induced fluorescence and provides a linear range from 1.0 nM to 0.1 pM.18,19 An amplification-free, hydrogel microparticle-based method for the determination of miRNA from cell lysates was also proposed by Lee et al. The authors demonstrated that miR-21, miR-145 and miR-146a could be detected from a sample containing less than 1000 cells.20 The methods mentioned above are mostly used for extracting the total RNA from a sample that is well known to be easily degraded by RNase contamination or hydrolysis during the sample preparation or storage, thus decreasing the method’s sensitivity.21 One of the best advantages of imaging technologies for miRNA examination is the fact that these methods not only reduce the risk of RNA loss or degradation during the RNA extraction, but also eliminate the procedure of RNA amplification. In other words, the direct examination of miRNA expression in cells using a simple highly sensitive imaging method will always be useful for miRNA-based diagnosis, prognosis, and studies on miRNA function in cells treated with a miRNA inhibitor or therapeutic miRNA transfection.22 The observation of miRNA from cells or tissue could be achieved via fluorescence in situ hybridization followed by tyramide signal amplification23 or enzyme-labeled fluorescence signal amplification.24 Recently, Gorska et al. developed a simple and sensitive method for the direct probing of miR-21 and miR-31 in different cancer cells via a template Staudinger reaction.25 Nevertheless, the direct observation of miRNAs inside the cells was demonstrated using atomic force microscopy,26 DNA-driven gold-upconversion nanoparticle pyramids,27 cascade hybridization reactions,28 and rolling circle amplification combined with a triple-helix probe.29 Furthermore, a sensitive method for visualizing individual miRNAs based on rolling circle amplification has been reported by Deng et al.30 In addition, a two-color imaging method for the simultaneous observation of miR-21 and let-7a via a hybridization chain reaction with graphene oxide as a carrier has been demonstrated.31 However, the loss of miRNA during cell treatment such as hybridization and washing limits the application of the imaging techniques for ultra-trace miRNA visualization. In this regard, fluorescence imaging has shown neither sensitivity nor suitability for observations of ultra-trace amounts of miRNA to date. Recently, a fluorescent in situ hybridization assay for probing the expression of 130 miRNAs from formaldehyde and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)-fixed tissues was proposed by Pena et al.32 This method revealed that EDC can immobilize a miRNA on the amino groups of proteins via an irreversible linkage between the 5′-phosphate of the miRNA and the amino group of the protein molecules, thereby preserving the miRNA quantity during the sample processing for the in situ hybridization.
Another limitation of the sensitivity of the fluorescence imaging for formaldehyde-fixed cells may be attributed to the strong autofluorescence that is either intrinsic or caused by the fixation reagent.33 On the other hand, a method for the sample irradiation may play an important role in the sensitivity improvement of the fluorescence imaging. For instance, one powerful fluorescence microscopy technique for single-molecule detection is well known: total internal reflection fluorescence (TIRF) microscopy.34,35 However, the radiant power of the evanescent waves exponentially decreases with penetration depth and therefore may limit the effective excitation volume. Subsequently, to increase the probing thickness within a cell, highly inclined and laminated optical sheet (HILO) microscopy for single-molecule imaging inside a cell was proposed by Tokunaga et al.36 The applications of HILO microscopy are mainly single molecule detection37 and single molecule tracking38 in living cells. Currently, there is no literature showing the suitability of HILO microscopy for trace miRNA visualization in formaldehyde or EDC-fixed cells with a high-contrast image quality. Moreover, there is also no research demonstrating that the imaging method may provide a better sensitivity and accuracy than traditional RT-qPCR. Therefore, in this work, we propose a simple and sensitive imaging method for the molecular probing of ultra-trace amounts of miRNA and its target mRNA in photobleached cells, formaldehyde fixed cells and EDC fixed cells by means of a series of molecular beacons followed by HILO microscopic illumination (Scheme 1). All of the images that were acquired using HILO microscopy were compared to the results obtained from conventional RT-qPCR, and the greatly improved sensitivity and the reproducibility of the proposed imaging method are demonstrated in this study.
 | ||
| Scheme 1 The workflow of the miRNA/mRNA visualization in hepatocellular carcinoma cells using HILO microscopy. | ||
Results and discussion
Optimization of the signal to noise ratio for miRNA imaging
The detection of the ultra-trace amounts of miRNA in the formaldehyde fixed cells may be extremely difficult if the miRNA cannot be fixed efficiently in the cells by covalent bonding between the miRNA and the molecules inside the cells. As shown in Fig. 1a, we show that additional EDC fixation may play an important role, especially for the visualization of ultra-trace amounts of miRNA in the formaldehyde fixed Huh-7 cells (81.9 ± 25.9 fM, see Table S1†). The formation of the phosphoramidate linkages39 between the miRNA and the amino group of the proteins is beneficial for preserving the ultra-trace amounts of miRNA for detection thereafter. | ||
| Fig. 1 The effect of EDC fixation on the visualization of the miR-10b* (white color) in the Huh-7 cells (a). DAPI staining (blue) was used for sample focusing before the miR-10b imaging. (b) The inhibitory effect of high-power (3 W) LED exposure on the backgrounds of the fixed cells (Huh-7) using HILO illumination at 640 nm and 488 nm. The coverslip backgrounds were 622 ± 26 (640 nm) and 674 ± 43 (488 nm), respectively. (c) A comparison of the results using epifluorescence, TIRF, and HILO illumination for the visualization of miR-10b in photobleached, formaldehyde fixed and EDC fixed hepatocellular carcinoma cells (Huh-7). | ||
On the other hand, we found that the autofluorescence of the formaldehyde fixed cells, and from the culture medium (especially for fetal calf serum) on the coverslip, can be minimized via exposure to a high-power light-emitting diode (LED) via the photobleaching of the intrinsically fluorescent molecules in the cells (Fig. 1b and S1†). The effective photobleaching by the LED irradiation greatly decreased the autofluorescence (∼6 fold) that was caused by the 640 nm HILO illumination (656 ± 24), which we subtracted as background from the coverslip and the dark current of the EMCCD (622 ± 26). Therefore, the photobleaching procedure could also play a key role in the improvement of the signal-to-background ratio for the ultra-sensitive imaging of the formaldehyde fixed cells. For the microscopic irradiation, Fig. 1c demonstrates that HILO microscopy (Fig. S2†) allowed us to obtain clearer fluorescence images from the thick probing volumes within a cell compared to the epifluorescence and TIRF imaging. The dashed squares in Fig. 1c also indicate that HILO microscopy provides thicker illumination than TIRF microscopy but still offers a higher signal-to-background ratio for the miRNA visualization compared to the other two illumination methods. In addition to the contributions of sample photobleaching by the LED exposure, HILO illumination and EDC fixation, we designed several molecular beacons by means of fluorescence resonance energy transfer in order to exclude the interference of the remaining molecular beacons. In other words, the quenching efficiency of the molecular beacons may also contribute to the improvement of the sensitivity. For example, the quenching efficiency for a molecular beacon of miR-10b reaches 95% in the absence of miR-10b and provides a 22-fold enhancement in the fluorescence intensity when miR-10b is hybridized to the molecular beacon if we take into account the wavelength range of the emission filters (Fig. S3†). Therefore, the image background that is caused by the remaining fluorescence probes may be neglected in the visualization of miRNA.
Molecular probing of miR-10b-3p in Huh7 and Hep3B cells
As shown in Fig. 2a and e, the greatly improved signal-to-background ratio helped us to directly visualize the ultra-trace amounts of miRNA in the hepatocellular carcinoma cells and to detect the difference in the expression levels of miR-10b between the Huh-7 and Hep3B cells (Fig. 2b and f). The average fluorescence intensity per pixel per Hep3B cytoplasm was 809 ± 58, and so the Hep3B cells could be easily discriminated from the Huh-7 cells (1003 ± 96) as well as from the background cells (659 ± 26). On the other hand, transfection with an artificial miRNA precursor or inhibitor is widely utilized for elucidating miRNA function. The determination of the amounts of miRNA after transfection is necessary to confirm the modulation efficiency. In this study, the transfection of a miR-10b precursor into both Huh-7 and Hep3B cells (Fig. 2c and g) showed mostly an increase in the fluorescence intensities; in contrast, the transfection of a miR-10b inhibitor caused obvious inhibition (Fig. 2d and h) compared to the native quantities of miR-10b (Fig. 2b and f) and the negative control (Fig. 2a and e), both with a p-value of < 0.0001 at the 95% confidence interval (n = 60). | ||
| Fig. 2 Visualization of miR-10b* in fixed Huh-7 cells (a–d) and Hep3B cells (e–h) and a comparison of the results using RT-qPCR (i and j). The negative control (a and e) without a molecular beacon indicated that slight autofluorescence and/or scattering may be observed after the HILO illumination. The baseline quantities of miR-10b* (b and f) were increased by the transfection of the miR-10b* precursor (c and g) and decreased by the transfection of the miR-10b* inhibitor (d and h). (i) The calibration curve of miR-10b* was constructed using RT-qPCR with a concentration range of 10 nM to 1.0 fM (n = 3 for each concentration). The comparison between RT-qPCR (open bars) and HILO microscopy (hatched bars) is shown in two-bar plots for the Huh-7 (j) and Hep3B cells (k). Sixty-cell samples were collected from each image set (a–h) for the calculation of the fluorescence intensities and standard deviations of the cells using HILO microscopy. The dashed line in panels j and k indicates the coverslip background during the 640 nm illumination (622 ± 26). The images were adjusted to the same scale for the convenient comparison of each image set. | ||
Comparison with RT-qPCR
To compare the sensitivity of our approach to a conventional method, a calibration curve for the miR-10b quantitation was constructed by stem-loop RT-qPCR (Fig. 2i). The linearity of RT-qPCR reached 0.9984 with the concentration range 1.0 fM to 10 nM (y = 3.44x − 15.56). The baseline expression levels of miR-10b in the Huh-7 and Hep3B cells are 29.5 ± 0.4 Ct (threshold cycles) [81.9 ± 25.9 fM] and 35.6 ± 1.2 Ct [1.75 ± 1.38 fM], respectively. The lower miR-10b level inside the Hep3B leads to a higher Ct value with a greater standard deviation and it is more difficult to discriminate them from the negative control (Table S1†).A comparison of the results using HILO microscopy and RT-qPCR in the bar plots of Fig. 2j and k shows that both of the methods detected the upregulation of miR-10b after precursor transfection. Nonetheless, the decrease in the miR-10b level to the subfemtomolar level became indistinguishable using RT-qPCR in the inhibitor-transfected cells (the measured concentrations obtained from the same total RNA sample via triplicate RT-qPCR are 0.70 fM, 5.78 fM, and 0.08 fM, respectively). Apparently, the measurement of ultra-trace amounts of miR-10b in the Hep3B cells using RT-qPCR after the transfection of an inhibitor is quite difficult; as a result, the coefficient of variation (CV) of RT-qPCR may become excessive (Table S1†). Consequently, RT-qPCR could not distinguish between similar quantities of the untreated Hep3B cells and inhibitor-transfected cells, where both concentrations were close to the cell background level (Fig. 2e and k). In contrast to RT-qPCR, HILO microscopy yielded more reliable and distinguishable data as a result of the stringent hybridization buffer and the higher reproducibility (lower standard deviation) at weaker fluorescence intensities. The linear correlation between the fluorescence intensities and the copy number obtained using RT-qPCR also indicates that the miRNA fluorescence intensities within the cells are approximately proportional to the copy number within a reaction tube (Fig. S4†).
The determination of the miRNA from the miRNA inhibitor transfected cells using RT-qPCR is difficult due to the fact that the chemically modified oligonucleotide (anti-miR) may block the binding site of the stem-loop primer (3′-end of mature miRNA) and reverse primers (5′-end of miRNA) during the RT and PCR reactions. Consequently, the ineffective annealing between miRNA and the two kinds of primers caused an unsuccessful RT-PCR reaction,40,41 and thus requires a special technique, miRNA polysome shift assay, for the evaluation of the anti-miR efficiency.42 In contrast, the sequence of molecular beacons used in this method are completely complementary to the mature miRNA with hybridization temperatures below the melting temperature of the miRNA inhibitor. In other words, the anti-miR still blocked the miRNA during the hybridization and it remained a double stranded duplex. Hence, only the unblocked miRNA could be detected by the designed molecular beacons without the interference of anti-miR.
Molecular probing of miR-10b-3p with reference gene
As a result of simultaneously probing U6 small noncoding RNA species (snRNA), which is internal standard, as housekeeping gene with miR-10b using two-color illumination (Fig. 3a and b), the U6 gene showed a constant expression level among the native Huh-7 cells (Fig. 3b), miR-10b precursor-transfected (Fig. 3c), and inhibitor-transfected cells (Fig. 3d). The CV of each individual U6 cell pool (Fig. 3m and n) may be up to 17.8%. We emphasize here that the CV of an individual bar in Fig. 2j, k, 3m and n does not indicate the reproducibility of the HILO microscopy results but represents the intrinsic heterogeneity among the individual cells. In this work, we also found that expression heterogeneity depended on the expression level of the analytes. For example, the CV of the miR-10b level in miR-10b precursor-transfected (Fig. 2c), native (Fig. 2b), and miR-10b inhibitor-transfected Huh-7 cells (Fig. 2d) is 12.8%, 9.5%, and 5.4%, respectively (Fig. 2j), indicating that a higher concentration of miRNA or mRNA may result in larger heterogeneity among the cells. Similarly, the expression heterogeneity may also be directly observed more easily in cell images of U6 (Fig. 3b to d). Nevertheless, the average intensities among six U6 pools in Fig. 3m and n is remarkably consistent (CV = 0.86%), revealing the excellent reproducibility of the direct probing of the miRNA, and already much better than that of ddPCR9 and the results of RT-qPCR, in which Ct is converted into units of molar concentration (Table S1†). These data reflect the fact that one unit of a PCR cycle ideally represents the fold change during the PCR amplification, and consequently even a 0.1 − Ct fluctuation may translate into a 10% alteration of the quantity, assuming an amplification efficiency of 1. The reproducibility of RT-qPCR does not only contain contributions from incomplete annealing or enzyme reactions but also an inherent limitation of the ultra-trace analysis. First, RT-PCR involves many liquid transfers with only a few microliter volume. The transfer probability of an occurrence of a molecule in a certain volume is governed by the Poisson distribution.43 For example, assume that the average number of molecules that were transferred to a vial for PCR reaction is 3 molecules per μl for a femtomolar miRNA sample with 200-fold dilution after reverse transcription (Table S2†). The probability that exactly three molecules are transferred via 1 μl pipetting would be only 22.4%. Consequently, the cases where anything other than three molecules are pipetted (77.6%) may attributed to the large standard derivation of the RT-PCR results. On the other hand, the adsorption of polyanionic nucleic acid on plastic containers have been demonstrated44 and may play an important role in the ultra-trace analysis. As shown in Fig. S5,† the standard deviation of the RT-qPCR increased, beginning at 10 pM when the miRNA molecules were dissolved in water without any blocking agent such as the yeast RNA we used in this study. This result also indicated that the miRNA adsorption may affect the reproducibility of RT-qPCR for ultra-trace quantitation. In fact, the RT-qPCR data from different miRNA expression platforms has been evaluated in a study of microRNA quality control, and the authors concluded that qPCR should not be used for the determination of small changes in the expression of miRNA.45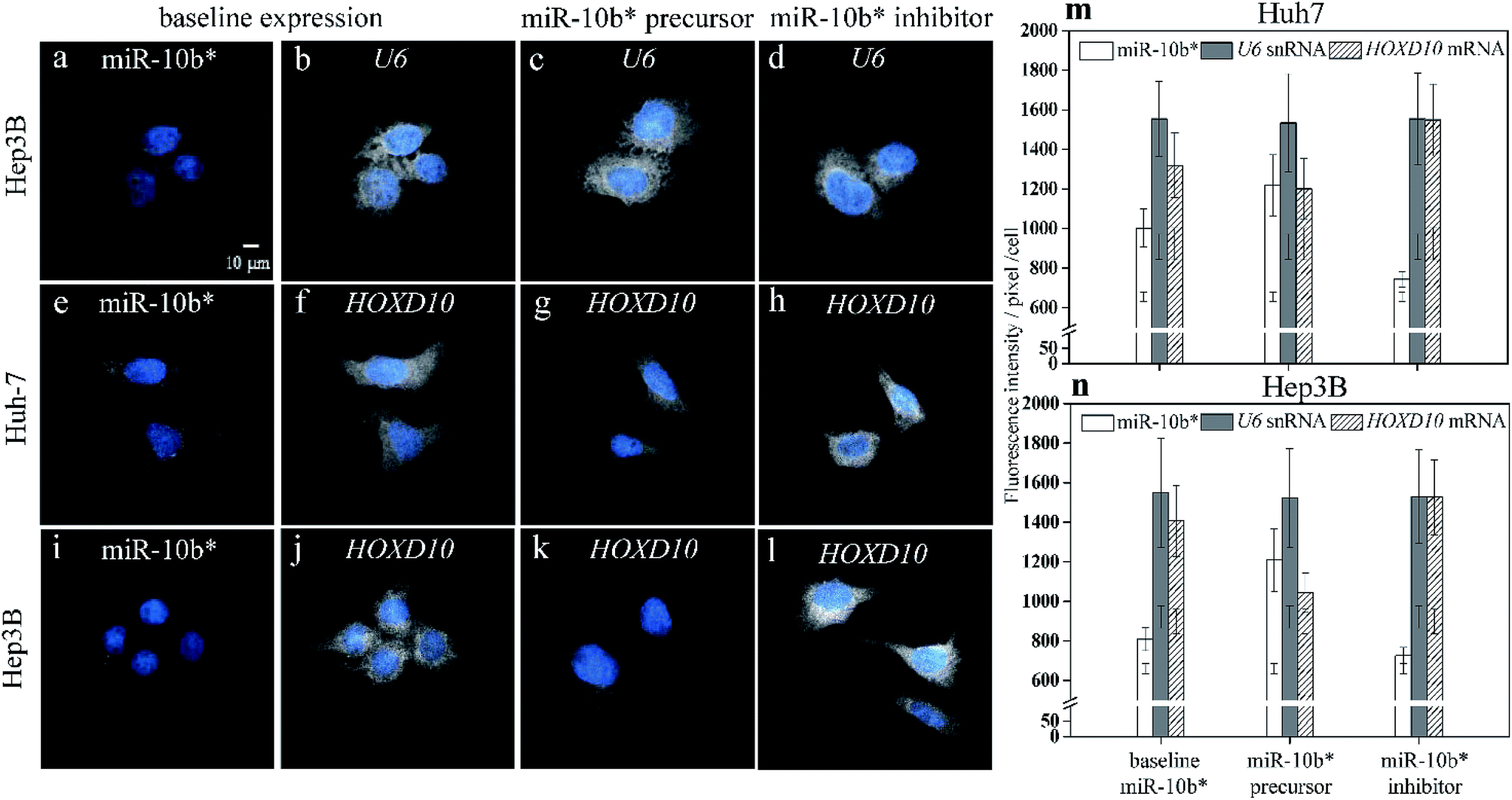 | ||
| Fig. 3 Simultaneous observations of miR-10b* (a) and U6 snRNA (b–d), miR-10b (e) and HOXD10 mRNA (f–h) in the Huh-7 cells, and miR-10b (i) and HOXD10 mRNA (j–l) in the Hep3B cells using two-color illumination during the HILO microscopy. The baseline expression of miR-10b* (a, e, i) is compared to the baseline expressions of U6 snRNA (b) and HOXD10 mRNA in the Huh-7 cells (f), and HOXD10 mRNA in the Hep3B cells (j). The expression level of U6 in miR-10b* precursor-transfected (c) and in miR-10b* inhibitor-transfected cells (d) remained similar to that of the untreated cell group (b). The HOXD10 mRNA quantities decreased with increased amounts of miR-10b* (g, k), and the upregulation of HOXD10 was observed after the inhibition of miR-10b* (h, l). The bar plots show the comparison among the expressions of miR-10b* (open bars), U6 (filled bars), and HOXD10 (hatched bars) in the Huh-7 (m) and Hep3B (n) cells. Thirty-cell samples were collected from each image set (a–l) for calculations of the fluorescence intensities and standard deviations obtained using HILO microscopy. The additional error bars inside the columns (m and n) indicate the cell backgrounds at 640 nm and 488 nm illumination, respectively. | ||
Molecular probing of miR-10b-3p and HOXD10
In addition to the U6 snRNA, the expression of the miR-10b target gene HOXD10 (Fig. 3f and j) can also be examined simultaneously with miR-10b-3p miRNA (Fig. 3e and i) using the proposed method. The expression level of HOXD10 is downregulated by miR-10b-5p miRNA, as has been reported previously,46,47 and the miR-10b-3p may also form a heteroduplex with the HOXD10 sequence (Table S3†).48 Hence, HOXD10 in the Hep3B cells is expressed more strongly than in the Huh-7 cells (Fig. 3m) as a result of the lower miR-10b expression in the Hep3B cells (Fig. 3n). Furthermore, the expression level of HOXD10 may decrease in the presence of an miR-10b precursor (Fig. 3g and k) and may increase after the miR-10b inhibitor is transfected into the cells (Fig. 3h and l). All of the trends of HOXD10 alteration before and after the miR-10b changes match the results of RT-qPCR (Fig. S6†). In other words, the causal relation between miR-10b miRNA and HOXD10 expression can be detected more easily using our proposed method with higher discriminatory power. For example, the extremely low miR-10b expression level in the Hep3B cells (Fig. 2f) could be further decreased via miR-10b inhibitor transfection (Fig. 2h), leading to a slight increase in the HOXD10 expression (Fig. 3l) as compared to the baseline level of the HOXD10 expression (Fig. 3j). The t-test shows that the fluorescence intensity differences between Fig. 3l and j are significant, with a p-value of 0.0163 at the 95% confidence interval. This result indicates that the direct imaging of miRNA and its target using HILO microscopy not only allows us to detect ultra-trace levels of miRNA but also the slight alterations in the HOXD10 expression caused by miRNA modulation at the subfemtomolar level.Experimental
Chemicals, materials and reagents
The chemicals including salts, buffers, Trolox, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, and formaldehyde were purchased from Sigma-Aldrich (St. Louis, MO, USA). The anti-fading mounting medium (Fluoro Gel with DABCO) was obtained from Electron Microscopy Sciences (PA, USA). The sequences of human miR-10b-3p (MIMAT0004556) and stem-loop hsa-miR-10b precursor (MI0000267) were acquired from miRbase.49 The synthetic RNA (miR-10b-3p), and Cy5-Iowa Black RQ-Sp-labelled molecular beacon used in this work were purchased from Integrated DNA Technologies (Coralville, IA, USA). The Alexa Fluor 488-BHQ1-labelled molecular beacon for HOXD10 mRNA probing was purchased from IBA Lifesciences (Goettingen, Germany), and the 6FAM-BHQ1-labelled molecular beacon for the hybridization of the U6 housekeeping gene was obtained from Sigma-Aldrich. All of the molecular beacons were designed for a given melting temperature at 37 °C in the stem portion in order to reach effective fluorescence resonance energy transfer at ambient temperature. All of the oligonucleotide sequences are listed in Table S3.† The molecular beacons were mixed with synthetic targets (DNA or RNA) to obtain the fluorescence ratios using a fluorospectrometer (Agilent, USA). The miR-10b-3p (miR-10b*) precursor (PM12387), miR-10b-3p inhibitor (AM12387), transfection agent (AM4510), TRIzol Reagent, 4′,6-diamidino-2-phenylindole (DAPI), yeast tRNA, RNase-free water, and diethyl pyrocarbonate (DEPC)-treated water were purchased from Life Technologies (Grand Island, NY, USA).Cell culture and modulation by a precursor or inhibitor of miR-10b
The HCC cell line, Hep3B, was purchased from the American Type Culture Collection (ATCC), and the Huh-7 cell line was acquired from the Health Science Research Resources Bank. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing fetal bovine serum (10%), penicillin (100 U ml−1), streptomycin (100 μg ml−1), L-glutamine (2 mM), and sodium pyruvate (1 mM) at 37 °C under a humidified atmosphere containing 5% CO2. For the modulation by a miRNA precursor and inhibitor, the cells were plated at 2 × 105 per well in 6-well plates and transfected with 100 nM miR-10b-3p precursor or miR-10b-3p inhibitor using the siPORT NeoFX transfection agent followed by 48 h of incubation. All of the harvested cells were collected into 15 ml tubes and centrifuged at 300 × g for 5 min, followed by the removal of the supernatants. The cells were then resuspended in 1× PBS containing one-tenth of the culture medium. Finally, 5000–8000 of the suspended cells were seeded on a (3-aminopropyl)triethoxysilane-coated no. 1.5H coverslip (Marienfeld, Germany) and then incubated for 6 h for cell adhesion. The prepared cells were then fixed using 4% formaldehyde and stored at −20 °C (in a freezer).RNA extraction and stem-loop RT-qPCR
The RNA extraction from the cultured cells was performed using the TRIzol reagent protocol provided by the manufacturer. Briefly, 3 ml of TRIzol reagent was directly added to the cells in a 30 mm culture dish. After pipetting several times, the homogenized samples of cell lysates were divided into 1 ml aliquots in autoclaved vials for phase separation. Then, 0.2 ml of chloroform was added into each vial, and we shook the vials vigorously for 30 s. After incubating the samples for 3 min at ambient temperature, we centrifuged the samples at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 15 min at 4 °C. The upper layer (aqueous phase) was carefully transferred to another vial that contained 0.5 ml of isopropanol. The samples were incubated for 10 min at room temperature, followed by centrifugation at 12000 × g for 10 min at 4 °C. After the removal of the supernatants from each vial, the RNA pellets were washed with 75% ethanol, followed by centrifugation at 7500 × g and vacuum drying for 10 min. Next, the RNA pellets were resuspended in DEPC water at 60 °C for 10 min, and we measured the RNA concentration on a microvolume spectrophotometer (Nanodrop, USA). For the reverse transcription, 1.0 μl of extracted total RNA (500 ng μl−1) or the synthetic miR-10b-3p RNA in the concentration range 10 nM to 1.0 fM (diluted using 10 ng μl−1 yeast tRNA in order to minimize the adsorption of miRNA) was mixed with 1.0 μl of stem-loop reverse-transcription (RT) primer (0.5 μM), 0.5 μl of dNTPs (10 mM) and 3.5 μl of double-deionized H2O (ddH2O). The sample mixture was heated at 65 °C for 5 min and added into an ice-water-precooled reaction mixture that consisted of 2 μl of RT buffer (5×), 0.5 μl of DTT (0.1 M), 0.1 μl of Superscript III reverse transcriptase (200 U μl−1), and 1.4 μl of ddH2O, to bring the final volume to 10 μl for the reverse transcription. The mixture was incubated at 25 °C for 10 min followed by ramping to 55 °C at a rate of 0.5 °C min−1. After additional incubation at 55 °C for 30 min, the resulting cDNA sample was heated at 85 °C for 5 min to inactivate the reverse transcriptase. The cDNA sample was diluted 20-fold with ddH2O for the reduction of qPCR interference caused by the excess RT primer. qPCR was then run in a mixture of 1.0 μl of diluted cDNA (0.05×), 2 μl of GS primer and reverse primer (1.0 μM each) as well as 5 μl of SYBR green mix (2×).50 The qPCR mixture was heated at 95 °C for 4 min followed by 2 cycles of 95 °C for 15 s, 45 °C for 30 s, and 63 °C for 30 s. Optical detection began for the next 50 cycles of amplification (95 °C for 15 s, 63 °C 30 s), and the reaction mixtures were returned to ambient temperature when the qPCR finished. For the U6 snRNA51 and HOXD10 mRNA,52 500 ng of oligo-dT primer (Integrated DNA Technologies, Iowa, USA) was mixed with 500 ng of total RNA for the RT reaction. The RT products were then diluted 20-fold, and we transferred 1 μl into the qPCR mixture subjected to the following temperature programme: 95 °C for 4 min, followed by 2 cycles of 95 °C for 15 s with 72 °C for 30 s. The qPCR amplification was then performed by means of 50 cycles of 95 °C for 15 s, 56 °C for 30 s, and 72 °C for 30 s. The theoretical lengths of the miR-10b-3p, U6, and HOXD10 amplicons are 59, 94, and 71 bp, respectively. All of the qPCRs were run on a spin-type real-time thermocycler (Rotor-Gene Q, Qiagen, Germany) with a slight modification of the optical detection. Briefly, bright 475 nm LEDs (Roithner Lasertechnik GmbH, Austria) with a 470 nm interference filter (Edmund, USA) were used for the excitation during PCR amplification. For the fluorescence collection, another 520 nm interference filter was mounted on a filter wheel immediately before the photomultiplier tube, to reject scattering from the excitation beam. We used the same optical settings for the hybridization curves of the molecular beacons except we used a 650 nm LED with a 647 nm excitation filter and a 676 nm emission filter for the Cy5 detection.
000 × g for 15 min at 4 °C. The upper layer (aqueous phase) was carefully transferred to another vial that contained 0.5 ml of isopropanol. The samples were incubated for 10 min at room temperature, followed by centrifugation at 12000 × g for 10 min at 4 °C. After the removal of the supernatants from each vial, the RNA pellets were washed with 75% ethanol, followed by centrifugation at 7500 × g and vacuum drying for 10 min. Next, the RNA pellets were resuspended in DEPC water at 60 °C for 10 min, and we measured the RNA concentration on a microvolume spectrophotometer (Nanodrop, USA). For the reverse transcription, 1.0 μl of extracted total RNA (500 ng μl−1) or the synthetic miR-10b-3p RNA in the concentration range 10 nM to 1.0 fM (diluted using 10 ng μl−1 yeast tRNA in order to minimize the adsorption of miRNA) was mixed with 1.0 μl of stem-loop reverse-transcription (RT) primer (0.5 μM), 0.5 μl of dNTPs (10 mM) and 3.5 μl of double-deionized H2O (ddH2O). The sample mixture was heated at 65 °C for 5 min and added into an ice-water-precooled reaction mixture that consisted of 2 μl of RT buffer (5×), 0.5 μl of DTT (0.1 M), 0.1 μl of Superscript III reverse transcriptase (200 U μl−1), and 1.4 μl of ddH2O, to bring the final volume to 10 μl for the reverse transcription. The mixture was incubated at 25 °C for 10 min followed by ramping to 55 °C at a rate of 0.5 °C min−1. After additional incubation at 55 °C for 30 min, the resulting cDNA sample was heated at 85 °C for 5 min to inactivate the reverse transcriptase. The cDNA sample was diluted 20-fold with ddH2O for the reduction of qPCR interference caused by the excess RT primer. qPCR was then run in a mixture of 1.0 μl of diluted cDNA (0.05×), 2 μl of GS primer and reverse primer (1.0 μM each) as well as 5 μl of SYBR green mix (2×).50 The qPCR mixture was heated at 95 °C for 4 min followed by 2 cycles of 95 °C for 15 s, 45 °C for 30 s, and 63 °C for 30 s. Optical detection began for the next 50 cycles of amplification (95 °C for 15 s, 63 °C 30 s), and the reaction mixtures were returned to ambient temperature when the qPCR finished. For the U6 snRNA51 and HOXD10 mRNA,52 500 ng of oligo-dT primer (Integrated DNA Technologies, Iowa, USA) was mixed with 500 ng of total RNA for the RT reaction. The RT products were then diluted 20-fold, and we transferred 1 μl into the qPCR mixture subjected to the following temperature programme: 95 °C for 4 min, followed by 2 cycles of 95 °C for 15 s with 72 °C for 30 s. The qPCR amplification was then performed by means of 50 cycles of 95 °C for 15 s, 56 °C for 30 s, and 72 °C for 30 s. The theoretical lengths of the miR-10b-3p, U6, and HOXD10 amplicons are 59, 94, and 71 bp, respectively. All of the qPCRs were run on a spin-type real-time thermocycler (Rotor-Gene Q, Qiagen, Germany) with a slight modification of the optical detection. Briefly, bright 475 nm LEDs (Roithner Lasertechnik GmbH, Austria) with a 470 nm interference filter (Edmund, USA) were used for the excitation during PCR amplification. For the fluorescence collection, another 520 nm interference filter was mounted on a filter wheel immediately before the photomultiplier tube, to reject scattering from the excitation beam. We used the same optical settings for the hybridization curves of the molecular beacons except we used a 650 nm LED with a 647 nm excitation filter and a 676 nm emission filter for the Cy5 detection.
Validation of the RT-qPCR products using capillary electrophoresis with laser-induced fluorescence
A capillary electrophoresis with laser induced fluorescence system was used for the validation of the RT-qPCR products by spiking samples with a 10-bp DNA ladder. Briefly, a high-voltage power supply (Gamma High Voltage Research Inc., Ormond Beach, FL, USA) was used for the electrophoresis. The entire detection system was enclosed within a black box with a high-voltage interlock. The high-voltage end of the separation system was placed in plastic housing for safety. A 532 nm solid-state laser from Tanyu (Kaohsiung, Taiwan) with a 5 mW output was used for the excitation. The fluorescence was monitored with a 10× objective (numerical aperture = 0.25), and a 610 nm interference filter was placed after the objective to reject the scattered light before the emitted light reached the photomultiplier tube (R3896, Hamamatsu Photonics, Hamamatsu, Japan). The amplified current was transferred directly through a 10 kΩ resistor to a 24-bit A/D interface at 10 Hz, controlled via Clarity software (DataApex, Prague, Czech Republic). The data were stored on a personal computer. Bare fused-silica capillary tubing (Polymicro Technologies, Phoenix, AZ, USA) with a 75 μm internal diameter was used as purchased for the separation without any further coating. The capillary length was 40 cm, and the effective length from the detector was 33 cm. Anodic and cathodic vials were filled with a 1.5% polyethylene oxide (PEO, Mw 8000000 Da) solution prepared in 100 mM Tris–boric acid buffer (pH 9.0) containing 2.5 μg ml−1 ethidium bromide. The capillary was filled with 1.5 M Tris–boric acid buffer (pH 10.0). The samples (DNA markers or markers mixed with the RT-qPCR products, Fig. S7†) were introduced into the capillary from the inlet (anodic) end via hydrodynamic injection at a height of 20 cm (the difference between the specimen vial and the cathodic vial) for 10 s. At an applied positive voltage of 15 kV, the DNA fragments migrating against the electro-osmotic flow entered the non-ionic PEO solution from the anodic end and were separated according to the sieving mechanism at ambient temperature.
Sample preparation for the specific molecular probing of miR-10b-3p, U6 snRNA, and HOXD10 mRNA
The coverslips with attached cells were gently rinsed with ddH2O followed by proteinase K (20 μg ml−1) digestion for 5 min at 37 °C. The coverslips were then washed with Tris–borate saline (TBS, pH 7.4) to remove the residual proteinase K. The digested cells were immersed in an EDC/imidazole solution for miRNA fixing according to another study.32 The EDC-fixed coverslips were then blocked by means of random-sequence oligonucleotides (∼20 nt, 1.0 μM) and 5% BSA in the presence of 0.3× SSC buffer (sodium chloride/sodium citrate, pH 7.0) with overnight illumination using a 3 W green/red LED (iCShop, Taiwan) (Fig. S1†). The overnight illumination was found to significantly reduce the autofluorescence from the fixed cells regardless of the wavelength of excitation. After the removal of the blocking materials via gentle rinsing with TBS, the molecular beacons were dissolved in 5% BSA and 0.3× SSC was added onto the coverslips for miRNA hybridization (1 h at 50 °C). The coverslips were then dipped 10 times into a 50% formamide solution in 2× SSC at 50 °C followed by washing on a thermo-shaker (60 rpm) three times with 50% formamide in 2× SSC at 50 °C for 10 min; three times with 50% formamide in 1× SSC at 50 °C for 10 min; three times with 0.3× SSC at 36 °C for 10 min; and one time with 1× SSC at 25 °C for 10 min. Finally, the coverslips were mounted with the DABCO mounting medium consisting of 1 μg ml−1 DAPI (4′,6-diamidino-2-phenylindole), 2 mM Trolox, and 1× SSC (pH 7.0) for the microscopic imaging. The DAPI counterstain (blue) of cell nuclear was used for sample focusing before each miRNA image.Optical setup of the HILO microscopy, image capturing and analysis
The lenses and reflection mirrors were acquired from Thorlabs (New Jersey, USA), and the dichroic mirrors and bandpass filters were purchased from Semrock (New York, USA). All of the imaging experiments were performed on a manual inverted microscope (Olympus IX71, Japan) with TIRF or HILO configuration. Briefly, two solid state lasers (405 and 640 nm, CNI optoelectronics technology, China) and one 488 nm Ar+ laser (Newport, CA, USA) were co-axially aligned into a beam expander. The expanded beams were ∼1 inch in diameter and were parallelly projected into the back port of the microscope. An achromatic lens with a focal length of 30 cm was used for focusing the laser beams to the back focal plane of the 60× TIRF objective (N.A. 1.49, Olympus, Japan). Either TIRF or HILO illumination can be achieved by adjusting the vertical direction of this focal lens (Fig. S2†). The measured laser power before the objective was 2.0 and 1.2 mW for 640 and 488 nm lasers, respectively. The incident angle of the HILO illumination was slightly less than the angle for the TIRF imaging, and caused the laser beam to come out of the objective. Three filter combinations (Semrock, USA) that involved the dichroic mirror and bandpass filter were installed on the microscope for both the excitation and fluorescence collection of DAPI (FF409-Di03-25 × 36, FF02-447/60-25), Alexa Fluor 488 and 6FAM (FF511-Di01-25 × 36, FF01-525/26-25) as well as Cy5 (FF650-Di01-25 × 36, FF01-680/30-25). All of the fluorescence images involving single-molecule detection were captured by means of an electron multiplier charge-coupled device (EMCCD; ProEM512, Princeton Instruments, USA) with an exposure time of 33 ms and a gain at 200 for all of the illumination settings. The background intensity of the EMCCD was 621.6 ± 26.2 and 673.8 ± 43.2 per pixel for the 640 and 488 nm illumination, respectively. The area (pixel number) of the cytoplasm and fluorescence intensity of each cell image were obtained using ImageJ software to obtain the average fluorescence intensity per pixel per cytoplasm of a single cell for a comparison between each cell group used in this study. Sixty-cell samples were collected from each image set for the calculation of the fluorescence intensities and standard deviations of the cells using HILO microscopy. All of the figures shown in this manuscript were adjusted either to the same brightness/contrast or autoscaled using Image J without any modifications.Conclusions
In this study, we developed a method that directly analyses miRNAs and their targets in fixed cells via molecular probing followed by HILO illumination. As a consequence of the combined EDC fixation, sample photobleaching, molecular probing, and HILO microscopy, the greatly improved signal-to-background ratio ensured the visualization of ultra-trace amounts of miRNA in the photobleached, formaldehyde fixed and EDC fixed cells. Furthermore, data in the resulting HILO microscopy images were compared to the results from conventional RT-qPCR. This comparison with RT-qPCR revealed that HILO microscopy offers better sensitivity and reproducibility for the measurement of ultra-trace amounts of miRNA. The ultra-sensitive detection of miRNA from aqueous solutions can also be achieved via miRNA capturing on a single microbead followed by isothermal exponential amplification.16 This proposed method may be potentially useful for miRNA analysis of bodily fluids, such as circulating miRNA biomarkers.21 However, the direct visualization method using HILO microscopy reduces the risk of RNA degradation during RNA extraction, liquid handling, storage, and amplification, thus raising the data reliability for the determination of miRNA or mRNA in low-level expression. The remarkable reproducibility of this method should increase the discriminatory power during analysis of that fluctuations in subfemtomolar amounts of miRNA caused by the transfection of a miRNA inhibitor. In addition, we also demonstrated that two-color HILO illumination allows us to simultaneously observe fluctuations in ultra-trace amounts of miRNA and its target mRNA more precisely. Consequently, we observed a slight alteration in mRNA quantity (miRNA targeting efficiency) after miRNA modulation. Therefore, this method not only offers a way to visualize ultra-trace amounts of miRNA (this visualization may be useful for direct diagnosis by means of circulating cancer cells from human bodily fluids) but also opens up the possibility of exploring the causal relation between a miRNA and the expression of its target gene at a single-cell, subfemtomolar level.Acknowledgements
The authors would like to thank the Ministry of Science and Technology, Taiwan for their partial funding support (NSC 100-2113-M-029-003-MY2, MOST 105-2113-M-029-004, MOST 103-2314-B-002-112-MY3).Notes and references
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS PubMed.
- W. C. Cho, Mol. Cancer, 2007, 6, 60 CrossRef PubMed.
- J. Hayes, P. P. Peruzzi and S. Lawler, Trends Mol. Med., 2014, 20, 460–469 CrossRef CAS PubMed.
- A. L. Kasinski and F. J. Slack, Nat. Rev. Cancer, 2011, 11, 849–864 CrossRef CAS PubMed.
- E. Wagner, Angew. Chem., Int. Ed., 2015, 54, 5824–5826 CrossRef CAS PubMed.
- D. D. Young, C. M. Connelly, C. Grohmann and A. Deiters, J. Am. Chem. Soc., 2010, 132, 7976–7981 CrossRef CAS PubMed.
- E. Callegari, L. Gramantieri, M. Domenicali, L. D’Abundo, S. Sabbioni and M. Negrini, Cell Death Differ., 2015, 22, 46–57 CrossRef CAS PubMed.
- C. Chen, D. A. Ridzon, A. J. Broomer, Z. Zhou, D. H. Lee, J. T. Nguyen, M. Barbisin, N. L. Xu, V. R. Mahuvakar, M. R. Andersen, K. Q. Lao, K. J. Livak and K. J. Guegler, Nucleic Acids Res., 2005, 33, e179 CrossRef PubMed.
- C. M. Hindson, J. R. Chevillet, H. A. Briggs, E. N. Gallichotte, I. K. Ruf, B. J. Hindson, R. L. Vessella and M. Tewari, Nat. Methods, 2013, 10, 1003–1005 CrossRef CAS PubMed.
- S. Bi, J. Zhang, S. Hao, C. Ding and S. Zhang, Anal. Chem., 2011, 83, 3696–3702 CrossRef CAS PubMed.
- N. Li, C. Jablonowski, H. Jin and W. Zhong, Anal. Chem., 2009, 81, 4906–4913 CrossRef CAS PubMed.
- Y. Cheng, X. Zhang, Z. Li, X. Jiao, Y. Wang and Y. Zhang, Angew. Chem., Int. Ed. Engl., 2009, 48, 3268–3272 CrossRef CAS PubMed.
- H. Jia, Z. Li, C. Liu and Y. Cheng, Angew. Chem., Int. Ed. Engl., 2010, 49, 5498–5501 CrossRef CAS PubMed.
- Q. Guo, F. Bian, Y. Liu, X. Qu, X. Hu and Q. Sun, Chem. Commun., 2017, 53, 4954–4957 RSC.
- H. Dong, X. Meng, W. Dai, Y. Cao, H. Lu, S. Zhou and X. Zhang, Anal. Chem., 2015, 87, 4334–4340 CrossRef CAS PubMed.
- X. Zhang, C. Liu, L. Sun, X. Duan and Z. Li, Chem. Sci., 2015, 6, 6213–6218 RSC.
- L. A. Neely, S. Patel, J. Garver, M. Gallo, M. Hackett, S. McLaughlin, M. Nadel, J. Harris, S. Gullans and J. Rooke, Nat. Methods, 2006, 3, 41–46 CrossRef CAS PubMed.
- R.-M. Jiang, Y.-S. Chang, S.-J. Chen, J.-H. Chen, H.-C. Chen and P.-L. Chang, J. Chromatogr. A, 2011, 1218, 2604–2610 CrossRef CAS PubMed.
- T.-H. Yang, D.-L. Ou, C. Hsu, S.-H. Huang and P.-L. Chang, Electrophoresis, 2012, 33, 2769–2776 CrossRef CAS PubMed.
- H. Lee, S. J. Shapiro, S. C. Chapin and P. S. Doyle, Anal. Chem., 2016, 88, 3075–3081 CrossRef CAS PubMed.
- J. Saikumar, K. Ramachandran and V. S. Vaidya, Clin. Chem., 2014, 60, 1158–1173 CAS.
- J. Kota, R. R. Chivukula, K. A. O’Donnell, E. A. Wentzel, C. L. Montgomery, H.-W. Hwang, T.-C. Chang, P. Vivekanandan, M. Torbenson and K. R. Clark, Cell, 2009, 137, 1005–1017 CrossRef CAS PubMed.
- A. N. Silahtaroglu, D. Nolting, L. Dyrskjøt, E. Berezikov, M. Møller, N. Tommerup and S. Kauppinen, Nat. Protoc., 2007, 2, 2520 CrossRef CAS PubMed.
- J. Lu and A. Tsourkas, Nucleic Acids Res., 2009, 37, e100 CrossRef PubMed.
- K. Gorska, I. Keklikoglou, U. Tschulena and N. Winssinger, Chem. Sci., 2011, 2, 1969–1975 RSC.
- H. Koo, I. Park, Y. Lee, H. J. Kim, J. H. Jung, J. H. Lee, Y. Kim, J.-H. Kim and J. W. Park, J. Am. Chem. Soc., 2016, 138, 11664–11671 CrossRef CAS PubMed.
- S. Li, L. Xu, W. Ma, X. Wu, M. Sun, H. Kuang, L. Wang, N. A. Kotov and C. Xu, J. Am. Chem. Soc., 2016, 138, 306–312 CrossRef CAS PubMed.
- Z. Cheglakov, T. M. Cronin, C. He and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 6116–6119 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, N. Zhang and S. Zhang, Chem. Sci., 2016, 7, 4184–4189 RSC.
- R. Deng, L. Tang, Q. Tian, Y. Wang, L. Lin and J. Li, Angew. Chem., Int. Ed., 2014, 53, 2389–2393 CrossRef CAS PubMed.
- L. Li, J. Feng, H. Liu, Q. Li, L. Tong and B. Tang, Chem. Sci., 2016, 7, 1940–1945 RSC.
- J. T. Pena, C. Sohn-Lee, S. H. Rouhanifard, J. Ludwig, M. Hafner, A. Mihailovic, C. Lim, D. Holoch, P. Berninger, M. Zavolan and T. Tuschl, Nat. Methods, 2009, 6, 139–141 CrossRef CAS PubMed.
- W. Baschong, R. Suetterlin and R. H. Laeng, J. Histochem. Cytochem., 2001, 49, 1565–1572 CrossRef CAS PubMed.
- M. Heilemann, S. van de Linde, M. Schuttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed. Engl., 2008, 47, 6172–6176 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- M. Tokunaga, N. Imamoto and K. Sakata-Sogawa, Nat. Methods, 2008, 5, 159–161 CrossRef CAS PubMed.
- A. S. Stender, K. Marchuk, C. Liu, S. Sander, M. W. Meyer, E. A. Smith, B. Neupane, G. Wang, J. Li, J.-X. Cheng, B. Huang and N. Fang, Chem. Rev., 2013, 113, 2469–2527 CrossRef CAS PubMed.
- A. Kusumi, T. A. Tsunoyama, K. M. Hirosawa, R. S. Kasai and T. K. Fujiwara, Nat. Chem. Biol., 2014, 10, 524–532 CrossRef CAS PubMed.
- G. T. Hermanson, in Bioconjugate techniques, Academic press, London, UK,2nd edn, 2008, ch. 27, pp. 969–1002 Search PubMed.
- J. Elmén, M. Lindow, A. Silahtaroglu, M. Bak, M. Christensen, A. Lind-Thomsen, M. Hedtjärn, J. B. Hansen, H. F. Hansen, E. M. Straarup, K. McCullagh, P. Kearney and S. Kauppinen, Nucleic Acids Res., 2008, 36, 1153–1162 CrossRef PubMed.
- R. Denzler, V. Agarwal, J. Stefano, D. P. Bartel and M. Stoffel, Mol. Cell, 2014, 54, 766–776 CrossRef CAS PubMed.
- J. R. Androsavich, D. J. Sobczynski, X. Liu, S. Pandya, V. Kaimal, T. Owen, K. Liu, D. A. MacKenna and B. N. Chau, Nucleic Acids Res., 2016, 44, e13 CrossRef PubMed.
- P.-L. Chang, W.-S. Hsieh, C.-L. Chiang, B. Yen-Liberman, G. W. Procop, H.-T. Chang and H.-T. Ho, Talanta, 2008, 77, 182–188 CrossRef CAS PubMed.
- C. Gaillard and F. Strauss, Tech. Tips Online, 1998, 3, 63–65 CrossRef.
- P. Mestdagh, N. Hartmann, L. Baeriswyl, D. Andreasen, N. Bernard, C. Chen, D. Cheo, P. D’Andrade, M. DeMayo, L. Dennis, S. Derveaux, Y. Feng, S. Fulmer-Smentek, B. Gerstmayer, J. Gouffon, C. Grimley, E. Lader, K. Y. Lee, S. Luo, P. Mouritzen, A. Narayanan, S. Patel, S. Peiffer, S. Ruberg, G. Schroth, D. Schuster, J. M. Shaffer, E. J. Shelton, S. Silveria, U. Ulmanella, V. Veeramachaneni, F. Staedtler, T. Peters, T. Guettouche, L. Wong and J. Vandesompele, Nat. Methods, 2014, 11, 809–815 CrossRef CAS PubMed.
- L. Ma, J. Teruya-Feldstein and R. A. Weinberg, Nature, 2007, 449, 682–688 CrossRef CAS PubMed.
- L. Ma, F. Reinhardt, E. Pan, J. Soutschek, B. Bhat, E. G. Marcusson, J. Teruya-Feldstein, G. W. Bell and R. A. Weinberg, Nat. Biotechnol., 2010, 28, 341–347 CrossRef CAS PubMed.
- K. C. Miranda, T. Huynh, Y. Tay, Y.-S. Ang, W.-L. Tam, A. M. Thomson, B. Lim and I. Rigoutsos, Cell, 2006, 126, 1203–1217 CrossRef CAS PubMed.
- A. Kozomara and S. Griffiths-Jones, Nucleic Acids Res., 2014, 42, D68–D73 CrossRef CAS PubMed.
- S.-J. Chen, G.-H. Chen, Y.-H. Chen, C.-Y. Liu, K.-P. Chang, Y.-S. Chang and H.-C. Chen, PLoS One, 2010, 5, e12745 Search PubMed.
- G. Xu, Y. Zhang, J. Wei, W. Jia, Z. Ge, Z. Zhang and X. Liu, BMC Cancer, 2013, 13, 469 CrossRef PubMed.
- Z. Liu, J. Zhu, H. Cao, H. Ren and X. Fang, Int. J. Oncol., 2012, 40, 1553–1560 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The LED device for the sample photobleaching, a schematic presentation of HILO microscopy, fluorescence spectra and hybridization curves of the molecular beacons, the linear correlation between the miRNA fluorescence intensity and the miRNA copy number, a validation of the miRNA adsorption and miRNA target gene expression via RT-qPCR, a validation of RT-qPCR using capillary electrophoresis, the reproducibility of RT-qPCR and Poisson distribution of the miRNA pipetting as well as a complete list of the oligonucleotides used in this study. See DOI: 10.1039/c7sc02701j |
| This journal is © The Royal Society of Chemistry 2017 |