
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preferential targeting of i-motifs and G-quadruplexes by small molecules†
Manish
Debnath
 a,
Shirsendu
Ghosh
b,
Ajay
Chauhan
a,
Rakesh
Paul
a,
Kankan
Bhattacharyya‡
b and
Jyotirmayee
Dash
*a
a,
Shirsendu
Ghosh
b,
Ajay
Chauhan
a,
Rakesh
Paul
a,
Kankan
Bhattacharyya‡
b and
Jyotirmayee
Dash
*a
aDepartment of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata-700032, India. E-mail: ocjd@iacs.res.in
bDepartment of Physical Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata-700032, India
First published on 8th September 2017
Abstract
i-Motifs and G-quadruplexes are dynamic nucleic acid secondary structures, which are believed to play key roles in gene expression. We herein report two peptidomimetic ligands (PBP1 and PBP2) that selectively target i-motifs and G-quadruplexes over double-stranded DNA. These peptidomimetics, regioisomeric with respect to the position of triazole/prolinamide motifs, have been synthesized using a modular method involving Cu(I)-catalyzed azide and alkyne cycloaddition. The para-isomer, PBP1 exhibits high selectivity for i-motifs while the meta-isomer PBP2 binds selectively to G-quadruplex structures. Interestingly, these ligands have the ability to induce G-quadruplex or i-motif structures from the unstructured single-stranded DNA conformations, as observed using single molecule Förster resonance energy transfer (smFRET) studies. The quantitative real-time polymerase chain reaction (qRT-PCR), western blot, and dual-luciferase assays indicate that PBP1 upregulates and PBP2 downregulates BCL-2 gene expression in cancer cells.
Introduction
Cytosine (C)-rich and guanine (G)-rich sequences can adopt stable nucleic acid secondary structures such as i-motifs1 and G-quadruplexes,2 respectively. The C-rich sequences form i-motif structures at acidic pH,3–6 whereas the G-rich sequences usually form G-quadruplexes at neutral pH in the presence of metal ions (Na+, K+). These sequences are prevalent in the promoter region of oncogenes like BCL-2 and c-MYC.7–10 It has been reported that small molecules bind G-quadruplexes11 and modulate the gene expression.12–17 Although i-motifs are hypothesized to play important roles in gene transcription,18–21 only a few ligands are known to selectively target i-motifs in biological systems.18–22 Furthermore, i-motifs and G-quadruplexes are highly dynamic and they can exist in equilibrium with unfolded DNA under physiological conditions.12,18–20 However, little is known about how small molecules can regulate the relative populations of these two dynamic secondary structures. In this context, we envisioned to develop small molecules that can discriminate between i-motif and G-quadruplex structures and modulate gene expression.The single molecule Förster resonance energy transfer (smFRET) technique provides key information about the structure, relative population distribution of folded or unfolded species, and the end-to-end distance of biomolecules.23–34 The smFRET technique has been used to elucidate the conformational dynamics of G-quadruplexes in the presence of metal ions (K+/Na+),28 protein,33 and small molecules.34 The population equilibrium of C-rich ILPR and BCL-2 promoter sequences has been studied using laser tweezer experiments.35,36 Majima and co-workers have used smFRET to quantitatively analyse the pH-induced intra-molecular folding dynamics of i-motif DNA.37 However, the use of smFRET to monitor the ligand induced change in the relative population distribution of i-motif and G-quadruplex structures present in oncogenic promoters is very limited.
Hurley and Hecht have reported that the steroid ligand IMC-48 folds the BCL-2 C-rich sequence into an i-motif, while the same sequence is folded into a hairpin duplex in the presence of the related ligand IMC-76.18,19 In this study, we describe the synthesis of two flexible peptidomimetic congeners, PBP1 and PBP2, which show structure-specific recognition for G-quadruplex and i-motif structures. The interaction of these ligands with BCL-2 or c-MYC i-motifs and G-quadruplexes has been evaluated using biophysical studies like melting analysis by Förster resonance energy transfer (FRET), thiazole-orange (TO) displacement assay, fluorescence quenching assay, and circular dichroism (CD) spectroscopy. In addition, the ability of these ligands to induce the formation of i-motif and G-quadruplex structures from the unfolded BCL-2 and c-MYC C-rich and G-rich promoter sequences has been investigated using smFRET and fluorescence lifetime studies at neutral pH. We have further demonstrated how ligand-dependent conformational changes of BCL-2 i-motif or G-quadruplex topologies can modulate the BCL-2 expression in cancer cells.
Results and discussion
Design and synthesis of peptidomimetic ligands
Peptidomimetics are designed to interact with specific biological targets as they exhibit enhanced proteolytic stability and improved cell permeability.38,39 We have anticipated that peptidomimetics containing the 2,6-pyridine dicarboxamide unit, linked to L-proline residues through triazole and arene motifs would be structurally flexible enough to adopt different conformations upon interacting with different DNA four stranded structures (i-motifs and G-quadruplexes). The proline residues play an important role in peptide conformation. The 2,6-pyridine dicarboxamide motif can adopt folded conformations due to the bifurcated H-bonding between the lone pair of pyridine nitrogen and amide –NH protons. The arene motif attached to the proline residues would provide additional flexibility to form topologically different positional isomers that could discriminate between different DNA structures such as i-motifs and G-quadruplexes (Fig. S1, ESI†). Furthermore, the triazole ring system could facilitate stacking interactions with the loop bases and, thus, could differentially interact with different DNA secondary structures with variations in the loop region.40 The triazole ring system, able to mimic the cis- or trans-conformations of amide bonds, would impart rigidity to the peptidomimetics. It has been reported that triazole containing ligands generated by “click” chemistry selectively bind G-quadruplexes.41–43The bis-triazole containing peptidomimetic type ligands PBP1 and PBP2 were assembled using a modular synthetic strategy involving a Cu(I)-catalyzed 1,3-dipolar azide–alkyne cycloaddition between azido prolinamides 1, 2 and pyridyl dialkyne 3 (Schemes 1 and S1, ESI†). The azido prolinamides 1 and 2 were obtained by amide coupling of N-Bocproline 4 with the para and meta-azido anilines 5 and 6. The dialkyne building block 3 was prepared from chelidamic acid 7. Chelidamic acid 7 was treated with oxalyl chloride to generate the corresponding acid chloride, which was subsequently coupled with propargyl amine 8, followed by alkylation of the resulting pyridyl dialkyne with 3-dimethylaminopropyl chloride 9, affording the dialkyne 3 in high overall yield. The Cu(I)-catalyzed Huisgen cycloaddition of azido prolinamide derivatives 1 and 2 with the dialkyne 3 and subsequent removal of the Boc group provided the bis-prolinamide derivatives PBP1 and PBP2 in high yields. The bis-prolinamide derivative PBP3 was similarly assembled from azido prolinamide 1 and pyridine-2,6-dicarboxylic acid.
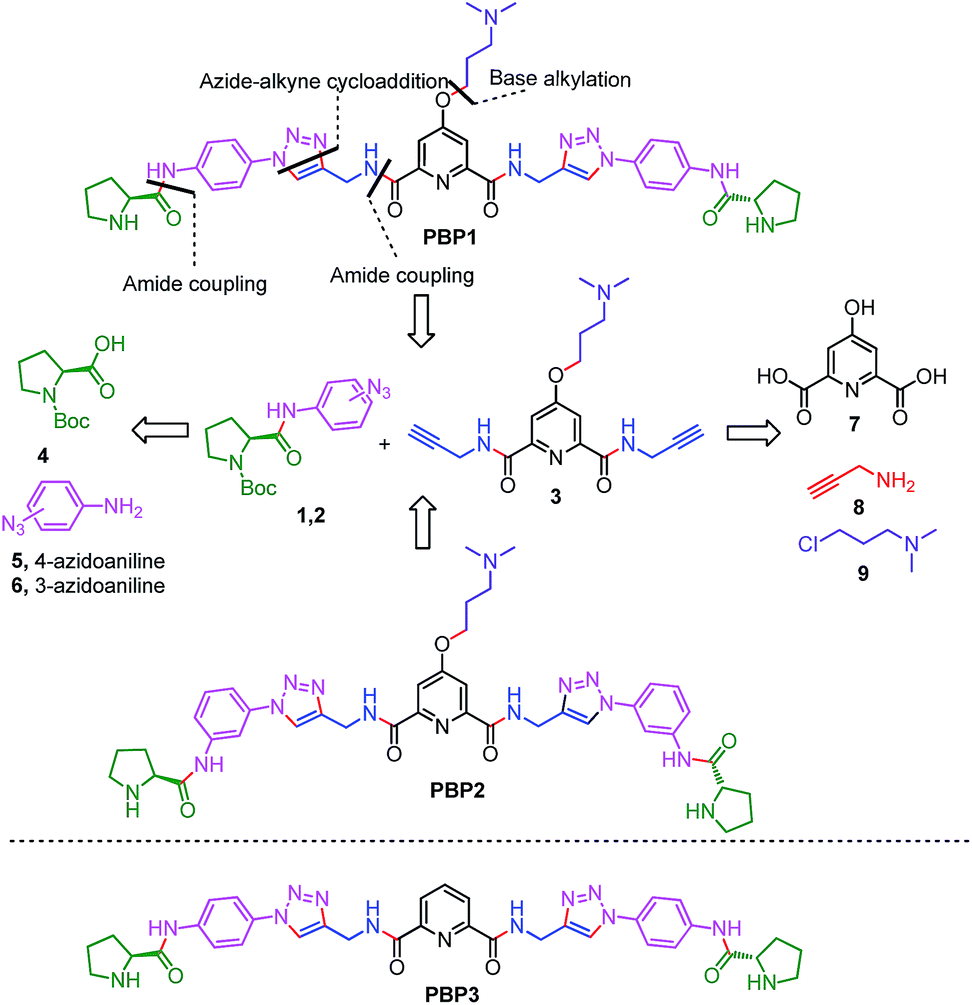 | ||
| Scheme 1 The synthesis of bis-prolinamide derivatives PBP1, PBP2 and the structure of PBP3. | ||
PBP1 and PBP2 exhibit differential binding between i-motifs and G-quadruplexes
The ability of these regioisomeric ligands to interact with G-quadruplexes and i-motifs was evaluated using biophysical assays. C-Rich sequences were folded into i-motifs by annealing in 60 mM K-cacodylate buffer, at pH 4.8 and then the pH was adjusted to 6 for biophysical analysis.18,22,44,45 | ||
| Fig. 1 The FRET melting and TO displacement assays. The FRET stabilization potential of bis-prolinamide derivatives PBP1 (1 μM), PBP2 (1 μM), and PBP3 (1 μM) upon interaction with (a) 100 nM folded i-motifs (c-MYC-C, BCL-2-C, and h-TELO-C) and ds DNA in 60 mM K-cacodylate buffer, (pH 6); (b) 100 nM folded G-quadruplexes (c-MYC-G, BCL-2-G, and h-TELO-G) and ds DNA in 60 mM K-cacodylate buffer, (pH 7); thermal shift profiles for (c) PBP1 (0–10 μM) and (d) PBP2 (0–15 μM) upon stabilizing i-motifs and ds DNA in 60 mM K-cacodylate buffer, (pH 6). The TO displacement from 250 nM BCL-2-C, c-MYC-C, and h-TELO-C i-motifs in 60 mM K-cacodylate buffer, (pH 6); BCL-2-G, c-MYC-G, and h-TELO-G G-quadruplexes and ds DNA in 60 mM K-cacodylate buffer, (pH 7) with increasing concentrations of (e) PBP1 (0–10 μM); (f) PBP2 (0–15 μM). | ||
Interestingly, the two positional isomers PBP1 and PBP2 exhibited a marked difference in increasing the Tm of folded G-quadruplexes and i-motifs at 1 μM ligand concentration (Table 1, Fig. 1a and b and S2, ESI†). Ligand PBP1, in which the prolinamide motifs are at the para position with respect to the triazole ring system, increased the Tm values of BCL-2-C and c-MYC-C i-motifs more effectively compared to ligand PBP2 at 1 μM ligand concentration (ΔTm = 16–29 °C for PBP1 and ΔTm = 8 °C for PBP2). In contrast, the meta regioisomer PBP2 increased the ΔTm value of c-MYC-G and BCL-2-G G-quadruplexes (ΔTm = 16 °C at 1 μM PBP2 and ΔTm = 5.2 °C at 1 μM PBP1) (Table 1). Ligand PBP3, which lacks the –NMe2 side chain in the central pyridine ring, showed low stabilization potential (ΔTm = 3–5 °C) for both G-quadruplex and i-motif structures (Table S1, ESI†). When BCL-2-C and c-MYC-C mutant C-rich sequences were used in the melting analysis, no melting curves were observed, thereby indicating their existence in the unfolded form (Fig. S3, ESI†).
| DNAa | DC50b (μM) | K d (μM) | ΔTmd (°C) | |||
|---|---|---|---|---|---|---|
| PBP1 | PBP2 | PBP1 | PBP2 | PBP1 | PBP2 | |
| a Unlabeled, single TAMRA labeled and dual FAM-TAMRA labeled sequences were used in the TO displacement, fluorescence quenching, and FRET melting experiments, respectively; HEG = hexaethylene glycol. b Error = ±5%. c K d values indicated for the 5′-labeled sequences (Kd = ± 5%). PBP1 (fold selectivity): BCL-2-C/c-MYC-C/BCL-2-G/c-MYC-G = 40/6/1.5/1; PBP2 (fold selectivity): BCL-2-C/c-MYC-C/BCL-2-G/c-MYC-G = 1.5/1/4.5/7. d ΔTm = ±1 °C; [PBP1] = [PBP2] = 1 μM. The Tm values of folded c-MYC-C, BCL-2-C, h-TELO-C i-motifs and ds DNA diluted in 60 mM K-cacodylate buffer at pH 6 are 48 ± 1 °C, 59 ± 1 °C, 43 ± 1 °C, and 60 ± 1 °C (Table S1, ESI). The Tm values of folded c-MYC-G, BCL-2-G, and h-TELO-G diluted in 60 mM K-cacodylate buffer at pH 7 are 69 ± 1 °C, 70 ± 1 °C, 55 ± 1 °C. | ||||||
| BCL-2-C: 5′-d(CAGC4GCTC3GC5T2C2TC3GCGC3GC4T)-3′ | 0.9 | 8.2 | 0.3 | 5.8 | 29 | 8 |
| BCL-2-G: 5′-d(AG4CG3CGCG3AG2A2G5CG3AGCG4CGT)-3′ | 5.7 | 2.4 | 7.2 | 1.9 | 5.2 | 16 |
| c-MYC-C: 5′-d(TC4AC2T2C4AC3TC4AC3TC4A)-3′ | 2.7 | 6.8 | 2.4 | 9.5 | 16 | 8 |
| c-MYC-G: 5′-d(TG4AG3TG4AG3TG4A2G2TG4A)-3′ | 8.5 | 1.3 | 12.5 | 1.3 | 5.2 | 16 |
| h-TELO-C: 5′-d(TA2C3TA2C3TA2C3TA2C3)-3′ | 4 | >15 | n.d. | n.d. | 9 | 5 |
| h-TELO-G: 5′-d(G3TTAG3TTAG3TTAG3)-3′ | 9.8 | 4.7 | n.d. | n.d. | 5 | 8 |
| ds DNA: 5′-d(TATAGCTATA-HEG-TATAGCTATA)-3′ | n.d. | n.d. | >25 | >25 | 0.94 | 1.1 |
Next, FRET melting experiments were carried out for BCL-2, c-MYC, and h-TELO i-motifs and G-quadruplexes using an increasing concentration of PBP1–2. PBP1 showed high ΔTm values for BCL-2-C, c-MYC-C, and h-TELO-C i-motifs while PBP2 exhibited high ΔTm values for the corresponding G-quadruplexes in a dose-dependent manner (Fig. 1c and d, S2–S4, ESI†). PBP1 showed a ΔTm value of 32 ± 1 °C (i.e., a Tm of 92 °C) for BCL-2-C at 1.3 μM concentration, whereas higher concentrations of PBP1 were required to attain a ΔTm value of 32 ± 1 °C for c-MYC-C (a Tm of 81 °C at 6.5 μM) and h-TELO-C (a Tm of 76 °C at 10 μM). These results indicate that PBP1 shows a preferential affinity for the BCL-2-C i-motif as it can obtain a maximum ΔTm value for the BCL-2-C i-motif at 5–8 fold lower concentrations than the c-MYC-C and h-TELO-C i-motifs. In contrast, BCL-2-G and c-MYC-G G-quadruplexes exhibited maximum ΔTm values at 3 fold lower concentrations of PBP2 than PBP1 (Fig. S2, ESI†). However, both PBP1 and PBP2 failed to alter the Tm of ds DNA even at high ligand concentrations (10–15 μM), indicating their selectivity for four stranded structures over double-stranded DNA.
The selectivity of PBP1 for i-motifs and PBP2 for G-quadruplexes was determined using the FRET competition assay with the competing G-quadruplex (TG5T)4 and double-stranded ds26 DNA (Fig. S2c and d, ESI†). The results show that no significant changes in the ΔTm values of PBP1 bound i-motifs and PBP2 bound G-quadruplexes were observed in the presence of 40 mol equivalent excess of the G-quadruplex and double-stranded DNA competitors.
PBP1 exhibited DC50 values of 0.9 μM, 2.7 μM and 4.0 μM for BCL-2-C, c-MYC-C and h-TELO-C i-motifs, respectively (Table 1). In comparison, the meta-isomer PBP2 displayed significantly lower affinity for BCL-2-C (DC50 = 8.2 μM), c-MYC-C (DC50 = 6.8 μM), and h-TELO-C (DC50 > 15 μM) i-motifs. On the other hand, PBP1 showed higher DC50 values for BCL-2-G, c-MYC-G, and h-TELO-G G-quadruplexes compared to PBP2 (Table 1). These results are in agreement with the FRET melting data suggesting the higher affinity of PBP1 for the BCL-2-C i-motif as compared to that of PBP2 and the preferential binding of PBP2 for c-MYC-G and BCL-2-G G-quadruplexes as compared to PBP1. However, ligand PBP3 exhibited high DC50 values for G-quadruplexes (DC50 = 8.4–10.2 μM) and i-motifs (DC50 = 7.9–10 μM), which indicates the weak affinity of PBP3 for both four stranded structures (Fig. S5, ESI†).
 | ||
| Fig. 2 The percentage fluorescence quenching observed upon titration of 250 nM of 5′-TAMRA labeled folded BCL-2-C and c-MYC-C i-motif structures in 60 mM K-cacodylate buffer, pH 6, and 250 nM 5′-TAMRA labeled folded BCL-2-G and c-MYC-G G-quadruplex structures in 60 mM K-cacodylate buffer, pH 7 by (a) 0–30 μM PBP1 and (b) 0–30 μM PBP2. | ||
When 5′-TAMRA labeled BCL-2-G and c-MYC-G G-quadruplexes were titrated with PBP1 and PBP2, a marked difference in their affinity was observed. PBP2 exhibited a 7 fold preference for the 5′-labeled c-MYC-G (Kd = 1.3 μM) G-quadruplex over the i-motif counterpart. Similarly, a 3 fold higher affinity of PBP2 was observed for the BCL-2-G G-quadruplex (Kd = 1.9 μM) over the BCL-2-C i-motif. It is intriguing to note that PBP1 showed a 24 fold higher selectivity for the BCL-2-C i-motif over BCL-2-G (Kd = 7.2 μM) and a 40 fold higher selectivity for the BCL-2-C i-motif over c-MYC-G (Kd = 12.5 μM) G-quadruplexes. To the best of our knowledge, this is one of the highest levels of selectivity reported by a small molecule ligand for i-motif over G-quadruplex structures.
Similar binding titrations with 3′-TAMRA labeled BCL-2 and c-MYC i-motifs and G-quadruplexes revealed that both PBP1 and PBP2 displayed a higher affinity (lower Kd value) for 5′-labeled G-quadruplexes and i-motifs over 3′-labeled structures (Fig. S6, Scheme S2, Tables S3 and S4, ESI†). Therefore, the 5′-end of G-quadruplex and i-motif structures is the preferred binding site for PBP1 and PBP2. In comparison, PBP3 induced a considerably lower level of fluorescence quenching (>40%) in TAMRA labeled G-quadruplexes and i-motifs (Fig. S7, ESI†), suggesting the weak affinity of PBP3 for these DNA structures. The weak affinity of PBP3 may be attributed to its poor solubility in aqueous buffer and the lack of a cationic side chain, and hence PBP3 was not selected for further studies. Importantly, PBP1 and PBP2 preferentially bind to the four stranded DNA structures over ds DNA, as control experiments with TAMRA labeled ds DNA showed no significant quenching upon addition of the ligands (Fig. 2 and S6, ESI†).
PBP1 and PBP2 induce the formation of i-motifs and G-quadruplexes, respectively
 | ||
| Fig. 3 The smFRET analysis of BCL-2-C and mutated BCL-2-C (BCL-2-C-mut). Photon bursts of donor/acceptor (background corrected) (a), and FRET efficiency distributions (b) of 100 pM dual fluorescent labeled BCL-2-C and BCL-2-C-mut under neutral (pH 7) and acidic (pH 4.8) conditions or in the presence of PBP1 (1 equiv.), PBP2 (1 equiv.). BCL-2-C-mut: 5′-FAM-d(CAGC2TCGCTC2TGC2TC2T2C2TC2TGCGC2TGC2TCG)-TAMRA-3′. | ||
The FRET histogram of the free c-MYC-C i-motif sequence at pH 7 showed two major population distributions having mean εFRET values of ∼0.55 and ∼0.8 with RDA ∼ 53.2 Å and ∼43.7 Å, respectively (Fig. S8, ESI†). Upon addition of PBP1 (1 equiv.), the εFRET distribution was shifted towards higher value (∼0.93) (RDA ∼ 35.7 Å), indicating the formation of a compact i-motif structure; whereas PBP2 (1 equiv.) did not significantly alter the FRET distribution pattern of the free c-MYC-C i-motif sequence at pH 7. Similar the BCL-2-C i-motif, the folded c-MYC-C i-motif at pH 4.8 showed a single population with a mean εFRET value ∼0.95, indicating the formation of a more compact structure with lower end-to-end distances (RDA ∼ 33.7 Å).
Similar to the folded i-motifs, the folded G-quadruplexes are known to exhibit lower RDA values compared to unstructured G-rich sequences.34 The free BCL-2-G G-quadruplex sequence showed a wide distribution centered at εFRET ∼ 0.6 with a corresponding RDA of ∼51.4 Å (Fig. S11a and b, ESI†). PBP2 (1 equiv.) could significantly shift the populations of free BCL-2-G sequence towards higher values (εFRET ∼ 0.95) with a low RDA value of ∼33.7 Å, indicating PBP2 could induce a compact G-quadruplex structure similar to the K+-folded G-quadruplex.34 However, the free BCL-2-G sequence showed two major populations with εFRET values of ∼0.6 (RDA ∼ 51.4 Å) and ∼0.85 (RDA ∼ 41.2 Å) in the presence of PBP1 (1 equiv.). The addition of PBP2 decreased the RDA value of free c-MYC-G from ∼57 Å to ∼41 Å in the absence of K+ ions (Fig. S11c, d and Table S6, ESI†). However, the non-G-quadruplex forming mutated BCL-2-G sequence did not exhibit any notable change in εFRET values upon the addition of ligands PBP1 and PBP2 (εFRET ∼ 0.6), suggesting that the mutated sequences are unstructured even in the presence of the ligands (Fig. S12, ESI†).
Collectively, the smFRET results suggest that ligand PBP1 can completely shift the dynamic equilibrium of C-rich BCL-2-C and c-MYC-C sequences towards the folded i-motifs from the unstructured form under physiologically relevant neutral pH conditions. However, PBP2 induces only a partial shift in the population distributions of free i-motif sequences at neutral pH but it has the ability to trigger the formation of G-quadruplexes from the unstructured G-rich sequences in the absence of K+ ions.
| System | τ avg | ε FRET | R DA (Å) | |
|---|---|---|---|---|
| a ±10%. b BCL-2-G-mut: 5′-FAM-d(AG2TGCG2TCGC G2AAG2A2G2 TG2 C GTAA GCG2TGCTG)-TAMRA-3′. | ||||
| BCL-2-C (pH 7) | D | 3.78 | 0.59 | 52 |
| DA | 1.56 | |||
| BCL-2-C-mut (pH 7)b | D | 3.62 | 0.53 | 54 |
| DA | 1.7 | |||
| BCL-2-C (pH 4.8) | D | 4.32 | 0.81 | 43.2 |
| DA | 0.84 | |||
| BCL-2-C + PBP1 (pH 7) | D | 3.24 | 0.87 | 40.1 |
| DA | 0.42 | |||
| BCL-2-C-mut + PBP1 (pH 7) | D | 2.05 | 0.55 | 53 |
| DA | 0.92 | |||
| BCL-2-C + PBP2 (pH 7) | D | 2.43 | 0.67 | 48.9 |
| DA | 0.79 | |||
| BCL-2-C-mut + PBP2 (pH 7) | D | 2.34 | 0.59 | 52 |
| DA | 0.96 | |||
| c-MYC-C (pH 7) | D | 3.32 | 0.52 | 54.3 |
| DA | 1.6 | |||
| c-MYC-C (pH 4.8) | D | 4.62 | 0.79 | 44.1 |
| DA | 0.97 | |||
| c-MYC-C + PBP1 (pH 7) | D | 3.04 | 0.72 | 47 |
| DA | 0.87 | |||
| c-MYC-C + PBP2 (pH 7) | D | 2.7 | 0.62 | 50.7 |
| DA | 1.02 | |||
Conversely, the addition of PBP2 decreased the RDA value of free BCL-2-G and c-MYC-G from ∼55 Å to ∼41 Å in the absence of K+ ions (Table S8, ESI†), which indicates that PBP2 folds single stranded BCL-2-G and c-MYC-G G-rich sequences into G-quadruplex structures. However, no significant changes in the RDA values of mutant BCL-2 C-rich and G-rich sequences were noted upon addition of PBP1 and PBP2 (Tables 2, S8 and Fig. S15, ESI†). These results suggest that the observed changes in RDA values of the investigated sequences are due to the formation of folded G-quadruplex or i-motif structures in the presence of ligands.
In agreement with the smFRET and lifetime analyses, the CD spectroscopy also supports the idea that the ligand PBP1 triggers the formation of BCL-2-C and c-MYC-C i-motif structures and PBP2 induces the formation of G-quadruplex structures (Fig. S17–S24, ESI†). Moreover, the change in CD intensity with the mole fraction of ligands (Job’s plot) suggests a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry of PBP1 and PBP2 with i-motifs and G-quadruplexes, respectively (Fig. S25 and S26, ESI†).
:1 binding stoichiometry of PBP1 and PBP2 with i-motifs and G-quadruplexes, respectively (Fig. S25 and S26, ESI†).
Growth inhibition assay
The growth-inhibitory activity of ligands PBP1 and PBP2 on human breast adenocarcinoma (MCF-7) cells, human colon cancer (HCT116) cells, and normal mouse myoblast (C2C12) cells were evaluated using MTT assay (Fig. S27 and S28, ESI†).52 After 24 h treatment of cells, PBP1 showed IC50 values of 17.9 ± 1.8 μM and 18.5 ± 1.9 μM in MCF-7 and HCT116 cells, respectively. Ligand PBP2 displayed IC50 values of 3.3 ± 0.7 μM and 3.9 ± 0.9 μM in MCF-7 cells and HCT116 cells, respectively, after 24 h (Table S9, ESI†). The IC50 values suggested a differential effect of PBP1 and PBP2 on cancer cells after a 48 h treatment. When the cells were treated with PBP1 for 48 h, no significant change in IC50 values (14.4 ± 1.4 μM for MCF-7 and 15.1 ± 1.5 μM for HCT116 cells) was observed (Fig. S28 and Table S10, ESI†). However, treatment of cells with PBP2 for 48 h caused a nearly 10 fold decrease in IC50 values (1.7 ± 0.2 μM for MCF-7 and 1.3 ± 0.15 μM for HCT116 cells) as compared to PBP1. This indicates that PBP2 can considerably inhibit the growth of cancer cells after a 48 h treatment, while PBP1 shows less potent cytotoxic activity. Importantly, both PBP1 and PBP2 exhibited negligible toxicity towards normal C2C12 cells after a 48 h treatment, even at >40 μM concentration.Ligand-dependent BCL-2 expression in cancer cells
 | ||
| Fig. 4 (a) Determination of the transcriptional regulation of BCL-2 mRNA in the presence of IC50 doses (24 h) of PBP1 or PBP2 in cancer cells (MCF-7 and HCT116) via qRT-PCR and quantified by double delta Ct analysis using GAPDH and 18S rRNA as reference genes. Data is presented in terms of fold change (the expression of control is 1 fold). The data are shown as mean ± SD. *P < 0.05, #P < 0.01, versus untreated cancer cells. (b) Immunoreactive bands of the BCL-2 protein were analyzed via western blot in MCF-7 and HCT116 cells. The data are shown as mean ± SD. *P < 0.05, #P < 0.01, versus untreated cancer cells. (c) The protein expression of the BCL-2 protein in the presence of IC50 doses (24 h) of PBP1 or PBP2 in MCF-7 and HCT116 cancer cells. (d) The relative luciferase expression in the LB322 BCL-2 promoter containing firefly plasmid normalized with pRL-TK Renilla plasmid (FF/RL) upon treatment with 5 μM of PBP1 and PBP2 in HCT116 cells, data shown here as mean ± SD. *P < 0.05, #P < 0.01, versus untreated cancer cells. (e) Flow cytometric analysis upon treatment with 5 μM of PBP1 and PBP2 in serum starved MCF-7 cells, Q3, Q4, Q2, and Q1 indicate healthy cells, early, late apoptotic, and necrotic cells, respectively. | ||
 | ||
| Scheme 2 A schematic representation of the working hypothesis: The BCL-2 GC-rich promoter region forming G-quadruplexes and i-motifs in opposite strands in the presence of the peptidomimetic ligands PBP1 and PBP2. | ||
Upon treatment with PBP2, the BCL-2 promoter-linked luciferase expression was decreased by 58% relative to the untreated control. In contrast, treatment with PBP1 exhibited a 42% increase in BCL-2 promoter-linked luciferase expression compared to the control. To further validate our results, we also investigated the effect of PBP1 and PBP2 on a firefly luciferase vector (pBV-Luc) containing non i-motif or G-quadruplex sequence (Fig. S31, ESI†). Interestingly, we did not observe any notable change in firefly luciferase expression in pBV-Luc treated HCT116 cells upon treatment of PBP1 and PBP2 compared to untreated control. In addition, ligand PBP1 did not show any significant change in the expression of reporter vector containing other promoter i-motif or G-quadruplex forming sequences such as c-MYC (Del 4 plasmid, Fig. S32, ESI†). These results indicate that PBP1 and PBP2 may regulate BCL-2 expression by targeting BCL-2 promoter i-motifs or quadruplexes in cancer cells.
Detection of apoptosis using Annexin V and caspases 3/7
To further investigate the influence of bis-prolinamides on cell survival, flow cytometry was employed using Annexin V and PI dual staining assays (Fig. 4e and S33, ESI†). Since PBP1 did not influence the healthy cancer cells in an apoptosis assay (unpublished data), we prepared model apoptotic cells via 48 h serum starvation in order to investigate the anti-apoptotic properties.53 MCF-7 and HCT116 cells were treated with 5 μM PBP1 and PBP2 after serum starvation. Control MCF-7 cells show a significant percentage of apoptotic cells (∼22%) due to serum starvation. Interestingly, treatment with PBP1 reduced the percentage of apoptotic MCF-7 cells to ∼14% whereas PBP2 efficiently increased the percentage of apoptotic MCF-7 cells to ∼60%. Similar results were obtained for PBP1 and PBP2 treated HCT116 cells (Fig. S33, ESI†).Since activation of caspases is an important process during apoptosis, the quantitative detection of executioner caspases 3 and 7 in HCT116 cells upon treatment with peptidomimetic ligands PBP1 and PBP2 was investigated using FLICA reagent based flow cytometry assay (Fig. S34 and Table S15, ESI†).54 The serum starved control cells exhibited moderate levels of active caspases 3 and 7 (∼18.4%). At 24 h post treatment with ligand PBP1 (5 μM), the level of active caspases 3/7 was significantly decreased (∼5.1%). However, cells incubated with ligand PBP2 (5 μM) for 24 h exhibited a higher level of active caspases 3/7 (∼44.7%). These results suggest that PBP1 decreases the level of active caspases 3 and 7 in cancer cells whereas PBP2 induced apoptosis is associated with the activation of caspases 3 and 7. However, the exact molecular mechanism of this behaviour is under investigation.
Conclusions
We have demonstrated that two flexible peptidomimetic congeners, PBP1 and PBP2, synthesized using ‘click chemistry’, can exhibit distinguishable recognition between i-motifs and G-quadruplexes. FRET melting and fluorescence spectroscopic studies reveal that both ligands show high selectivity for i-motifs and G-quadruplexes over duplex DNA. These studies also indicate that PBP1 preferentially binds to the BCL-2-C i-motif over G-quadruplexes and PBP2 selectively binds to G-quadruplexes over i-motifs. In addition, smFRET studies indicate that PBP1 folds the unstructured BCL-2 and c-MYC C-rich DNA sequences into i-motif structures at neutral pH; whereas PBP2 promotes G-quadruplex formation from single stranded BCL-2 and c-MYC G-rich sequences in the absence of metal ions. Cellular studies revealed that PBP1 upregulates BCL-2 gene expression while PBP2 inhibits BCL-2 gene expression. Furthermore, PBP2 triggers apoptosis via activation of caspases 3 and 7; whereas PBP1 reduces the level of active caspases 3/7 and decreases the percentage of apoptotic cancer cells. These results indicate that a small change in the ligand structure can have a dramatic effect on the molecular recognition properties, providing a new platform to achieve differential recognition of G-quadruplexes and i-motifs. These observations further suggest that ligand induced folding of i-motifs or G-quadruplexes may provide an attractive way to control gene expression and to develop therapies for cancer and neurodegenerative diseases.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Professor Shankar Balsubramanian for useful suggestions. We thank DST, India and the DST-Center for Ultrafast Spectroscopy and Microscopy for funding. KB thanks DST for a JC Bose fellowship. JD thanks DST for a SwarnaJayanti fellowship and DBT for funding. MD and RP thank DST for an INSPIRE fellowship and SG thanks CSIR-India for research fellowship. We thank Dr Tania Das, Dr Y. Pavan Kumar, and Gargi Chakraborti for their help during the preparation of this manuscript.References
- K. Gehring, J. L. Leroy and M. Gueron, Nature, 1993, 363, 561–565 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- K. Guo, A. Pourpak, K. Beetz-Rogers, V. Gokhale, D. Sun and L. H. Hurley, J. Am. Chem. Soc., 2007, 129, 10220–10228 CrossRef CAS PubMed.
- C. Chen, M. Li, Y. Xing, Y. Li, C.-C. Joedecke, J. Jin, Z. Yang and D. Liu, Langmuir, 2012, 28, 17743–17748 CrossRef CAS PubMed.
- S. Dhakal, Z. Yu, R. Konik, Y. Cui, D. Koirala and H. Mao, Biophys. J., 2012, 102, 2575–2584 CrossRef CAS PubMed.
- Y. Cui, D. Kong, C. Ghimire, C. Xu and H. Mao, Biochemistry, 2016, 55, 2291–2299 CrossRef CAS PubMed.
- T. A. Brooks and L. H. Hurley, Nat. Rev. Cancer, 2009, 9, 849–861 CAS.
- G. F. Salgado, C. Cazenave, A. Kerkour and J.-L. Mergny, Chem. Sci., 2015, 6, 3314–3320 RSC.
- K. M. Felsenstein, L. B. Saunders, J. K. Simmons, E. Leon, D. R. Calabrese, S. Zhang, A. Michalowski, P. Gareiss, B. A. Mock and J. S. Schneekloth, ACS Chem. Biol., 2016, 11, 139–148 CrossRef CAS PubMed.
- D. Sun and L. H. Hurley, J. Med. Chem., 2009, 52, 2863–2874 CrossRef CAS PubMed.
- R. Rodriguez, S. Müller, J. A. Yeoman, C. Trentesaux, J.-F. Riou and S. Balasubramanian, J. Am. Chem. Soc., 2008, 130, 15758–15759 CrossRef CAS PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef CAS PubMed.
- B. Maji and S. Bhattacharya, Chem. Commun., 2014, 50, 6422–6438 RSC.
- D. Panda, M. Debnath, S. Mandal, I. Bessi, H. Schwalbe and J. Dash, Sci. Rep., 2015, 5, 13183 CrossRef CAS PubMed.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867–5892 RSC.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- J. Alzeer, B. R. Vummidi, P. J. C. Roth and N. W. Luedtke, Angew. Chem., Int. Ed., 2009, 48, 9362 CrossRef CAS PubMed.
- S. Kendrick, H.-J. Kang, M. P. Alam, M. M. Madathil, P. Agrawal, V. Gokhale, D. Yang, S. M. Hecht and L. H. Hurley, J. Am. Chem. Soc., 2014, 136, 4161–4171 CrossRef CAS PubMed.
- H.-J. Kang, S. Kendrick, S. M. Hecht and L. H. Hurley, J. Am. Chem. Soc., 2014, 136, 4172–4185 CrossRef CAS PubMed.
- B. Roy, P. Talukder, H.-J. Kang, S. S. Tsuen, M. P. Alam, L. H. Hurley and S. M. Hecht, J. Am. Chem. Soc., 2016, 138, 10950–10962 CrossRef CAS PubMed.
- H. A. Day, P. Pavlou and Z. A. E. Waller, Bioorg. Med. Chem., 2014, 22, 4407–4418 CrossRef CAS PubMed.
- E. P. Wright, H. A. Day, A. M. Ibrahim, J. Kumar, L. J. E. Boswell, C. Huguin, C. E. M. Stevenson, K. Pors and Z. A. E. Waller, Sci. Rep., 2016, 6, 39456 CrossRef CAS PubMed.
- P. V. Jena, P. S. Shirude, B. Okumus, K. Laxmi-Reddy, F. Godde, I. Huc, S. Balasubramanian and T. Ha, J. Am. Chem. Soc., 2009, 131, 12522–12523 CrossRef CAS PubMed.
- S. Weiss, Science, 1999, 283, 1676–1683 CrossRef CAS PubMed.
- A. A. Deniz, M. Dahan, J. R. Grunwell, T. Ha, A. E. Faulhaber, D. S. Chemla, S. Weiss and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3670–3675 CrossRef CAS.
- L. Ying, J. J. Green, H. Li, D. Klenerman and S. Balasubramanian, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14629–14634 CrossRef CAS PubMed.
- T. Ha, Nat. Meth., 2014, 11, 1015–1018 CAS.
- P. S. Shirude, B. Okumus, L. Ying, T. Ha and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 7484–7485 CrossRef CAS PubMed.
- J. Choi and T. Majima, Chem. Soc. Rev., 2011, 40, 5893–5909 RSC.
- J. Choi and T. Majima, Photochem. Photobiol., 2013, 89, 513–522 CrossRef CAS PubMed.
- S. L. Noer, S. Preus, D. Gudnason, M. Aznauryan, J.-L. Mergny and V. Birkedal, Nucleic Acids Res., 2016, 44, 464–471 CrossRef CAS PubMed.
- T. Otosu, K. Ishii and T. Tahara, Nat. Commun., 2015, 6, 7685 CrossRef PubMed.
- A. Tanaka, J. Choi, S. K. Kim and T. Majima, J. Phys. Chem. B, 2013, 117, 6711–6717 CrossRef CAS PubMed.
- M. Debnath, S. Ghosh, D. Panda, I. Bessi, H. Schwalbe, K. Bhattacharyya and J. Dash, Chem. Sci., 2016, 7, 3279–3285 RSC.
- S. Dhakal, J. D. Schonhoft, D. Koirala, Z. Yu, S. Basu and H. Mao, J. Am. Chem. Soc., 2010, 132, 8991–8997 CrossRef CAS PubMed.
- Y. Cui, D. Koirala, H. Kang, S. Dhakal, P. Yangyuoru, L. H. Hurley and H. Mao, Nucleic Acids Res., 2014, 42, 5755–5764 CrossRef CAS PubMed.
- J. Choi, S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2011, 133, 16146–16153 CrossRef CAS PubMed.
- M. Debnath, A. Chauhan, R. Paul and J. Dash, Indian Patent Application No.: 201731028836, 14 August 2017.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- A. H. El-Sagheer and T. Brown, Chem. Sci., 2014, 5, 253–259 RSC.
- A. D. Moorhouse, A. M. Santos, M. Gunaratnam, M. Moore, S. Neidle and J. E. Moses, J. Am. Chem. Soc., 2006, 128, 15972–15973 CrossRef CAS PubMed.
- W. C. Drewe and S. Neidle, Chem. Commun., 2008, 5295–5297 RSC.
- A. D. Moorhouse, S. Haider, M. Gunaratnam, D. Munnur, S. Neidle and J. E. Moses, Mol. BioSyst., 2008, 4, 629–642 RSC.
- E. P. Wright, J. L. Huppert and Z. A. E. Waller, Nucleic Acids Res., 2017, 45, 2951–2959 CrossRef PubMed.
- A. M. Fleming, Y. Ding, R. A. Rogers, J. Zhu, J. Zhu, A. D. Burton, C. B. Carlisle and C. J. Burrows, J. Am. Chem. Soc., 2017, 139, 4682–4689 CrossRef CAS PubMed.
- J.-L. Mergny, Biochemistry, 1999, 38, 1573–1581 CrossRef CAS PubMed.
- H. A. Day, C. Huguin and Z. A. E. Waller, Chem. Commun., 2013, 49, 7696–7698 RSC.
- J. Dai, E. Hatzakis, L. H. Hurley and D. Yang, PLoS ONE, 2010, 5, e11647 Search PubMed.
- D. Monchaud and M.-P. Teulade-Fichou, Methods Mol. Biol., Humana Press, Totowa, NJ, 2010, 257–271 CrossRef CAS.
- Q. Sheng, J. C. Neaverson, T. Mahmoud, C. E. M. Stevenson, S. E. Matthews and Z. A. E. Waller, Org. Biomol. Chem., 2017, 15, 5669–5673 CAS.
- D. D. Le, M. Di Antonio, L. K. M. Chan and S. Balasubramanian, Chem. Commun., 2015, 51, 8048–8050 RSC.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. J. Raffo, H. Perlman, M.-W. Chen, M. L. Day, J. S. Streitman and R. Buttyan, Cancer Res., 1995, 55, 4438–4445 CAS.
- C. Slator, N. Barron, O. Howe and A. Kellett, ACS Chem. Biol., 2016, 11, 159–171 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, characterization data of compounds, 1H NMR and 13C NMR spectra, FRET melting, TO displacement, smFRET, lifetime data, CD spectra, western blot, dual luciferase, caspase assay. See DOI: 10.1039/c7sc02693e |
| ‡ Present address: Department of Chemistry, Indian Institute of Science Education & Research, Bhopal, Bhopal-462066, India. |
| This journal is © The Royal Society of Chemistry 2017 |