
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining the catalytic enantioselective reaction of visible-light-generated radicals with a by-product utilization system†
Xiaoqiang
Huang‡
 a,
Shipeng
Luo‡
a,
Olaf
Burghaus
a,
Richard D.
Webster
b,
Klaus
Harms
a and
Eric
Meggers
*a
a,
Shipeng
Luo‡
a,
Olaf
Burghaus
a,
Richard D.
Webster
b,
Klaus
Harms
a and
Eric
Meggers
*a
aFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, 35043 Marburg, Germany. E-mail: meggers@chemie.uni-marburg.de
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
First published on 1st September 2017
Abstract
We report an unusual reaction design in which a chiral bis-cyclometalated rhodium(III) complex enables the stereocontrolled chemistry of photo-generated carbon-centered radicals and at the same time catalyzes an enantioselective sulfonyl radical addition to an alkene. Specifically, employing inexpensive and readily available Hantzsch esters as the photoredox mediator, Rh-coordinated prochiral radicals generated by a selective photoinduced single electron reduction are trapped by allyl sulfones in a highly stereocontrolled fashion, providing radical allylation products with up to 97% ee. The hereby formed fragmented sulfonyl radicals are utilized via an enantioselective radical addition to form chiral sulfones, which minimizes waste generation.
Introduction
The conversion of prochiral carbon-centered radicals into stereocenters in a catalytic and enantioselective fashion is extremely challenging owing to the inherent high reactivity and conformational flexibility of such radical species.1 Over the past few years, photoinduced electron transfer (PET) has emerged as a powerful tool to access radical species in a mild and economic way,2 thus spurring on the discovery of novel asymmetric catalytic systems involving radical processes.3 However, the catalytic asymmetric PET-processes developed to date mainly deal with achiral radicals reacting with a prochiral C(sp2)-center bound to a chiral catalyst.4 In contrast, strategies for the direct stereocontrol of photo-generated carbon radical centers are underdeveloped.5–7 In this respect, some elegant protocols have been disclosed in enantioselective transformations of photo-generated allylic carbon radicals. In 2013, MacMillan and co-workers introduced a radical–radical recombination process by the combination of a chiral amine and photoredox catalyst in which only one example with moderate enantioselectivity was reported showing the challenge of stereocontrol over such radical intermediates (Fig. 1a).8 After that, Yoon and co-workers described a dual Lewis acid and photoredox catalyst for enantioselective radical [2 + 2] cycloadditions (Fig. 1b).9 Very recently, Melchiorre and co-workers reported that a single chiral iminium ion enabled the enantioselective coupling of β-enaminyl radicals with alkyl radicals (Fig. 1c).10 Despite these significant processes, novel transformations with new mechanisms are still highly desirable.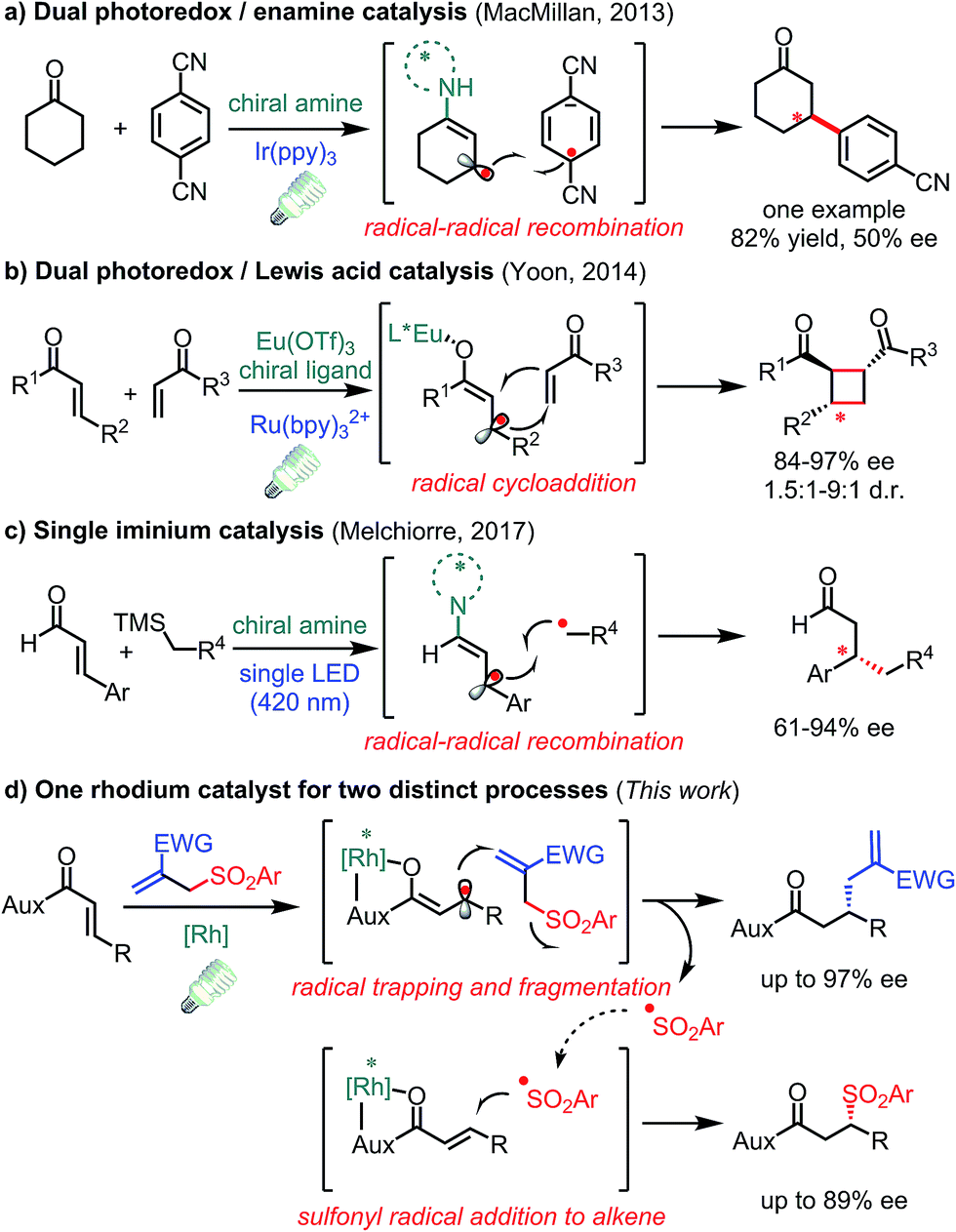 | ||
| Fig. 1 Visible-light-activated asymmetric transformations with allylic C(sp2) radical intermediates. | ||
Herein, we demonstrate that a rhodium-based Lewis acid bearing exclusive metal-centered chirality can effectively control the stereochemistry of visible-light-generated prochiral radicals for the asymmetric radical allylation reaction with allyl sulfones as radical traps (Fig. 1d). Notably, the leaving sulfonyl radicals can be utilized providing β-sulfonyl carbonyl compounds which is without precedence in the chemistry of these well-developed sulfone-based radical trap reagents.11,12
Reaction design
Our design is based on our recently introduced bis-cyclometalated chiral rhodium-based Lewis acids (LAs)13,14 for the electronic activation of substrates through two-point binding and employs Hantzsch esters (HEs)15 as a photoredox mediator for the generation of radical species under mild conditions (Fig. 2). Initially, the bidentate coordination of substrate 1 with the Lewis acid forms the LA/substrate complex A which is a much better electron acceptor than the free substrate 1.14c Therefore, the selective SET reduction of A by the visible-light-excited HE (HE*) generates the key Rh-coordinated radical intermediate B, which is trapped by an electron-deficient allyl sulfone 2 delivering the secondary radical intermediate C. The subsequent fragmentation11 of C provides the sulfonyl radical E and enolate intermediate D, the latter of which yields the C–C bond formation product 3 upon protonation. Meanwhile, the sulfonyl radical E undergoes a stereocontrolled radical addition16 to A in a reversible fashion11 and a subsequent HAT followed by ligand exchange provides the C–S bond formation product 4.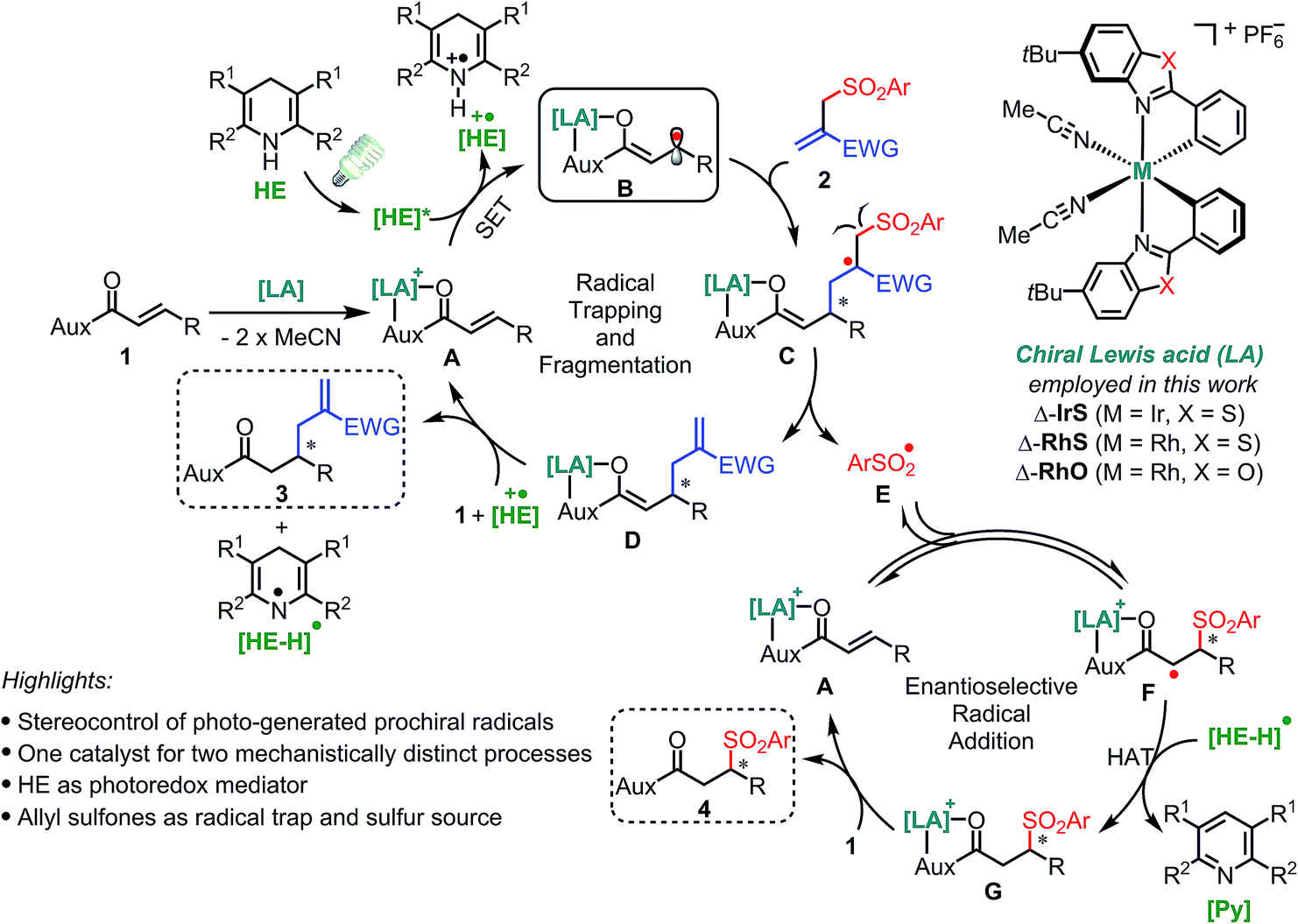 | ||
| Fig. 2 Reaction design and mechanistic proposal. One chiral rhodium catalyst for both enantioselective transformation of a carbon-centered radical and sulfonyl radical addition to an alkene. HE = Hantzsch ester, SET = single electron transfer, HAT = hydrogen atom transfer. | ||
Two key challenges are needed to be solved to achieve a high asymmetric induction. Firstly, a robust and effective chiral Lewis acid catalyst is required to control the stereochemistry of two mechanistically distinct processes as well as reduce the reduction potential of substrate 1 to ensure a highly chemoselective reduction. Secondly, the radical trapping and subsequent fragmentation process should be fast enough to compete with the protonation of intermediate B which would generate undesirable free β-carbonyl carbon radicals. The reaction of such free radicals with 2 would compromise the enantioselectivity of product 3. Therefore, this design with the utilization of the leaving sulfonyl radicals, which otherwise would lead to by-products, is very attractive not only from the perspective of green and sustainable chemistry17 but also for suppressing side reactions of the sulfonyl radical and shifting the equilibrium of a potentially reversible radical fragmentation.11
Results and discussion
We commenced this proposed process by the reaction of α,β-unsaturated N-acylpyrazole 1a with allyl sulfone 2a under visible light irradiation employing a stoichiometric amount of the Hantzsch ester HE-1 as the photoredox mediator and reductant.15 Although our well-established iridium catalyst Δ-IrS18 could not give any detectable product (Table 1, entry 1), the rhodium analog Δ-RhS14b enabled the transformation affording the expected allylation product 3a and C–S formation product 4a in excellent yields with moderate enantioselectivities (entry 2). Interestingly, the related Δ-RhO14a,19 provided 3a in much higher ee (96% ee, entry 3) indicating that the mechanism differs from our previous reports14c,d about Giese-type radical reactions in which RhS, which features a higher steric congestion, works better than RhO. Notably, our recently developed chiral rhodium complexes show a unique reactivity for this reaction. Other Lewis acids such as Sc(OTf)3 gave very low efficiency while LiBF4 could not even catalyze the process (entries 4 and 5) and no conversion was observed without catalyst (entry 6). Other substituted HE species also worked very well (entries 7 and 8), whereas DIPEA, which is widely used as a sacrificial reductant in photoredox catalysis, could not accomplish the transformation (entry 9). These results highlight the multiple functions of the HE in our system acting as a photoredox mediator as well as an electron donor and proton source. Furthermore, on illumination with blue LEDs, which do not emit any UV light (see ESI for the emission spectra†), the reaction gave comparable results (entry 10). Together with the control experiments in darkness (entry 11), this confirms that the reaction is activated by visible light.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Lewis acidb | HE | 3a | 4a | ||
| Yieldc (%) | eed (%) | Yieldc (%) | eed (%) | |||
| a Reaction conditions: 1a (0.20 mmol), 2a (0.10 mmol), Lewis acid and Hantzsch ester (0.15 mmol) in 1,4-dioxane (1.0 mL) were stirred at room temperature for 24 h and irradiated with a 21 W CFL. b Loadings in mol% provided in brackets. c Isolated yields. d Determined by HPLC on a chiral stationary phase. e 0.15 mmol of N,N-diisopropylethylamine (DIPEA) was employed. f Blue LEDs (24 W) were used instead of a CFL (21 W). g Performed in darkness. n.a. = not applicable. | ||||||
| 1 | Δ-IrS (8.0) | HE-1 | <5 | n.a. | <5 | n.a. |
| 2 | Δ-RhS (8.0) | HE-1 | 92 | 83 | 95 | 84 |
| 3 | Δ-RhO (8.0) | HE-1 | 85 | 96 | 92 | 85 |
| 4 | Sc(OTf)3 (20) | HE-1 | 10 | n.a. | 10 | n.a. |
| 5 | LiBF4 (200) | HE-1 | 0 | n.a. | 0 | n.a. |
| 6 | None | HE-1 | 0 | n.a. | 0 | n.a. |
| 7 | Δ-RhO (8.0) | HE-2 | 78 | 96 | 82 | 80 |
| 8 | Δ-RhO (8.0) | HE-3 | 80 | 94 | 85 | 85 |
| 9e | Δ-RhO (8.0) | None | 0 | n.a. | 0 | n.a. |
| 10f | Δ-RhO (8.0) | HE-1 | 77 | 92 | 80 | 80 |
| 11g | Δ-RhO (8.0) | HE-1 | 0 | n.a. | 0 | n.a. |

With the optimized conditions at hand, we next investigated the substrate scope with respect to radical acceptors (Table 2). A wide range of allyl sulfones 2 with different leaving groups works well, delivering the radical allylation product 3a in good yields and excellent ee (up to 97% ee) along with the recycled C–S formation products 4a–h in good yields and ee (up to 89% ee) (entries 1–8). Intriguingly, a lower yield and slightly lower ee were observed for the radical functionalization product 3b when allyl sulfone bearing a less electron deficient ester group was employed (compare entries 9 with 1). It is noteworthy that functional groups including a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond, a C
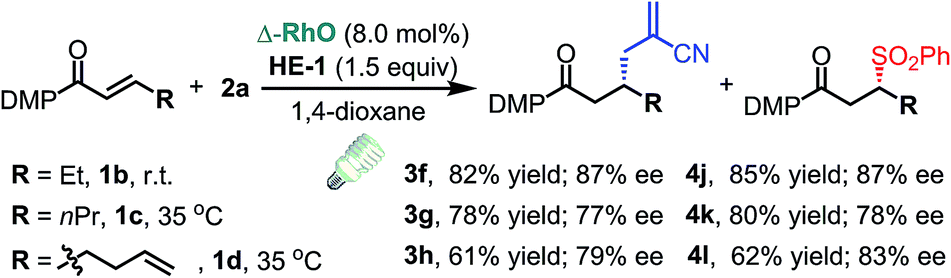
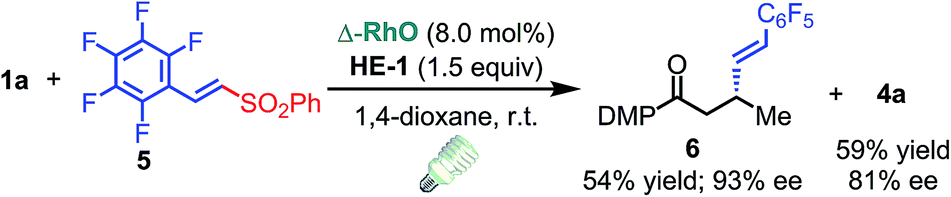
C triple bond, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and an imide are well tolerated under these mild conditions (entries 11–13). As a limitation, substrates with a long chain at the β-position (1b–d) produced the radical allylation products 3f–h with decreased ee (eqn (1)) and β-aryl α,β-unsaturated N-acylpyrazole could not afford any expected product. Furthermore, the alkenyl sulfone 5 was proven to be competent, providing the radical alkenylation product 6 in 54% yield with 93% ee (eqn (2)).
C double bond and an imide are well tolerated under these mild conditions (entries 11–13). As a limitation, substrates with a long chain at the β-position (1b–d) produced the radical allylation products 3f–h with decreased ee (eqn (1)) and β-aryl α,β-unsaturated N-acylpyrazole could not afford any expected product. Furthermore, the alkenyl sulfone 5 was proven to be competent, providing the radical alkenylation product 6 in 54% yield with 93% ee (eqn (2)).
|
|
||||
|---|---|---|---|---|
| Entry | EWG | Ar | 3, Yieldb, eec | 4, Yieldb, eec |
| a Reaction conditions: 1a (0.20 mmol), 2 (0.10 mmol), Δ-RhO (0.008 mmol) and HE-1 (0.15 mmol) in 1,4-dioxane (1.0 mL) were stirred at room temperature and irradiated with a 21 W CFL. b Isolated yields. c Determined by HPLC on a chiral stationary phase. d 35 °C. | ||||
| 1 | CN | C6H5 | 3a, 85%, 96% ee | 4a, 92%, 85% ee |
| 2 | CN | 4-MeC6H4 | 3a, 68%, 96% ee | 4b, 70%, 79% ee |
| 3 | CN | 4-BrC6H4 | 3a, 81%, 97% ee | 4c, 88%, 80% ee |
| 4 | CN | 4-CF3C6H4 | 3a, 78%, 95% ee | 4d, 78%, 76% ee |
| 5 | CN | 2-MeC6H4 | 3a, 71%, 95% ee | 4e, 72%, 86% ee |
| 6d | CN | 2,4,6-Me3C6H2 | 3a, 57%, 94% ee | 4f, 60%, 89% ee |
| 7d | CN | 2-Naphthyl | 3a, 82%, 94% ee | 4g, 88%, 83% ee |
| 8d | CN | 1-Naphthyl | 3a, 78%, 91% ee | 4h, 84%, 80% ee |
| 9 | COOEt | C6H5 | 3b, 65%, 94% ee | 4a, 68%, 84% ee |
| 10d | COOEt | 4-MeOC6H4 | 3b, 65%, 92% ee | 4i, 63%, 81% ee |
| 11d |
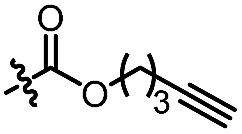
|
C6H5 | 3c, 60%, 92% ee | 4a, 69%, 82% ee |
| 12d |
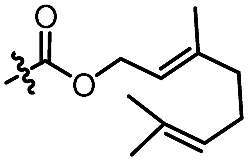
|
C6H5 | 3d, 62%, 92% ee | 4a, 72%, 83% ee |
| 13d |

|
C6H5 | 3e, 73%, 92% ee | 4a, 78%, 82% ee |

| (1) |
| (2) |
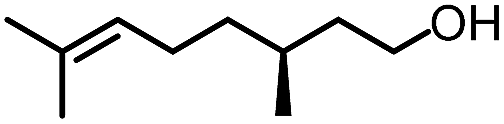
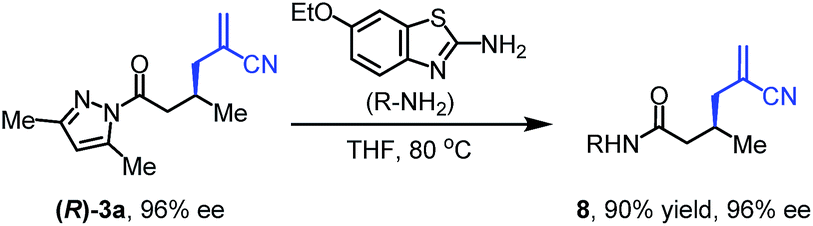
To further evaluate the functional group tolerance and robustness of this catalytic system, we conducted the reaction 1a + 2a → 3a + 4a in the presence of a series of common chemical functionalities (Table 3 and ESI†).20 To our satisfaction, this reaction shows a high chemo-selectivity towards the Lewis acid coordinated substrate (intermediate A) as additives containing azido, cyano, and carbonyl groups that are vulnerable to reductive conditions can be recovered in high yields under the standard conditions (entries 1–4). Importantly, several heterocycles which might competitively coordinate to the catalyst did not erode the enantiomeric excess of products (entries 3–9). Several natural products, including coumarin, caffeine, and (−)-citronellol, were found to have little influence on the reaction outcomes (entries 7–10). Furthermore, N-acylpyrazoles known as a useful and reactive synthetic building block can be easily converted into other compounds, such as alcohols or amides (eqn (3) and (4)). Overall, these results highlight the potential of this protocol for further applications in the synthesis of complex molecules.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | Additive recoveredb | (R)-3a | (S)-4a | ||
| Yieldb | eec | Yieldb | eec | |||
| a Reaction conditions: 1a (0.20 mmol), 2a (0.10 mmol), Λ-RhO (0.008 mmol), HE-1 (0.15 mmol) and additives (0.10 mmol) in 1,4-dioxane (1.0 mL) were stirred at room temperature for 24 h and irradiated with a 21 W CFL. b Isolated yields. c Determined by HPLC on a chiral stationary phase. | ||||||
| 1 |
|
88% | 86% | 96% ee | 85% | 86% ee |
| 2 |
|
94% | 86% | 96% ee | 85% | 83% ee |
| 3 |

|
99% | 86% | 96% ee | 92% | 83% ee |
| 4 |
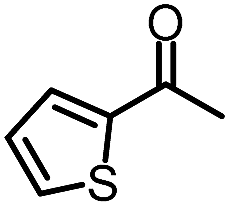
|
88% | 82% | 96% ee | 88% | 83% ee |
| 5 |

|
95% | 86% | 95% ee | 92% | 85% ee |
| 6 |
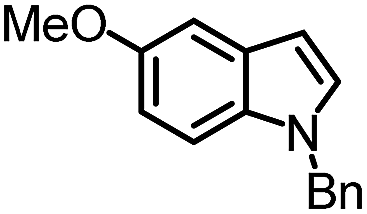
|
99% | 86% | 96% ee | 85% | 83% ee |
| 7 |
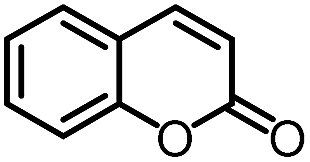
|
96% | 82% | 96% ee | 92% | 85% ee |
| 8 |

|
99% | 82% | 95% ee | 92% | 83% ee |
| 9 |
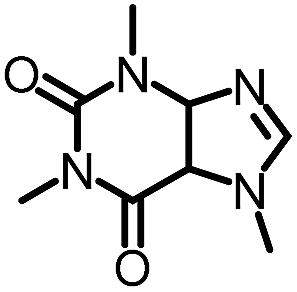
|
99% | 86% | 96% ee | 92% | 85% ee |
| 10 |
|
94% | 86% | 96% ee | 92% | 85% ee |
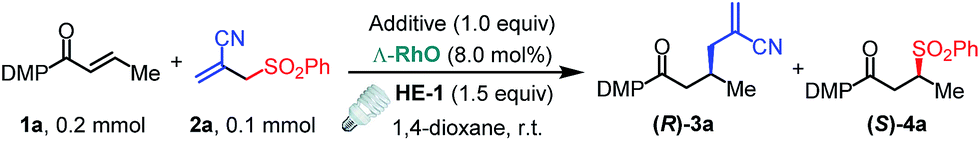
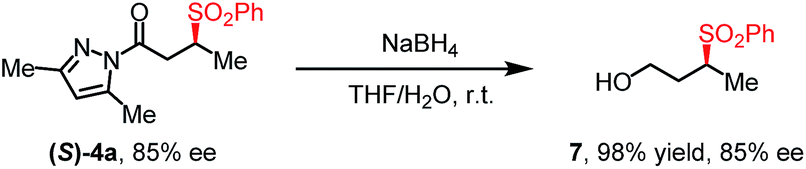
| (3) |
| (4) |
A number of experiments support the proposed mechanism (Fig. 2). Firstly, UV/vis spectra and luminescence spectra support the role of the HE as a visible light harvesting antenna being consistent with recent reports (Fig. S3 in ESI†).15 Secondly, Stern–Volmer quenching experiments demonstrate that the rhodium coordinated substrate RhO-1a21 but not the free substrates 1 or 2 can effectively quench the luminescence of HE-1, suggesting that a selective SET process between the photoexcited HE* and intermediate A might occur (Fig. S4 and S5 in ESI†). This is further confirmed by cyclic voltammetry, in which a large difference in the reduction peak potential by almost 1 V was observed between 1a (−2.59 V vs. Fc/Fc+) and RhO-1a (−1.62 V vs. Fc/Fc+), thus making highly selective SET between RhO-1a and the excited state of HE-1 (E(HE˙+/HE*) = −2.23 V vs. Fc/Fc+) feasible (Fig. S6 in ESI†). Furthermore, the title reaction was greatly inhibited upon adding 2,6-di-tert-4-methylphenol (BHT) or 2,2,6,6-tetramethyl-piperidinooxy (TEMPO) as radical scavengers. When the reaction was monitored by electron paramagnetic resonance (EPR) using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a free radical spin-trapping agent, mixed signals containing two radical species were observed, one of which was identified as a phenyl sulfonyl radical (g = 2.006, αN = 9.5 G, αβH = 12.9 G) (Fig. S8 in ESI†).22 Finally, a quantum yield of 0.09 was determined for the reaction 1a + 2a → 3a + 4a, which is consistent with the proposed mechanism being devoid of any chain process (see ESI for details†).
Conclusions
In conclusion, we here have introduced an unusual reaction scheme in which a chiral rhodium complex enables the catalytic enantioselective functionalization of a photo-generated carbon radical employing cheap and readily available Hantzsch esters as a photoredox mediator and reductant. Intriguingly, in this radical allylation reaction using allyl sulfones as reagents, the generated sulfonyl radical by-product can be trapped by electron deficient alkenes and transformed into valuable enantioenriched S-containing building blocks thereby minimizing waste generation. The simple reaction setup and the mild reaction conditions as well as the demonstrated compatibility with a wide range of functionalities render this robust catalytic system an appealing process. Further investigations on the stereocontrolled chemistry of prochiral radicals are ongoing in our laboratory.23Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for funding from the Deutsche Forschungsgemeinschaft (ME 1805/13-1).Notes and references
- (a) D. P. Curran, N. A. Porter and B. Giese, Stereochemistry of Radical Reactions, VCH, Weinheim, 1996 Search PubMed; (b) M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263 CrossRef CAS PubMed; (c) J. Zimmerman and M. P. Sibi, Top. Curr. Chem., 2006, 263, 107 CrossRef CAS; (d) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- For selected reviews on PET, see: (a) G. Pandey, Top. Curr. Chem., 1993, 168, 175 CrossRef CAS; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (d) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874 CrossRef CAS PubMed.
- For recent catalytic asymmetric PET processes, see: (a) A. G. Amador, E. M. Sherbrook and T. P. Yoon, J. Am. Chem. Soc., 2016, 138, 4722 CrossRef CAS PubMed; (b) X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636 CrossRef CAS PubMed; (c) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS PubMed; (d) E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679 CrossRef CAS PubMed; (e) Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694 CrossRef CAS PubMed; (f) G. Filippini, M. Silvi and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 4447 CrossRef CAS PubMed.
- For a review, see: R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed.
- For asymmetric nickel catalysis, see: (a) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed; (b) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS PubMed.
- For asymmetric copper catalysis, see: Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681 CrossRef CAS PubMed.
- For chiral Brønsted/Lewis acid catalysis, see: (a) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735 CrossRef CAS PubMed; (b) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768 CrossRef CAS PubMed; (c) C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685 CrossRef CAS PubMed; (d) T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953 CrossRef CAS PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef CAS PubMed.
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868 CrossRef CAS PubMed.
- For a review on the reaction of sulfonyl radicals, see: M. P. Bertrand, Org. Prep. Proced. Int., 1994, 26, 257 CrossRef CAS.
- For selected reports on sulfone-based radical traps with the leaving of sulfonyl radicals as waste, see: (a) B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 1996, 118, 1209 CrossRef CAS; (b) A.-P. Schaffner and P. Renaud, Angew. Chem., Int. Ed., 2003, 42, 2658 CrossRef CAS PubMed; (c) H. Zhang, E. B. Hay, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 16610 CrossRef CAS PubMed; (d) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602 CrossRef CAS PubMed; (e) J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872 CrossRef CAS PubMed; (f) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312 CrossRef CAS PubMed; (g) Y. Zhu, X. Wen, S. Song and N. Jiao, ACS Catal., 2016, 6, 6465 CrossRef CAS.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320 CrossRef CAS PubMed.
- (a) C. Wang, L.-A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094 RSC; (b) J. Ma, X. Shen, K. Harms and E. Meggers, Dalton Trans., 2016, 45, 8320 RSC; (c) H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936 CrossRef CAS PubMed; (d) C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495 CrossRef CAS PubMed.
- For PET with direct excitation of the Hantzsch ester, see: (a) X.-Q. Zhu, Y.-C. Liu and J.-P. Cheng, J. Org. Chem., 1999, 64, 8980 CrossRef CAS; (b) J. Jung, J. Kim, G. Park, Y. You and E. J. Cho, Adv. Synth. Catal., 2016, 358, 74 CrossRef CAS; (c) W. Chen, H. Tao, W. Huang, G. Wang, S. Li, X. Cheng and G. Li, Chem.–Eur. J., 2016, 22, 9546 CrossRef CAS PubMed.
- For a review, see: (a) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753 CrossRef; For selected asymmetric examples, see: (b) M. P. Sibi, J. Ji, J. H. Wu, S. Gürtler and N. A. Porter, J. Am. Chem. Soc., 1996, 118, 9200 CrossRef CAS; (c) M. P. Sibi and J. Ji, J. Org. Chem., 1997, 62, 3800 CrossRef CAS; (d) L. R. Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452 CrossRef PubMed.
- B. M. Trost, Science, 1991, 254, 1471 CAS.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed.
- We modified the resolution process of RhO using a chiral auxiliary, namely (R)-3-fluoro-2-(4-phenyl-4,5-dihydrooxazol-2-yl)phenol, instead of a chiral proline. The corresponding rhodium-auxiliary complexes are stable and both could be obtained by flash chromatography, thus improving the atom economy of the catalyst synthesis (see ESI for details†).
- For selected applications of a robustness screen, see: (a) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed; (b) K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619 CrossRef CAS PubMed.
- We determined a very rapid substrate binding to bis-cyclometalated rhodium-based Lewis acids in our previous work. See: X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120 CrossRef CAS PubMed ; and ref. 14a.
- (a) V. Cholvad, K. Szaboova, A. Staško, O. Nuyken and B. Voit, Magn. Reson. Chem., 1991, 29, 402 CrossRef CAS; (b) H. Wang, Q. Lu, C.-W. Chiang, Y. Luo, J. Zhou, G. Wang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 595 CrossRef CAS PubMed.
- For examples, see: (a) Z. Zhou, Y. Li, B. Han, L. Gong and E. Meggers, Chem. Sci., 2017, 8, 5757 RSC; (b) W. Yuan, Z. Zhou, L. Gong and E. Meggers, Chem. Commun., 2017, 53, 8964 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. CCDC 1547314–1547316. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02621h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |