
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular substitution uncages fluorogenic probes for detection of metallo-carbapenemase-expressing bacteria†
Aiguo
Song
 a,
Yunfeng
Cheng
a,
Jinghang
Xie
a,
Niaz
Banaei
b and
Jianghong
Rao
*a
a,
Yunfeng
Cheng
a,
Jinghang
Xie
a,
Niaz
Banaei
b and
Jianghong
Rao
*a
aMolecular Imaging Program at Stanford, Departments of Radiology and Chemistry, Stanford University, 1201 Welch Road, Stanford, CA 94305-5484, USA. E-mail: jrao@stanford.edu
bDepartment of Pathology, Clinical Microbiology Laboratory, Stanford Hospital and Clinics, Palo Alto, CA 94304, USA
First published on 21st September 2017
Abstract
This work reports a novel caging strategy for designing fluorogenic probes to detect the activity of β-lactamases. The caging strategy uses a thiophenyl linker connected to a fluorophore caged by a good leaving group—dinitrophenyl. The uncaging proceeds in two steps through the sulfa-releasing and subsequent intramolecular substitution. The length of the linker has been examined and optimized to maximize the rate of intramolecular reaction and thus the rate of fluorescence activation. Finally based on this strategy, we prepared a green fluorogenic probe CAT-7 and validated its selectivity for detecting metallo-carbapenemases (VIM-27, IMP-1, NDM-1) in carbapenem-resistant Enterobacteriaceae (CRE) lysates.
Introduction
Fluorescent probes have been widely used for bioanalytical and bioimaging applications because of high sensitivity, ease of use and low expense.1–4 Among them fluorogenic probes offer the feature of signal “turn-on” in contact with the analyte without the need to remove or wash off the unreacted probes, and are thus particularly attractive.5 A number of strategies have been reported to develop fluorogenic probes, such as the popular fluorescence resonance energy transfer (FRET) mechanism,6–9 the modulation of the pi-conjugation10–12 or conformation13 of the fluorophore, and photo electron transfer.14–16 Another common strategy is to chemically cage the fluorophore with a group that can be specifically removed by the analyte (Fig. 1).17 The caging group may be directly coupled to the fluorophore and provide the “off-state” probe. When this direct caging strategy cannot be implemented or a fluorogenic probe with the direct caging performs poorly, for example, slow kinetics due to the steric effect of the bulky fluorophore, a linker is often designed to connect the two units to facilitate the design. Such linkers are designed to be immolative to enable the generation of the free fluorophore and restoration of the fluorescence emission. In this work, we report a new linker design for indirect caging of fluorophore: instead of self-immolation, the fluorophore regeneration proceeds through intramolecular substitution reaction after uncaging. In demonstrating this design, we successfully applied it to develop turn-on fluorogenic probes for detecting β-lactamases. | ||
| Fig. 1 Comparison of direct and indirect caging strategies. | ||
β-Lactamases are a class of bacterial hydrolase responsible for the microbial resistance to lactam antibiotics through enzymatic degradation of the lactam rings.18 Since the first β-lactamase was discovered in 1940,19 hundreds of β-lactamases have been identified. Among them, carbapenemases20 can hydrolyse broad-spectrum carbapenem antibiotics that are often considered the last option for effective therapy of infections with resistant organisms, and are responsible for the emergence of carbapenem-resistant Enterobacteriaceae (CRE)21 over the past decade. CRE infection has left clinicians with few treatment options, and the global spread of CRE has created a serious threat to public health.
Carbapenemases have been classified into two major groups based on their active sites: (i) serine carbapenemases belonging to the class A penicillinases and class D oxacillinases, which possess a serine residue active-site as a nucleophile to cleave the β-lactam ring; and (ii) metallo-β-lactamases (MBLs) belonging to the class B carbapenemases, which require a Zn2+–OH for nucleophilic attack on the β-lactam bond.21–23 Among the members of the metallo-carbapenemase family, Verona integron-encoded MBLs (VIM), “active on imipenem” (IMP), and New Delhi metallo-β-lactamase-1 (NDM-1) are the most common.21,24 Antibiotic resistance due to acquired metallo-carbapenemase-expressing bacteria is considered to be more serious because of their strong hydrolysis ability and rapid dissemination.25 Inexpensive and simple operational fluorescent carbapenemase imaging assay is thus requisite to monitor the occurrence of carbapenemase-expressing bacteria.
There have been a number of fluorescent probes reported for the detection of β-lactamases based on cephalosporin where fluorophores are often attached to the 3′ carbon position.26–33 The hydrolysis of the lactam ring by β-lactamases results in the release of attached fluorophores through fragmentation. In designing specific carbapenemase-responsive probes, carbapenem has been used as recognition moiety and conjugated a fluorophore BODIPY via an alkenyl linkage, though it is unclear whether this approach can be generalized for other fluorophores.34 We have previously converted the R configuration of C7 in the cephalosporin to S,35 and directly attach a blue fluorophore coumarin to the 3′ position. Herein, we conceptualized an indirect caging strategy using an intramolecular substitution and designed a probe specific for metallo-carbapenemases like IMP-1, VIM-27 and NDM-1 with green fluorescent emission.
Results and discussion
We began our caging strategy design from the observations that many of the fluorophores carry a phenolic group and that caged phenolic groups lead to the decrease, sometimes even loss, of fluorescence emission. The direct coupling of the phenolic group of the fluorophore to the 3′ position of cephalosporin affords an allyl phenyl ether structure, which is prone to spontaneous hydrolysis in water. In comparison, allyl phenyl thioether would offer better stability. However, in order to take advantage of the stability of allyl phenyl thioether in the linker, a fluorophore-releasing strategy would have to be in place since the thiophenyl group is generally not present in fluorophores. We envision a design that the released thiophenyl group can act through an intramolecular substitution reaction to produce fluorescence activation. In this design, the fluorophore caging takes place through a separate group, unlike in the direct caging strategy that uses the linker to cage the fluorophore. Fig. 2 shows examples of such a design based on cephalosporin with a thiophenyl linker to a green fluorescence emission fluorophore Tokyo green caged by dinitrophenyl arylation. The choice of dinitrophenyl caging is motivated by reported high efficiency in fluorescence quenching and stability in water toward endogenous nucleophiles such as cysteine and glutathione (GSH).36–38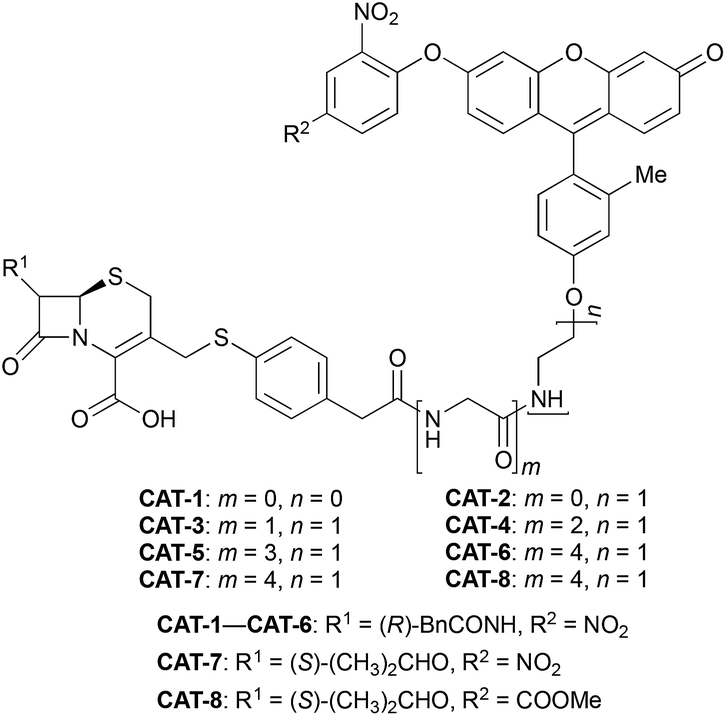 | ||
| Fig. 2 Structure of cephalosporin-arylated Tokyo green probes. | ||
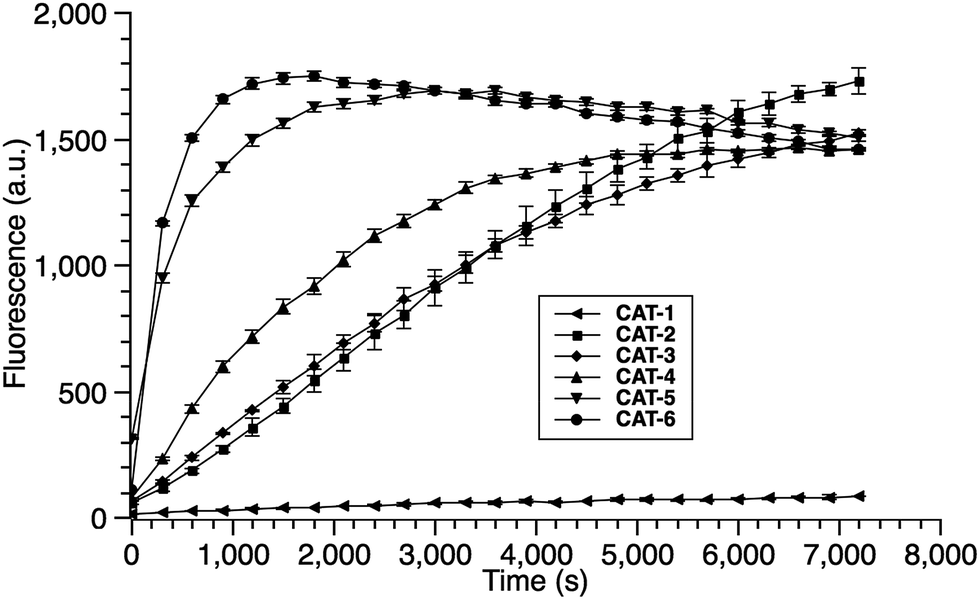
Cephalosporin-arylated Tokyo green (CAT) probes (Fig. 2) were prepared by conjugating arylated Tokyo green to cephalosporin through a thiophenyl linker (Scheme 1). Probe CAT-1 was obtained by direct conjugation of thiophenylacetic acid to arylated Tokyo green and cephalosporin. As expected, CAT-1 was slowly responsive upon treatment with 20 nM or even 200 nM BlaC to produce only 2-3-fold increase in fluorescence emission in the phosphate-buffered saline (PBS, pH = 7.4), likely due to the short linker that prevents intramolecular substitution from occurring (Fig. 3 and S1†). To our delight, the kinetics was significantly improved when probe CAT-2 bearing a longer linkage was tested under the same condition to give a 19-fold of increase in fluorescence emission within 2 h (Fig. 3 and S1†).
 | ||
| Scheme 1 Synthesis of cephalosporin-arylated Tokyo green probes and conditions: (i) 2,4-dinitro-1-fluorobenzene, K2CO3, DMF, rt, 1 h; (ii) TFA, TIPS, DCM, rt, 3 h; (iii) Boc-(Gly)m-OH, HBTU, DIPEA, DMF, rt, 1 h; (iv) 4-mercaptophenylacetic acid, DIPEA, DCM, rt, 0.5 h; (v) B or C, HBTU, DIPEA, DMF, rt, 1 h; (vi) TFAA, NaI, acetone, −20 °C, 2 h. | ||
 | ||
| Fig. 3 Time dependence of CAT probes (10 μM in PBS, pH = 7.4) upon incubation with BlaC (20 nM) at 22 °C. Fluorescence data were collected with excitation at λ = 490 nm (bandwidth 5 nm) and emission at λ = 510 nm (bandwidth 5 nm). Error bars are ± SD. | ||
We further examined the correlation between enzymatic reaction rate and the linker length and synthesized probes CAT-3 to CAT-6. It was found that probes bearing a longer linker generally showed faster fluorescence enhancement upon the treatment of β-lactamase (Fig. 3 and S1†). For example, treatment of CAT-6 with 20 nM BlaC in PBS (pH = 7.4) at 22 °C yielded a maximal fluorescence increase within 20 min. CAT-5 was slightly slower in reaching the maximal fluorescence increase than CAT-6, and both were much faster than CAT-4 under the same condition. These observations were confirmed by the catalytic efficiency data of the CAT probes summarized in Table S1.† Moreover, these cephalosporin-arylated Tokyo green scaffolds were proved to be stable in PBS (pH = 7.4) at 22 °C within 24 h (kuncat ≈ 10−8 s−1).
Based on these results, we selected CAT-6 as the design for metallo-carbapenemase detection. In addition to the conversion of the R configuration of C7 in the cephalosporin to S, an isopropyloxy substitution was incorporated to the 7-position to produce metallo-carbapenemase specific probe CAT-7 (Fig. 2). The enzymatic activation process of CAT-7 by metallo-carbapenemase IMP-1 was analysed by plate reader, fluorometer and HPLC (Fig. 4–6), revealing the formation of the expected product CAT-7-P confirmed by HRMS (Fig. S2†). Probe CAT-7 showed a typical broad absorption band at 460 nm; upon treatment with IMP-1, the absorption peak shifted to 490 nm, and its fluorescent intensity around 510 nm increased by 37-fold in PBS (pH = 7.4) at 22 °C within 1 h (Fig. 6).
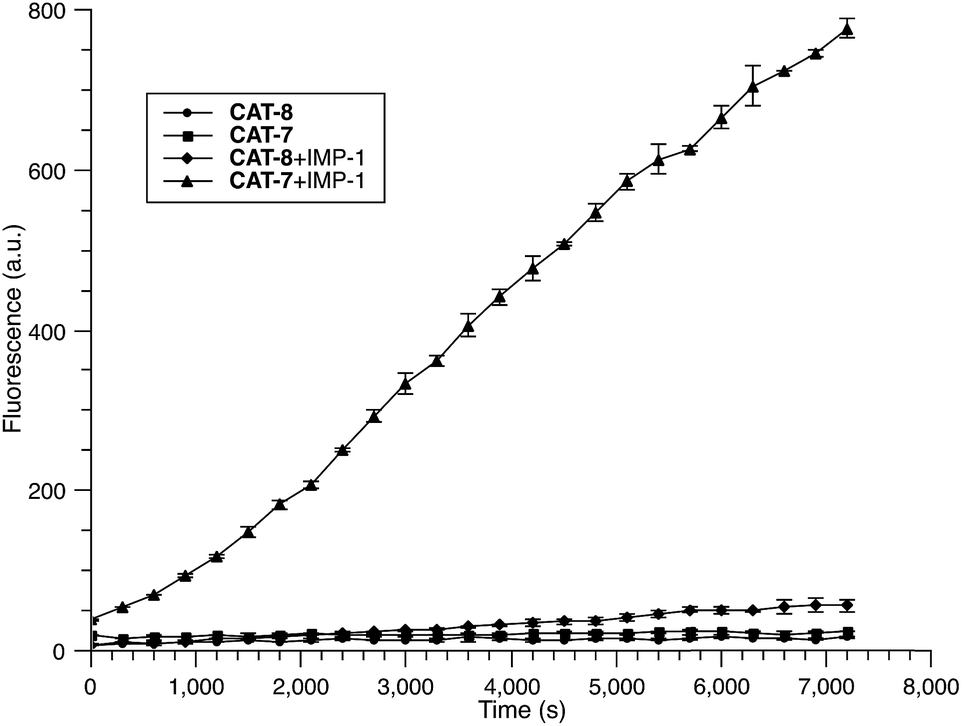 | ||
| Fig. 4 Time-dependent fluorescence response of CAT-7 (10 μM) and CAT-8 (10 μM) upon incubation with IMP-1 (0.3 nM) in PBS (pH = 7.4) at 22 °C. Fluorescence intensity was measured on a plate reader with excitation at λ = 490 nm (bandwidth 5 nm) and emission at λ = 510 nm (bandwidth 5 nm). Error bars are ± SD. | ||
 | ||
| Fig. 5 HPLC traces of CAT-7 (5 μM) in the absence or presence of IMP-1 (5 nM) in PBS (pH = 7.4) at 22 °C for 2 h. | ||
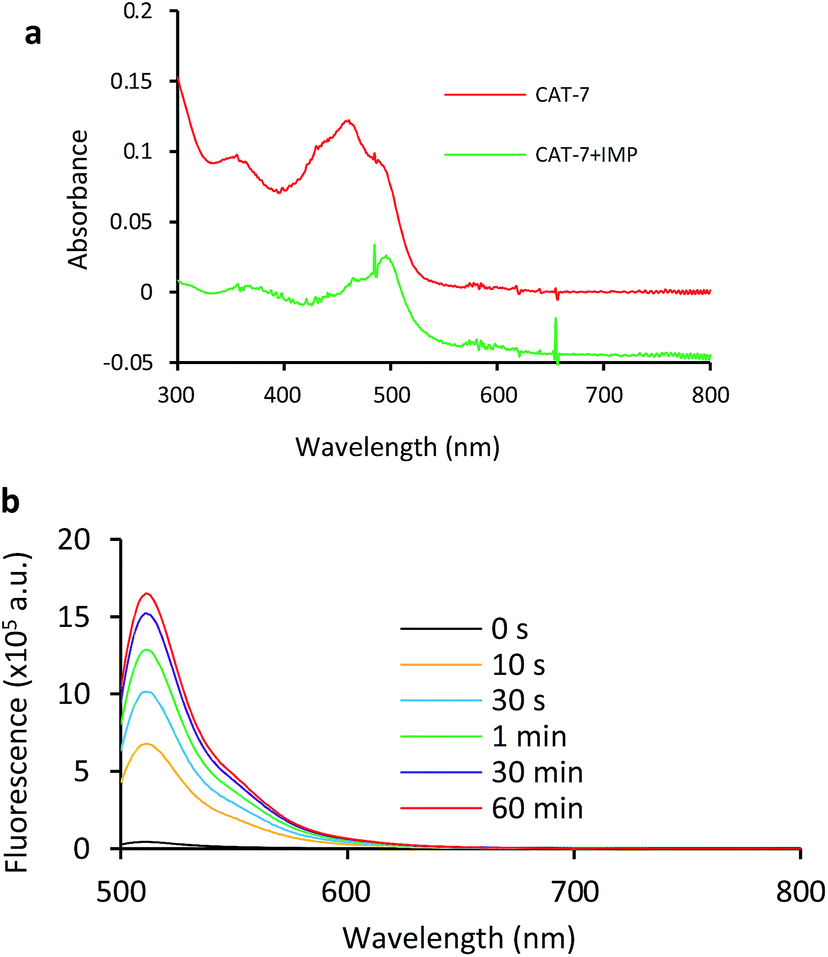 | ||
| Fig. 6 UV-vis (a) and fluorescence (b) spectra of CAT-7 incubated with IMP-1 (2.5 nM) for 2 h in PBS (pH = 7.4) at 22 °C. (a) CAT-7 (10 μM); (b) CAT-7 (1 μM). Fluorescence data were collected with excitation at λ = 490 nm and emission at λ = 510 nm on a fluorometer. | ||
To further validate the intramolecular substitution mechanism, we replaced the dinitrophenyl caging group by a mono-nitrophenyl group to make probe CAT-8. This substitution would expect to affect the intramolecular reaction due to the enhanced stability of the mono-nitrophenyl ether. As shown in Fig. 4, CAT-8 displayed a much lower fluorescent response than CAT-7 and yielded just 3-fold increase in fluorescence emission within 2 h.
The specificity of CAT-7 was investigated by measuring the fluorescence response after exposure to a variety of β-lactamases in PBS (pH = 7.4) at 22 °C. As shown in Fig. 7, there is a concentration-dependent fluorescent response to metallo-carbapenemases VIM-27, IMP-1 and NDM-1 but not other β-lactamases. These results demonstrated the excellent selectivity of CAT-7 for metallo-carbapenemases over other β-lactamases. Treatment of CAT-7 with 5 nM IMP-1 in aqueous buffer (PBS, pH 7.4) resulted in a 77-fold increase in fluorescence over 2 h (Fig. 7a). The kinetic parameters of CAT-7 upon incubation with IMP-1, VIM-27 and NMD-1 were determined (Table 1). In comparison to the coumarin based CRE probe (S)-CC-3,35 the kinetic efficiency for IMP-1, VIM-27 and NDM-1 is similar, but the chemical stability of CAT-7 was significantly improved (the spontaneous hydrolysis rate decreased by 30-fold).
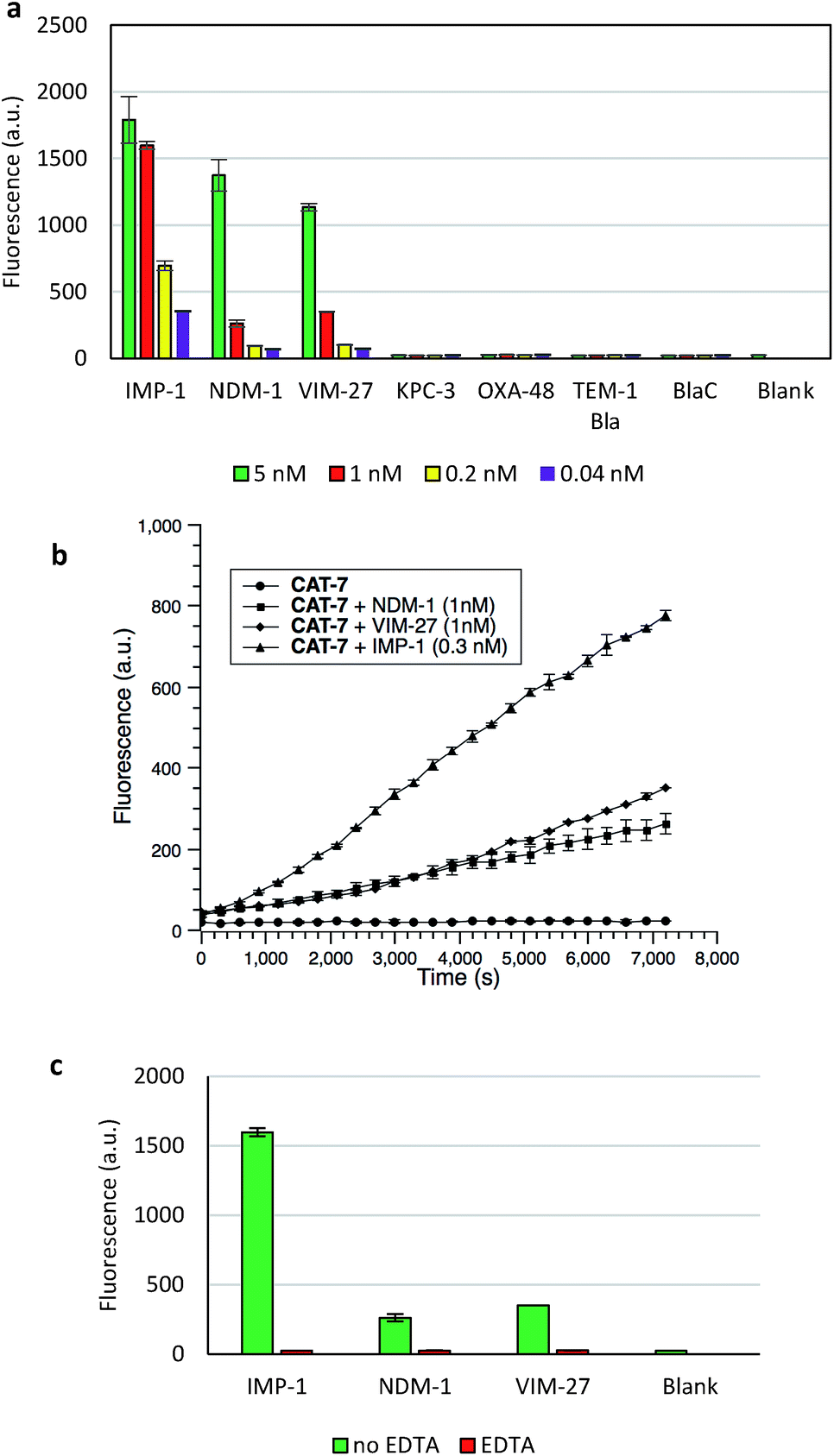 | ||
| Fig. 7 Fluorescent assay of metallo-carbapenemases by CAT-7 (10 μM). (a) Fluorescence response of CAT-7 upon incubation with various concentrations of indicated β-lactamases within 2 h in PBS (pH 7.4) at 22 °C. (b) Time dependence of CAT-7 in the absence or presence of VIM-27, IMP-1 and NDM-1. (c) Inhibition of VIM-27, IMP-1 or NDM-1 (1 nM) by EDTA (100 μM) upon incubation with CAT-7. Fluorescence intensity was measured on a plate reader with excitation at λ = 490 nm (bandwidth 5 nm) and emission at λ = 510 nm (bandwidth 5 nm). Error bars are ± SD. | ||
| Name | VIM-27 | IMP-1 | NDM-1 | Spontaneous | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K m [μM] | k cat [s−1] | k cat/Km [×104 s−1 M−1] | K m [μM] | k cat [s−1] | k cat/Km [×105 s−1 M−1] | K m [μM] | k cat [s−1] | k cat/Km [×104 s−1 M−1] | Hydrolysis rate [×10−8 s−1] | |
| a Kinetic data were measured in PBS buffer (100 mM, pH = 7.4) at room temperature (22 °C). Fluorescence intensity was measured on a plate reader with excitation at λ = 490 nm (bandwidth 5 nm) and emission at λ = 510 nm (bandwidth 5 nm). All data indicate averages of three replicate experiments. | ||||||||||
| CAT-7 | 13.0 ± 2.5 | 0.8 ± 0.01 | 6.4 ± 1.1 | 52.7 ± 23 | 10.3 ± 3.5 | 2.0 ± 0.22 | 8.8 ± 0.73 | 0.4 ± 0.16 | 4.5 ± 0.23 | 1.5 ± 0.1 |
To confirm that the fluorescence enhancement was dependent on the metallo-carbapenemase catalysed process, EDTA was added to inhibit the enzyme activity (Fig. 7c). No detectable signal increase was observed after 2 h, confirming that the fluorescence increase is directly related to the activity of metallo-carbapenemases. Furthermore, the inhibition of IMP-1 with a thiol analogue captopril (100 μM) was also investigated and confirmed using probe CAT-7, as shown in Fig. S4.†
Finally, we evaluated if CAT-7 could detect metallo-carbapenemases in clinical bacteria samples. As shown in Fig. 8, a variety of bacterial lysates (4 × 105 cfu in 100 μL PBS, pH = 7.4) was incubated with CAT-7 for 2 hours, and bacteria samples VIM-27, IMP-1, and NDM-1 expressing showed a greater enhancement in fluorescence upon incubation with over control β-lactamases, demonstrating the ability of CAT-7 to detect metallo-carbapenemases in clinical CRE strains. As a comparison, commercially available fluorogenic probe Fluorocillin showed fluorescence enhancement when incubated with all bacterial strains without any specificity (Fig. S5†).
 | ||
| Fig. 8 Fluorescence response of CAT-7 (10 μM) upon incubation with various bacteria lysates (4 × 105 cfu in 100 μL PBS, pH = 7.4) at 22 °C for 2 h. Fluorescence intensity was measured on a plate reader with excitation at λ = 490 nm (bandwidth 5 nm) and emission at λ = 510 nm (bandwidth 5 nm). Error bars are ± SD. | ||
Conclusions
In summary, a novel platform for caging fluorophores and designing fluorogenic probes is presented, which uses a thiophenyl linker connected to a fluorophore caged by a good leaving group—dinitrophenyl. Upon the reaction with the analyte, the thiophenyl group is released and intramolecularly substitutes the dinitrophenyl caging group on the fluorophore. While we demonstrated this caging platform for designing Tokyo green (CAT) based fluorogenic probes to detect metallo-carbapenemases in carbapenem-resistant Enterobacteriaceae (CRE) lysates, we expect that it can be generally applied to other analytes and fluorophores.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health grant NIH 1R01AI125286-01A1.Notes and references
- A. R. Lippert, G. C. Van De Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed.
- A. Fernández and M. Vendrell, Chem. Soc. Rev., 2016, 45, 1182–1196 RSC.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- O. Kocaoglu and E. E. Carlson, Nat. Chem. Biol., 2015, 12, 472–478 CrossRef PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- L. Yuan, F. Jin, Z. Zeng, C. Liu, S. Luo and J. Wu, Chem. Sci., 2015, 6, 2360–2365 RSC.
- J. Hatai, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2017, 139, 2136–2139 CrossRef CAS PubMed.
- T. Pinkert, D. Furkert, T. Korte, A. Herrmann and C. Arenz, Angew. Chem., Int. Ed., 2017, 56, 2790–2794 CrossRef CAS PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- R. Zhang, J. Zhao, G. Han, Z. Liu, C. Liu, C. Zhang, B. Liu, C. Jiang, R. Liu, T. Zhao, M. Han and Z. Zhang, J. Am. Chem. Soc., 2016, 138, 3769–3778 CrossRef CAS PubMed.
- M. Gao, H. Su, Y. Lin, X. Ling, S. Li, A. Qin and B. Z. Tang, Chem. Sci., 2017, 8, 1763–1768 RSC.
- A. F. Chaudhry, S. Mandal, K. I. Hardcastle and C. J. Fahrni, Chem. Sci., 2011, 2, 1016–1024 RSC.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- J. A. Cotruvo Jr, A. T. Aron, K. M. Ramos-Torres and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC.
- D. Asanuma, M. Sakabe, M. Kamiya, K. Yamamoto, J. Hiratake, M. Ogawa, N. Kosaka, P. L. Choyke, T. Nagano, H. Kobayashi and Y. Urano, Nat. Commun., 2015, 6, 6463 CrossRef CAS PubMed.
- K. Bush and R. B. Sykes, in Antimicrobial Drug Resistance, ed. L. E. Bryan, Academic Press, New York, 1984, pp. 1–31 Search PubMed.
- E. P. Abraham and E. Chain, Nature, 1940, 46, 837 CrossRef.
- A. M. Queenan and K. Bush, Clin. Microbiol. Rev., 2007, 20, 440–458 CrossRef CAS PubMed.
- P. Nordmann, T. Naas and L. Poirel, Emerging Infect. Dis., 2011, 17, 1791–1798 CrossRef CAS PubMed.
- S. M. Diene and J. M. Rolain, Clin. Microbiol. Infect., 2014, 20, 831–838 CrossRef CAS PubMed.
- H. Yang, H. Young, S. Yu, L. Sutton and M. W. Crowder, Biochem. J., 2014, 464, 271–279 CrossRef CAS PubMed.
- S. Dahiya, P. Singla, U. Chaudhary and B. Singh, Int. J. Adv. Health Sci., 2015, 2, 11–17 Search PubMed.
- J.-M. Rodriguez-Martinez, P. Nordmann, N. Fortineau and L. Poirel, Antimicrob. Agents Chemother., 2010, 54, 1471–1476 Search PubMed.
- W. Gao, B. Xing, R. Y. Tsien and J. Rao, J. Am. Chem. Soc., 2003, 125, 11146–11147 CrossRef CAS PubMed.
- B. Xing, A. Khanamiryan and J. Rao, J. Am. Chem. Soc., 2005, 127, 4158–4159 CrossRef CAS PubMed.
- H. Yao, M. So and J. Rao, Angew. Chem., Int. Ed., 2007, 46, 7031–7034 CrossRef CAS PubMed.
- Y. Kong, H. Yao, H. Ren, S. Subbian, S. L. G. Cirillo, J. C. Sacchettini, J. Rao and J. D. Cirillo, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12239–12244 CrossRef CAS PubMed.
- H. Xie, J. Mire, Y. Kong, M. Chang, H. A. Hassounah, C. N. Thornton, J. C. Sacchettini, J. D. Cirillo and J. Rao, Nat. Chem., 2012, 4, 802–809 CrossRef CAS PubMed.
- J. Zhang, Y. Shen, S. L. May, D. C. Nelson and S. Li, Angew. Chem., Int. Ed., 2012, 51, 1865–1868 CrossRef CAS PubMed.
- Q. Shao, Y. Zheng, X. Dong, K. Tang, X. Yan and B. Xing, Chem.–Eur. J., 2013, 19, 10903–10910 CrossRef CAS PubMed.
- Y. Cheng, H. Xie, P. Sule, H. Hassounah, E. A. Graviss, Y. Kong, J. D. Cirilo and J. Rao, Angew. Chem., Int. Ed., 2014, 126, 9514–9518 CrossRef.
- W. Mao, L. Xia and H. Xie, Angew. Chem., Int. Ed., 2017, 56, 4468–4472 CrossRef CAS PubMed.
- H. Shi, Y. Cheng, K. H. Lee, R. F. Luo, N. Banaei and J. Rao, Angew. Chem., Int. Ed., 2014, 53, 8113–8116 CrossRef CAS PubMed.
- W. Jiang, Q. Fu, H. Fan, J. Ho and W. Wang, Angew. Chem., Int. Ed., 2007, 46, 8445–8448 CrossRef CAS PubMed.
- W. Lin, L. Long and W. Tan, Chem. Commun., 2010, 46, 1503–1505 RSC.
- D. Yu, Q. Zhai, S. Yang and G. Feng, Anal. Methods, 2015, 7, 7534–7539 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02416a |
| This journal is © The Royal Society of Chemistry 2017 |