
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-functional bis(alkynyl)gold(III) N⁁C complexes with distinct mechanochromic luminescence and electroluminescence properties†
Ben Yiu-Wing
Wong
,
Hok-Lai
Wong
,
Yi-Chun
Wong
,
Vonika Ka-Man
Au
 ,
Mei-Yee
Chan
* and
Vivian Wing-Wah
Yam
*
,
Mei-Yee
Chan
* and
Vivian Wing-Wah
Yam
*
Institute of Molecular Functional Materials [Areas of Excellence Scheme, University Grants Committee (Hong Kong)] and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk; chanmym@hku.hk
First published on 23rd August 2017
Abstract
A new class of donor–acceptor type luminescent bis(alkynyl)gold(III) N⁁C complexes has been synthesized and characterized. These gold(III) complexes not only exhibit high photoluminescence quantum yields of up to 0.81, but also interesting mechanochromic luminescence behaviors that are reversible. Upon grinding, a dramatic luminescence color change from green to red can be observed in solid samples of the gold(III) complexes, and the mechanochromic luminescence can be readily tuned via a judicious selection of substituents on the pyridine ring. In addition, solution-processable OLEDs based on this class of complexes with EQE values of up to 4.0% have been realized, representing the first demonstration of bis(alkynyl)gold(III) N⁁C complexes as emissive materials in solution-processable OLEDs.
Introduction
The development of smart materials that exhibit changes in response to external perturbations, such as radiation, pressure or temperature, has recently attracted immense interest, as the materials hold great promise for applications in various optical storage devices, pressure sensors, memory chips and security inks.1 Mechanochromic luminescence materials are an important class of these responsive materials, in which their emission color changes upon the application of appropriate pressure or mechanical forces, such as crushing, rubbing and grinding.2 With the continuous advancement of these mechanochromic materials, the dyes can also exhibit mechanochromic luminescence behavior in various solid state substrates, such as doped polymers, spin coated films or casted paper.3 The mechanochromic behavior is believed to arise from chemical or micro-environmental changes in the compounds. The former usually involves the modification of the molecular structure, including interconversion between the open and closed forms of the cyclic structures as well as the stereochemistry of the double bonds in alkene moieties, i.e. E and Z configurations.4 Due to the involvement of bond breakage and formation during the transition, there are only a limited number of examples with outstanding performances on the reversibility, reproducibility and complete conversion of solid-state chemical reactions. On the other hand, materials that are subject to micro-environmental transformations originating from a change in the molecular packing, intermolecular interactions and conformation usually show more promising performances with distinct mechanochromic luminescence properties.5 The systematic manipulation of the molecular packing as well as the morphology could therefore be an effective tool in the dynamic control of the reversible solid-state luminescence in response to mechanical stimuli.Recently, a number of different types of materials, including organic dyes, liquid crystals and polymers that can alter the degree of aggregation in response to mechanical stimuli, were demonstrated.3b,6 The mechanical deformation leads to alterations in the intermolecular distances, and results in a change in the extent of aggregation, as exemplified by the changes in their absorption and emission properties. However, most of these materials are organic compounds. Although examples of coordination metal complexes, especially those of platinum(II)7 and gold(I)8 metal systems, are known, they have not been extensively studied. It is anticipated that the presence of non-covalent interactions, such as metal–metal interactions, in these coordination complexes accounts for their rich polymorphism behavior and interesting luminescence mechanochromism.7,8 The synthetic challenge in coordinating both electron-donating and electron-accepting moieties into the metal center to generate donor–acceptor (D–A) type molecules may also be one of the key reasons why the library of mechanochromic metal complexes is limited.
A good mechanochromic material should be capable of having a dramatic color change upon the application of a mechanical force. Square-planar metal polypyridine complexes are believed to be promising candidates for mechanochromic metal complexes, where their emission color can be readily tuned via the modification of the polypyridine ligands.9 Among them, square-planar platinum(II) complexes are the most studied.9 However, there are no known corresponding studies on isoelectronic gold(III) complexes. Despite the rich photophysical properties of alkynylgold(III) complexes and the ready tunability of their emission color via the modification of the cyclometalating ligands, the mechanochromic properties of cyclometalated gold(III) complexes have not been reported previously.10 As first demonstrated by Yam and co-workers, the introduction of a strong σ-donating ligand greatly enhances the luminescence of the gold(III) metal complexes by raising the energy of their d–d states, leading to a higher possibility of populating their low-lying emissive state.11 The resultant stable gold(III) alkyl and aryl complexes exhibit rich photoluminescence properties, even at room temperature.10,11 An extension of this concept had also been successfully demonstrated by incorporating alkynyl ligands into gold(III) complexes,10a,b,c and more recently into dendritic structures,10e,g in which efficient solution-processable OLEDs with external quantum efficiencies (EQEs) of up to 10.0% and small efficiency roll-offs of less than 1% at a luminance of 1000 cd m−2 can be realized.10g Very recently, a new class of gold(III) complexes with tetradentate C⁁N⁁C⁁C ligands that exhibit extraordinarily high photoluminescence quantum yields (PLQYs) of up to 80% has been developed.12 Herein, we report the design and synthesis of a new class of D–A type cyclometalated bis(alkynyl)gold(III) N⁁C complexes that exhibit intense photoluminescence at both room temperature and low temperature, as well as interesting mechanochromic properties. Taking advantage of the highly luminescent nature of the N⁁C bis(alkynyl)gold(III) core,11b the incorporation of twisted donor moieties13 into the cyclometalating ligands creates a D–A type molecule with distinctive mechanochromic luminescence behavior. Upon grinding the solid sample, a dramatic luminescence color change from green to red can be clearly distinguished with the naked eye. Such emission color variation can be easily reversed to its original state via solvent annealing for a few seconds. This work represents the first demonstration of high-contrast mechanochromic luminescent gold(III) complexes containing cyclometalating N⁁C ligands, and the first report on gold(III) N⁁C complexes as emissive materials in solution-processable OLEDs, demonstrating the multi-functional properties of this new class of D–A type organometallic complexes and their potential for various optoelectronic and stimuli-responsive functions.
Synthesis and characterization
The ligands were synthesized according to modifications of literature procedures for amine- or fluorene-containing N⁁C ligands.14,15 The transmetalation of the mercury(II) to gold(III) metal centers was performed according to a modified literature method,11b and the bis(alkynyl)gold(III) complexes were synthesized via a dehydrohalogenation reaction of dichlorogold(III) complexes with their corresponding alkynyls in the presence of a catalytic amount of copper(I) iodide.10a The resulting gold(III) complexes are stable enough to be purified using column chromatography on silica gel, and were isolated as pale yellow to orange crystals after subsequent recrystallization from the layering of methanol onto a concentrated ethyl acetate solution of the complexes. All of the N⁁C gold(III) complexes are found to have good solubility in common organic solvents, such as ethyl acetate, toluene and dichloromethane. The identities of the complexes were confirmed using 1H and 19F{1H} NMR spectroscopy, fast atom bombardment mass spectrometry, as well as satisfactory elemental analysis. The synthetic pathways of these gold(III) complexes are shown in Scheme 1.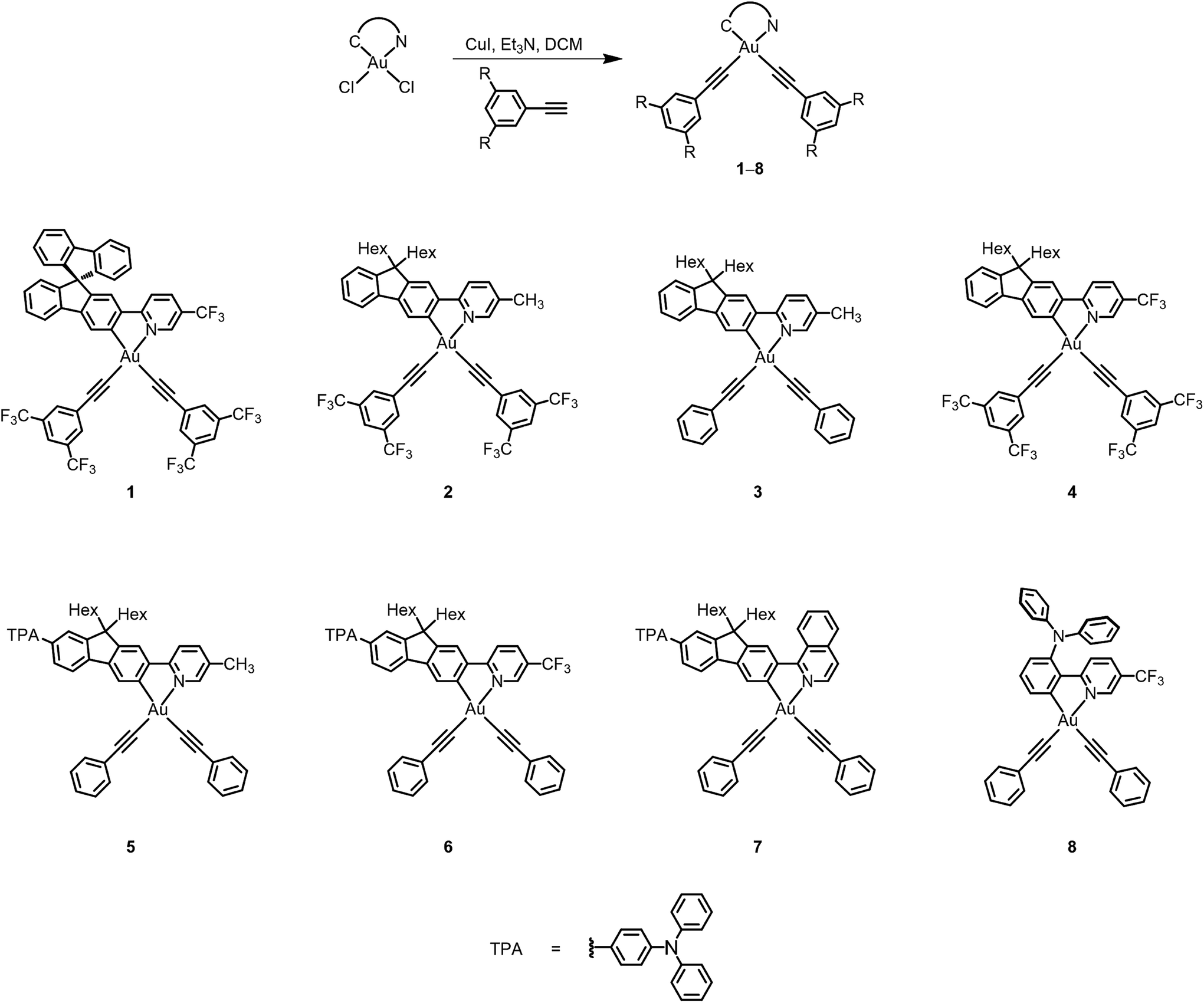 | ||
| Scheme 1 Synthetic scheme and chemical structures of the bis(alkynyl)gold(III) N⁁C complexes 1–8. | ||
X-ray crystal structures
Single crystals of complex 8 were obtained via the layering of methanol onto a concentrated ethyl acetate solution of the complex, and the structure has been confirmed using X-ray crystallography. The crystal structure determination data are tabulated in Table S1 (in the ESI†), and selected bond lengths and bond angles are summarized in Table S2.†The gold(III) center of 8 adopts a square-planar geometry in the crystal structure. A perspective view of the crystal structure of 8 is shown in Fig. 1. The angles of the N–Au–C(phenyl) bond are found to be 80.8°, probably ascribed to the small bite angle of the phenylpyridine ligand. Meanwhile, the N–Au–C(alkynyl), C(phenyl)–Au–C(alkynyl) and C(alkynyl)–Au–C(alkynyl) angles are found to be slightly larger than 90°. Similar bond angles were also observed in the related bis(alkynyl)gold(III) complexes.11b The phenylpyridine moiety is observed to be co-planar. The C(phenyl)–Au and N–Au bond lengths are found to be 2.046 and 2.060 Å, respectively, which are in good agreement with those of other related complexes.11b The Au–C(alkynyl) bond distance is in the range of 1.949–2.042 Å, which is typical for an Au–C bond of terminal alkynyl coordination.
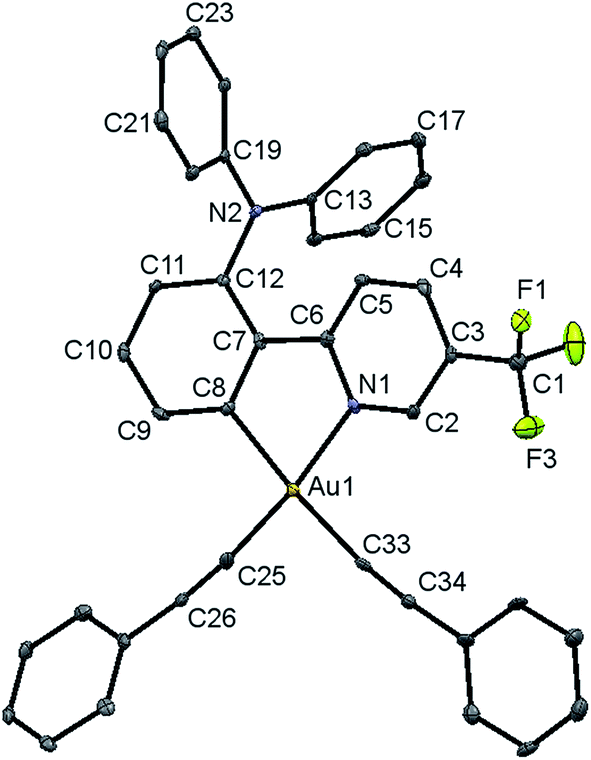 | ||
| Fig. 1 Perspective view of 8 with an atomic numbering scheme. The hydrogen atoms have been omitted for clarity. The thermal ellipsoids are drawn at the 30% probability level. | ||
Electrochemistry
The electrochemical properties of these bis(alkynyl)gold(III) N⁁C complexes have been studied using cyclic voltammetry in dichloromethane (0.1 mol dm−3 nBu4NPF6). In general, all of the gold(III) complexes exhibit an irreversible reduction wave between −1.23 and −1.64 V versus the saturated calomel electrode (SCE), depending on the substituent on the pyridine ring. The electrochemical data are depicted in Table 1. Complexes 1, 4, 6 and 8 with electron-withdrawing trifluoromethyl groups display a reduction potential of ca. −1.30 V, whereas complexes 2–3 and 5 bearing electron-donating methyl groups on the pyridine units show a shift in the reduction potential to ca. −1.60 V. The occurrence of the reduction at a less negative potential in 7 compared to 5 is probably due to the more extended π-conjugation of the isoquinoline moiety, resulting in a lower-lying lowest unoccupied molecular orbital (LUMO) level. Together with the observation of similar reduction potential values for complexes 3 and 5, the reduction waves are tentatively assigned to a pyridine- or isoquinoline-based reduction.| Complex | Oxidation E1/2/V vs. SCEb (ΔEp/mV)c [Epa/V vs. SCE]d | Reduction Epc/V vs. SCEe |
|---|---|---|
| a In dichloromethane solution with Bu4NPF6 (0.1 M) as the supporting electrolyte at room temperature; the scan rate was 100 mV s−1. b Quasi-reversible reduction couple, E1/2 = (Epa + Epc)/2, where Epa and Epc are the peak anodic and peak cathodic potentials, respectively. c The difference between the anodic peak and cathodic peak potentials, ΔEp = |Epa − Epc|. d E pa refers to the anodic peak potential for the irreversible oxidation. e E pc refers to the cathodic peak potential for the irreversible reduction. | ||
| 1 | [+1.87] | −1.23 |
| 2 | [+1.80] | −1.60 |
| 3 | [+1.76] | −1.57 |
| 4 | [+1.90] | −1.28 |
| 5 | +0.93 (95) | −1.64 |
| 6 | +0.94 (92) | −1.33 |
| 7 | +0.92 (93) | −1.33 |
| 8 | [+1.32] | −1.39 |
Complexes 1–4 exhibit an irreversible oxidation wave ranging from +1.76 to +1.90 V (vs. SCE). The potential for the first oxidation wave of complex 4 is found to occur at a more positive value than that of 2, showing that the introduction of the electron-withdrawing trifluoromethyl group to the pyridine moiety would lead to a lower-lying highest occupied molecular orbital (HOMO) level, implying a reduced ease of oxidation. The potential value of the oxidation wave is found to be insensitive to the substituents at the 9-position of the fluorene moiety, as revealed by the observation of similar potential values for 1 and 4. With reference to the previous studies on bis(alkynyl)gold(III) N⁁C complexes,11b the oxidation processes of 1–4 were therefore tentatively assigned as alkynyl ligand-centered oxidation, probably with some perturbation of the gold(III) N⁁C moiety. Complexes 5–7 with electron-donating N,N-diphenyl-4-(fluoren-2-yl)aniline units exhibit a quasi-reversible oxidation couple at ca. +0.93 V (vs. SCE), which originates from the oxidation of the N,N-diphenyl-4-(fluoren-2-yl)aniline framework. An irreversible oxidation wave at +1.32 V is observed in 8, which is tentatively assigned as triphenylamine-based oxidation. The irreversible nature of the oxidation wave in 8 is in line with the rapid formation of benzidine that is commonly found in triphenylamine oxidation, but not in diphenylamine-type oxidation, as in complexes 5–7.
Electronic absorption studies
The electronic absorption spectra of bis(alkynyl)gold(III) N⁁C complexes 1–8 in dichloromethane solution at 298 K exhibit intense absorption bands at ca. 310–460 nm with extinction coefficients in the order of 104 dm−3 mol−1 cm−1. Table 2 summarizes the photophysical data of 1–8, and Fig. 2 depicts the electronic absorption spectra of 1–8. The lowest-energy absorption bands are found to be dependent on the substituents on the pyridine moiety as well as the substituents at the 7-position of the fluorene unit. Complexes 1–4 display low-energy vibronic-structured absorption at ca. 375–405 nm, with vibrational progressional spacings of ca. 1100 cm−1. Changing the 9,9-spirobifluorene unit in 1 to the 9,9-dihexylfluorene unit in 4 was found to have very little influence on the electronic absorption spectra. On the other hand, the replacement of the electron-donating methyl group with an electron-withdrawing trifluoromethyl group leads to a bathochromic shift of the low-energy absorption band from 392 nm in 2 to 409 nm in 4. In addition, the lower-energy absorption band is found to change slightly from 392 nm in 2 to 388 nm in 3, in which the strongly electron-withdrawing bis-3,5-(trifluoromethyl)phenylethynyl is replaced by the phenylethynyl moiety. It is suggested that the lowest-energy absorption band of these N⁁C gold(III) complexes should be assigned as a metal-perturbed intraligand (IL) [π → π*] transition of the cyclometalating ligand, with the mixing of some charge-transfer character from the fluorene framework to the pyridine moiety.| Complex | Absorption | Emission | ||
|---|---|---|---|---|
| λ max/nm (εmax/dm3 mol−1 cm−1) | Medium | λ max/nm (τo/μs) | Φ lum | |
| a The relative photoluminescence quantum yields of the solutions were measured at room temperature using [Ru(bpy)3]Cl2 as a standard. The thin film absolute PLQY values were measured using a Hamamatsu C9920-03 Absolute PLQY Measurement System. | ||||
| 1 | 309 (31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 745), 317 (26685), 355 (7926), 390 (15070), 408 (15990) 745), 317 (26685), 355 (7926), 390 (15070), 408 (15990) |
CH2Cl2 | 451 (<0.1), 552, 594, 644 (100.0) | 0.37 |
| 2 | 307 (25925), 324 (11190), 338 (10780), 375 (15340), 392 (14775) |
CH2Cl2 | 432 (<0.1), 543, 568, 630 (104.5) | 0.16 |
| 2% in MCP | 536, 578, 628 | 0.13 | ||
| 5% in MCP | 536, 579, 629 | 0.13 | ||
| 10% in MCP | 535, 577, 628 | 0.11 | ||
| 20% in MCP | 538, 578, 628 | 0.09 | ||
| 3 | 310 (25295), 326 (20020), 338 (15355), 372 (18905), 388 (17110) |
CH2Cl2 | 432 (<0.1), 540, 568, 630 (103.4) | 0.14 |
| 4 | 317 (21030), 348 (7990), 391 (17750), 409 (18400) |
CH2Cl2 | 453 (<0.1), 557, 597, 647 (99.4) | 0.41 |
| 2% in MCP | 547, 590, 642 | 0.19 | ||
| 5% in MCP | 546, 588, 640 | 0.19 | ||
| 10% in MCP | 547, 590, 643 | 0.17 | ||
| 20% in MCP | 548, 591, 643 | 0.13 | ||
| 5 | 315 (48900), 349 (29695), 405 (42930) |
CH2Cl2 | 580, 615 (60.0) | 0.24 |
| 2% in MCP | 481, 569 | 0.19 | ||
| 5% in MCP | 484, 573 | 0.22 | ||
| 10% in MCP | 487, 575 | 0.22 | ||
| 20% in MCP | 498, 574 | 0.18 | ||
| 6 | 335 (47475), 436 (39515) |
CH2Cl2 | 680 (31.6) | 0.13 |
| 2% in MCP | 528 | 0.33 | ||
| 5% in MCP | 544 | 0.33 | ||
| 10% in MCP | 550 | 0.31 | ||
| 20% in MCP | 564 | 0.26 | ||
| 7 | 336 (42350), 452 (32690) |
CH2Cl2 | 683 (38.5) | 0.07 |
| 8 | 321 (12950), 463 (3365) |
CH2Cl2 | 615 (7.3) | 0.30 |
| 5% in MCP | 556 | 0.84 | ||
| 10% in MCP | 563 | 0.84 | ||
| 15% in MCP | 565 | 0.84 | ||
| 20% in MCP | 571 | 0.81 | ||
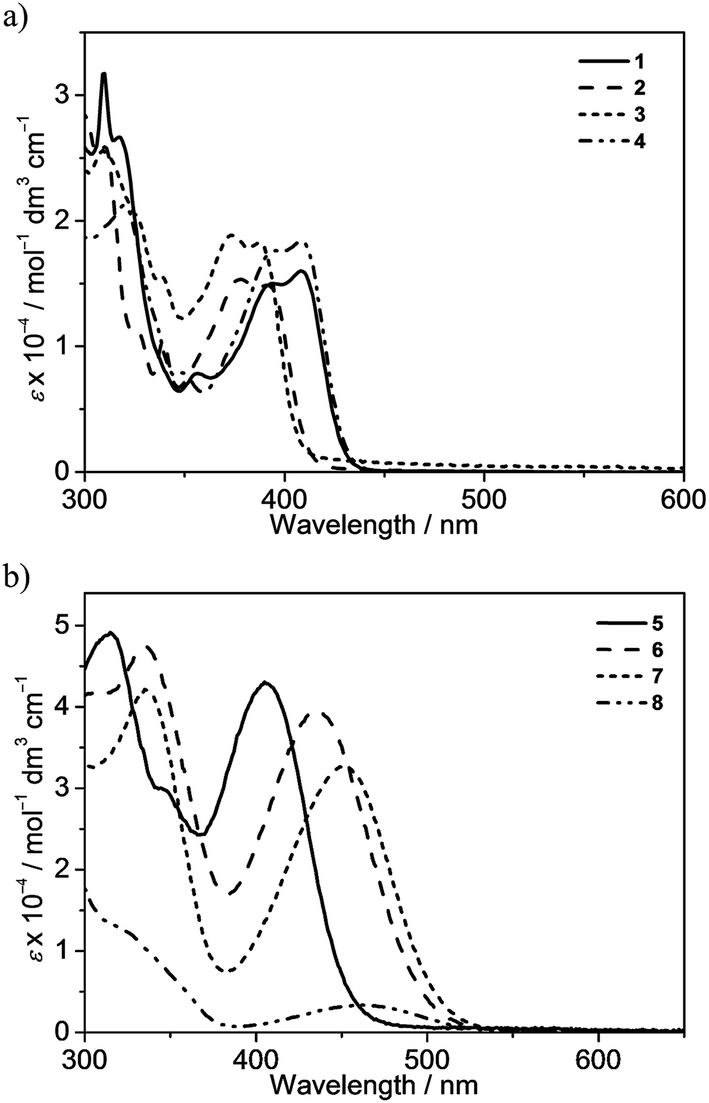 | ||
| Fig. 2 Electronic absorption spectra of (a) 1–4 and (b) 5–8 in dichloromethane at 298 K. | ||
The attachment of electron-donating triphenylamine to the 7-position of the fluorene unit shows a large bathochromic shift of the lowest-energy absorption band. In addition, as the substituents on the pyridine change from methyl in 5 to trifluoromethyl in 6, the absorption maximum shifts from 405 nm to 436 nm. Modification of the pyridine moiety to the more π-conjugated isoquinoline unit in 7 further red-shifts the absorption band to 452 nm. Together with the observation of the structureless absorption band, the lowest-energy absorption band of 5–7 is assigned as an intraligand charge transfer (ILCT) [π(arylamine) → π*(N⁁C core)] transition. Complex 8 also exhibits a structureless absorption band at 463 nm. In view of the presence of an electron-donating diphenylamine moiety on the cyclometalating N⁁C ligand, the lower-energy absorption band would be assigned as an ILCT [π(diphenylamine) → π*(N⁁C core)] transition.
Luminescence studies
All of the gold(III) N⁁C complexes exhibit satisfactory PLQY values between 0.07 and 0.41 in dichloromethane solution. Notably, interesting luminescence properties have been observed in this class of gold(III) N⁁C complexes. Upon photo-excitation, complexes 1–4 exhibit dual emission at ca. 430–450 nm and 540–560 nm. The higher-energy emission could be ascribed to the spin-allowed 1IL excited [π → π*] state of the fluorene-pyridine core, supported by the fact that these emissions could be observed under aerobic conditions with short emission lifetimes. Similar emission behavior has also been observed in other gold(III) complexes.16 The lower-energy vibronic emission band at ca. 545 nm with vibrational progressional spacings of about 1200 cm−1 was found to have a long emission lifetime of ca. 100 μs. The emission spectra of all of the complexes are shown in Fig. 3 and S1.† Similar to the absorption studies, a bathochromic-shifted emission band from 543 nm in 2 to 557 nm in 4 is observed, which can be attributed to the inductive effect of the substituents on the pyridine ring. Complex 1 with 9,9-spirobifluorene is found to show a similar emission energy as 4 with the 9,9-dihexylfluorene unit. In addition, only a very small blue shift in the emission energy is observed from 543 nm in 2 to 540 nm in 3, indicating that the emission energies are almost insensitive to the alkynyl ligands. Together with the large Stokes shifts, the luminescence of these low-energy emission bands is tentatively assigned to derive from the metal-perturbed spin-forbidden IL excited [π → π*(N⁁C)] state, mixed with some charge-transfer character from the fluorene moiety to the pyridine ring. This assignment shows good agreement with the results of related cyclometalated bis(alkynyl)gold(III) N⁁C and biscyclometalated alkynylgold(III) C⁁N⁁C complexes.10–12,16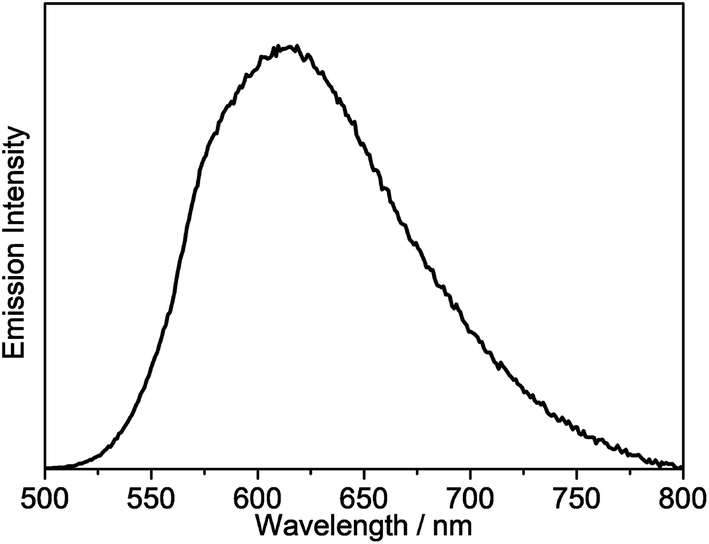 | ||
| Fig. 3 Emission spectrum of 8 in dichloromethane at 298 K. | ||
Similar to the absorption studies, the attachment of the electron-donating triphenylamine group to the 7-position of the fluorene unit in 5–7 exhibits a large bathochromic shift of the emission band. A vibronic-structured emission band with a band maximum at 580 nm is observed in complex 5. The vibrational progressional spacings of approximately 1250 cm−1 are indicative of the C![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) N and CC stretching modes of the cyclometalating N⁁C ligand.11b These emissions possibly originate from the metal-perturbed 3IL [π → π*(N⁁C)] state, probably with some charge-transfer character from the electron-donating amine moiety to the pyridine ring. The presence of the electron-withdrawing trifluoromethyl group at the pyridine ring in 6 results in a structureless emission band at 680 nm, whereas changing the pyridine ring to isoquinoline moiety in 7 results in a structureless emission band at 683 nm. In view of the presence of the electron-donating triphenylamine framework as well as the lower-lying π* orbital energy of the trifluoromethylpyridine and isoquinoline units, the emission is tentatively assigned to the 3ILCT [π(triphenylamine) → π*(N⁁C core)] excited state. Complex 8 displays intense photoluminescence at 615 nm, which is tentatively assigned to the 3ILCT [π(triphenylamine) → π*(N⁁C core)] excited state.
N and CC stretching modes of the cyclometalating N⁁C ligand.11b These emissions possibly originate from the metal-perturbed 3IL [π → π*(N⁁C)] state, probably with some charge-transfer character from the electron-donating amine moiety to the pyridine ring. The presence of the electron-withdrawing trifluoromethyl group at the pyridine ring in 6 results in a structureless emission band at 680 nm, whereas changing the pyridine ring to isoquinoline moiety in 7 results in a structureless emission band at 683 nm. In view of the presence of the electron-donating triphenylamine framework as well as the lower-lying π* orbital energy of the trifluoromethylpyridine and isoquinoline units, the emission is tentatively assigned to the 3ILCT [π(triphenylamine) → π*(N⁁C core)] excited state. Complex 8 displays intense photoluminescence at 615 nm, which is tentatively assigned to the 3ILCT [π(triphenylamine) → π*(N⁁C core)] excited state.
Solvent-dependent absorption and emission studies have been performed for 6 to further investigate the spectroscopic origin. The related photophysical data are summarized in Table S3.† In general, the lowest-energy absorption band exhibits negative solvatochromism, whereas the emission band displays large positive solvatochromism from 505 nm in cyclohexane to 680 nm in dichloromethane. The emission spectra of 6 in different solvents and the Lippert–Mataga plot are depicted in Fig. 4. The linear relationship between the energy of the Stokes shift and the orientation polarization parameter of the solvents in the plot suggests the involvement of charge-transfer character in the excited state, further substantiating the idea that the origin of the emission derives from the 3ILCT [π(triphenylamine) → π*(N⁁C core)] excited state.
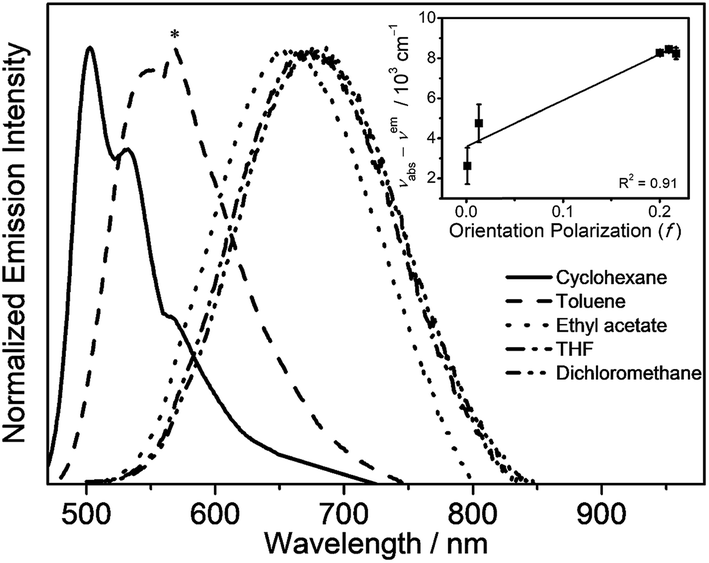 | ||
| Fig. 4 Emission spectra of 6 in different solvents at room temperature (the asterisk indicates an instrumental artifact). The inset shows the Lippert–Mataga plot of 6. | ||
The emission properties of the gold(III) N⁁C complexes in solid-state thin films have also been investigated, in which the gold(III) complexes were doped in 1,3-bis(carbazol-9-yl)-benzene (MCP) to prepare the thin films. All of the selected complexes display similar emission behavior to those in dichloromethane solution. For instance, complex 4 exhibits vibronic emission bands at ca. 548 nm with vibrational progressional spacings of about 1350 cm−1, as shown in Fig. S2a.† These emission bands are also found to be independent of the dopant concentration, which ranges from 2 wt% to 20 wt%. On the contrary, the thin films doped with complexes 6 and 8 show concentration-dependent emission, and a bathochromic shift in the emission maximum is observed with increasing dopant concentration (Fig. S2b† and Fig. 5). It should be highlighted that complex 8 exhibits a very high PLQY value of up to 0.84 in the thin films, and the PLQY values remain almost unchanged, even when the dopant concentration increases up to 20% (PLQY = 0.81). Such high PLQY values at high dopant concentrations are highly desirable for the preparation of OLEDs.
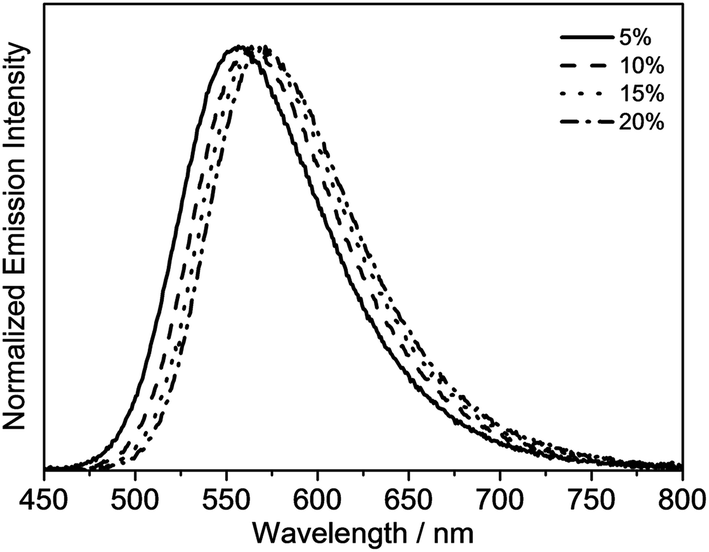 | ||
| Fig. 5 Normalized emission spectra of thin films of 8 doped in MCP at different concentrations at 298 K. | ||
Electroluminescence properties
Taking advantage of the high PLQY value of 8 in the solid-state thin films, solution-processable OLEDs based on this complex as the phosphorescent dopant have been fabricated. In particular, solution-processable OLEDs with the configuration of indium-tin oxide (ITO)/poly(ethylene-dioxythioxythiophene):poly(styrene sulfonic acid) (PEDOT:PSS; 70 nm)/x% 8:MCP (60 nm)/tris[2,4,6-trimethyl-3-(pyridine-3-yl)-phenyl]borane (3TPYMB) (5 nm)/1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB; 30 nm)/LiF (0.8 nm)/Al (100 nm) were prepared, in which 3TPYMB and TmPyPB were used as the hole-blocking and electron-transporting layers, respectively. The emissive layer was prepared via the spin-coating of a chloroform solution of x% 8:MCP blend. As depicted in Fig. 6, the electroluminescence (EL) spectra of all of the devices resemble those of the photoluminescence spectra of the thin films, which suggests efficient energy transfer from the MCP host to 8. In good agreement with the luminescence studies, the EL maximum is dependent on the dopant concentration (Fig. 6), in which the emission maximum has been red-shifted from 544 nm to 564 nm with an increasing dopant concentration from 10 wt% to 20 wt%. This corresponds to a change of the CIE coordinates from (0.40, 0.57) to (0.47, 0.52). In addition, a satisfactory performance with a maximum EQE value of 4.0%, current efficiency of 12.6 cd A−1 and power efficiency of 4.0 lm W−1 is obtained (Table 3). To the best of our knowledge, this is the first successful demonstration of solution-processable OLEDs based on gold(III) complexes with bidentate ligands.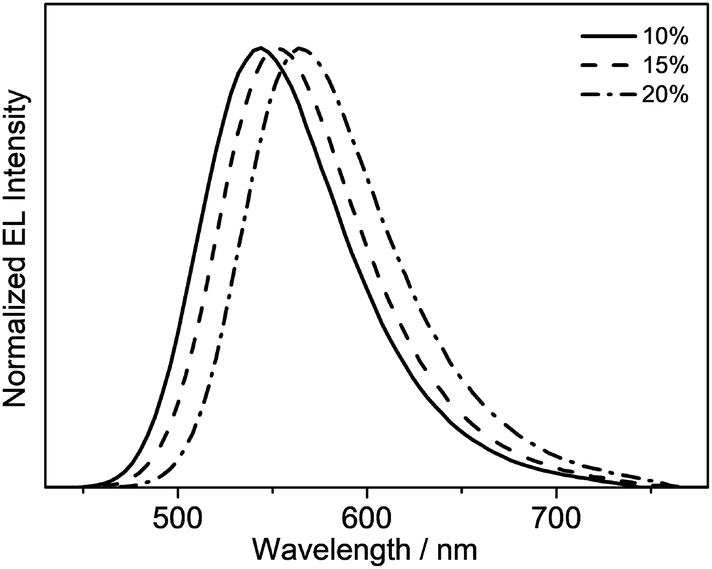 | ||
| Fig. 6 Normalized EL spectra of solution-processable devices made from 8. | ||
| Dopant conc. | CE/cd A−1 | PE/lm W−1 | EQE/% | λ max/nm (CIE (x, y)) |
|---|---|---|---|---|
| a Dopant conc., CE, PE and EQE refer to the dopant concentration, current efficiency, power efficiency and external quantum efficiency, respectively. | ||||
| 10 wt% | 12.1 | 4.22 | 3.5 | 544 (0.40, 0.57) |
| 15 wt% | 11.0 | 2.88 | 3.3 | 552 (0.43, 0.55) |
| 20 wt% | 12.6 | 3.95 | 4.0 | 564 (0.47, 0.52) |
Mechanochromic properties
In addition to the high PLQY values, interesting mechanochromic properties have been observed in complexes 5–8, which contained electron-donating arylamine groups. The luminescence data and emission spectra of different forms of complexes 5–8 are shown in Table 4, and Fig. 7 and S3,† respectively. For complex 5, the pale yellow pristine solids display a greenish-blue emission band that peaked at 504 nm, while the pristine samples of 6–8 exhibit yellow photoluminescence. Upon grinding using a pestle, orange solids of 5 with yellow emission at 553 nm are observed. Both the ground solid samples of 6 and 7 show red emission at ca. 635 nm, and that of 8 displays reddish orange emission. Surprisingly, the color of the solid sample also changes upon grinding, such as in 7 where it changed from yellow to red (Fig. S4†). Thus, the bathochromic-shifted emission of the ground solid may be a result of ground state aggregation rather than excimer formation. It is also worth noting that the mechanochromic change in these complexes is significant, as the color variation between the pristine/recovered solid and the ground solid could be readily and clearly distinguished with the naked eye. More importantly, the mechanochromic luminescence behaviors of these complexes are reversible and can be tuned by varying the substituent on the pyridine moiety. The luminescence of these ground solids can be restored to their original color by annealing them in ether vapor for a few seconds. In addition, such mechanochromic behaviors can be clearly observed when these complexes are dispersed on other solid substrates, such as weighing paper. The application of a mechanical force eventually leads to a transformation of the luminescence color from greenish blue to yellow for 5, while the emission color changes from yellow to red for 7 (Fig. 8).| Complex | Form | Emission λmax/nm |
|---|---|---|
| 5 | Pristine | 504 |
| Ground | 553 | |
| Recovered | 502 | |
| 6 | Pristine | 555 |
| Ground | 630 | |
| Recovered | 549 | |
| 7 | Pristine | 554 |
| Ground | 640 | |
| Recovered | 542 | |
| 8 | Pristine | 562 |
| Ground | 610 | |
| Recovered | 567 |
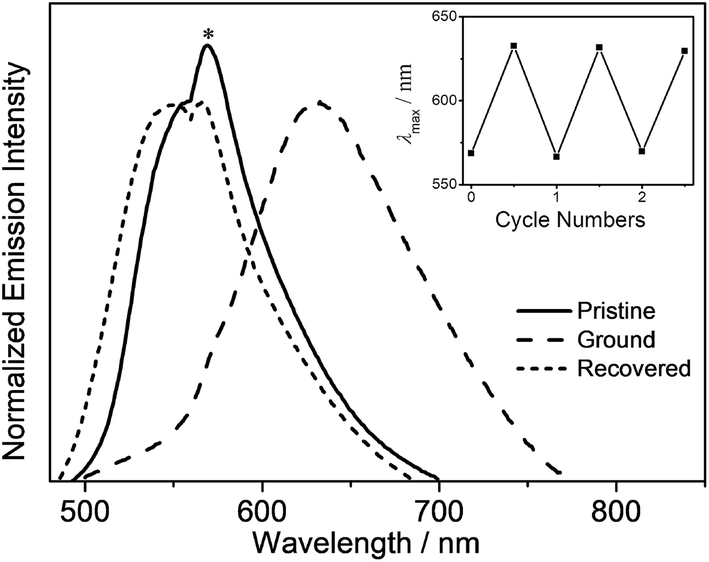 | ||
| Fig. 7 Normalized emission spectra of 6 in powder, ground and fumed forms (the asterisk indicates an instrumental artifact). The inset shows the emission wavelength of the repeated mechanochromic behavior of 6. | ||
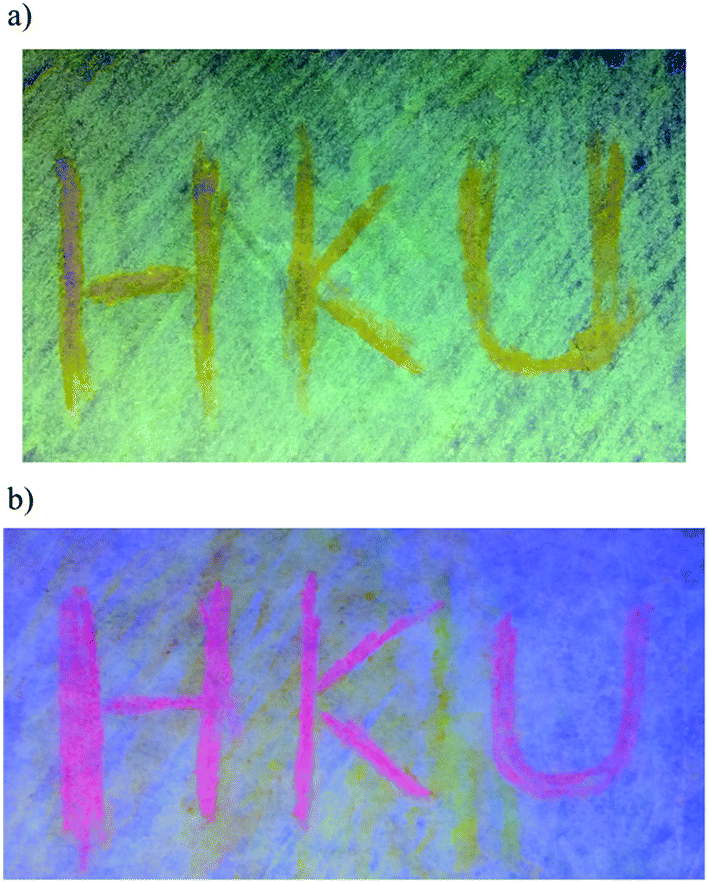 | ||
| Fig. 8 An image of the weighing paper impregnated with (a) 5 and (b) 7 under UV irradiation, where “HKU” was written with a glass rod. | ||
X-Ray diffraction (XRD) studies have been performed on these complexes to decipher the different spectroscopic properties of the various solid forms, as shown in Fig. 9 and S5.† All of the pristine solids display sharp and intense diffraction peaks in their spectra, which is suggestive of a well-ordered crystalline phase. The disappearance of the diffraction peaks upon grinding reveals the amorphous nature of the solid form. Mechanical grinding is believed to break the well-ordered microcrystalline nature, leading to random molecular packing with no regular intermolecular distance. After the exposure of the ground powders to ether vapor, the solid samples exhibit sharp and intense diffraction peaks again. The recovered sample of complex 8 shows a similar XRD pattern to those of the pristine and simulated ones. It suggests that the solvent annealing process would lead to the recovery of an ordered packing structure that is similar to the pristine one, as reflected by the distinct signals in the XRD studies. These results indicate that the application of the mechanical grinding can effectively transform the well-ordered crystalline structure to an amorphous phase, leading to emission variation upon grinding the solid. Meanwhile, the solvent annealing process can promote the transformation of the amorphous state to an ordered crystalline state with the recovery of the majority of the diffraction peaks, as shown in the PXRD patterns of the pristine solids.17 This, together with the fact that 1–4 show no mechanochromic behavior, suggests that the bathochromic shift in both the absorption and emission spectra in the ground phase is probably due to the reduction of the interplanar angle between the donor and acceptor units, resulting in better π-conjugation and a more planarized conformation in these metastable amorphous states.13f,18 Differential scanning calorimetry (DSC) studies on 8 further reveal that the pristine samples display no peaks until the decomposition temperature has been reached (Fig. S6†), while an additional exothermic peak at ca. 103 °C is observed in the ground solid, which is indicative of a phase transition at that temperature.
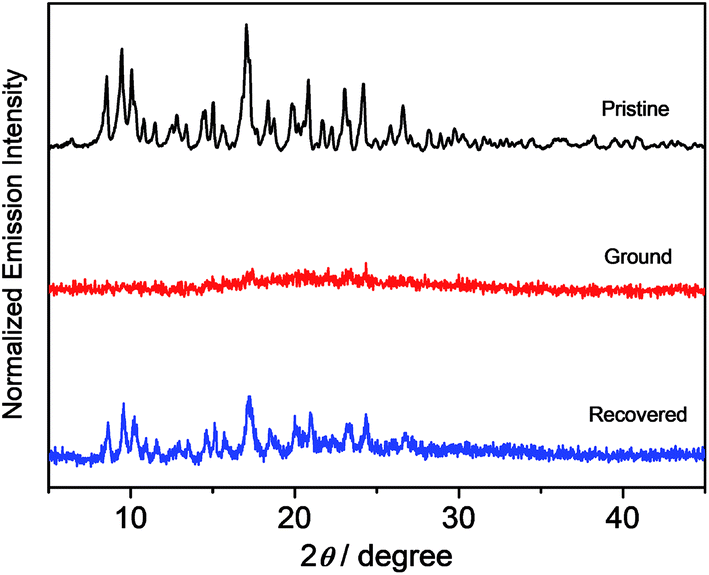 | ||
| Fig. 9 XRD patterns of 6 in pristine, ground and recovered forms. | ||
Conclusions
A new class of highly emissive bis(alkynyl)gold(III) N⁁C complexes with rich mechanochromic luminescence has been synthesized and characterized. These gold(III) complexes not only exhibit high PLQY values of up to 0.84 in solid-state thin films, but also interesting mechanochromic behavior with sharp luminescence color changes from green to red upon grinding. The XRD analysis suggests that the mechanochromic response may result from the morphological changes from the crystalline to the amorphous state. Solution-processable OLEDs based on this complex with a maximum EQE value of up to 4.0% can also be realized, representing the first successful demonstration of the use of bis(alkynyl)gold(III) N⁁C complexes as emissive materials in OLEDs.Experimental section
Materials and reagents
Mercury(II) acetate was purchased from Sigma-Aldrich Co. Ltd. Potassium tetrachoroaurate(III) was purchased from ChemPur. Phenylacetylene was purchased from Acros Organics. Tetrahydrofuran and dichloromethane for the reactions were purified using an Innovative Technology, Inc. PureSolv MD 5 Solvent Purification System before use. All of the other solvents and commercially available reagents were of analytical grade and were used as received. Tetra(n-butyl)-ammonium hexafluorophosphate (Aldrich) was recrystallized three times from absolute ethanol before use. All of the reactions were performed under inert and anhydrous conditions using standard Schlenk techniques unless otherwise specified.Physical measurements and instrumentation
1H and 19F{1H} NMR spectra were recorded on a Bruker DPX-400 Fourier transform NMR spectrometer with the chemical shifts (δ, ppm) recorded relative to tetramethylsilane (Me4Si) and trichlorofluoromethane (CFCl3), respectively. Positive EI and FAB mass spectra were recorded using a Thermo Scientific DFS High Resolution Magnetic Sector Mass Spectrometer. Elemental analyses were performed using the Carlo Erba 1106 elemental analyzer at the Institute of Chemistry, Chinese Academy of Sciences (Beijing, China). UV-Vis absorption spectra were recorded on a Varian Cary 50 spectrophotometer equipped with a Xenon flash lamp. Steady-state emission spectra were recorded using a Spex Fluorolog-3 model FL3-211 fluorescence spectrofluorometer equipped with an R2658P PMT detector. Relative luminescence quantum yields were measured using the optical dilute method reported by Demas and Crosby,19 whereas the absolute luminescence quantum yields of the thin films were measured on a Hamamatsu C9920-03 Absolute PLQY Measurement System. A degassed aqueous solution of [Ru(bpy)3]Cl2 (Φ = 0.042, with the excitation wavelength at 436 nm)20 was used as the reference and corrected for the refractive index of the solution for the relative luminescence quantum yield measurements. All of the solution samples for the photophysical studies were freshly prepared under high vacuum. A round-bottomed flask equipped with a side-arm 1 cm fluorescence cuvette was used and was sealed from the atmosphere using a Rotaflo HP6/6 quick-release Teflon stopper. The solutions were rigorously degassed on a high-vacuum line in a two-compartment cell with no less than four successive freeze–pump–thaw cycles. Cyclic voltammetric measurements were performed using a CH Instruments, Inc. model CHI 620A electrochemical analyzer. Electrochemical measurements were performed in dichloromethane solutions with 0.1 M nBu4NPF6 as the supporting electrolyte at room temperature. The reference electrode was an Ag/AgNO3 (0.1 M in acetonitrile) electrode, and the working electrode was a glassy carbon electrode (CH Instruments, Inc.), with a platinum wire as the counter electrode. The working electrode surface was first polished with 1 μm alumina slurry (Linde) on a microcloth (Buehler Co.) and then with 0.3 μm alumina slurry. It was then rinsed with ultrapure deionized water and sonicated in a beaker that contained ultrapure water for 5 min. The polishing and sonicating steps were repeated twice, and then the working electrode was finally rinsed under a stream of ultrapure deionized water. The ferrocenium/ferrocene couple (Fc+/Fc) was used as the internal reference.21 All of the solutions for the electrochemical studies were deaerated with prepurified argon gas prior to the measurements. Emission lifetime measurements were performed using a conventional pulsed laser system. The excitation source used was the 355 nm (third harmonic, 8 ns) output of a Spectra-Physics Quanta-Ray Q-switched GCR-150-10 pulsed Nd-YAG laser (10 Hz). Luminescence decay signals were detected using a Hamamatsu R928 PMT and recorded on a Tektronix Model TDS-620A (500 MHz, 2 GS per s) digital oscilloscope and analyzed using a program for exponential fits. Solution-processable devices based on 8 with the configuration of indium-tin oxide (ITO)/poly(ethylenedioxythioxythiophene):poly(styrenesulfonic acid) (PEDOT:PSS) (70 nm)/x% 8:MCP (60 nm)/tris[2,4,6-trimethyl-3-(pyridine-3-yl)-phenyl]-borane (3TPYMB) (5 nm)/1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) (30 nm)/LiF (0.8 nm)/Al (100 nm) had been prepared, in which 3TPYMB and TmPyPB were used as the hole blocking layer and electron-transporting layer, respectively. The emissive layer was prepared by spin-coating a chloroform solution of 8:MCP blend at different concentrations. OLEDs were fabricated on patterned ITO-coated glass substrates with a sheet resistance of 30 Ω per square. The substrates were cleaned with Decon 90, rinsed with deionized water then dried in an oven, and finally treated in an ultraviolet ozone chamber. A 70 nm thick PEDOT:PSS was spin-coated onto the ITO-coated glass substrates as hole-transporting layer. After that, emissive layer was formed by mixing 8 with MCP to prepare a 10 mg cm−3 solutions in chloroform and spin-coating onto PEDOT:PSS layer to give 60 nm thick uniform thin film. Onto this, a 5 nm thick 3TPYMB and a 30 nm thick TmPyPB were evaporated as a hole-blocking layer and an electron-transporting layer, respectively; while a LiF/Al was used as the metal cathode. All films were sequentially deposited at a rate of 0.1–0.2 nm s−1 without vacuum break. A shadow mask was used to define the cathode and to make four 0.1 cm2 devices on each substrate. Current density–voltage–luminance characteristics and EL spectra were measured simultaneously with a programmable Keithley model 2420 power source and a Photoresearch PR-655 spectrometer.Synthesis and characterization
:1 v/v) as eluent. Subsequent recrystallization via the layering of methanol onto the concentrated ethyl acetate solution of the complex gave the resulting complex as pale yellow to orange crystals.
(2-(5-(Trifluoromethyl)pyridin-2-yl)-9,9′-spirobi[fluoren]-3-yl)bis((3,5-bis(trifluoro-methyl)phenyl)ethynyl)gold(III) (1). Pale yellow crystals. Yield: 0.045 g (66%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 6.72–6.80 (m, 3H), 7.10 (s, 1H), 7.11–7.23 (m, 3H), 7.38–7.46 (m, 3H), 7.76 (s, 2H), 7.83–7.93 (m, 4H), 7.97 (s, 2H), 8.07–8.12 (m, 1H), 8.17 (s, 2H), 8.76 (s, 1H), 9.90 (s, 1H). 19F{1H} NMR (376 MHz, CDCl3, δ/ppm): δ −62.78 (s), −62.98 (s), −63.16 (s). Positive FAB-MS: m/z 1131 ([M]+). Elemental analysis calcd (%) for C51H23AuF15N·H2O: C, 53.28; H, 2.19; N, 1.22; found: C, 53.02; H, 2.24; N, 1.39.
(9,9-Dihexyl-2-(5-methylpyridin-2-yl)-9H-fluoren-3-yl)bis((3,5-bis(trifluoromethyl)-phenyl)ethynyl)gold(III) (2). Pale yellow crystals. Yield: 0.038 g (58%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 0.57–0.66 (m, 4H), 0.74 (t, J = 7.0 Hz, 6H), 0.99–1.12 (m, 12H), 1.96–2.03 (m, 4H), 2.50 (s, 3H), 7.33–7.36 (m, 3H), 7.59 (s, 1H), 7.66–7.71 (m, 1H), 7.75 (s, 1H), 7.81 (s, 1H), 7.92–7.95 (s, 2H), 8.00 (s, 2H), 8.11 (s, 2H), 8.57 (s, 1H), 9.39 (s, 1H). 19F{1H} NMR (376 MHz, CDCl3, δ/ppm): δ −63.05 (s), −63.01 (s). Positive FAB-MS: m/z 1095 ([M]+). Elemental analysis calcd (%) for C51H44AuF12N·0.5H2O: C, 55.44; H, 4.11; N, 1.27; found: C, 55.44; H, 4.36; N, 1.16.
(9,9-Dihexyl-2-(5-methylpyridin-2-yl)-9H-fluoren-3-yl)bis(phenylethynyl)gold(III) (3). Pale yellow crystals. Yield: 0.028 g (56%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 0.53–0.70 (m, 4H), 0.74 (t, J = 7.0 Hz, 6H), 1.01–1.12 (m, 12H), 1.95–2.02 (m, 4H), 2.48 (s, 3H), 7.26–7.34 (m, 7H), 7.35–7.40 (m, 2H), 7.55–7.63 (m, 3H), 7.67–7.75 (m, 3H), 7.85–7.92 (m, 2H), 8.70 (s, 1H), 9.57 (s, 1H). Positive FAB-MS: m/z 823 ([M]+). Elemental analysis calcd (%) for C47H48AuN·H2O: C, 67.05; H, 5.99; N, 1.66; found: C, 66.73; H, 5.81; N, 1.66.
(9,9-Dihexyl-2-(5-(trifluoromethyl)pyridin-2-yl)-9H-fluoren-3-yl)bis((3,5-bis(tri-fluoromethyl)phenyl)ethynyl)gold(III) (4). Pale yellow crystals. Yield: 0.049 g (71%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 0.53–0.65 (m, 4H), 0.74 (t, J = 7.0 Hz, 6H), 0.95–1.13 (m, 12H), 1.96–2.08 (m, 4H), 7.33–7.43 (m, Hz, 3H), 7.68 (s, 1H), 7.70–7.74 (m, 1H), 7.77 (s, 1H), 7.83 (s, 1H), 7.98 (s, 2H), 8.12 (s, 2H), 8.18 (d, J = 8.6 Hz, 1H), 8.34–8.27 (m, 1H), 8.59 (s, 1H), 9.97 (s, 1H). 19F{1H} NMR (376 MHz, CDCl3, δ/ppm): δ −62.70 (s), −62.98 (s), −63.12 (s). Positive FAB-MS: m/z 1149 ([M]+). Elemental analysis calcd (%) for C51H41AuF15N C, 53.27; H, 3.59; N, 1.22; found: C, 53.17; H, 3.57; N, 1.11.
(7-(4-(Diphenylamino)phenyl)-9,9-dihexyl-2-(5-methylpyridin-2-yl)-9H-fluoren-3-yl)bis(phenylethynyl)gold(III) (5). Yellow crystals. Yield: 0.048 g (75%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 0.62–0.70 (m, 4H), 0.74 (t, J = 6.9 Hz, 6H), 1.00–1.12 (m, 12H), 1.97–2.06 (m, 4H), 2.48 (s, 3H), 7.04 (t, J = 7.3 Hz, 2H), 7.13–7.19 (m, 6H), 7.26–7.34 (m, 8H), 7.38 (t, J = 7.5 Hz, 2H), 7.51–7.54 (m, 1H), 7.54–7.58 (m, 4H), 7.60 (d, J = 7.5 Hz, 2H), 7.71 (d, J = 7.5 Hz, 2H), 7.75 (d, J = 7.3 Hz, 1H), 7.86–7.92 (m, 2H), 8.70 (s, 1H), 9.57 (s, 1H). Positive FAB-MS: m/z 1066 ([M]+). Elemental analysis calcd (%) for C65H61AuN2·H2O C, 71.94; H, 5.85; N, 2.58; found: C, 71.65; H, 5.80; N, 2.52.
(7-(4-(Diphenylamino)phenyl)-9,9-dihexyl-2-(5-(trifluoromethyl)pyridin-2-yl)-9H-fluoren-3-yl)bis(phenylethynyl)gold(III) (6). Orange crystals. Yield: 0.050 g (74%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 0.60–0.69 (m, 4H), 0.74 (t, J = 7.0 Hz, 6H), 0.98–1.13 (m, 12H), 1.97–2.10 (m, 4H), 7.05 (t, J = 7.3 Hz, 2H), 7.12–7.20 (m, 6H), 7.26–7.31 (m, 6H), 7.31–7.36 (m, 2H), 7.37–7.42 (m, 2H), 7.53–7.56 (m, 2H), 7.56–7.61 (m, 4H), 7.66 (s, 1H), 7.72 (d, J = 7.3 Hz, 2H), 7.78 (d, J = 7.3 Hz, 1H), 8.14 (d, J = 8.6 Hz, 1H), 8.25 (d, J = 8.6 Hz, 1H), 8.73 (s, 1H), 10.14 (s, 1H). Positive FAB-MS: m/z 1120 ([M]+). Elemental analysis calcd (%) for C65H58AuF3N2·0.5H2O C, 69.08; H, 5.26; N, 2.48; found: C, 68.80.; H, 5.28; N, 2.52.
(7-(4-(Diphenylamino)phenyl)-9,9-dihexyl-2-(isoquinolin-1-yl)-9H-fluoren-3-yl)-bis(phenylethynyl)gold(III) (7). Orange crystals. Yield: 0.050 g (76%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 0.72–0.79 (m, 10H), 1.05–1.15 (m, 12H), 2.03–2.10 (m, 4H), 7.05 (t, J = 7.5 Hz, 2H), 7.13–7.20 (m, 6H), 7.26–7.31 (m, 6H), 7.31–7.35 (m, 2H), 7.36–7.41 (m, 2H), 7.55–7.59 (m, 3H), 7.59–7.63 (m, 3H), 7.69 (d, J = 6.4 Hz, 1H), 7.72 (d, J = 7.6 Hz, 2H), 7.80–7.86 (m, 2H), 7.92 (t, J = 7.5 Hz, 1H), 7.99 (d, J = 7.6 Hz, 1H), 8.08 (s, 1H), 8.81–8.86 (m, 2H), 9.62 (d, J = 6.4 Hz, 1H). Positive FAB-MS: m/z 1102 ([M]+). Elemental analysis calcd (%) for C68H61AuN2·0.5H2O C, 73.43; H, 5.62; N, 2.52; found: C, 73.62; H, 5.56; N, 2.63.
(3-(Diphenylamino)-2-(5-(trifluoromethyl)pyridin-2-yl)phenylbis(phenylethynyl))-gold(III) (8). Orange crystals. Yield: 0.038 g (80%). 1H NMR (400 MHz, CDCl3, δ/ppm): δ 6.98–7.04 (m, 6H), 7.11 (d, J = 8.0 Hz, 1H), 7.22–7.25 (m, 3H), 7.26–7.31 (m, 4H), 7.30–7.37 (m, 3H), 7.49 (d, J = 7.7 Hz, 1H), 7.53 (d, J = 7.7 Hz, 2H), 7.60–7.65 (m, 2H), 8.00–8.08 (m, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.72 (d, J = 8.0 Hz, 1H), 10.16 (s, 1H). 19F{1H} NMR (376 MHz, CDCl3, δ/ppm): δ −62.69 (s). Positive FAB-MS: m/z 788 ([M]+). Elemental analysis calcd (%) for C40H26AuF3N2·0.5H2O C, 60.23; H, 3.41; N, 3.51; found: C, 60.33; H, 3.25; N, 3.60.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. W.-W. Y. acknowledges the support from The University of Hong Kong and the URC strategic Research Theme on New Materials. This work was supported by the University Grands Committee Areas of Excellence Scheme (AoE/P-03/08) and the Research Grants Council General Research Fund (HKU17304715) of Hong Kong Special Administrative Region, P. R. China. B. Y.-W. W. and Y.-C. W. acknowledge the receipt of postgraduate studentships from The University of Hong Kong. Dr K.-H. Low, Dr C.-T. Poon and Dr Eugene Y.-H. Hong are gratefully acknowledged for the X-ray crystal structure data collection and determination, XRD, TGA and DSC measurements.References
- (a) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS PubMed; (b) M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed; (c) F. B. L. Gougnon and J. K. M. Sanders, Acc. Chem. Res., 2011, 45, 2211 CrossRef PubMed; (d) P. Lavalle, J.-C. Voegel, D. V. Vautier, B. Senger, P. Schaaf and V. Ball, Adv. Mater., 2011, 23, 1191 CrossRef CAS PubMed; (e) B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621 RSC; (f) C. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161 RSC; (g) J. Thévenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099 RSC; (h) J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421 RSC; (i) Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222 CrossRef CAS PubMed.
- (a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed; (b) M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755 CrossRef CAS PubMed; (c) A. Pucci and G. Ruggeri, J. Mater. Chem., 2011, 21, 8282 RSC; (d) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; (e) K. Ariga, T. Mori and J. P. Hill, Adv. Mater., 2012, 24, 158 CrossRef CAS PubMed; (f) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376 RSC; (g) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed.
- (a) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520 CrossRef CAS PubMed; (b) Y. Sagara, S. Tamane, T. Mutai, K. Araki and T. Kato, Adv. Funct. Mater., 2009, 19, 1869 CrossRef CAS; (c) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed; (d) H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556 CrossRef CAS PubMed; (e) Y. Matsunaga and J.-S. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985 CrossRef CAS PubMed.
- (a) A. Schönberg, M. Elkaschef, M. Nosseir and M. M. Sidky, J. Am. Chem. Soc., 1958, 80, 6312 Search PubMed; (b) D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. V. Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. R. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed; (c) J. M. Lenhardt, W. J. Ogle, M. T. Ong, R. Choe, T. J. Martinez and S. L. Craig, J. Am. Chem. Soc., 2011, 133, 3222 CrossRef CAS PubMed; (d) M.-J. Teng, X.-R. Jia, X.-F. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 51, 6398 CrossRef CAS PubMed; (e) C. G. Schäfer, M. Gallei, J. T. Zahn, J. Engelhardt, G. P. Hellmann and M. Rehahn, Chem. Mater., 2013, 25, 2309 CrossRef.
- (a) C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625 CrossRef; (b) M. Sase, S. Yamaguchi, Y. Sagara, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2011, 21, 8347 RSC; (c) A. Pucci, R. Bizzarri and G. Ruggeri, Soft Matter, 2011, 7, 3689 RSC; (d) Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372 RSC; (e) X. Zhang, J. Wang, J. Ni, L. Zhang and Z. Chen, Inorg. Chem., 2012, 51, 5569 CrossRef CAS PubMed.
- (a) B. R. Crenshaw and C. Weder, Chem. Mater., 2003, 15, 4717 CrossRef CAS; (b) M. Kinami, B. R. Crenshaw and C. Weder, Chem. Mater., 2006, 18, 946 CrossRef CAS; (c) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS PubMed; (d) X. Chen, R. Wei, Y. Xiang, Z. Zhou, K. Li, P. Song and A. Tong, J. Phys. Chem. C, 2011, 115, 14353 CrossRef CAS.
- (a) V. N. Kozhenvnikov, B. Donio and D. W. Brue, Angew. Chem., Int. Ed., 2008, 47, 6286 CrossRef PubMed; (b) Y. Nishiuchi, A. Takayama, T. Suzuki and K. Shinozaki, Eur. J. Inorg. Chem., 2011, 11, 1815 CrossRef; (c) J. Ni, X. Zhang, N. Qiu, Y. Wu, L. Zhang, J. Zhang and Z. Chen, Inorg. Chem., 2011, 50, 9090 CrossRef CAS PubMed; (d) X. Zhang, J. Wang, J. Ni, L. Zhang and Z. Chen, Inorg. Chem., 2012, 51, 5569 CrossRef CAS PubMed; (e) S. J. Choi, J. Kuwabara, Y. Nishimura, T. Arai and T. Kanbara, Chem. Lett., 2012, 41, 65 CrossRef CAS.
- (a) Y.-A. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778 CrossRef CAS PubMed; (b) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044 CrossRef CAS PubMed; (c) M. Osawa, I. Kawata, S. Igawa, M. Hoshino, T. Fukunaga and D. Hashizume, Chem.–Eur. J., 2010, 16, 12114 CrossRef CAS PubMed; (d) K. Kawaguchi, T. Seki, T. Karatsu, A. Kitamura, H. Ito and S. Yagai, Chem. Commun., 2013, 49, 11391 RSC; (e) T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187 RSC; (f) S. Yagai, T. Seki, H. Aonuma, K. Kawaguchi, T. Karatsu, T. Okura, A. Sakon, H. Uekusa and H. Ito, Chem. Mater., 2016, 28, 234 CrossRef CAS.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107 CrossRef CAS PubMed; (b) K. M.-C. Wong, X. Zhu, L.-L. Hung, W. H. Lam, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 4350 CrossRef CAS PubMed; (c) V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 9076 CrossRef CAS PubMed; (d) J. A. Garg, O. Blacque and K. Venkatesan, Inorg. Chem., 2011, 50, 5430 CrossRef CAS PubMed; (e) M.-C. Tang, D. P.-K. Tsang, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446 CrossRef CAS PubMed; (f) W.-P. To, K.-T. Chan, G. S.-M. Tong, C. Ma, W.-M. Kwok, X. Guan, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648 CrossRef CAS PubMed; (g) M.-C. Tang, D. P.-K. Tsang, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17861 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, S. W.-K. Choi, T.-F. Lai and W.-K. Lee, J. Chem. Soc., Dalton Trans., 1993, 1001 RSC; (b) V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem.–Eur. J., 2011, 17, 130 CrossRef CAS PubMed.
- B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2017, 56, 302 CrossRef CAS PubMed.
- (a) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed; (b) P. Xue, P. Chen, J. Jia, Q. Xu, J. Sun, B. Yao, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569 RSC; (c) Z.-H. Guo, Z.-X. Jin, J.-Y. Wang and J. Pei, Chem. Commun., 2014, 50, 6088 RSC; (d) Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005 CrossRef CAS; (e) P. Xue, J. Sun, P. Chen, P. Wang, B. Yao, P. Gong, Z. Zhang and R. Lu, Chem. Commmun., 2015, 51, 10381 RSC; (f) Y. Zhang, K. Wang, G. Zhuang, Z. Xie, C. Zhang, F. Cao, G. Pan, H. Chen, B. Zou and Y. Ma, Chem.–Eur. J., 2015, 21, 2474 CrossRef CAS PubMed.
- C.-L. Ho, W.-Y. Wong, G.-J. Zhou, B. Yao, Z. Xie and L. Wang, Adv. Funct. Mater., 2007, 17, 2925 CrossRef CAS.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed.
- F.-F. Hung, W.-P. To, J.-J. Zhang, C. Ma, W.-Y. Wong and C.-M. Che, Chem.–Eur. J., 2014, 20, 8604 CrossRef CAS PubMed.
- M. Mizoshita, T. Tani and S. Inagaki, Adv. Mater., 2012, 24, 3350 CrossRef PubMed.
- (a) M. Maus and W. Rettig, Chem. Phys., 1997, 218, 151 CrossRef CAS; (b) F. Chen, J. Zhang and X. Wan, Chem.–Eur. J., 2012, 18, 4558 CrossRef CAS PubMed; (c) P. Gautam, R. Maragni, S. M. Mobin and R. Misra, RSC Adv., 2014, 4, 52526 RSC.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- L. Wallace and D. P. Rillema, Inorg. Chem., 1993, 32, 3836 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1552808. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02410j |
| This journal is © The Royal Society of Chemistry 2017 |