
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatially-resolved soft materials for controlled release – hybrid hydrogels combining a robust photo-activated polymer gel with an interactive supramolecular gel†
Phillip R. A.
Chivers
and
David K.
Smith
 *
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk; Web: http://www.york.ac.uk/chemistry/staff/academic/o-s/dsmith/
First published on 12th September 2017
Abstract
Hybrid hydrogels based on self-assembling low-molecular-weight gelator (LMWG) DBS-CONHNH2 (DBS = 1,3;2,4-dibenzylidene-D-sorbitol) and crosslinked polymer gelator (PG) PEGDM (poly(ethyleneglycol) dimethacrylate) are reported, and an active pharmaceutical ingredient (naproxen, NPX) is incorporated. The use of PEGDM as PG enhances the mechanical stiffness of the hybrid gel (G′ increases from 400 to 4500 Pa) – the LMWG enhances its stability to very high frequency. Use of DBS-CONHNH2 as LMWG enables interactions with NPX and hence allows pH-mediated NPX release – the PG network is largely orthogonal and only interferes to a limited extent. Use of photo-activated PEGDM as PG enables spatially-resolved photo-patterning of robust hybrid gel domains within a preformed LMWG network – the presence of the LMWG enhances the spatial resolution. The photo-patterned multi-domain gel retains pH-mediated NPX release properties and directionally releases NPX into a compartment of higher pH. The two components within these hybrid PG/LMWG hydrogels therefore act largely independently of one another, although they do modify each others properties in subtle ways. Hybrid hydrogels capable of spatially controlled unidirectional release have potential applications in tissue engineering and drug-delivery.
Introduction
Gels are well-known colloidal soft materials with a wide range of commercial applications – from personal care and lubrication industries to drug formulation and biotechnology. Historically, many gels are based on polymer networks where either entanglement/interactions or covalent cross-linking provide the resulting polymer gel (PG) with the desired physical properties – these two types of PG are referred to as physical gels and chemical gels respectively.1 In recent times, polymer chemists have begun to look to combining two different interpenetrating polymer networks (IPNs) to yield gels in which the new material is endowed with properties from each individual gel network.2 Polymer gel IPNs are of great current interest, amongst other applications, for their ability to achieve controlled release of bioactive therapeutics.3An alternative to the PG approach to gels relies on low-molecular-weight gelators (LMWGs) that self-assemble into highly responsive, tunable and versatile nanoscale supramolecular gel networks, with potential for high-tech applications.4 Such LMWG-based materials are physical gels, and attention has recently begun to focus on using them in drug delivery and tissue engineering applications5 – although their exploitation lags far behind that of PGs.1b,6 In the absence of drug–gel interactions, such materials show Fickian release kinetics with the size of the analyte modifying release rate depending on network density,7 but if the drug directly interacts with the gel, release can be significantly modified.8 If the drug molecule is part of the LMWG itself, then chemical breakdown of the gelator prodrug is required to release the active drug.9
Just as PG chemists have turned to combining different gel networks within a single material to achieve synergistic results, the area of hybrid PG/LMWGs, has recently begun to emerge.10 In principle, orthogonal assembly of the two gel networks allows each of them to confer its respective properties on the bulk material without damaging the desirable features of the other. In practical terms, this most commonly means that a PG network is used for its robustness in order to provide additional mechanical strength to a self-assembled LMWG network, which is weak, but highly tunable and responsive to external stimuli such as light, pH, ultrasound and temperature.11 For example, we combined PG agarose with a pH-responsive LMWG and demonstrated that the PG provided mechanical stability while the LMWG maintained its ability to assemble/disassemble within the overall gel via pH cycling.12 Other groups have employed an enzyme-trigger to switch-on an LMWG network.13 Yang and co-workers demonstrated the ability of hybrid hydrogels to remain physically robust while extracting and releasing dyes from aqueous solutions,14 then showed that a polymer additive could boost the anticancer activity of supramolecular LMWG nanofibres based on a taxol derivative.15 Qi and co-workers developed hybrid hydrogels which incorporated taxol and achieved sustained release.16 Feng and co-workers reported the preliminary use of such materials in tissue engineering.17 In addition to the impact of the PG on the mechanical stability of hybrid gels,18 it has also been demonstrated that, inversely, an LMWG network can impact on the mechanical performance of a PG.19 Intriguingly, however, in contrast to IPNs formed from PGs, which have seen an explosion of interest, and a large number of high-impact publications,2,3 reports of hybrid gels based on PG/LMWG combinations remain surprisingly limited. The PG/LMWG strategy has seen little detailed study or wider exploitation, in spite of the fact it clearly has tremendous potential in a wide range of settings.
Recent work in our group has focused on the versatile family of industrially-relevant gelators based on 1,3;2,4-dibenzylidene-D-sorbitol (DBS), which can be synthesised on large scale.20 Of relevance here, we recently demonstrated the photo-initiation of a poly(ethylene glycol) dimethacrylate (PEGDM) gel network within a carboxylic acid-functionalised DBS framework (DBS-COOH).21 We also reported 1,3;2,4-dibenzylidene-D-sorbitol-p,p′-dihydrazide (DBS-CONHNH2),22 capable of stoichiometric non-covalent interaction with active pharmaceutical ingredients (APIs), and pH-dependent drug release.23 In this work we aimed to combine the desirable properties of LMWG (DBS-CONHNH2) and PG (PEGDM) (Fig. 1) in a hybrid hydrogel for the first time, and show that each has an impact on its performance. In this way, the innovative hybrid PG/LMWG hydrogel would exhibit new synergistic forms of behaviour.
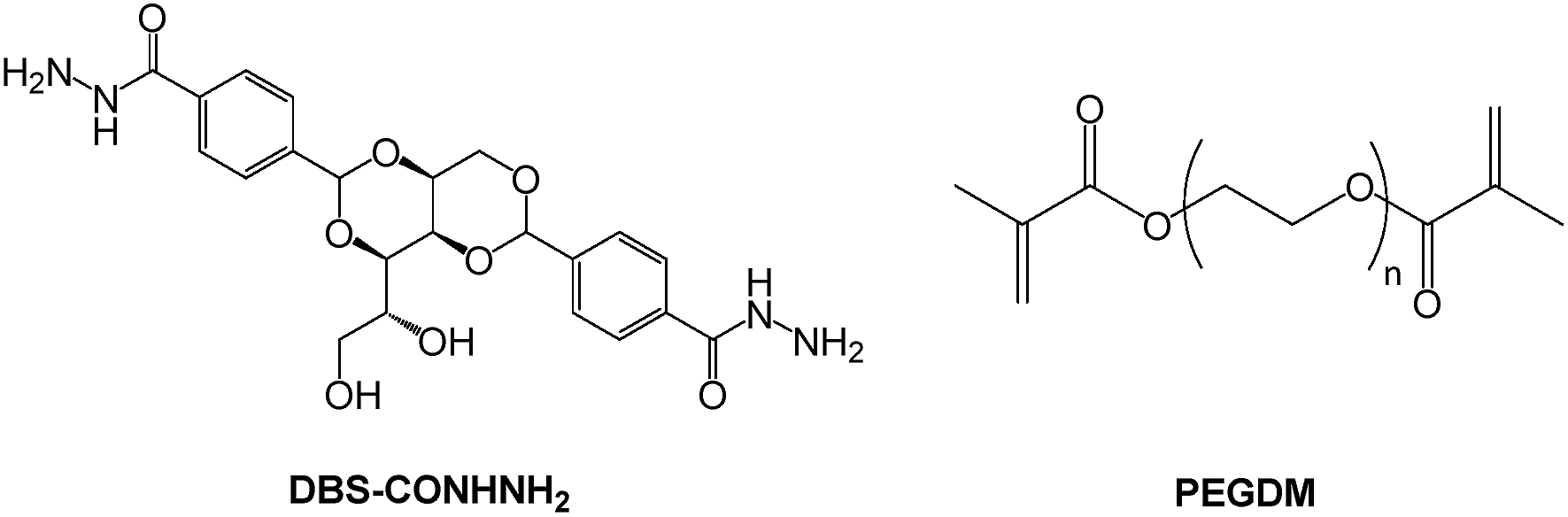 | ||
| Fig. 1 Structures of LMWG (DBS-CONHNH2) and PG (PEGDM) used in this study. | ||
Results and discussion
Synthesis of gelators and gel preparation
LMWG (DBS-CONHNH2) and PG (PEGDM) were synthesised according to previously reported methods.22,24 In brief, DBS-CONHNH2 was prepared in two steps; acid-catalysed condensation of D-sorbitol with two equivalents of methyl-4-formyl benzoate, followed by reaction of the resulting methyl ester with hydrazine monohydrate. Self-assembly of DBS-CONHNH2 in water (6–8 mM) was stimulated by sonication (15 min) followed by a heat/cool cycle, yielding translucent hydrogels. PEGDM was formed by reaction of poly(ethylene glycol) (average Mw = 8000 Da) with two equivalents of methacrylic anhydride in the presence of triethylamine, followed by precipitation in diethyl ether. PEGDM hydrogels were fabricated by UV-photo-polymerisation of an aqueous solution containing a known weight/volume (wt/vol) of PEGDM (minimum gelation concentration = 3% wt/vol) and the photoinitiator (PI) 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (0.05% wt/vol). Gels were photo-polymerised for 30 min under a long-wavelength UV light source (λ = 315–405 nm, PI activation = 365 nm), which gives rise to a covalently-crosslinked, robust chemical gel.25As a result of the orthogonal gel formation methods, it was envisaged that a hybrid hydrogel comprising two separate gel networks could easily be fabricated. Indeed, this would be a much simpler fabrication method than our previously reported hybrid PEGDM/DBS-CO2H system, which required the presence of an additional component to control pH. Hybrid gels were prepared by gelling a 6 mM solution (1 mL) of DBS-CONHNH2, then carefully loading a solution (1 mL) of PEGDM (known wt/vol) and PI (0.05% wt/vol) on top of the LMWG. Diffusion of PEGDM/PI into the gel was then allowed to proceed. When performed in NMR tubes and followed by NMR spectroscopy, this was achieved with ca. 85% efficiency over 3 days (see ESI†) – loading is faster in systems with larger contact areas, but samples were left for 3 days to ensure complete diffusion. Formation of the PG network within the pre-formed LMWG network was initiated by UV-curing (30 min) to yield the hybrid hydrogel. By forming gels in vials with removable bases, it was possible to extract gel cylinders. The presence of PG in these gels allows them to be handled and manipulated – not possible for self-assembled LMWG DBS-CONHNH2 in the absence of PG. Hybrid gels are referred to as x%, where x is the loading (wt/vol) of PEGDM in the supernatant.‡ All hybrid gels contain 6 mM DBS-CONHNH2.
Given the clinical importance of the non-steroidal anti-inflammatory drug naproxen (NPX),26 there has been developing literature interest in its formulation with LMWGs.27 Building on our own previous work,23 NPX was therefore selected for incorporation into these gels by mixing stoichiometric amounts of NPX with DBS-CONHNH2 before gelation via sonication and heating as described above. The PEGDM/PI solution was then pipetted on top of the NPX-loaded DBS-CONHNH2 hydrogel and left for 3 days. Comparable diffusion of PEGDM into the gel was seen (by NMR) as in the absence of NPX. Additionally, little-to-no release of the drug into the supernatant was observed – we suggest NPX is bound to the gel fibres under these conditions. UV-curing, as above, generated self-standing drug-loaded hybrid hydrogels (Fig. 2).
 | ||
| Fig. 2 NPX-loaded free-standing hybrid hydrogels with (a) 5%, (b) 7%, and (c) 10% PEGDM loading. | ||
NMR studies were used to determine how much NPX was bound to the gel fibres. To form gels in NMR tubes, an equimolar mixture of DBS-CONHNH2 and NPX was sonicated and heated to dissolution in D2O containing DMSO as an internal standard (0.028 mol dm−3) before transferring to the NMR tube. After gelation, any NPX signals visible in the NMR spectrum were taken to represent unbound API, the concentration of which was calculated by comparison of the integrals of relevant peaks to that of DMSO (δ = 2.5 ppm). This experiment indicated that in these samples, >92% of the NPX in the gel matrix was bound to the LMWG gel nanofibres (see ESI†) and in this ‘solid-like’ environment could not be observed in the NMR spectrum. Similar results were observed in the hybrid gel.
We employed IR spectroscopy on dried xerogels to learn more about the interactions present within these gels. We focussed initially on key DBS-CONHNH2 IR stretches: O–H (3296 cm−1), N–H (3184 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1639 cm−1). Intriguingly, there were some shifts to the O–H stretch in the presence of the PEGDM gel network (−7 cm−1), while the N–H stretch was broadened. This suggests that there may be some degree of interaction between the two gel networks, and is discussed in more detail below. However, it should also be noted that the hybrid gel was more challenging to dry fully and this may also play a role. Importantly, we then followed the CO stretch of naproxen. In pure solid NPX, the CO stretch appears at 1725 cm−1. In the DBS-CONHNH2 gel, this is shifted slightly to 1727 cm−1 indicative of non-covalent interactions with the gel network, whereas in a PEGDM gel, the NPX CO stretch was unaffected at 1725 cm−1, in-line with the view that NPX does not interact with the PEGDM network. Importantly, in the hybrid gel, the CO stretch was shifted to 1732 cm−1, which suggests non-covalent interactions with DBS-CONHNH2 still take place within the hybrid gel.
O (1639 cm−1). Intriguingly, there were some shifts to the O–H stretch in the presence of the PEGDM gel network (−7 cm−1), while the N–H stretch was broadened. This suggests that there may be some degree of interaction between the two gel networks, and is discussed in more detail below. However, it should also be noted that the hybrid gel was more challenging to dry fully and this may also play a role. Importantly, we then followed the CO stretch of naproxen. In pure solid NPX, the CO stretch appears at 1725 cm−1. In the DBS-CONHNH2 gel, this is shifted slightly to 1727 cm−1 indicative of non-covalent interactions with the gel network, whereas in a PEGDM gel, the NPX CO stretch was unaffected at 1725 cm−1, in-line with the view that NPX does not interact with the PEGDM network. Importantly, in the hybrid gel, the CO stretch was shifted to 1732 cm−1, which suggests non-covalent interactions with DBS-CONHNH2 still take place within the hybrid gel.
We then performed scanning electron microscopy (SEM) of different gels in order to gain more detailed insight into the nanoscale structuring (Fig. 3). Samples were prepared by freeze-drying of the gels with low temperatures being used in order to minimise structural change during drying. DBS-CONHNH2 showed the expected nanofibrillar structure, with fibre dimensions of 25 ± 11 nm. In contrast, PEGDM exhibited a more sheet-like structure, as expected in agreement with the covalently-crosslinked nature of this polymer gel. Imaging of the hybrid gel indicated both sheet-like structures and nanofibres (ca. 20 nm diameter) in agreement with the hypothesis that both networks are present in the hybrid material. Intriguingly, the DBS-CONHNH2 nanofibres appeared to be somewhat aggregated on the sheet-like structures of the PEGDM network – of course, this may be an artefact of the drying process, but it would also be consistent with the observation from IR that there may be interactions between the two networks.
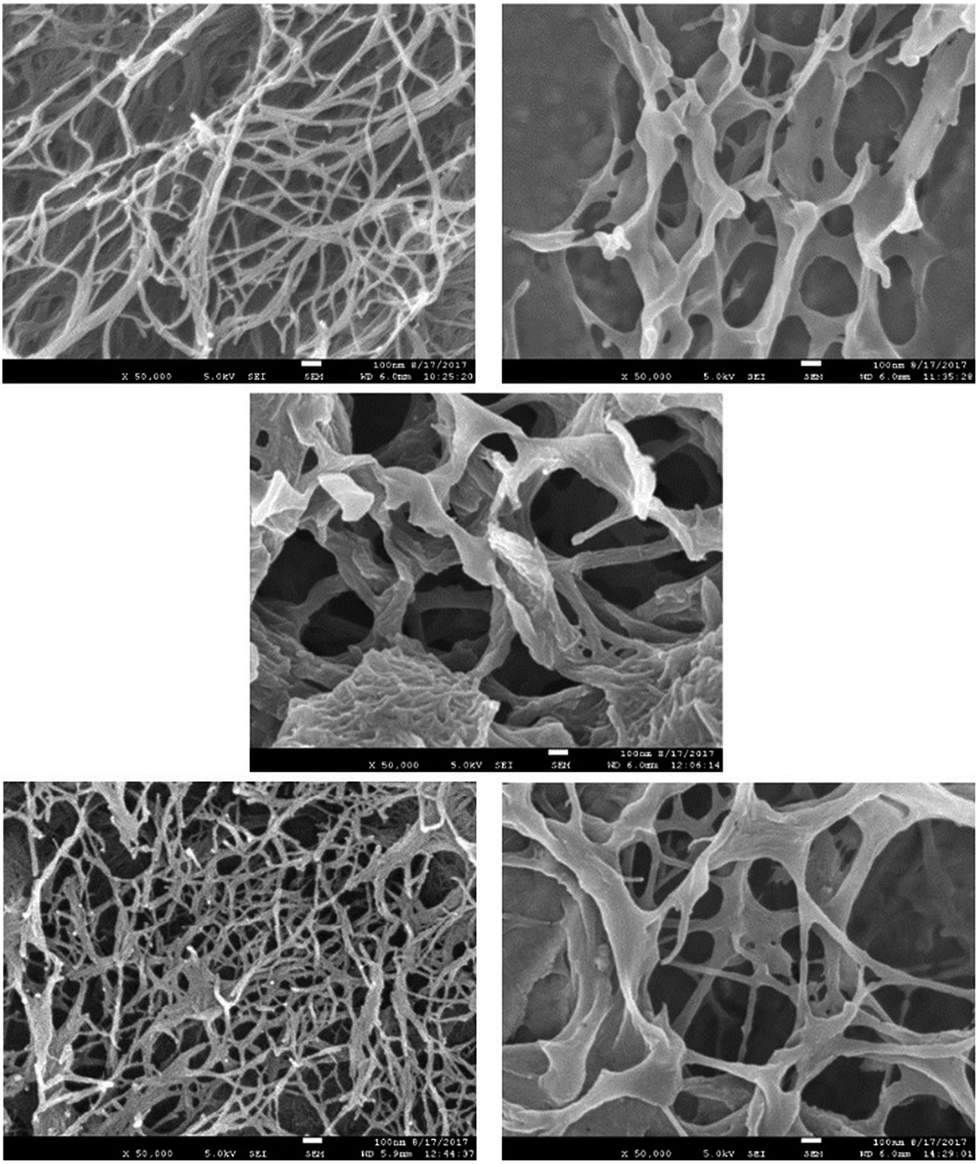 | ||
| Fig. 3 SEM images of freeze-dried xerogels of (top left) DBS-CONHNH2 (top right) 10% PEGDM, (centre) 10% hybrid, (bottom left) DBS-CONHNH2 + NPX, (bottom right) 10% hybrid + NPX. | ||
In the presence of naproxen, the SEM image of DBS-CONHNH2 once again indicated the presence of nanofibres, with diameters of 23 ± 10 nm, and appeared very similar to the SEM image of DBS-CONHNH2 alone. This suggests NPX binds to the DBS-CONHNH2 nanofibres without significant structural perturbation. When the hybrid material was interacted with NPX, nanofibres and sheets could once again both be observed, corresponding to DBS-CONHNH2 and PEGDM. The nanofibre structure appeared more open, and less aggregated onto sheets than in the absence of NPX, which suggests that as the NPX interacts with DBS-CONHNH2, it may compete for interactions between DBS-CONHNH2 and PEGDM and hence open up the structure. We discuss this in further detail below. We did not observe much evidence of NPX crystal formation in these gel samples by SEM.
Macroscopic properties and rheological characterisation
The macroscopic effects of incorporating PEGDM and NPX within the DBS-CONHNH2 network were studied with respect to gel–sol transition temperature (Tgel) and rheological performance. The Tgel value was determined using a simple but reproducible tube-inversion method.28 Gels (0.5 mL) were prepared in vials and placed in a thermostatted water bath. The temperature was raised slowly and the vials inverted at regular intervals. The Tgel was recorded as the temperature at which the gel no longer adhered to the glass surface (Table 1).| LMWG DBS-CONHNH2 | PG PEGDM | Drug NPX | T gel |
|---|---|---|---|
| 6 mM | — | — | 80 °C |
| 8 mM | — | — | 94 °C |
| 6 mM | 5% | — | 85 °C |
| 6 mM | 7% | — | 91 °C |
| 6 mM | 10% | — | >100 °C |
| 6 mM | — | 6 mM | 90 °C |
| 6 mM | 5% | 6 mM | 94 °C |
| 6 mM | 7% | 6 mM | 99 °C |
| 6 mM | 10% | 6 mM | >100 °C |
As expected, increasing LMWG concentration from 6 mM to 8 mM improves thermal stability, as does increasing the concentration of the PG within the hybrid gel matrix from 5% to 7–10%. Interestingly, incorporation of NPX (6 mM) into the gel network also raises Tgel by ca. 10 °C – both for LMWG alone, and in the hybrid PG/LMWG gels. We suggest this may be a result of the relatively high partition coefficient of NPX (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 3.34)29 which means the hydrophobicity of the DBS-CONHNH2:NPX complex is greater than DBS-CONHNH2 alone, hence encouraging more effective self-assembly and network formation.
P = 3.34)29 which means the hydrophobicity of the DBS-CONHNH2:NPX complex is greater than DBS-CONHNH2 alone, hence encouraging more effective self-assembly and network formation.
Rheology using parallel plate geometry was used to characterize mechanical properties. The response to changes in shear strain at constant frequency was probed (see ESI†). The loading of PEGDM has a very significant effect on the rheological properties of the gels. Increasing the PEGDM loading increased the stiffness of the gel in both PG and hybrid gels, indicated by the significant increase in G′ (Fig. 4). In general terms, the hybrid gels behave similarly to the gels of PEGDM alone, suggesting that the PG network dominates the rheological behaviour of the hybrid materials in terms of stiffness. It is worth noting that at 5% PEGDM loading, the hybrid gel is actually less stiff than DBS-CONHNH2, but at 10% PEGDM, the gel becomes significantly stiffer (three times) than DBS-CONHNH2. Reflecting this, the 10% hybrid hydrogel can also be manipulated manually – unlike the other softer materials.
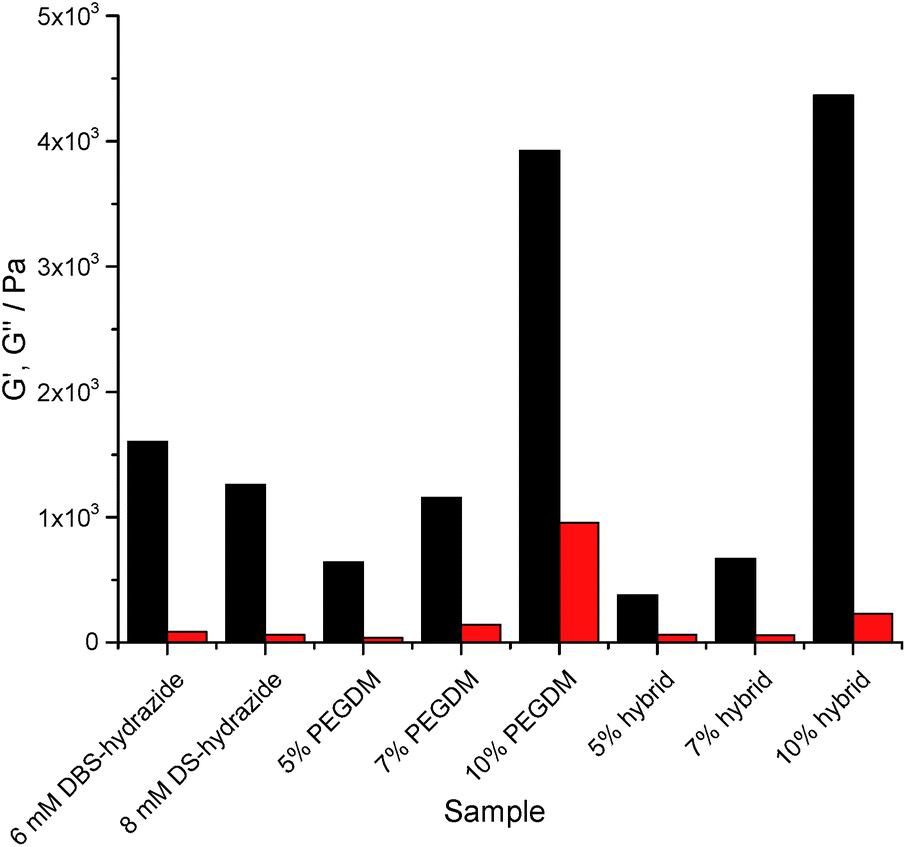 | ||
| Fig. 4 Storage (G′, black) and loss (G′′, red) moduli of hydrogels in the absence of NPX at 0.25% shear strain and 1 Hz. Errors in moduli are ca. ±10% based on repeat experiments. Trends are maintained, and the impact of PEGDM on increasing stiffness of the gel is clearly significant, especially at 10% loading. | ||
In the presence of NPX, the LMWG gel network roughly doubled its stiffness (see ESI†). This supports the view outlined earlier that NPX encourages self-assembly as a result of its relative hydrophobicity. In PEGDM alone (see ESI†), the presence of NPX had little-to-no impact on gel stiffness – as would be expected given that there are no specific interactions proposed between NPX and the PG network. Furthermore, the hybrid gels behave similarly to those of PEGDM alone. This confirms that in NPX-loaded hybrid hydrogels, the PEGDM network remains dominant in determining gel stiffness.
The response of gels to increasing frequency at constant shear strain was also studied (see ESI†). We note that in our frequency sweep experiments, frequency was increased to (unusually) high values (ca. 100 Hz) and this leads to an increase in G′ and G′′, indicative of hardening being induced by these high frequencies, under which conditions gel dynamics are being studied over very short timescales – similar effects have been reported previously.30 Interestingly, the DBS-CONHNH2 gels harden only at ca. 40 Hz, whilst the PEGDM hydrogels harden at only ca. 15 Hz. The hybrid gels incorporating both networks harden at intermediate values of ca. 25–30 Hz. Incorporation of NPX had no significant impact on frequency stability. This suggests that frequency stability is greater for the LMWG DBS-CONHNH2 network – presumably its lower stiffness makes it better able to flex and withstand increasing frequency. Furthermore, the presence of DBS-CONHNH2 within the hybrid gel appears to endow it with some of this flexibility.
This study demonstrates that both components of the hybrid hydrogel provide the material with some favourable rheological characteristics. PEGDM enhances stiffness, overall robustness and ability to be handled, while DBS-CONHNH2 provides the hybrid gels with greater frequency resistance.
Release of NPX from hybrid hydrogels controlled by pH
DBS-CONHNH2 is stable both in molecular terms and as a gel across the pH range ca. 3–11,22 and has also been shown to demonstrate pH-dependent NPX release.23 At pH 4, NPX remained largely bound to gel nanofibres, while at pH 8, NPX was released, as deprotonation of NPX disrupted the acid–base interactions formed with DBS-CONHNH2. Controlled release into a pH 8 medium has potential benefits for oral administration, as releasing NPX under intestinal rather than gastric conditions should enhance drug uptake, maximising the effect of each dose, limiting adverse effects and providing more effective pain relief.31In this work, NPX-loaded gels were covered with buffer, incubated at 37 °C and NPX release into the supernatant was monitored by UV-vis absorbance at 329 nm – negative control experiments were performed on the same gels without NPX. A similar pH-dependence of NPX release from 6 mM DBS-CONHNH2 was observed as previously reported. Over 24 h ca. 25–30% of the total loaded drug was released into pH 4 buffer (Fig. 5 (top)). In contrast, ca. 80–90% was released when using pH 7 and pH 8 buffers. A pH 5.5 buffer led to release of a total amount of NPX which was similar to pH 7 and 8 buffers, and suggests the feasibility of this gel as a topical release medium, particularly useful for pain medications (the surface of skin is pH 5.4–5.9).32 Changing DBS-CONHNH2 concentration from 6 mM to 8 mM had little impact on NPX release.
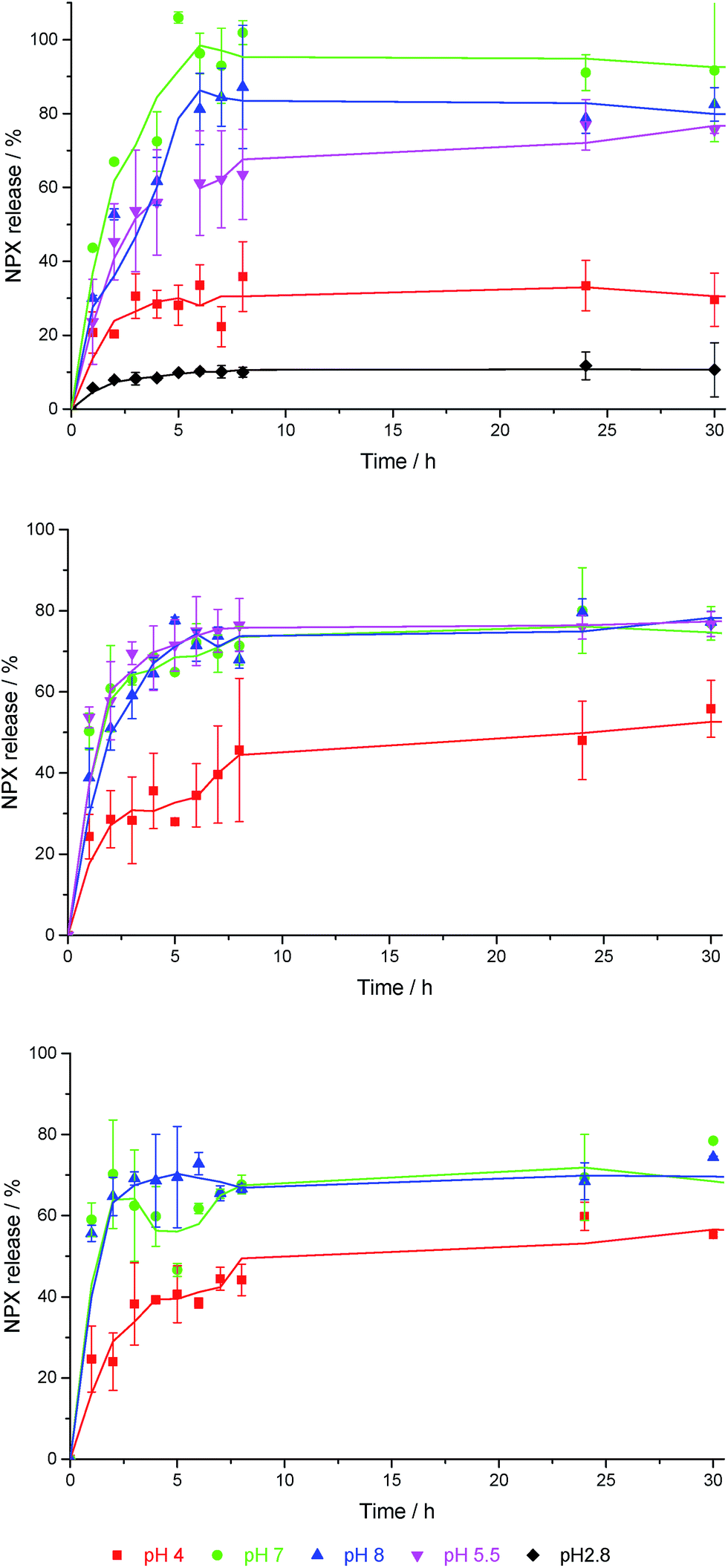 | ||
| Fig. 5 NPX release profiles for (top) 6 mM DBS-CONHNH2, (centre) 5% hybrid gel, and (bottom) 10% hybrid gel, into buffer solutions with different pH values in each case. | ||
Differences in release of NPX at different pH values can be rationalised using the Henderson–Hasselbalch equation:
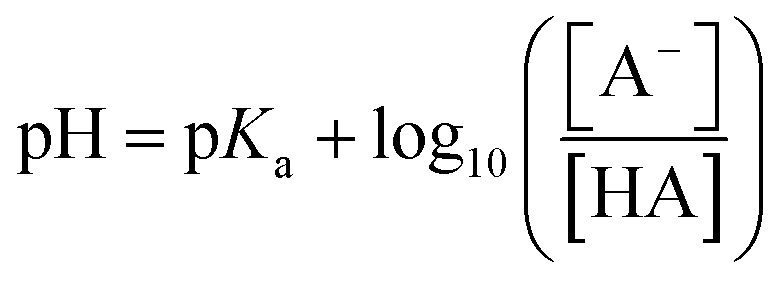 | (1) |
Despite the excellent release properties demonstrated by DBS-CONHNH2,23 the LMWG is much too weak to be manipulated. Although this may be useful for transdermal delivery,32 it limits potential for oral delivery or other applications where mechanical integrity is desirable. We reasoned that the PG PEGDM network may solve the rheological problems, whilst the presence of the LMWG would continue to endow the gel with pH-mediated release characteristics. In this way, the two components in the hybrid hydrogel would cooperate synergistically, playing active roles to control performance.
Both 5% and 10% hybrid hydrogels demonstrate a good degree of pH-dependent NPX release. For samples incubated in pH 7 and 8 buffer, there was a slight decrease in the amount of NPX released at equilibrium from 80–90% to ca. 70% (Fig. 5 (centre) and (bottom)). We suggest this is the result of a greater diffusional barrier to release, especially for NPX distant from the gel–sol interface (see below). Conversely, there is a slight increase in release at pH 4 from 25–30% to ca. 50%. We suggest this is the result of the PEGDM network somewhat disrupting NPX:DBS-CONHNH2 interactions, increasing the proportion of unbound NPX and hence percentage release. A mechanism for this would result from a degree of interaction between PEGDM and DBSCONHNH2, as suggested by the IR and SEM studies described above. Such interactions would mean NPX has to compete for interactions with the DBS-CONHNH2 nanofibres, and hence give rise to a slightly greater amount of free NPX in the hybrid gel. The change in release at each pH value becomes more significant as the PEGDM loading increases, suggesting the PG network is indeed responsible for causing these changes. Once again, SEM imaging (see ESI†) after soaking these hybrid gels in pH 4 buffer did not indicate significant morphological change.
Importantly, when using 5% and 10% PEGDM in the absence of DBS-CONHNH2, NPX release over 24 hours was completely independent of pH (see ESI†), confirming that specific interactions between DBS-CONHNH2 and NPX are essential for pH-dependent release in the hybrid hydrogels and that this is not simply a feature of NPX formulated within any gel in this manner. At 10% loading, the PEGDM PG alone prevented complete release of NPX, with only ca. 80% being released in all conditions, presumably this is due to some NPX becoming trapped in the crosslinked PG network. This agrees with the effect of PEGDM loading on NPX release from the hybrid gels described above in which the presence of PEGDM somewhat decreased the total release at pH 7/8. We therefore conclude that DBS-CONHNH2 partly retains its properties when mixed with PEGDM, and endows the hybrid hydrogel with its own pH-control characteristics – albeit somewhat mediated by the PEGDM loading.
We also considered the initial kinetics of NPX release (see ESI for details of method†). Assuming zero-order kinetics it was found that for DBS-CONHNH2 the initial release rate when the pH of the receiving solution was less than the pKa of NPX was ca. 6 × 10−9 mol min−1. At pH values greater than the pKa, the release rate increased two-fold to ca. 2.5–3.5 × 10−8 mol min−1. A small decrease in release rate was calculated for the 5% hybrid gel, consistent with an increase in resistance to diffusion. However, incorporating 10% PEGDM significantly increased NPX release rate under all conditions. We propose, as described above, that at high PG concentrations interactions between DBS-CONHNH2 and PEGDM mean some NPX is ‘free’ within the hybrid gel, increasing initial release kinetics, but diffusional effects then limit the total equilibrium release of NPX as some of it remains trapped. For gels made from PEGDM alone, the release rate was essentially independent of pH – demonstrating that interactions with the LMWG nanofibres are responsible for controlling NPX release kinetics.
Photopatterning of multidomain gels
Importantly, the choice of photo-initiated PEGDM as the PG network in our hybrid gels introduces the exciting possibility of an additional level of control over these materials – spatial control. By performing photo-irradiation through a mask it is, in principle, possible to pattern regions of PG network within a pre-formed LMWG matrix. We reasoned this may allow us to ‘write’ patterns into gels loaded with a bioactive component – in this case NPX. Although photo-patterning is relatively common within hybrid IPN polymer gels,33 the only example in which a PG has been photo-patterned within an LMWG matrix is that published by us based on pH-responsive DBS-COOH.21To demonstrate that spatial resolution could be achieved for the simple DBS-CONHNH2 and PEGDM system, the LMWG was pre-dissolved in DMSO (0.4 mL) and added to boiling water (9.6 mL). The concentration of DBS-hydrazide in this final solution was 6 mM. The hot solution was transferred to a square glass mould (width = 50 mm, depth = 10 mm) and left to cool, upon which a translucent gel formed (depth = 4 mm). A solution of PEGDM (10% wt/vol) and PI (0.05% wt/vol) was then loaded onto this gel and left for 3 days to diffuse in, after which the supernatant was removed. Acetate photomasks were printed and fixed over the mould – the use of laser-printed acetate photomasks allows for simple preparation of a very wide range of 2D-photo-patterned geometries, as they can simply be printed with any desired pattern. The mould was placed in ice and the sample photo-polymerised under a UV lamp for 30 min. Performing the experiment in this geometry allows for effective UV penetration and prevents heating effects that can disrupt the pre-formed LMWG DBS-CONHNH2 gel. Areas of the gel shielded from the UV light by the photomask were not photopolymerised and remained as a weak LMWG material composed of self-assembled DBS-CONHNH2 nanofibres and unpolymerised PEGDM. However, in those areas that UV light could access through the mask, the PEGDM network was crosslinked and the material was much more robust, containing both PG and LMWG networks. Photopatterning was achieved with excellent spatial resolution, in stark contrast to control experiments carried out in the absence of the supporting DBS-CONHNH2 LMWG matrix, in which all of the PEGDM polymerised regardless of whether or not the area was masked. This highlights the vital role of the pre-formed LMWG network in controlling the formation of a spatially-resolved multi-domain gel system. This is presumably because performing the ‘patterning-in’ step within a gel limits diffusion and convection, hence allowing photo-crosslinking polymerisation to proceed with much greater spatial control.
The above methodology was then adapted for fabrication of NPX-loaded multidomain gels by dissolving the drug in DMSO alongside DBS-CONHNH2. In this way, we could pattern the hybrid gel, pre-loaded with bioactive NPX, into any shape we chose – to exemplify, we generated a photo-‘patterning in’ a ring-shaped NPX-loaded hybrid gel domain, with LMWG domains inside and outside the ring (Fig. 6). These LMWG domains could then be easily removed from the multi-domain gel by washing as they are rheologically very weak, hence leaving behind the free-standing NPX-loaded ring-shaped PG/LMWG object. This simple ‘pattern exposure’ washing step was achieved using a low pressure jet of water from a ‘squeezy bottle’ which fully breaks down and removes the soft LMWG, while the robust hybrid PG/LMWG domain remains completely undamaged. Other elegant approaches to free-standing supramolecular hydrogels have also very recently been reported.34
 | ||
| Fig. 6 (a) A ring-shaped photomask was applied over (b) the NPX-loaded LMWG gel and the pattern transferred to the gel by UV-photopatterning. The weak LMWG interior and exterior were easily removed to leave (c) the more robust hybrid hydrogel ring. | ||
In summary, the use of PEGDM as the PG component endows the materials with the capacity to be photo-patterned. Spatial resolution is enhanced by the presence of the LMWG network – once again illustrating how the two components subtly influence one another's performance within these hybrid PG/LMWG gels. Importantly, the patterning step still takes place successfully in the presence of a UV-stable bioactive component present within the gel, in this case, NPX.
Directional release of NPX from a photopatterned gel matrix
The ability to pattern robust PG domains of any shape into an interactive LMWG afforded us a unique opportunity to achieve spatially-resolved and directional NPX release. To demonstrate the potential of this material to preferentially release an active ingredient in one direction over another, a 10 mL sample of NPX-loaded (6 mM) 10% hybrid gel was prepared in a mould as described above. An acetate photomask was fixed over the mould and the sample photo-polymerised with UV light for 15 min. This ‘patterned-in’ a vertical band of hybrid hydrogel (width = 20 mm) down the centre of the mould, from which the surrounding LMWG was easily removed using the subsequent ‘pattern exposure’ washing step (Fig. 7). Buffer solutions with pH values of 2.8 and pH 7 (1.5 mL) were then pipetted onto either side of this band and the solutions stirred using magnetic fleas. The release of NPX into each compartment at room temperature was monitored by UV-vis spectroscopy over 3 hours. At each time point, 100 μL of buffer solution was removed and diluted to 2 mL in a UV cuvette, with fresh buffer being added to maintain constant volume – we allowed for this systematic removal of some NPX in our release calculations. | ||
| Fig. 7 (Centre) a vertical band of hybrid gel was photopatterned into the LMWG. Release of NPX into (left) pH 2.8 buffer and (right) pH 7 buffer (right) was monitored over a 3 hour period. | ||
A significant difference between release rates into the two compartments was observed over the duration of the experiment – after 3 hours the amount of NPX released into pH 7 buffer was 6 times greater than into pH 2.8 buffer (Fig. 7). The initial release rate at pH 7 was ca. 10 times greater (1.15 × 10−8 mol min−1) than under acidic conditions (1.18 × 10−9 mol min−1). Interestingly, as incorporation of NPX somewhat clouds the gel, the gradual differences in release from either side of hybrid gel band could even be visually observed (Fig. 8). The left-hand side of the gel in contact with pH 2.8 buffer remained largely cloudy, while the right hand side became increasingly transparent as the NPX is released into the pH 7 buffer. The initial release rate of NPX from this photo-patterned hybrid gel at pH 7 is slower than in the release studies described above. This is believed to be due to the lower experimental temperature and decreased contact surface area of the gel:buffer interface in this geometry.§ The separate pH values of the two receiving buffer solutions were maintained throughout the 3 hour experiment, illustrating the ability of the patterned hybrid gel to act as an effective membrane between compartments on this timescale. If this experiment was left for much longer (e.g. days/months), we anticipate it would eventually come to equilibrium of pH and drug concentration, but importantly, the gel significantly retards this process. Further, if the released drug was being used up at a faster rate, for example in a biological process, removing the system from equilibrium, this system would offer an effective way of achieving directional control over outcomes. Experiments moving gels away from equilibrium are an emerging interest,35 and focussing on coupling biological processes in specific compartments is a key future target.
 | ||
| Fig. 8 The increase in NPX loss from the hybrid gel preferentially into pH 7 buffer (to the right of the gel band) can be seen as the gel gradually becomes optically transparent on the right hand side over time: (top left) 0.5 h (top right) 3 h. The lower image represents a summary of the controlled, directional release experiment. | ||
In the literature, previous examples of unidirectional release from gels achieve this by either protection36 or deprotection of one face of a drug reservoir37 or application of an external field.38 Our report constitutes an innovative approach to the important goal of unidirectional release, driven by pH differences between compartments. Importantly, the process is not just controlled by drug solubility – the PG alone releases NPX equally into solutions of all pH values. Directional release is controlled by the interactions between DBS-CONHNH2 and NPX in different pH regimes.
Crucially, controlled directional release without the need for any further modification of the gel matrix is a result of combining (i) the robust self-supporting nature of these materials endowed by PEGDM, with (ii) the pH-dependence of the NPX:DBS-CONHNH2 interactions, and (iii) the shape control induced by PEGDM photo-patterning. In this way, both components of the hybrid gel actively contribute to the observed innovative function. This approach suggests the development of spatially-defined materials which only release their cargoes when brought into contact with a suitable environment. Such systems could have considerable value for applications in spatially-targeted drug delivery from implanted gels or tissue engineering with spatially-resolved growth factor release depending on the growing tissue in contact with the gel surface. The value of shape-specific release of drugs/bioactives is well understood in the PG field,39 and we suggest many of the principles developed there can be transferred to the new field of hybrid PG/LMWG hydrogels explored here.
Conclusions
In conclusion, we have developed a simple, scalable approach to hybrid hydrogels based on a self-assembling LMWG (DBS-CONHNH2) and a covalently cross-linked PG (PEGDM). The components combine synergistically to endow their own properties onto the overall material. We have carefully characterised the impact of both components and characterised the extent to which they are orthogonal:• The PG endows the hybrid hydrogels with stiffness and at 10% loading good ability to be handled. This is not adversely affected by the LMWG, but the softer LMWG network has greater resistance to higher frequencies, which is also partly passed on to the hybrid gel.
• The LMWG controls NPX release depending on pH, based on the pKa value of the drug. In the absence of LMWG, the PG cannot control drug release, but increasing amounts of PG in the hybrid gel limit overall release and pH selectivity, potentially as a result of competitive interactions of the LMWG nanofibres with the PG network.
• The PEGDM PG can be formed with shape-specificity using photo-patterning trough simple design-printed acetate masks – however, the presence of the LMWG is essential for this to be achievable with spatial resolution.
• The LMWG endows the spatially-defined hybrid gels with pH-selective release properties, as demonstrated by preferential directional release into a compartment of pH 7 buffer in competition with a compartment of pH 2.8 buffer.
In summary, we conclude that the PG and LMWG both clearly express their functionality in the hybrid hydrogel and are largely orthogonal, but note that when both networks are present they do impact on each other in subtle ways.
These spatially-resolved hybrid hydrogels have potential for a wide range of applications, from more evident ones such as drug delivery and tissue engineering, to more speculative ones including nano-electronics and microfluidics. We suggest that using using two-photon methods to achieve detailed 3D pattering of such hybrid hydrogels will be of particular future interest. Given the intense interest in IPNs incorporating two different PGs, we propose that the surprisingly over-looked synergistic combination of LMWGs and PGs – a field still in its infancy – offers great potential for future exploitation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank EPSRC and University of York for funding (PRAC).Notes and references
- (a) Polymer Gels: Fundamentals and Applications, ed. H. B. Bohidar, P. Dubin and Y. Osada, American Chemical Society, Washington DC, 2002 Search PubMed; (b) J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS.
- (a) C. W. Peak, J. J. Wilker and G. Schmidt, Colloid Polym. Sci., 2013, 291, 2031–2047 CrossRef CAS; (b) Z. Lin, C. Qiang and X. Kun, Prog. Inorg. Chem., 2014, 26, 1032–1038 Search PubMed; (c) E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- (a) A. Lohani, G. Singh, S. S. Bhattacharya and A. Verma, J. Drug Delivery, 2014, 583612 Search PubMed; (b) T. M. Aminabhavi, M. N. Nadagouda, U. A. More, S. D. Joshi, V. H. Kulkami, M. N. Noolvi and P. V. Kulkami, Expert Opin. Drug Delivery, 2015, 12, 669–688 CrossRef CAS PubMed.
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed; (c) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed; (d) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed; (e) D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Rev., 2017, 46, 2404–2420 RSC.
- (a) K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC; (b) J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed; (c) M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- (a) T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS; (b) W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- (a) S. Cao, X. Fu, N. Wang, H. Wang and Y. Yang, Int. J. Pharm., 2008, 357, 95–99 CrossRef CAS PubMed; (b) S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef CAS PubMed; (c) S. Koutsopoulos, L. D. Unsworth, Y. Nagaia and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623–4628 CrossRef CAS PubMed.
- (a) Y. Nagai, L. D. Unsworth, S. Koutsopoulos and S. Zhang, J. Controlled Release, 2006, 115, 18–25 CrossRef CAS PubMed; (b) S. Soukasene, D. J. Toft, T. J. Moyer, H. Lu, H.-K. Lee, S. M. Standley, V. L. Cryns and S. I. Stupp, ACS Nano, 2011, 5, 9113–9121 CrossRef CAS PubMed; (c) D. Limón, E. Amirthalingam, M. Rodrigues, L. Halbaut, B. Andrade, M. L. Garduño-Ramirez, D. B. Amabilino, L. Pérez-Garcia and A. C. Campo, Eur. J. Pharm. Biopharm., 2015, 96, 421–436 CrossRef PubMed; (d) S. Gupta, M. Singh, A. Redy, P. S. Yavvari, A. Srivasatava and A. Bajaj, RSC Adv., 2016, 6, 19751–19757 RSC; (e) D. Limón, C. Jiménez-Newman, A. C. Calpena, A. González-Campo, D. B. Amabilino and L. Pérez-Garcia, Chem. Commun., 2017, 53, 4509–4512 RSC; (f) R. Parveen, B. Sravanthi and P. Dastidar, Chem.–Asian J., 2017, 12, 792–803 CrossRef CAS PubMed.
- (a) K. J. C. van Bommel, M. C. A. Stuart, B. L. Feringa and J. van Esch, Org. Biomol. Chem., 2005, 3, 2917–2920 RSC; (b) S. Bhuniya, Y. J. Seo and B. H. Kim, Tetrahedron Lett., 2006, 47, 7153–7156 CrossRef CAS; (c) P. K. Vemula, G. A. Cruikshank, J. M. Karp and G. John, Biomaterials, 2009, 30, 383–393 CrossRef CAS PubMed; (d) Y. Gao, Y. Kuang, Z.-F. Guo, Z. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS PubMed; (e) H. Wang, C. Yang, L. Wang, D. Kong, Y. Zhang and Z. Yang, Chem. Commun., 2011, 47, 4439–4441 RSC; (f) Y. Zhou, H. Cui, C. Shu, Y. Ling, R. Wang, H. Li, Y. Chen, T. Lu and W. Zhang, Chem. Commun., 2015, 51, 15294–15296 RSC.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC.
- (a) J. Wang, X. Miao, Q. Fengzhao, C. Ren, Z. Yang and L. Wang, RSC Adv., 2013, 3, 16739–16746 RSC; (b) Y. Mao, T. Su, Q. Wu, C. Liao and Q. Wang, Chem. Commun., 2014, 50, 14429–14432 RSC; (c) Q. Wei, W. Xu, M. Liu, Z. Wu, L. Cheng and Q. Wang, J. Mater. Chem. B, 2016, 4, 6302–6306 RSC.
- (a) J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC; (b) J. Wang, H. Wang, Z. Song, D. Kong, X. Chen and Z. Yang, Colloids Surf., B, 2010, 80, 155–160 CrossRef CAS PubMed.
- C. Yang, M. Bian and Z. Yang, Biomater. Sci., 2014, 2, 651–654 RSC.
- R. Huang, W. Qi, L. Feng, R. Su and Z. He, Soft Matter, 2011, 7, 6222–6230 RSC.
- P. Li, X.-Q. Dou, C.-L. Feng and D. Zhang, Soft Matter, 2013, 9, 3750–3757 RSC.
- (a) B. O. Okesola, S. K. Suravaram, A. Parkin and D. K. Smith, Angew. Chem., Int. Ed., 2016, 55, 183–187 CrossRef CAS PubMed; (b) Y. Xie, J. Zhao, R. Huang, W. Qi, Y. Wang, R. Su and Z. He, Nanoscale Res. Lett., 2016, 11, 184 CrossRef PubMed; (c) X. Gong, C. Branford-White, L. Tao, S. Li, J. Quan, H. Nie and L. Zhu, Mater. Sci. Eng., C, 2016, 58, 478–486 CrossRef CAS PubMed; (d) E. Çelik, C. Bayram, R. Akçapınar, M. Türk and E. B. Denkbaş, Mater. Sci. Eng., C, 2016, 66, 221–229 CrossRef PubMed; (e) Y. Yu, Y. Wang and C. Feng, Chem. Res. Chin. Univ., 2016, 32, 872–876 CrossRef CAS.
- (a) Y. Ohsedo, M. Taniguchi, K. Saruhashi and H. Watanabe, RSC Adv., 2015, 5, 90010–90013 RSC; (b) J. Zhang, W. Ji, T. Liu and C. Feng, Macromol. Chem. Phys., 2016, 217, 1197–1204 CrossRef CAS.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 CAS.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- E. J. Howe, B. O. Okesola and D. K. Smith, Chem. Commun., 2015, 51, 7451–7454 RSC.
- S. Lin-Gibson, S. Bencherif, J. A. Cooper, S. J. Wetzel, J. M. Antonucci, B. M. Vogel, F. Horkay and N. R. Washburn, Biomacromolecules, 2004, 5, 1280–1287 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- (a) F. L. Lanza, F. K. L. Chan and E. M. M. Quigley, Am. J. Gastroenterol., 2009, 104, 728–738 CrossRef PubMed; (b) P. McGettigan and D. Henry, PLoS One, 2011, e1001098 CAS; (c) N. Bhala, J. Emberson, A. Merhi, S. Abramson, N. Arber, J. A. Baron, C. Bombardier, C. Cannon, M. E. Farkouh and G. A. FitzGerald, et al. , Lancet, 2013, 382, 769–779 CrossRef CAS PubMed.
- (a) J. Mamunder, J. Deb, M. R. Das, S. S. Jana and P. Dastidar, Chem. Commun., 2014, 50, 1671–1674 RSC; (b) N. Goyal, H. P. R. Mangunuru, B. Parikh, S. Shrestha and G. Wang, Beilstein J. Org. Chem., 2014, 10, 3111–3121 CrossRef PubMed; (c) H. Vilaca, A. C. L. Hortelao, E. M. S. Castanheira, M. J. R. P. Queiroz, L. Hilliou, I. W. Hamley, J. A. Martins and P. M. T. Ferreira, Biomacromolecules, 2015, 16, 3562–3573 CrossRef CAS PubMed; (d) X. Li, G. Pu, X. Yu, S. Shi, J. Yu, W. Zhao, Z. Luo, Z. He and H. Chen, RSC Adv., 2016, 6, 62434–62438 RSC.
- S. R. Raghavan and B. H. Cipriano, in Molecular Gels, Materials with Self-Assembled Fibrillar Networks, ed. R.G. Weiss and P. Terech, Springer, Dordrecht, Netherlands, 2006, pp. 235–236 Search PubMed.
- F. Barbato, M. I. La Rotonda and F. Quaglia, J. Pharm. Sci., 1997, 86, 225–229 CrossRef CAS PubMed.
- (a) E. R. Draper, L. L. E. Mears, A. M. Castilla, S. M. King, T. O. McDonald, R. Akhtar and D. J. Adams, RSC Adv., 2015, 5, 95369–95378 RSC; (b) Y. Abidine, V. M. Laurent, R. Michel, A. Duperray, L. I. Palade and C. Verdier, Europhys. Lett., 2015, 109, 38003 CrossRef.
- C. Bombardier, L. Laine, A. Reicin, D. Shapiro, R. Burgos-Vargas, B. Davis, R. Day, M. B. Ferraz, C. J. Hawkey, M. C. Hochberg, T. K. Kvien and T. J. Schnitzer, N. Engl. J. Med., 2000, 343, 1520–1528 CrossRef CAS PubMed.
- K. S. Paudel, M. Milewski, C. L. Swadley, N. K. Brogden, P. Ghosh and A. L. Stinchcomb, Ther. Delivery, 2010, 1, 109 CrossRef CAS.
- J. W. Nichol, S. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef CAS PubMed.
- M. Lovrak, W. E. J. Hendriksen, C. Maity, S. Mytnyk, V. Van Steijn, R. Eelkema and J. H. van Esch, Nat. Commun., 2017, 15317 CrossRef CAS PubMed.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017 10.1039/c7cs00246g.
- (a) K. M. Ainslie, C. M. Kraning and T. A. Desai, Lab Chip, 2008, 8, 1042–1047 RSC; (b) M. McGinity, J. Floyd, J. McGinity and F. Zhang, Drug Dev. Ind. Pharm., 2017, 43, 1–28 CrossRef PubMed; (c) P. Govindasamy, B. R. Kesavan and J. K. Narasimha, Asian Pac. J. Trop. Biomed., 2013, 3, 995–1002 CrossRef CAS PubMed.
- (a) P. Marizza, S. S. Keller, A. Müllertz and A. Boisen, J. Controlled Release, 2014, 173, 1–9 CrossRef CAS PubMed; (b) M. F. Bédard, B. G. De Geest, H. Möhwald, G. B. Sukhorukov and A. G. Skirtach, Soft Matter, 2009, 5, 3927–3931 RSC.
- X. Ge, J. Huang, J. Xu and G. Luo, Lab Chip, 2014, 14, 4451–4454 RSC.
- (a) D. A. Edwards, J. Hanes, G. Caponetti, J. Hrkach, A. Ben-Jebria, M. L. Eskew, J. Mintzes, D. Deaver, N. Lotan and R. Langer, Science, 1997, 276, 1868–1871 CrossRef CAS PubMed; (b) V. A. Liu and S. N. Bhatia, Biomed. Microdevices, 2002, 4, 257–266 CrossRef CAS; (c) T. Halil, T. Tsinman, J. G. Sanchez, B. J. Jones, G. Camci-Unal, J. W. Nichol, R. Langer and A. Khademhosseini, J. Am. Chem. Soc., 2011, 133, 12944–12947 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental methods and further data from assays. See DOI: 10.1039/c7sc02210g |
| ‡ The actual wt/vol PEGDM in the 5%, 7% and 10% hybrid gels were estimated from NMR studies to be 4.4, 5.8 and 8.6% respectively (see ESI†). |
| § The interface contact area for the ‘gel-in-vial’ release experiments was ca. 10 cm2 compared to 2 cm2 per interface for the directional release experiment. |
| This journal is © The Royal Society of Chemistry 2017 |