
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorometric probing of the lipase level as acute pancreatitis biomarkers based on interfacially controlled aggregation-induced emission (AIE)†
Jie
Shi
 a,
Qianchun
Deng
a,
Chuyun
Wan
a,
Mingming
Zheng
a,
Fenghong
Huang
*a and
Bo
Tang
*b
a,
Qianchun
Deng
a,
Chuyun
Wan
a,
Mingming
Zheng
a,
Fenghong
Huang
*a and
Bo
Tang
*b
aHubei Key Laboratory of Lipid Chemistry and Nutrition, Oil Crops and Lipids Process Technology National & Local Joint Engineering Laboratory, Key Laboratory of Oilseeds Processing, Ministry of Agriculture, Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan 430062, China. E-mail: shij@whu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan 250014, China. E-mail: tangb@sdnu.edu.cn
First published on 23rd June 2017
Abstract
As a sudden inflammation of the pancreas, acute pancreatitis presents severe complications and a high mortality rate, despite treatment. Lipase in serum serves as an essential biomarker of acute pancreatitis and even pancreatic cancer. Therefore, developing robust, convenient and sensitive probing of lipase levels is greatly needed. In this work, we present glutamate functionalized tetraphenylethylene (TPE) as a “turn-on” fluorescent probe (S1) based on the aggregation-induced emission (AIE) mechanism for lipase levels with new recognition units. In heterogeneous media, the hydrophilic amino and carboxyl groups in the probe were specifically introduced to facilitate its full access to lipase at the oil–water interface and achieve an interfacially controlled AIE process. The linear response of fluorescence ranging from 0 to 80 U L−1, which included the concentration range of the lipase level in human serum, considering the dilution factor if necessary, the limit of detection as low as 0.13 U L−1, and the fast response time (7 min) were determined. The value of the apparent Michaelis–Menten constant (Km) was obtained as 4.23 μM, which indicated superior affinity between lipase and the probe molecule. The selectivity, photostability, dynamic monitoring of the enzymatic reaction, and preliminary commercial enzyme activity screening were summarized. As far as we know, this is the fastest, easiest and most sensitive method for lipase level probing in the reported literature. Finally, probing the lipase level for the first time in real human serum samples was also conducted successfully.
Introduction
Acute pancreatitis is a sudden inflammation of the pancreas.1 In severe cases, acute pancreatitis can result in bleeding into the gland, serious tissue damage, infection, and cyst formation.2 The mortality for severe acute pancreatitis can be 10 to 30%, sufferers often endure pain and malnutrition and are most likely left with a higher risk of pancreatic cancer (mortality rate > 99%).3 The diagnosis of pancreatic problems is often difficult because the organ is relatively inaccessible and treatments are therefore delayed.4 However, lipase in serum, which is an exocrine enzyme from the pancreas, has been employed as an essential biochemical indicator for the early diagnosis and condition assessment of acute pancreatitis.5–13 Also, measuring the serum lipase level associated with pancreatic tumor markers CA 19–9 can diagnose pancreatic cancer.14,15 Under the conditions of pancreatic injury, pancreatic autolysis induces significant elevation of the serum lipase level, and a high level of lipase strongly suggests acute pancreatitis.16,17 Therefore, the development of a robust, convenient and sensitive detection of the lipase level is of great significance in terms of both clinical diagnoses and a pathological analysis towards acute pancreatitis.So far, to our knowledge, very few methods have been established for lipase probing. Traditional methods, such as colorimetric or turbidimetric methods, usually require sample processing and complex instrumentation and have various limitations. Therefore, some new methods have emerged to overcome these obstacles. For instance, Tian’s group developed some gold nanoparticle (GNP) based biosensors for lipase assays and colorimetric probes using Tween 20 functionalized GNPs, and they reported one-pot nanoparticle growth.18,19 Moreover, some drawbacks still exist for the reported lipase assays, and some specific substrates and indicators have been used as the main screening tools, however, all of these possess relatively complicated production processes and suffer from lower sensitivity. Due to this, their application in the detection of the lipase level in real biological samples and as early warnings of acute pancreatitis cannot be implemented. Up until now, fluorometric assays have received remarkable attention due to their convenience, unparalleled sensitivity, simplicity, rapid implementation, noninvasive monitoring capability and usability in biological samples.20–37 Nevertheless, fluorescent probes for lipase level detection was a virgin area until our work was published.38 In our previous work, an aggregation-induced emission (AIE)-based fluorescence probe bearing characteristics to rapidly visualize the lipase activities was constructed with compound P1. Unfortunately, however, the sensitivity and detection limit were quite poor and the range of detection was outside the concentration range of the lipase level in human serum. Therefore, in order to obtain a probe with excellent performance towards lipase, the affinity between the probe and enzyme must be greatly improved by considering the interfacial reaction characteristics of lipase, and the improvement of the interfacial catalytic efficiency is a key factor in the limitation of detection and sensitivity of the probe molecule.
Herein, we report a new fluorometric “turn-on” detection of the lipase level by manipulating the deaggregation and aggregation of probe S1. The whole design rationale is illustrated in Scheme 1 and explained as follows: (1) probe S1 is derived from a tetraphenylethylene (TPE) core with two glutamate units because TPE derivatives are known for their fantastic AIE behavior; (2) lipase is a heterogeneous catalytic enzyme, and it is well known that the best catalytic performance of lipase can be achieved at the oil–water interface. Therefore, by coupling the glutamate units onto the TPE core, we could notably improve the solubility of the AIE-active probe in a hydrophobic medium with the existence of alkyl chains. In contrast, the hydrophilicity of S1 was significantly increased by the absence of amino and carboxyl groups, facilitating full access to the lipase in the aqueous phase at the oil–water interface, thus an interfacial control effect can be conducted simply via the hydrophilic group; (3) as shown in Scheme 1, the lipase-catalyzed cleavage of the ester group in probe S1 will induce the transformation of S1 into 4,4′-dihydroxy tetraphenylethylene (see Scheme S1,†TPE-2OH), and the glutamate units will be cut off, which may release an insoluble TPE residue (TPE-2OH) into the solvent. Accordingly, the aggregation of 4,4′-dihydroxy tetraphenylethylene in solvent will occur and result in strong blue emission. That is to say, it will turn on the fluorescence; (4) due to the presence of the interfacial control effects, the very low lipase level in some real biological samples can be probed, and as the glutamate units are the specific substrates for the lipase hydrolysis reaction, S1 could serve as a selective probe for the lipase level in real biological samples like serum. To this end, through this pathway, a simple fluorescence turn-on assay for lipase probing can be established with S1, and the catalytic rate of reaction can be significantly improved by the AIE-based interfacial control effect, and therefore, the detection of lipase with higher sensitivity and a lower detection limit is achieved. As far as we know, no reports have been published to date on the application of a fluorescent probe in testing the lipase level of real human serum samples. This work solved the drawbacks of the high detection limit and low sensitivity of the lipase probe and built an analytical method for the early warning of acute pancreatitis. We believe that research into the screening of the lipase level would not only broaden the spectrum of AIE luminogen-based enzyme assays but also provide a new approach to improving the catalytic efficiency of the interface.
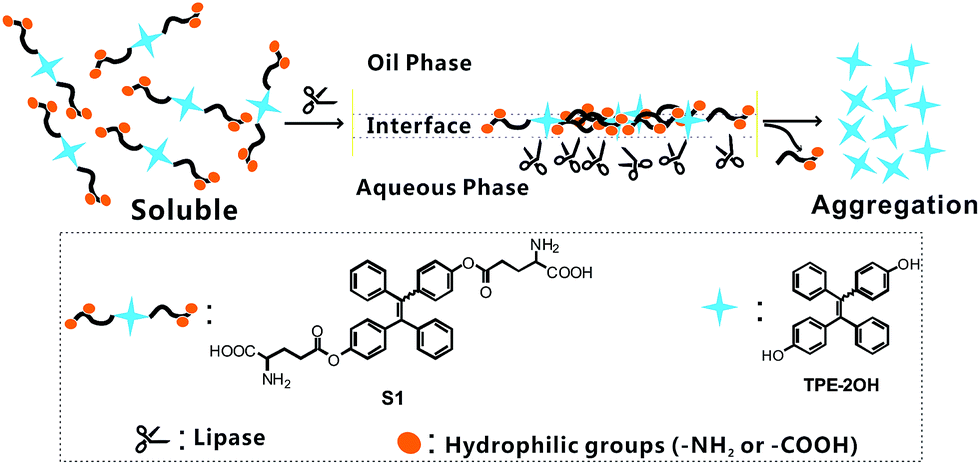 | ||
| Scheme 1 Schematic illustration of the sensing mechanisms and structure of probe S1 in lipase detection. | ||
Experimental
Materials
All the chemicals and reagents were commercially available and used as received without further purification. Superdry tetrahydrofuran (THF) was purchased from J&K Scientific. Titanium tetrachloride (TiCl4), 4-dimethylaminopyridine (DMAP), 4-hydroxybenzophenone, and n-butyllithium were purchased from Aldrich. Dicyclohexylcarbodiimide (DCC), p-toluene sulfonic acid (PTSA) monohydrate, and Boc-L-glutamic acid 1-tert-butyl ester (Boc-Glu-OtBu) were purchased from Acros Organics. Lipases from Pseudomonas cepacia (PCL), Candida rugosa (CRL), Candida Antarctica B (CALB), Pseudomonas fluorescens (PFL), Porcine pancreas (PPL), and Aspergillus niger (ANL) were all ordered from Amano Enzymes. Glucose oxidase, alkaline phosphatase, acylase, laccase, and α-amylase were all purchased from Sigma-Aldrich. The Lipase Assay Kit was purchased from the Nanjing Jiancheng Bioengineering Institute. The human serum samples were supplied by Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, and the serum samples were from both healthy people and acute pancreatitis patients. The water used in the tests was triple distilled, treated with an ion exchange column, and then treated with a Milli-Q water purification system.Instrumentation
1H and 13C NMR spectroscopy was conducted with a Bruker Avance 400 MHz NMR spectrometer using tetramethylsilane (TMS; δ = 0 ppm) as the internal standard. Fluorescence spectroscopy was performed on a Hitachi F-7000 fluorescence spectrophotometer with a Xe lamp as the excitation source. ESI-MS spectra were recorded with a Thermo TSQ QUANTUM ULTRA EMR system. Dynamic laser scattering (DLS) measurements were performed on a commercial laser light scattering instrument (Malvern ZEN3690, Malvern Instruments) at 25 °C.Synthesis
Preparation of the solution of probe S1 and lipase
A stock solution of S1 (1 × 10−3 M) was prepared by dissolving the S1 solid (6.22 mg) in hexane (10 mL). The solution of S1 (10 μM) in hexane was prepared by taking 0.1 mL of S1 stock solution, then diluting the solution to 10 mL with hexane. A series of commercially purchased lipase sample solutions with concentration gradients (5 to 90 U L−1) and other commercial lipases for preliminary screening (80 U L−1) were dissolved in 0.1 M potassium phosphate buffer (pH 7.4), which was prepared from K2HPO4 and KH2PO4.Lipase detection and dynamic monitoring of enzymatic hydrolysis
Different concentration lipase stock solutions (5–90 U L−1, 3 mL) were mixed with 10 μM S1 solution (3 mL) in a 10 mL flask and stirred at 37 °C for 20 min. Then the organic layer was taken to proceed fluorescence measurements with an excitation wavelength of 360 nm. To examine the kinetics of the enzyme reaction, the enzymatic hydrolysis process was monitored using fluorescence spectral measurements that scanned at 1 min intervals from 0 to 10 min after incubation with 0, 5, 10, 30, 50, 70 and 80 U L−1 of lipase. To investigate the photobleaching of the reaction product aggregates, the enzymatic product mixture solution was exposed to an ultraviolet lamp (254 and 365 nm simultaneously) for up to 150 min.Lipase level detection in real samples
For lipase detection in serum, human blood samples were obtained from Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. Written informed consent was obtained from the human subjects in compliance with the relevant laws and institutional guidelines. Human intravenous blood samples were obtained through venipuncture and allowed to clot for 1 h at room temperature. After removing the clotting part by centrifuging at 3000 rpm for 5 min, the obtained serum samples were ten-fold diluted with 0.1 M potassium phosphate buffer (pH 7.4). Standard recovery experiments were performed by adding a known quantity of lipase to the diluted human serum samples of healthy people. The percent recovery referred to the difference in the lipase level determined before and after the addition of lipase in comparison with the added amount. Fluorescence assay was performed as above to determine the amount of lipase. For comparison, the lipase level was determined using a commercial Lipase Assay Kit.Results and discussion
Design, synthesis and sensing mechanism of probe S1
The sensing mechanism is illustrated in Scheme 1. As is well known, lipases can catalyze the hydrolysis of ester bonds, hence this mode of stimuli can activate the AIE characteristic, giving a fluorescent response through a simple reaction. Soluble glutamate groups were attached onto the TPE core, so as to make the probe molecules dissolve well in certain media. The hydrolytic cleavage of the ester bond in S1 was catalyzed by lipase and compound TPE-2OH was produced, and then the rapid aggregation of TPE-2OH was triggered due to its poor solubility, which was caused by the loss of glutamate units, thus the strong blue fluorescence can “turn on” due to the AIE effect. Probe S1 was synthesized using the synthetic protocol described in Scheme S1†. Starting from 4-hydroxybenzophenone, via a McMurry coupling reaction, compound TPE-2OH was produced, which was the product of an esterification of two equivalents of Boc-Glu-OtBu to yield the intermediate. After hydrogen chloride was bubbled into the DCM solution of compound TPE-BGO for 1 h, the desired product S1 was obtained. The structure and purity of S1 were confirmed by 1H NMR and 13C NMR spectroscopy and ESI-MS.Fluorescent response toward lipase
To check the feasibility of our design, firstly we studied the photophysical properties of TPE-2OH when it aggregates. The results in Fig. S1† suggest that TPE-2OH is an AIE-active substance. Secondly, we investigated the fluorescence change when probe S1 was treated with a lipase-buffer solution in hexane. First, the effects of pH and temperature on the reaction were investigated, with the results summarized in Fig. S2 and S3,† respectively. A pH value of 7.4 (PBS buffer) and a temperature of 37 °C were chosen in order to ensure sensitive and rapid detection. A hexane solution of probe S1 (10 μM) was prepared for the fluorescence spectral studies and porcine pancreas lipase (PPL) was selected to evaluate the application of probe S1. In Fig. 1, as we expected, the fluorescence intensity of probe S1 (10 μM) had almost no emission under the testing conditions and nearly no photoluminescence could be seen upon excitation, even in a dark room. Next, we added a series of fixed concentration lipase to a solution of probe S1 and, excitingly, after the incubation, the blue fluorescence emission displayed apparent enhancement. As shown in Fig. 1A, when the concentration of lipase was increased in the test system, the intensity centered at about 453 nm increased correspondingly. In fact, even at a concentration of lipase as low as 20 U L−1, the emission intensity at about 453 nm increased to about 15 times the original one. When the concentration of lipase reached 80 U L−1, the fluorescence intensity increased to a maximum of 42-fold compared to the blank. At last, the lipase concentration reached 90 U L−1, and the fluorescence depicted no significant enhancement and the intensity reached a plateau. This phenomenon verified our design idea mentioned above: by pairing the leakage of the alkoxy chain with the hydrophilic groups of the glutamate group linked to the TPE core after incubation with lipase, the aggregation behavior changed to show a detectable photophysical signal in response to the analytes. On the other hand, MS analysis (Fig. S4†) also suggests that the product of the enzymatic reaction has the same molecular structure as TPE-2OH. Ideally, the change of the fluorometric probe should be from one extreme case to another, just like in our case, from the “off” state to the “on” state. According to the calibration curve (Fig. 1B), it was also remarkable that the relative fluorescence intensity at about 453 nm gave a linear increase with the concentration of lipase in the range from 0 to 80 U L−1 (R2 = 0.9985). The photograph shows the solution of probe S1 alone and the probe after incubation with lipase in hexane solution under UV illumination at 365 nm. It is obvious that blue fluorescence appeared after treatment with lipase, so a distinct fluorescence “turn on” was observed. More importantly, the limit of detection (LOD) is as low as 0.13 U L−1, which is much better compared with previous reports and sensitive enough for determining the lipase level in biological assays considering the dilution factor, given that for normal adults aged 60 and younger, the normal lipase level in blood is 10–140 U L−1, and that for an adult older than aged 60 it is 18–180 U L−1.39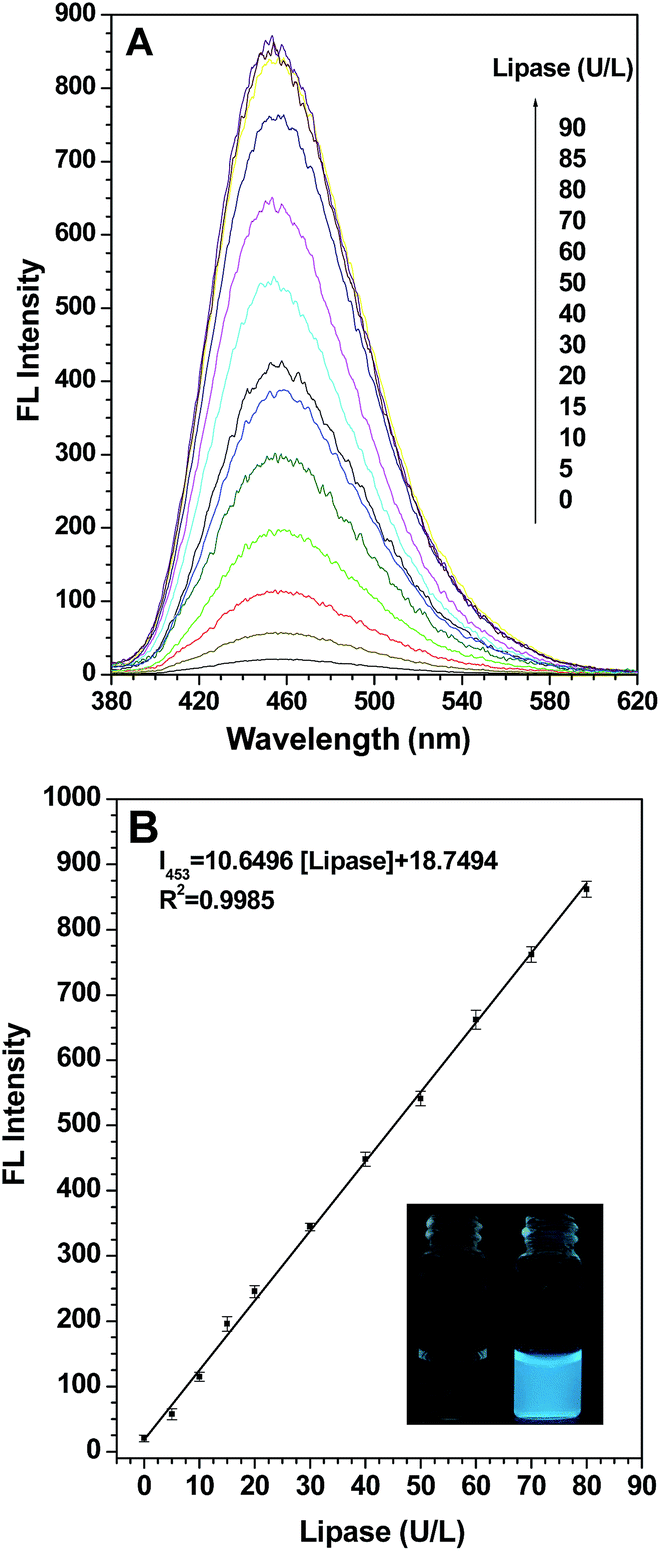 | ||
| Fig. 1 (A) The fluorescence emission spectra of probe S1 (10 μM) after incubation with different concentration lipase-buffer (0.1 M PBS, pH 7.4) solutions at 37 °C; (B) the calibration curve versus the different concentrations (0–80 U L−1) of lipase; the inset shows the corresponding solutions of probe S1 (10 μM) without (left) and with (right) lipase (90 U L−1) after incubation at 37 °C for 15 min under UV light (365 nm) illumination. Excitation wavelength: 360 nm. | ||
To further verify that the formation of aggregates of probe S1 was caused by lipase, the particle sizes were analysed by DLS (Dynamic Laser Scattering) analysis, and the results are displayed in Fig. S5.† In the stock solution of probe S1 (10 μM) with no lipase added, the nanoparticles could be observed in the range of 3–20 nm, while the mean diameter of S1 was approximately 210 nm after incubation with a definite concentration of lipase (80 U L−1), which uncovered that aggregates are indeed occurring in the solution. Transmission Electron Microscopy (TEM) also confirms that compound TPE-2OH was produced in the reaction mixture (Fig. S6†), and the length of the aggregates formed appeared to be 100–400 nm, which was well consistent with DLS analysis. It is suggested that the aggregate size significantly influenced the fluorescence of the solution because the AIE effect could be triggered by larger aggregates.
To investigate the possibility of quantitative analysis of the lipase level, we carried out kinetic studies of the enzymatic catalytic reaction. Fig. 2A displays the variation of the emission intensity at 453 nm of probe S1 (10 μM) during incubation with different concentrations of lipase (0–80 U L−1) for different times at 37 °C. Evidently, the fluorescence intensity increased gradually by prolonging the incubation time, it was also found that the rate of fluorescence enhancement was directly related to the concentration of lipase, the intensity of the probe increased gradually with different magnitudes, and more rapid fluorescence enhancement was observed when more lipase was used. This is understandable when considering the fact that a high concentration of lipase would facilitate the hydrolysis of probe S1. The fluorescence changes more clearly in the initial stage for all lipase concentrations but becomes slower in the later period. The fluorescence emission reaches a plateau after about 7 min, illustrating that the test could be finished within 7 min, which is more convenient and faster than conventional turbidimetric methods. As displayed in Fig. 2A, such emission enhancement was still detectable even when the concentration of lipase was as low as 30 U L−1 within 1 min. This reveals that the lipase turn-on mode could be applied potentially for the real-time assay of lipase levels.
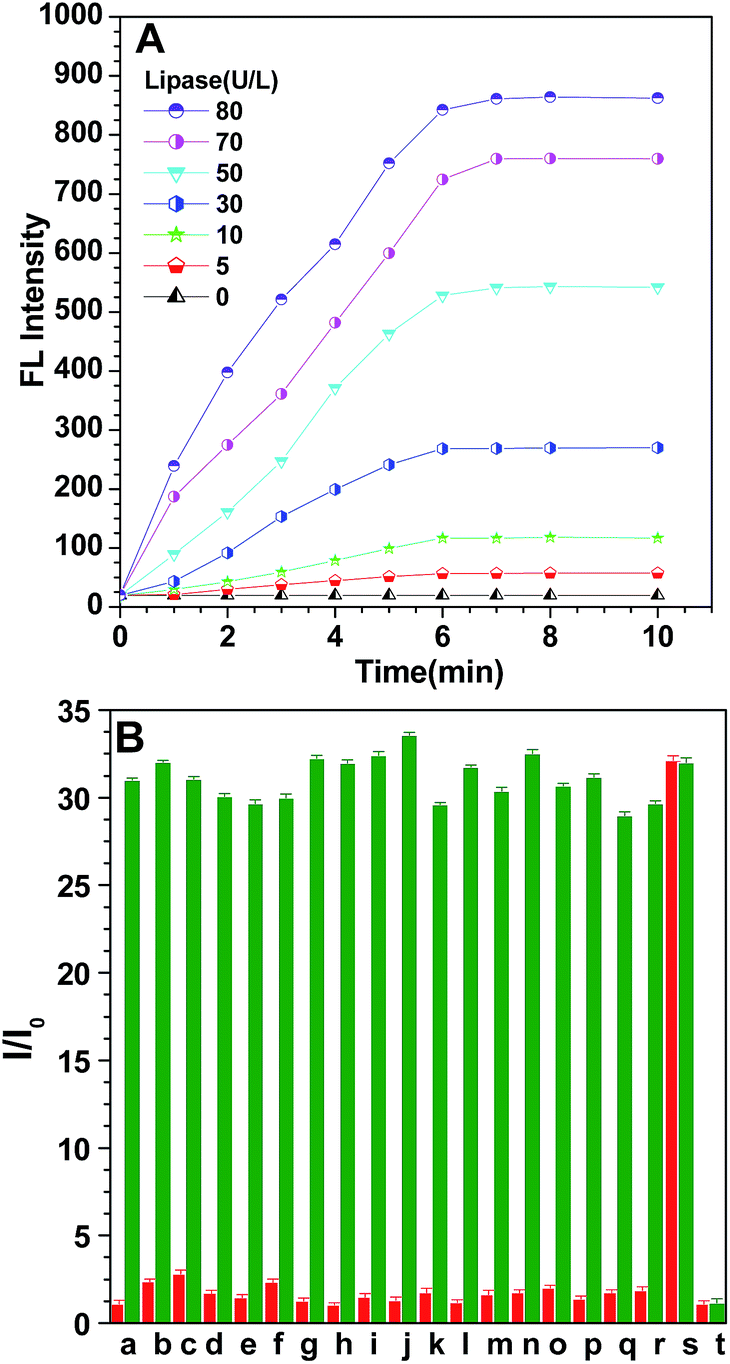 | ||
| Fig. 2 (A) Time-dependent fluorescence intensity of probe S1 (10 μM) in hexane solution at 453 nm versus the hydrolysis reaction time in the presence of different concentrations of lipase in PBS (pH 7.4, 0.1 M) at 37 °C. Excitation wavelength: 360 nm. (B) Fluorescence response of a 10 μM probe in PBS-buffered water (pH 7.4, 0.1 M) at 37 °C for 15 min in the presence of different biologically-relevant substances (red bars); the fluorescence response of a 10 μM probe in PBS-buffered water (pH 7.4, 0.1 M) at 37 °C for 15 min in the presence of 80 U L−1 lipase and with the addition of different biological substances respectively (green bars). (a) NaCl; (b) KCl; (c) MgCl2; (d) CaCl2; (e) AlCl3; (f) NaNO3; (g) Na2CO3; (h) Na2SO4; (i) arginine; (j) leucine; (k) glutamic acid; (l) phenylalanine; (m) cysteine; (n) glucose oxidase; (o) alkaline phosphatase; (p) acylase; (q) laccase; (r) α-amylase; (s) lipase; (t) blank; the concentration of the ions and amino acids is 100 μM, while the concentration of the protein enzymes is 80 U L−1. Excitation wavelength: 360 nm. | ||
As mentioned above, in view of the fact that lipase catalytic reactions only occurred at the oil–water interface, compared to the reported probe P1,38 herein, carboxyl and amino groups that will improve the affinity of the probe towards the lipase in the aqueous phase were specifically introduced into probe S1. Firstly, the substrate dependence of the initial degradation velocities for lipase were measured (Fig. S7†). The data of P1 and S1 for the enzyme kinetics follow the Michaelis–Menten equation, and the kinetic parameters have been determined as Vmax = 1.35 μM min−1 and Km = 28.66 μM for P1 and Vmax = 1.42 μM min−1 and Km = 4.23 μM for S1. The Km value of S1 is much smaller, indicating a high affinity between lipase and the probe S1 molecule. In addition, as depicted in Fig. S8,† the fluorescence response of P1 began to appear when the lipase level rose to 120 U L−1, and the limit of detection was up to 44.23 U L−1, which is 338 times higher than that of probe S1 (LOD = 0.13 U L−1). Also, the test could be finished within 7 min, which was faster than that of probe P1 (13 min). Therefore, this hydrophilic group modified interfacial control can successfully bring high sensitivity, a low detection limit and a short response time.
Furthermore, to evaluate the selectivity of probe S1, the specific nature of this fluorometric assay for lipase was also tested, and a control experiment with other nonspecific biological substances including ions (such as Na+, K+, Mg2+, Ca2+, Al3+, NO3−, CO32− and SO42−), amino acids (such as leucine, glutamic acid, cysteine, arginine and phenylalanine) and other enzymes (such as alkaline phosphatase, acylase, glucose oxidase, α-amylase and laccase) was performed with a fixed concentration. From the red bars in Fig. 2B, it can be seen that only lipase induces a prominent intensity enhancement, which brings about a 32-fold fluorescence enhancement of the probe that was recorded with the effect of lipase, while nearly no rise in the emission intensity was observed for other biological substances. Moreover, we also tested the interference of these biologically-relevant substances towards the probe. The red bars tell us that the presence of other background substances did not produce any obvious disturbance, indicating that S1 had a superior selective response toward lipase and these potentially interfering substances show negligible interference on the enzymatic reaction between lipase and probe. To further reveal that fluorescence enhancement was caused by lipase-catalyzed hydrolysis rather than other factors, a control experiment between lipase and inactivated lipase was performed (Fig. S9†). Herein, deactivated lipase, which was deactivated by treatment at a high temperature (100 °C), was selected. It is well known that high temperature environments can denature the protein. According to our hypothetical sensing mechanism, inactivated lipase could not hydrolyze probe S1 and no fluorescence “turn on” is expected. The results in Fig. S9† indicate that inactivated lipase cannot bring enhancement of the fluorescence, which reveals that it is the catalysis effect of lipase that is acting on this process, excluding other interference effects such as electrostatic interaction and hydrophobic interaction between the probes and proteins. Additionally, the resistance to photobleaching for the aggregates of the enzymatic products was also discussed after direct exposure to ultraviolet lamps (Fig. S10†). The relative fluorescence intensity change ratio (It/I0) remained almost unchanged upon continuous illumination and the fluorescence intensity of the reaction product aggregates was only reduced by 2.9%, which indicate that the reaction product aggregates have good photostability.
We carried out preliminary screening using the sensing mechanism to evaluate the activity of some commercial lipase samples (ANL, CALB, RFL, PPL, RNL and PCL). The fluorescence intensity in the presence of fixed concentration (80 U L−1) lipase samples was monitored after a certain time incubation (20 min). As shown in Fig. 3, the variety of fluorescence intensity was dependent on the different lipase activities. We concluded that the lipase activity increases in the following order: PCL < RFL < CRL < CALB < ANL < PPL. This sequence is consistent with the results detected with a pH-stat method (Table S1†), demonstrating that this fluorometric assay protocol can be used as an alternative to conventional high throughput screening technologies for lipase activity. The inserted photograph in Fig. 3 shows probe S1 after incubation with different commercial lipases under UV illumination at 365 nm. It is clear that the different activities of the lipases lead to strong or weak blue fluorescence, namely, it can be easily distinguished by the naked eye under UV light.
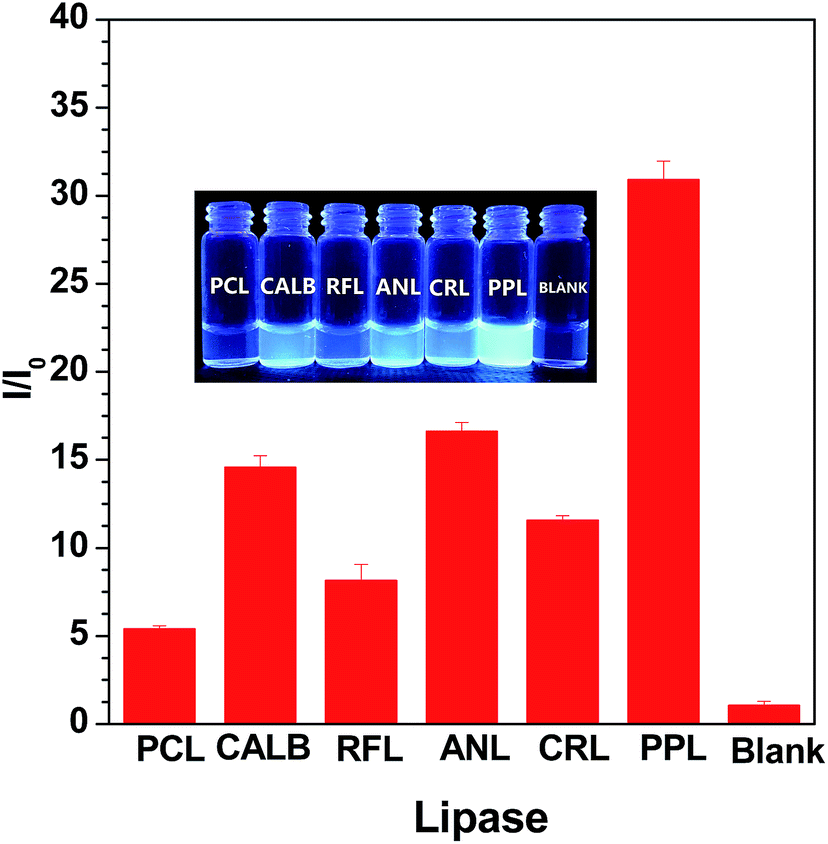 | ||
| Fig. 3 Plot of the I/I0 intensity of probe S1 upon the addition of a fixed concentration (80 U L−1) of commercial lipase samples, respectively, and within an interval of 20 min. The inset shows the corresponding visual fluorescence under UV light illumination at 365 nm. Excitation wavelength: 360 nm. | ||
Lipase detection in real human serum samples
Afterwards, in order to explore the operability and efficacy of the probe system in complex biological fluids, the probe was applied to detect lipase levels in real samples, such as human serum. Herein, we tried to detect the endogenous lipase level in human serum samples using the calibration curve (Fig. 1B) as a standard. Firstly, a standard recovery experiment was performed by adding fixed amounts of lipase to human serum, as depicted in Table S2,† and the recovery value was between 95.97% and 102.44%; the SD values from these two methods are also similar, suggesting the reliability and accuracy of our sensing system for lipase detection. For real serum samples, the procedures of detection are illustrated in Fig. 4A, and since the linear response of the probe is ranging from 0 to 80 U L−1, the dilution factor of the serum sample must be taken into account. The serum samples were obtained from human venous blood via centrifugation, and then the diluted serum was incubated with the probe solution for a while. The fluorescence emission of the S1 probe will experience off-to-on change, which can be monitored using the FL spectra, as well as the naked eye under UV light illumination at 365 nm. In the serum, the endogenous lipase level of both healthy people (sample no. 1–no. 6) and acute pancreatitis (AP) patients (sample no. 7–no. 12) measured by this probe are close to those measured by the Lipase Assay Kit. The results from probe S1 in Table 1 indicate that the lipase levels in the serum samples of no. 1 to no. 6 were in the range of 96.51–171.35 U L−1, which is a normal level of serum lipase, however, for AP patients, the serum lipase levels were significantly higher in the range of 425.59–581.05 U L−1. The inserted photograph (Fig. 4B) shows probe S1 after incubation with diluted real human serum samples under UV illumination at 365 nm to conduct naked eye detection. It is clear that, for serum samples from healthy people (no. 1–no. 6), very weak fluorescence is observed, and that for AP patient serum samples (no. 7–no. 12) significant fluorescence emission enhancement can be observed. In addition, the determined results were consistent with those obtained by the Lipase Assay Kit and, moreover, when compared with the commercial Lipase Assay Kit, our probe has the advantages of a much lower limit of detection (0.13 U L−1), which is 38-fold lower than the assay kit (5.0 U L−1), a relatively simple test process of samples except for the pre-treatment, and a shorter test time (7 min) than the assay kit (25 min). It’s indicated that our fluorescence probe could act as a quite convenient one-step assay for the accurate detection of the lipase level in human serum and has great potential as an effective detection method for the lipase level in diagnostic applications.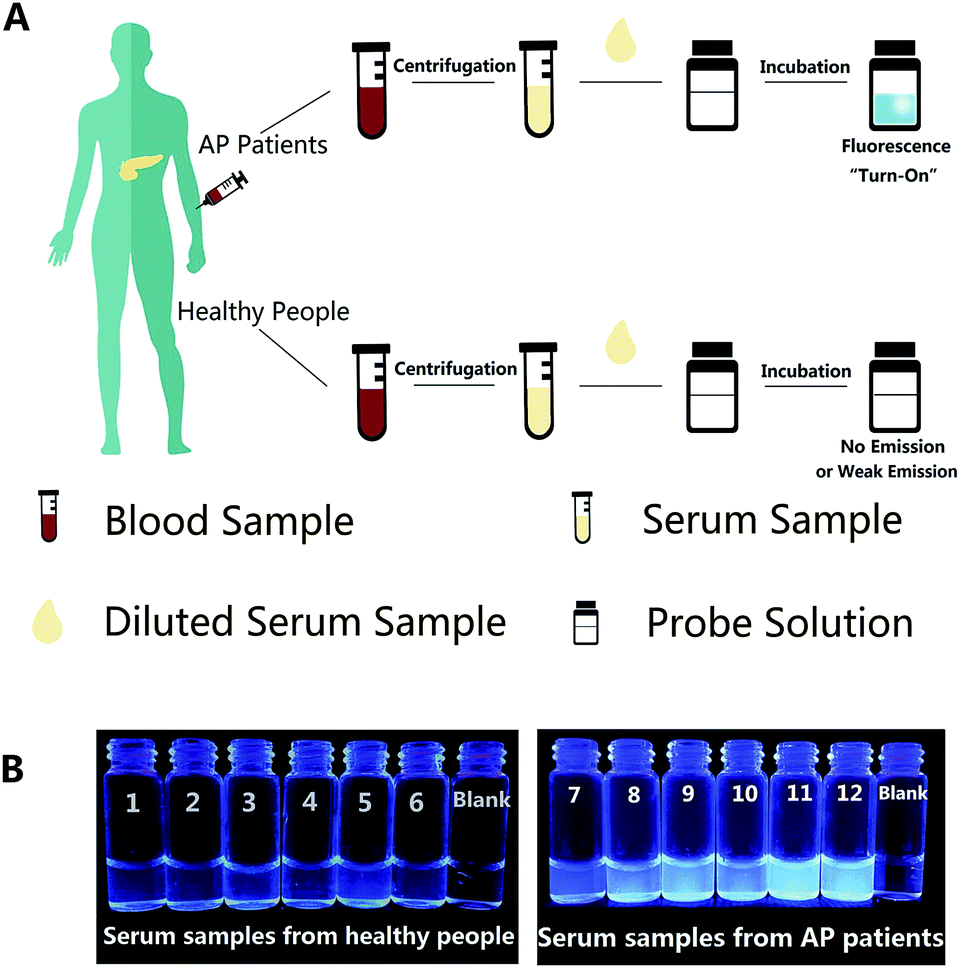 | ||
| Fig. 4 (A) Schematic illustration of this simple technique for lipase level detection in real human serum samples. (B) Corresponding visual fluorescence of probe S1 after incubation with 10-fold diluted real human serum samples under UV light illumination at 365 nm. | ||
| People | Serum no. | Detected by assay kit (U L−1) | Detected by probe S1 (U L−1) |
|---|---|---|---|
| a Note: the serum samples were 10-fold diluted for measurement, and the values (mean ± SD) herein represent the lipase level in undiluted serum samples, which are calculated based on the measurements for the diluted samples. Samples no. 1 to no. 6 were obtained from healthy people; samples no. 7 to no. 12 were obtained from patients with acute pancreatitis. | |||
| Healthy people | 1 | 129.79 ± 2.09 | 134.06 ± 1.38 |
| 2 | 119.36 ± 3.17 | 123.64 ± 2.67 | |
| 3 | 152.18 ± 1.63 | 157.97 ± 1.85 | |
| 4 | 87.81 ± 2.44 | 96.51 ± 2.41 | |
| 5 | 176.87 ± 2.59 | 171.35 ± 3.62 | |
| 6 | 106.01 ± 3.81 | 104.83 ± 2.74 | |
| AP Patients | 7 | 422.69 ± 1.68 | 425.59 ± 3.26 |
| 8 | 471.14 ± 5.06 | 479.87 ± 3.82 | |
| 9 | 517.07 ± 3.47 | 510.20 ± 2.11 | |
| 10 | 441.89 ± 2.84 | 443.43 ± 4.97 | |
| 11 | 584.41 ± 3.31 | 581.05 ± 2.06 | |
| 12 | 549.62 ± 4.01 | 557.03 ± 3.45 | |
Conclusions
In summary, for lipase fluorescent probes, the interfacial catalytic efficiency determines its sensitivity. We designed a novel fluorescence turn-on probe for lipase detection, which functions through the lipase-induced transformation of soluble to insoluble features and the corresponding AIE effect. We adjusted the reaction behavior of the AIE probe in the interface by the introduction of hydrophilic groups, and the highest sensitivity, lowest detection limit and shortest response time in reported literature were obtained. The probe is more suitable for the linear detection of the lipase level ranging from 0 to 80 U L−1, which fits into the concentration range of lipase in human serum, considering the dilution factor if necessary. The aggregate products also show a good photostability against direct exposure to ultraviolet, and there was less interference by other biological components and good agreement with the assay kit results. To the best of our knowledge, this is the first report on the detection of lipase level in real human serum samples using a fluorescent probe. Therefore, the probe would provide a helpful approach for further elucidating the physiological roles of lipase as well as conducting pathological analysis for diseases involving lipase.Acknowledgements
We are grateful to the National Science Foundation of China (no. 31501423 and no. 31671820), Director Fund of Oil Crops Research Institute of CAAS (no. 1610172015006) and Agricultural Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI) for financial support.Notes and references
- L. Sommermeyer, Am. J. Nurs., 1935, 35, 1157–1161 Search PubMed.
- P. Hastier, M. J. M. Buckley, E. P. Peten, N. Demuth, R. Dumas, J. F. Demarquay, F. X. Caroli-Bosc and J. P. Delmont, Am. J. Gastroenterol., 2000, 95, 3295–3298 CrossRef CAS PubMed.
- J. P. Neoptolemos, M. Raraty, D. M. Finch and R. Sutton, Gut, 1998, 42, 886–891 CrossRef CAS PubMed.
- E. J. Balthazar, Radiology, 2002, 223, 603–613 CrossRef PubMed.
- Y. Dhiraj, N. Agarwal and C. S. Pitchumoni, Am. J. Gastroenterol., 2002, 97, 1309–1318 CrossRef PubMed.
- R. Catanzaro, B. Cuffari, A. Italia and F. Marotta, World J. Gastroenterol., 2016, 22, 7660–7675 CrossRef PubMed.
- R. C. Smith, J. Southwell-Keely and D. Chesher, ANZ Journal of Surgery, 2005, 75, 399–404 CrossRef PubMed.
- J. Treacy, A. Williams, R. Bais, K. Willson, C. Worthley, J. Reece, J. Bessell and D. Thomas, ANZ Journal of Surgery, 2001, 71, 577–582 CrossRef CAS PubMed.
- W. M. Steinberg, S. S. Goldstein, N. D. Davis, J. Shamma’a and K. Anderson, Ann. Intern. Med., 1985, 102, 576–580 CrossRef CAS PubMed.
- X. Z. Lin, S. S. Wang, Y. T. Tsai, S. D. Lee, S. C. Shiesh, H. B. Pan, C. H. Su and C. Y. Lin, J. Clin. Gastroenterol., 1989, 11, 47–52 CrossRef CAS PubMed.
- V. Keim, N. Teich, F. Fiedler, W. Hartig, G. Thiele and J. Mössner, Pancreas, 1998, 16, 45–49 CrossRef CAS PubMed.
- S. Ignjatović, N. Majkić-Singh, M. Mitrović and M. Gvozdenović, Clin. Chem. Lab. Med., 2000, 38, 1141–1144 CrossRef PubMed.
- B. Sternby, J. F. O’Brien, A. R. Zinsmeister and E. P. Dimagno, Mayo Clin. Proc., 1996, 71, 1138–1144 CrossRef CAS PubMed.
- D. P. Ryan, T. S. Hong and N. Bardeesy, N. Engl. J. Med., 2014, 371, 1039–1049 CrossRef CAS PubMed.
- A. Vincent, J. Herman, R. Schulick, R. H. Hruban and M. Goggins, Lancet, 2011, 378, 607–620 CrossRef.
- R. Smith, J. Southwell-Keely and D. Chesher, ANZ Journal of Surgery, 2005, 75, 399–404 CrossRef PubMed.
- J. Corsetti, C. Cox, T. Schulz and D. Arvan, Clin. Chem., 1993, 39, 2495–2499 CAS.
- W. Zhang, Y. Tang, J. Liu, L. Jiang, W. Huang, F. Huo and D. Tian, J. Agric. Food Chem., 2015, 63, 39–42 CrossRef CAS PubMed.
- W. Zhang, Y. Tang, J. Liu, Y. Ma, L. Jiang, W. Huang, F. Huo and D. Tian, J. Mater. Chem. B, 2014, 2, 8490–8495 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 45, 4332–4353 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Hou, J. Peng, F. Zeng, C. Yu and S. Wu, Materials Chemistry Frontiers, 2017, 1, 660–667 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Shi, Q. Li, X. Zhang, M. Peng, J. Qin and Z. Li, Sens. Actuators, B, 2010, 145, 583–587 CrossRef CAS.
- Y. Yuan, C. Zhang, S. Xu and B. Liu, Chem. Sci., 2016, 7, 1862–1866 RSC.
- Y. Yuan, C. Zhang, R. T. K. Kwok, D. Mao, B. Z. Tang and B. Liu, Chem. Sci., 2017, 8, 2723–2728 RSC.
- W. Zhang, Y. Chen, T. Shao and L. Fan, J. Mater. Chem. B, 2014, 2, 5249–5255 RSC.
- X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC.
- Y. Jia, X. Zuo, X. Lou, M. Miao, Y. Cheng, X. Min, X. Li and F. Xia, Anal. Chem., 2015, 87, 3890–3894 CrossRef CAS PubMed.
- Y. Ding, L. Shi and H. Wei, Chem. Sci., 2015, 6, 6361–6366 RSC.
- L. Zong, Y. Song, Q. Li and Z. Li, Sens. Actuators, B, 2016, 226, 239–244 CrossRef CAS.
- Z. Liu, S. Chen, B. Liu, J. Wu, Y. Zhou, L. He, J. Ding and J. Liu, J. Am. Chem. Soc., 2014, 86, 12229–12235 CAS.
- L. Yan, H. Shi, X. He, K. Wang, J. Tang, M. Chen, X. Ye, F. Xu and Y. Lei, Anal. Chem., 2014, 86, 9271–9277 CrossRef CAS PubMed.
- W. Zhang, W. Liu, P. Li, H. Xiao, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2014, 126, 12697–12701 CrossRef.
- X. Xie, M. Li, F. Tang, Y. Li, L. Zhang, X. Jiao, X. Wang and B. Tang, Anal. Chem., 2017, 89, 3015–3020 CrossRef CAS PubMed.
- F. Kong, Y. Zhao, Z. Liang, X. Liu, X. Pan, D. Luan, K. Xu and B. Tang, Anal. Chem., 2017, 89, 688–693 CrossRef CAS PubMed.
- Y. Ding, L. Shi and H. Wei, Chem. Sci., 2015, 6, 6361–6366 RSC.
- J. Shi, S. Zhang, M. Zheng, Q. Deng, C. Zheng, J. Li and F. Huang, Sens. Actuators, B, 2017, 238, 765–771 CrossRef CAS.
- D. C. Whitcomb, N. Engl. J. Med., 2006, 354, 2142–2150 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterisation, test conditions and other supplementary data. See DOI: 10.1039/c7sc02189e |
| This journal is © The Royal Society of Chemistry 2017 |