
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh-catalyzed aerobic oxidative cyclization of anilines, alkynes, and CO†
Xinyao
Li
a,
Jun
Pan
a,
Hao
Wu
a and
Ning
Jiao
 *ab
*ab
aState Key Laboratory of Natural and Biomimetic Drugs, Peking University, School of Pharmaceutical Sciences, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: jiaoning@pku.edu.cn
bState Key Laboratory of Elemento-organic Chemistry, Nankai University, Weijin Rd. 94, Tianjin 300071, China
First published on 3rd July 2017
Abstract
Transition-metal-catalyzed oxidative C–H cyclization of anilines has been an attractive and powerful strategy for the efficient construction of N-heterocycles. However, primary and tertiary anilines are rarely employed in this strategy due to the relative instability with strong oxidants or the presence of three C–N bonds. We describe here a novel Rh-catalyzed C–H cyclization of a wide range of anilines with alkynes and CO, using an aerobic oxidative protocol. Particularly, the simple primary anilines and readily prepared tertiary anilines could be easily converted to quinolin-2(1H)-ones, which are high value-added, biologically significant N-heterocycles, via C–N bond cleavage.
Introduction
N-heterocyclic compounds are of great importance in pharmaceuticals and are also versatile building blocks for natural products, bioactive compounds, and drug synthesis.1 Transition metal-catalyzed oxidative C–H cyclization of anilines and their derivatives has recently emerged as one of the most attractive and powerful strategies for the efficient construction of N-heterocycles in terms of atom and step economy.2–6 In the past decades, N-monosubstituted secondary (2° amine) anilines and alkynes have been extensively used in this field with the assistance of stoichiometric oxidants in most cases (i, Scheme 1a).3,4 However, despite this significance, the preparation of 2° anilines often requires tedious multi-steps because the tertiary (3°) amines would be mainly obtained by a traditional substitution reaction. In addition, the difficulty in removing the R group from the formed N-heterocycles also limits their further transformation.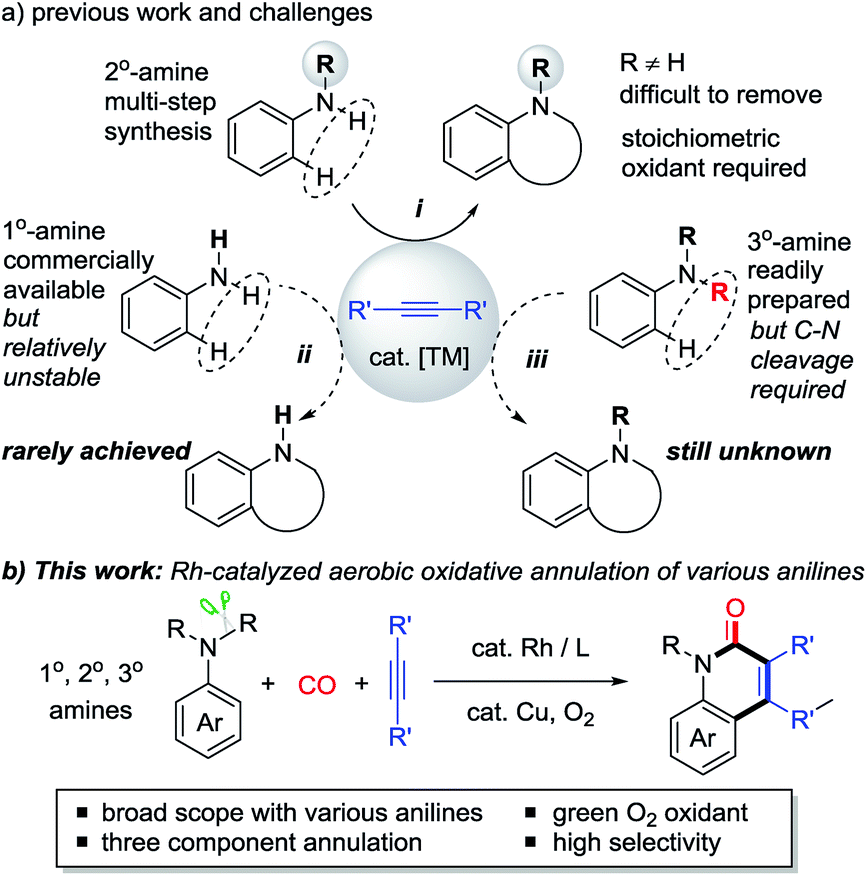 | ||
| Scheme 1 Aerobic oxidative annulation strategies to construct heterocycles. | ||
In contrast, primary (1°) and tertiary (3°) anilines are more readily available. However, only a few examples of oxidative cyclization reactions with 1° anilines have been reported (ii, Scheme 1a),6 because of the instability of these primary anilines under strong oxidative conditions, and because they usually prefer to undergo acylation, hydroamination, and homocoupling.7 Moreover, the C–H cyclization of alkynes with 3° anilines which are easily prepared is still unknown because cleavage of one C–N bond must be overcome in this process (iii, Scheme 1a).8 Most efforts in the field of C–N bond cleavage of 3° anilines have been focused on amination, alkylation, and arylation transformations.9 Therefore, the practical oxidative C–H cyclization of 1° and 3° anilines as stable chemicals to construct N-heterocycles is very desirable but still a challenging issue. In addition, the transition metal-catalyzed C–H cyclization of anilines with green and mild O2 as the oxidant10,11 is also very desirable.
Herein, using an aerobic oxidative strategy, we report an efficient Rh-catalyzed C–H cyclization of simple anilines, alkynes, and CO (Scheme 1b). The advantages of this protocol are threefold: (1) a wide range of anilines are significantly employed in this cyclization protocol and this enables new direct routes to N–H and N–R quinolin-2(1H)-ones, which represent an important class of fused heterocyclic compounds found in many alkaloids.12,13 To the best of our knowledge, this chemistry is the first straightforward transformation of primary anilines to N–H quinolin-2(1H)-ones, whose synthesis usually suffers from multi-step synthesis including protection and deprotection processes.13 In addition, the intermolecular C–H cyclization of tertiary anilines with alkynes through C–N cleavage was still unknown until this case. (2) O2 is successfully utilized as the sacrificial oxidant for the aerobic oxidative cyclizations in efficient ways to fulfill the requirements of green chemistry.10,11 (3) In contrast to the well-known two component aerobic oxidative cyclizations for N-heterocycles such as indoles,3 this chemistry provides a three component cyclization approach to N-heterocycles with CO, which has been used as a prominent C1 synthon in organic synthesis.14,15
Results and discussion
We initially chose N,N-dimethylaniline (1a), simple aniline (2a), N-methylaniline (3a), and dec-5-yne (4a) as model substrates for the investigation of C–H cyclization with CO (Table 1). Considering the simple anilines could not survive well with strong oxidant, we started the screening with O2 as the oxidant under atmospheric pressure. Interestingly, we were excited that the reaction of 1a catalyzed by [Rh(CO)2Cl]2 with a catalytic amount of Cu(OPiv)2 (10 mol%) afforded N-methyl-quinolin-2(1H)-one 5a in 20% yield (entry 1, Table 1). Changing the mixed gas ratio and screening ligands demonstrated that dppb performed best under a mixed gas of CO/O2 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, giving the product 5a in 60% yield (entries 2–8). Finally, 5a was obtained in 72% yield under the optimized conditions with a catalytic amount of PivOH as the additive (condition A, entry 9, Table 1). Control experiments indicated that the Cu and Rh catalysts were both essential for this transformation (see entry 10, Tables 1 and S1 in the ESI† for details). Meanwhile, the C–H cyclization with the primary aniline 2a was also investigated. To our delight, under similar conditions (condition B, entry 11, Table 1), aniline (2a) was smoothly converted into the corresponding N–H quinolin-2(1H)-one 6a in 67% yield. Finally, under the optimized conditions (condition B, entry 12, Table 1), the reaction of N-methylaniline (3a) produced 5a in 85% yield.
:1, giving the product 5a in 60% yield (entries 2–8). Finally, 5a was obtained in 72% yield under the optimized conditions with a catalytic amount of PivOH as the additive (condition A, entry 9, Table 1). Control experiments indicated that the Cu and Rh catalysts were both essential for this transformation (see entry 10, Tables 1 and S1 in the ESI† for details). Meanwhile, the C–H cyclization with the primary aniline 2a was also investigated. To our delight, under similar conditions (condition B, entry 11, Table 1), aniline (2a) was smoothly converted into the corresponding N–H quinolin-2(1H)-one 6a in 67% yield. Finally, under the optimized conditions (condition B, entry 12, Table 1), the reaction of N-methylaniline (3a) produced 5a in 85% yield.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aniline | Ligand | x | y | CO/O2 | Yieldb (%) |
| a Reaction conditions: aniline 1a–3a (0.3 mmol), alkyne 4a (0.45 mmol), Rh catalyst (2 mol%), ligand (5 mol%), Cu catalyst (x mol%), and additive (y mol%) in PhCl (2 mL) under 1 atm CO/O2 (v/v, 1 atm) at 130 °C for 36 h. b Yields were determined by GC analysis. c Isolated yields. | ||||||
| 1 | 1a | — | 10 | 100 | 4/1 | 5a, 20 |
| 2 | 1a | — | 10 | 100 | 1/2 | 5a, 34 |
| 3 | 1a | SIMes | 10 | 100 | 1/2 | 5a, 36 |
| 4 | 1a | Xantphos | 10 | 100 | 1/2 | 5a, 41 |
| 5 | 1a | BINAP | 10 | 100 | 1/2 | 5a, 46 |
| 6 | 1a | dppp | 10 | 100 | 1/2 | 5a, 46 |
| 7 | 1a | dppb | 10 | 100 | 1/2 | 5a, 49 |
| 8 | 1a | dppb | 10 | 100 | 2/1 | 5a, 60 |
| 9 | 1a | dppb | 20 | 30 | 2/1 | 5a, 74(72)c |
| 10 | 1a | dppb | 0 | 30 | 2/1 | 5a, trace |
| 11 | 2a | — | 10 | 100 | 4/1 | 6a, (67)c |
| 12 | 3a | — | 10 | 100 | 4/1 | 5a, (85)c |
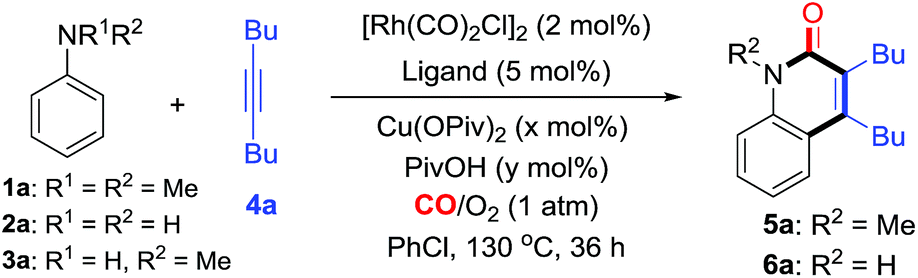
With the established aerobic oxidative conditions in hand, we initially examined the scope of tertiary anilines 1 with alkynes 4 under condition A (Table 2). The reaction proved to be tolerant of different substitution patterns on the aromatic core. In general, both electron-donating and electron-withdrawing substituents of anilines were well tolerated under these conditions (5a–h). Anilines substituted with halogens (F, Cl, and Br) afforded the corresponding quinolin-2(1H)-ones in moderate to good yields (5d–f), which could be used for further functionalization. In addition, other alkynes were compatible to give the expected products under condition A in moderate efficiencies (5i and j). The asymmetrical aryl alkyl alkynes were also suitable for this reaction, affording the corresponding products in good yields with moderate regioselectivity (5k and l).
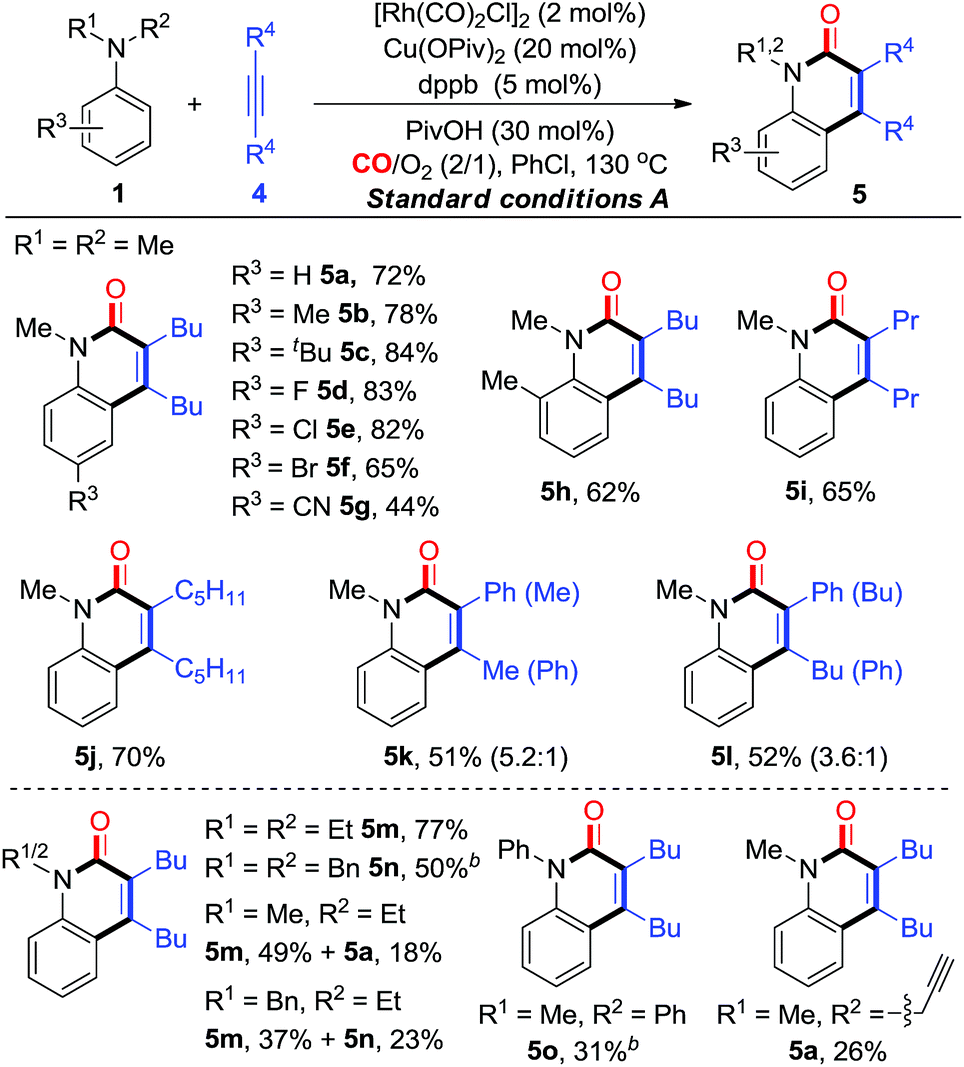
Furthermore, we investigated the scope of N,N-dialkylanilines to determine whether N-ethyl and N-benzyl groups were compatible (5m and n). Interestingly, when N-ethyl-N-methylaniline was treated with 4a, two products 5m and 5a were obtained in 49% and 18% yields, respectively. N-benzyl-N-ethylaniline was smoothly converted into the products 5m and 5n in 37% and 23% yields, respectively. It is worth noting that N-methyl-N-(prop-2-yn-1-yl)-aniline provided 5a, while N-methyl-N-phenylaniline afforded 5o as the sole product in 31% yield, which could not be produced from diphenylamine under condition B. The activation of N groups has the order of Me > Bn > Et > Ph, indicating that the less sterically hindered alkyl group is much more facile for cleavage.
On the basis of the optimization studies, we then evaluated the scope and generality of primary anilines in this transformation (Table 3). In general, primary anilines containing electron-rich/deficient functional groups reacted smoothly to give N–H quinolin-2(1H)-ones 6a–k in moderate to good yields. Furthermore, a series of o-substituents can be compatible and the corresponding quinolin-2(1H)-ones were obtained in 40–79% yields (6l–q). The annulation proceeded well with disubstituted aniline (6r). The protocol could also be applied to olefin-containing aniline, thereby allowing further derivatization of the formed quinolin-2(1H)-one 6s. When the reaction was carried out using a heterocyclic amine, the desired product 6t was obtained in 20% yield. Additionally, a diarylalkyne reacted smoothly to give the corresponding product 6u in moderate yield. For the unsymmetrical aryl alkyl alkyne, its annulations gave the desired compounds (6v and w) in 35–40% yields with moderate regioselectivity. Interestingly, multi-substituted pyridin-2(1H)-one 6x could be constructed from the enamine through this synthetic strategy albeit in low yield (15%, Table 3).
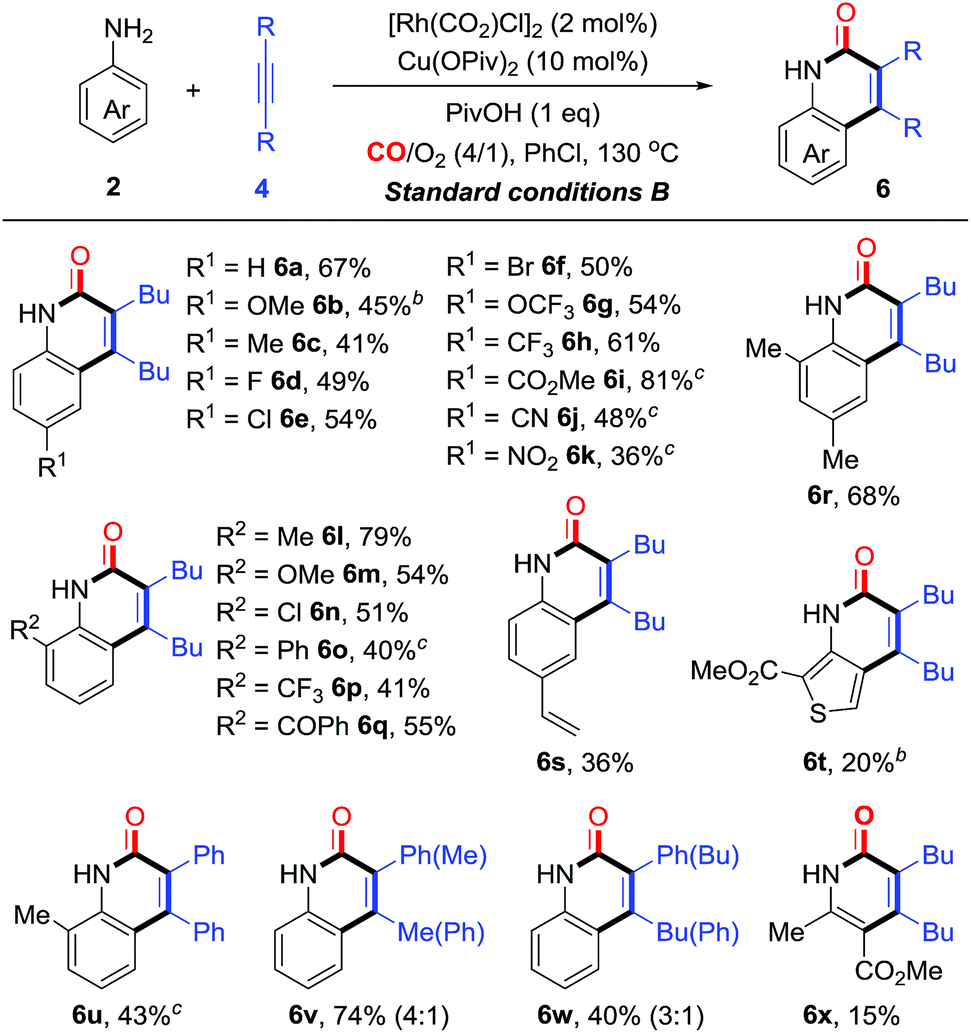
At this point, a range of secondary anilines 3 were also fully compatible under the conditions B (Table 4). A variety of N-methylanilines bearing electron-donating or electron-withdrawing substituents on the phenyl ring underwent the annulation to afford the corresponding quinolin-2(1H)-ones (5c–g and 5p–s) in moderate to excellent yields. For more broadly synthetic interests, various functionalized groups, such as halides, free carboxylic acid, esters, and nitriles were tolerated. In the case of N-alkyl substituted anilines with a PMB group, even with a cyclopropyl group, the desired quinolin-2(1H)-ones (5t and u) were obtained successfully. Interestingly, tetrahydroquinoline, tetrahydro-1H-benzo[b]azepine, and dihydrodibenzooxazepines smoothly led to the more complex poly-heterocyclic compounds (5v–x) in moderate to good yields, which are usually found in natural products as the core structural motif and show possible bioactivity. Diaryl-substituted alkynes also were proved to be very suitable to such annulations, thus yielding the corresponding products 5y–z in 62% yields. The unsymmetrical alkynes were also suitable for this reaction, affording the corresponding products in high yields with moderate regioselectivity (5k and l).
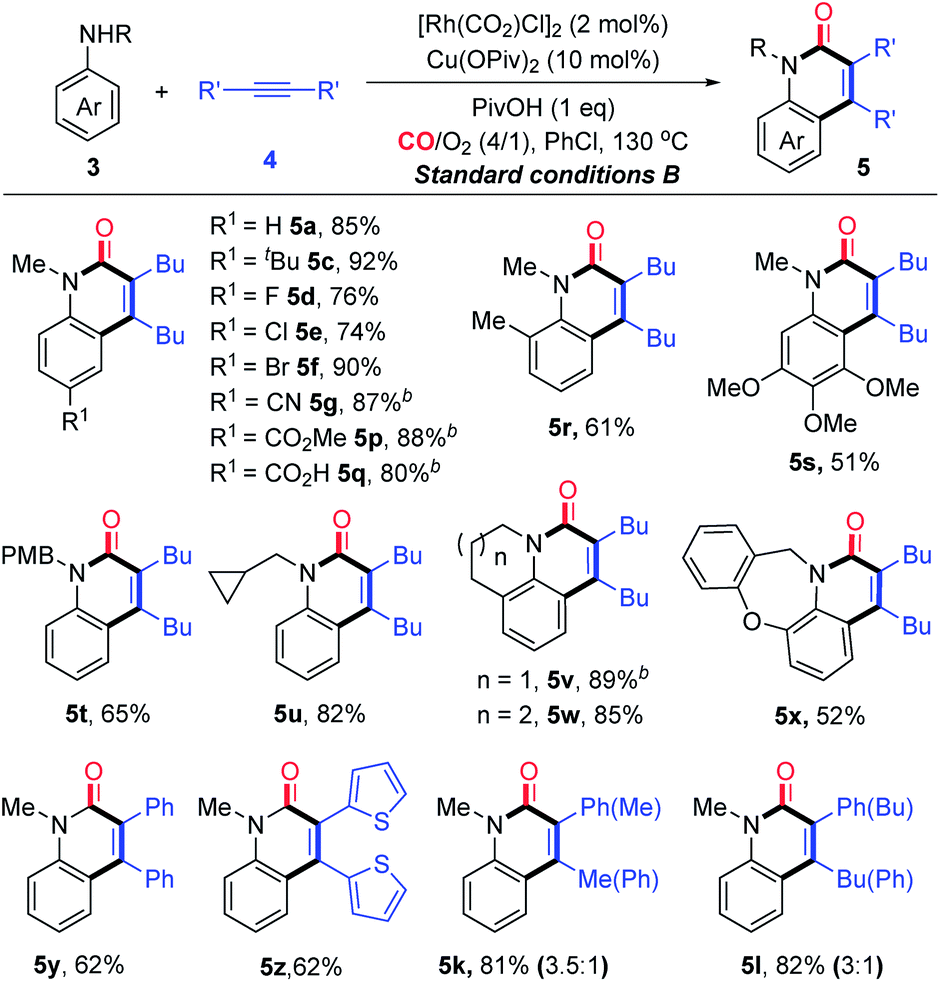
After having proven a wide substrate scope tolerance, this protocol was further applied to the synthesis of significant intermediates and complex bioactive molecules. To be specific, a mixture of 1a and 3a can be converted into the sole product 5a in good yields (Scheme 2A), which demonstrates that the secondary aniline could also be tolerated by the protocol. Notably, compound 7, known as a potential p38aMAP kinase inhibitor,16 can be directly constructed in satisfactory yield through the protocol (Scheme 2B). Additionally, the significant intermediate 9, which can undergo further derivatization to obtain supramolecular catalysts, can be directly obtained through only one step from the corresponding primary aniline 8 under the current standard condition B (Scheme 2C). In contrast, 9 was reported to be prepared through six steps through Larock’s method including the intermediacy of o-iodoamide.17 The current protocol could be applied to the late-stage construction of α 4 integrin inhibitory 15 from aniline 3m and the prepared alkyne 14, in 40% total yield with four steps (Scheme 2D), which has potential utility in medicinal chemistry.18
 | ||
Scheme 2 Further application of the protocol to synthesize significant intermediates and complex bioactive molecules. (a) Et3N, DCM, RT, 2 h, 99%; (b) Tf2O, Py, DCM, RT, 1 h, 62%; (c) MeC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CTMS, Pd(dppf)Cl2, CuI, iPr2NEt, DMF, 90 °C, 24 h, 98%. CTMS, Pd(dppf)Cl2, CuI, iPr2NEt, DMF, 90 °C, 24 h, 98%. | ||
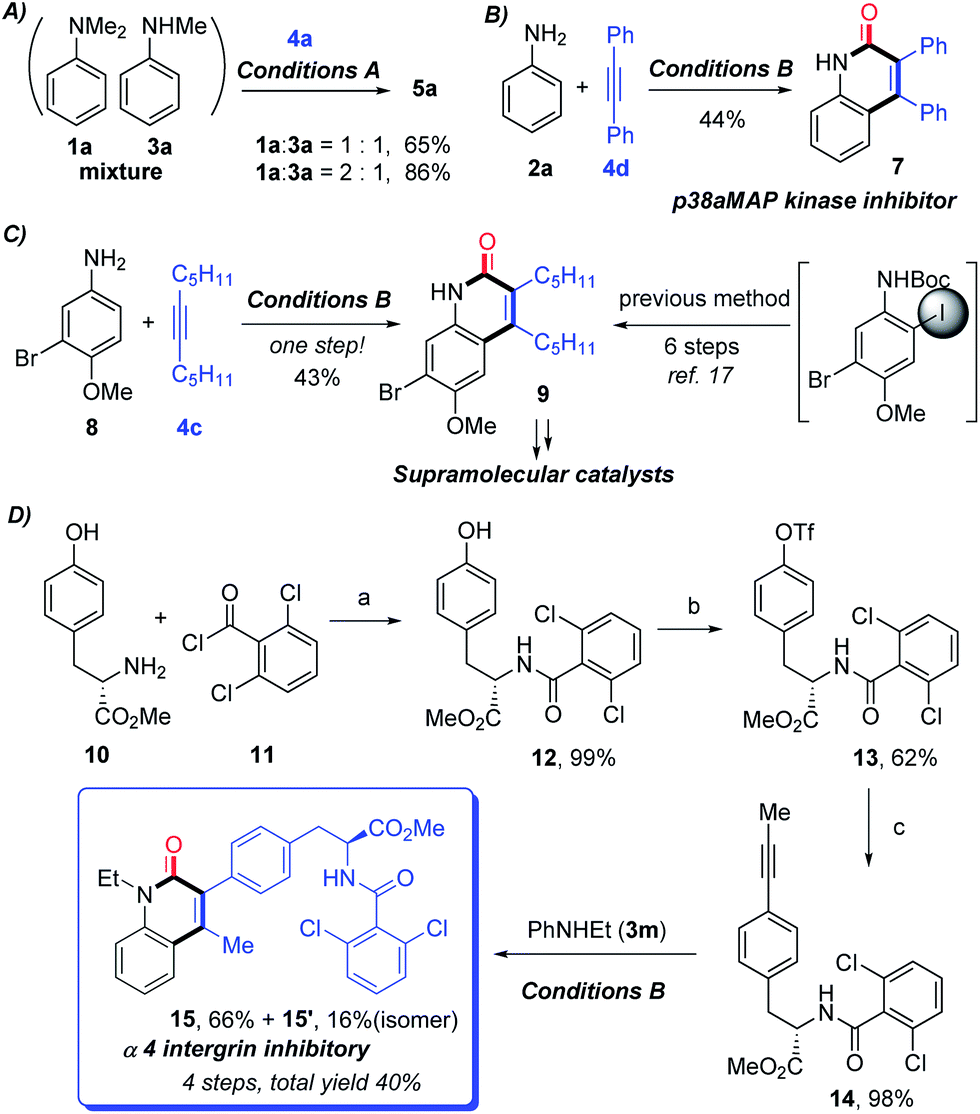
To gain some preliminary understanding of the reaction mechanism, several experiments were carried out under the standard conditions. An isotopic labelling experiment was carried out on the model reaction of 1a or 3a with 4a under standard conditions, and a H/D exchange of 6% in product 5a was found with the addition of deuterated water, thereby implicating a reversible cyclometalation mode [eqn (1)]. In addition, the intermolecular KIE for the annulation reaction was determined to be kH/kD = 1.13 for 1a and 1.04 for 3a, indicating that C(sp2)–H bond cleavage should not be involved in the rate-determining step of the catalytic cycle [eqn (2)].
Control experiments showed that no desired product was observed in the absence or presence of CO, when formylated aniline 16 was treated with 4a [eqn (3)]. These results indicate that the formation of formylated aniline is not involved in this process. In addition, carbamic chloride 17 also did not give the expected product 5a through oxidative addition in the presence of Rh catalyst [eqn (4)]. It is worth noting that deuteration of N-methyl groups results in a significant KIE, kH/kD = 2.53 for intramolecular KIE and kH/kD = 2.00 for intermolecular KIE, indicating that C(sp3)–H bond cleavage might be involved in the rate-determining step for tertiary anilines [eqn (5) and (6)]. Besides, when N,N′-dibenzylaniline was employed, a 5% yield of benzaldehyde (18) was detected along with the expected product [eqn (7)]. This by-product is likely to be generated from the imine intermediate formed in situ through the oxidation of C(sp3)–H at the ortho position of the N atom. The kinetic experiments show that the initial reaction rate of 3a is about three times that of 1a (Fig. 1), implying that the C–N bond cleavage of 1a was the key step.
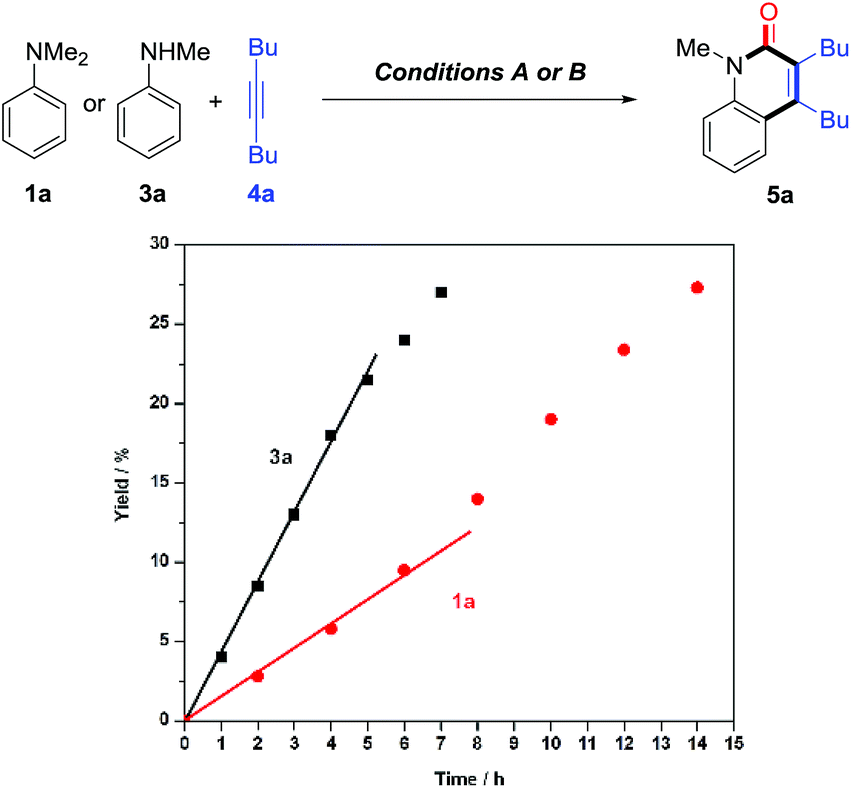 | ||
| Fig. 1 Annulation profile under conditions A or B: red circles for 1a; black squares for 3a. | ||
Based on our preliminary mechanistic studies, we conducted a density functional theory (DFT) investigation into the Rh-catalyzed assembly of anilines, alkynes, and CO for quinolin-2(1H)-one construction to better understand the mechanism of this transformation.19 The tBu groups in acid and nBu groups in alkyne were replaced by methyl groups to save computational time but without sacrificing credibility. Initially, the direct aerobic oxidation of Rh(I) to Rh(III) is calculated to be endergonic by 31.1 kcal mol−1 and the generation of the intermediate INT2 requires a free energy of 34.2 kcal mol−1, indicating that the direct aerobic oxidation of Rh(I) is disfavored (Fig. 2). However, Rh(I) can be easily oxidized to Rh(III) by Cu(II), which is slightly endergonic by 11.6 kcal mol−1. The formed Cu(I) can also be oxidized to Cu(II) using O2 in the presence of carboxylic acid, with a free energy of 19.9 kcal mol−1. This is consistent with the experimental observation that the Cu catalyst plays a pivotal role in the transformation.
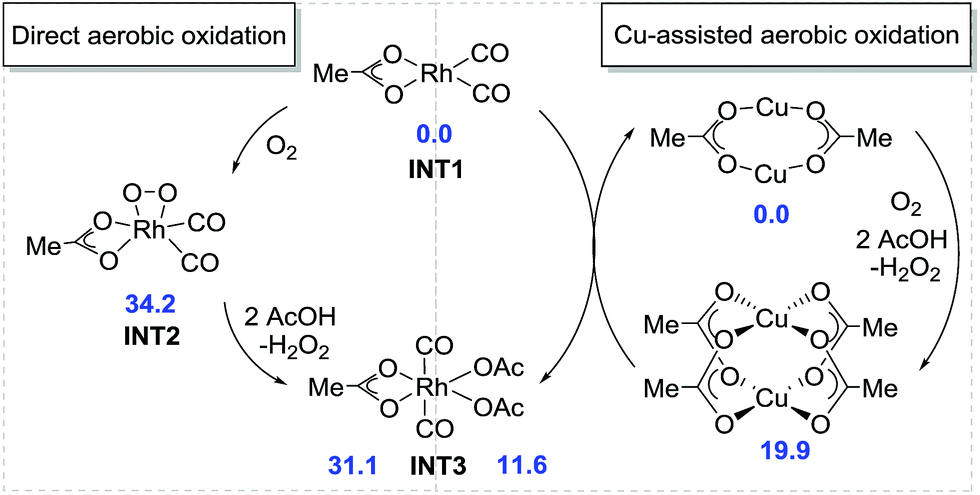 | ||
| Fig. 2 Direct and Cu-assisted aerobic oxidation of Rh(I). | ||
The C–N bond cleavage process with tertiary aniline is interesting. On the basis of the previous results and our primary experiments,9,20 the calculations showed that the Cu(II) species are capable of oxidizing tertiary aniline through single electron transfer (SET) and hydrogen atom transfer (HAT) pathways to give the iminium-type intermediate, which requires a free energy of 8.8 kcal mol−1 (Fig. 3). The formed iminium-type intermediate is then facilely hydrolyzed to the secondary aniline by the elimination of aldehyde as an exergonic process by 34.9 kcal mol−1. The Cu(I) catalyst is then reoxidized by oxygen to form the Cu(II) species in the presence of carboxylic acid, with a free energy of 19.9 kcal mol−1.
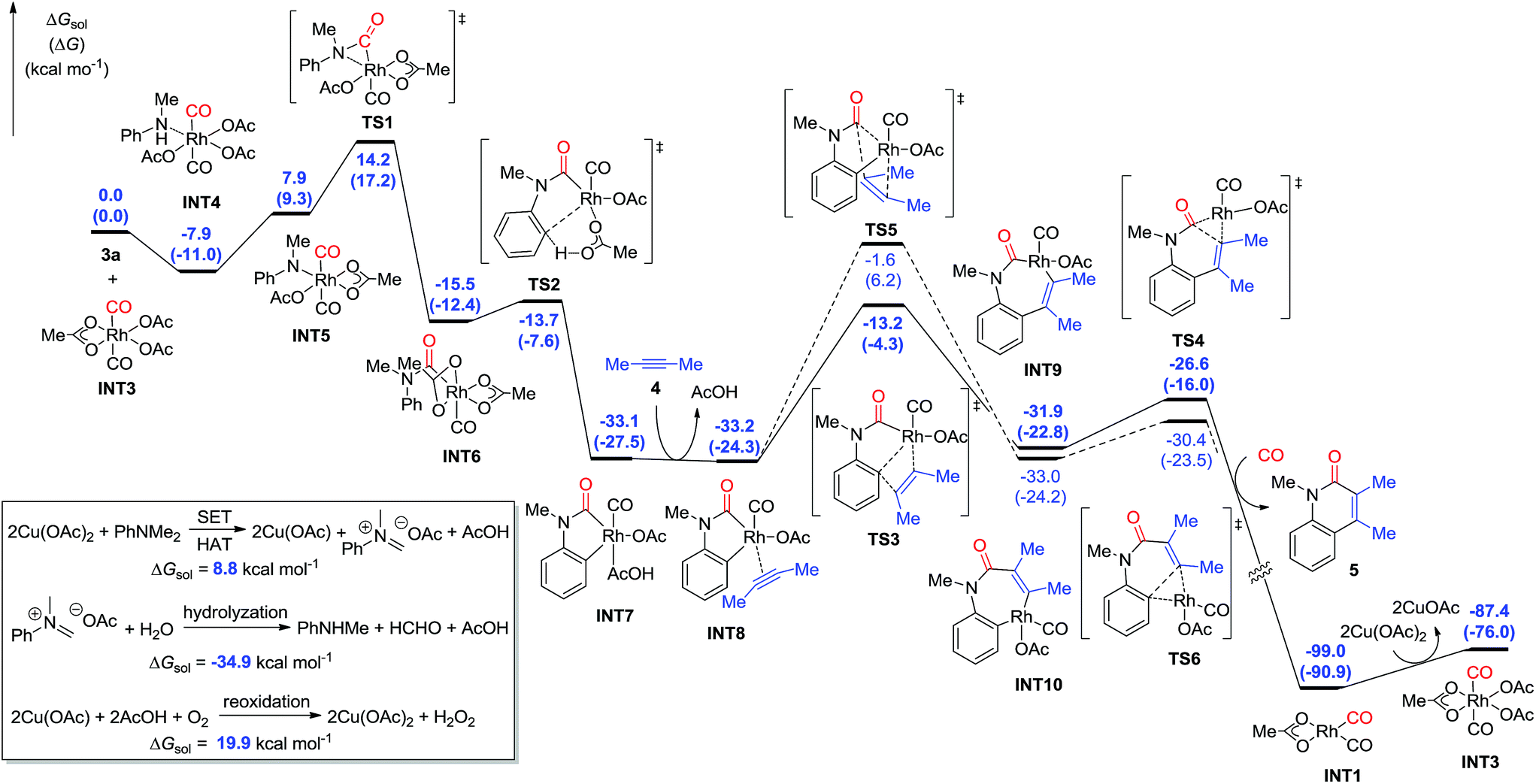 | ||
| Fig. 3 DFT-computed energy profiles for quinolin-2(1H)-one construction through Rh-catalyzed assembly of anilines, alkynes, and CO. The energies discussed are the Gibbs free energies in PhCl (ΔGsol). The numbers in the parentheses are the Gibbs free energies in the gas-phase (ΔG). | ||
When the Rh(III) species INT3 is formed, the coordination of the secondary aniline 3a to Rh(III) species is exergonic by 7.9 kcal mol−1 to give the stable complex INT4 that undergoes further dehydrogenation with the release of HOAc to furnish INT5 endergonicly (Fig. 3). Then, the consequent insertion of CO from Rh to aniline takes place through the transition state TS1 with an activation free energy of 22.1 kcal mol−1 to afford INT6. The following H-abstraction through the transition state TS2 requires an activation free energy of only 1.8 kcal mol−1 to afford the much more stable INT7. After the ligand exchange of INT7 with alkyne 4, the formed rhodacycle complex INT8 undergoes alkyne insertion through two stepwise pathways. When the C(Ar)–C(alkyne) bond was formed preferentially, it required an activation free energy of 20.0 kcal mol−1 to afford the seven-membered rhodacycle INT9. Finally, the reductive elimination is facile through TS4 with an energetic barrier of only 5.3 kcal mol−1, delivering the product 5 exogonicly, whilst regenerating the Rh(I) catalyst. Alternatively, when the C(CO)–C(alkyne) bond was formed preferentially, the barrier to the transition state TS5 was high at 31.6 kcal mol−1, which is 11.6 kcal mol−1 higher than TS3, although the consequent reductive elimination occurs almost without a barrier to provide the product 5. The delivered Rh(I) species INT1 is reoxidized to the Rh(III) complex INT3 in the presence of Cu(II). Reviewing the whole energy profile, we found that the CO insertion and alkyne insertion in the Rh(III) species are the key processes for this transformation.
This synthetic strategy can be efficiently catalyzed by the Rh-catalysis system. However, a Pd-catalysis system proved ineffective experimentally. Accordingly, the energy profile of the unreactive Pd-catalysis system was also calculated for comparison (Fig. 4, see the ESI† for details). It was found that the Pd-catalysis system suffers from a high energetic barrier of 38.3 kcal mol−1 (TS9) in the alkyne insertion process due to the unstable quadridentate fashion of the Pd complex, although the CO insertion is facile with an energetic barrier of only 13.6 kcal mol−1 (TS7). This is qualitatively consistent with the experimental observation. In addition, an Ir-catalysis system has been employed in a recent report.4c The current calculation study indicated that the CO insertion catalyzed by the Ir complex suffers from an activation free energy of 28.3 kcal mol−1, which is 6.2 kcal mol−1 higher than that of the Rh-catalysis, once again in agreement with the experimental demand of 30 atm CO for Ir-catalysis.
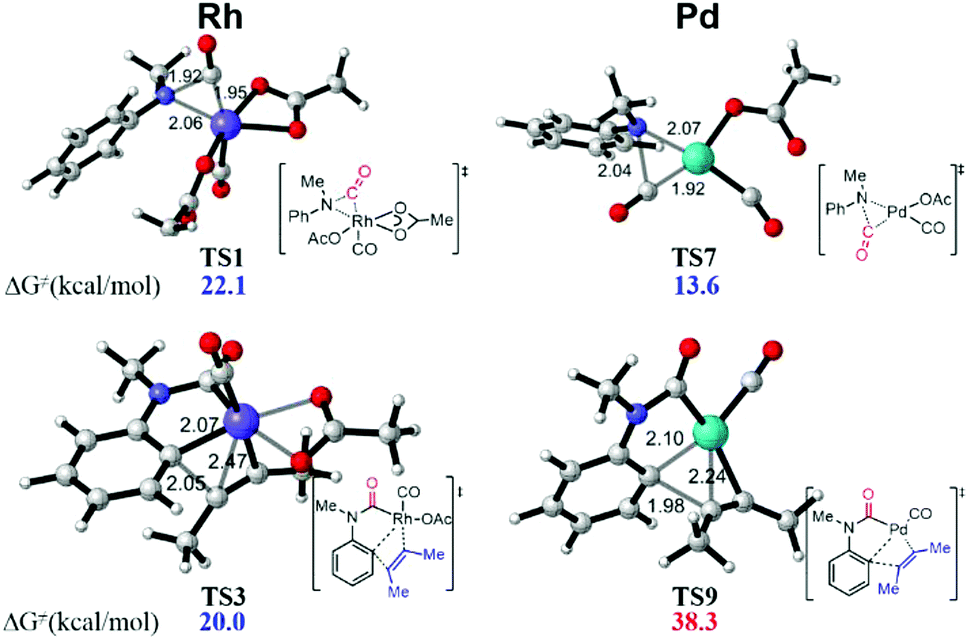 | ||
| Fig. 4 DFT-computed transition state structures of CO insertion and alkyne insertion in Rh and Pd catalysis systems. | ||
Conclusions
In conclusion, a wide variety of anilines, especially simple primary and tertiary anilines, were successfully employed in intermolecular C–H cyclization with alkynes and CO. This novel protocol using Rh-catalysis provides a direct approach to N–H/N-substituted quinolin-2(1H)-ones. The readily available anilines and the operationally simple conditions under 1 atm mixture of CO and O2 make this protocol very practical, green, and sustainable. This chemistry provides a direct route to quinolin-2(1H)-ones, which are biologically significant N-heterocycles and useful building blocks in organic synthesis. DFT calculations suggest that the relay of CO insertion and alkyne insertion in Rh-catalysis are disclosed as the key processes, while the unreactive Pd-catalysis suffers from a high energetic barrier in the alkyne insertion process due to the unstable quadridentate fashion of the Pd complex. Further applications of this transformation are ongoing in our laboratory.Acknowledgements
Financial support from National Basic Research Program of China (973 Program) (grant No. 2015CB856600), National Natural Science Foundation of China (No. 21325206, 21632001), National Young Top-notch Talent Support Program, and Peking University Health Science Center (No. BMU20160541) is greatly appreciated. We thank Lingsheng Ai in this group for reproducing the results for 5m, 6f, and 5v.Notes and references
- (a) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, 2nd edn, Wiley-VCH, Weinheim, 2003 Search PubMed; (b) G. Jones, Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, New York, 1996 Search PubMed.
- For reviews on C–H annulations, see: (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (c) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (d) M. Gulñas and J. L. MascareÇas, Angew. Chem., Int. Ed., 2016, 55, 11000 CrossRef PubMed; (e) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS.
- For examples of N-heterocycle synthesis from anilines and derivatives with alkynes, see: (a) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS PubMed; (b) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed; (c) M. P. Huestis, L. Chan, D. R. Stuart and K. Fagnou, Angew. Chem., Int. Ed., 2011, 50, 1338 CrossRef CAS PubMed; (d) C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS PubMed; (e) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed; (f) L. Ackermann and A. V. Lygin, Org. Lett., 2012, 14, 764 CrossRef CAS PubMed; (g) J. Chen, G. Song, C.-L. Pan and X. Li, Org. Lett., 2010, 12, 5426 CrossRef CAS PubMed; (h) S. Cai, K. Yang and D. Z. Wang, Org. Lett., 2014, 16, 2606 CrossRef CAS PubMed; (i) P. Tao and Y. Ji, Chem. Commun., 2014, 50, 7367 RSC; (j) Y. Kim and S. Hong, Chem. Commun., 2015, 51, 11202 RSC.
- For examples of N-heterocycle synthesis from the three-component cyclization of secondary anilines with alkynes and CO (1–30 atm), using a stoichiometric amount of BQ or copper salt as the oxidant, see: (a) J. Chen, K. Natte, A. Spannenberg, H. Neumann, M. Beller and X.-F. Wu, Chem.–Eur. J., 2014, 20, 14189 CrossRef CAS PubMed; (b) X. Y. Li, X. W. Li and N. Jiao, J. Am. Chem. Soc., 2015, 137, 9246 CrossRef CAS PubMed; (c) F. Zhu, Y. Li, Z. Wang and X.-F. Wu, Adv. Synth. Catal., 2016, 358, 3350 CrossRef CAS; (d) J. Chen, J.-B. Feng, K. Natte and X.-F. Wu, Chem.–Eur. J., 2015, 21, 16370 CrossRef CAS PubMed.
- For examples of N-heterocycle synthesis from other N-containing substrates: (a) R. Bernini, G. Fabrizi, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2009, 48, 8078 CrossRef CAS PubMed; (b) S. Würtz, S. Rakshit, J. J. Neumann, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 7230 CrossRef PubMed; (c) X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS PubMed; (d) T. K. Hyster and T. Rovis, Chem. Sci., 2011, 2, 1606 RSC; (e) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS PubMed; (f) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed; (g) L. Ackermann, A. V. Lygin and N. Hofmann, Org. Lett., 2011, 13, 3278 CrossRef CAS PubMed; (h) Z. Shi, B. Zhang, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 4036 CrossRef CAS PubMed; (i) Y. Wei, I. Deb, N. Yoshikai, J. Am. Chem. Soc., 2012, 134, 9098 Search PubMed; (j) L. Ackermann, L. Wang and A. V. Lygin, Chem. Sci., 2012, 3, 177 RSC; (k) L. Wang and L. Ackermann, Org. Lett., 2013, 15, 176 CrossRef CAS PubMed; (l) S. Y. Hong, J. Jeong and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 2408 CrossRef CAS PubMed; (m) K. Alex, A. Tillack, N. Schwarz and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2304 CrossRef CAS PubMed; (n) M. Shen, B. E. Leslie and T. G. Driver, Angew. Chem., Int. Ed., 2008, 47, 5056 CrossRef CAS PubMed; (o) Á. M. Martínez, J. Echavarren, I. Alonso, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Sci., 2015, 6, 5802 RSC.
- (a) Z. Shi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572 CrossRef CAS PubMed; (b) G. Zhang, H. Yu, G. Qin and H. Huang, Chem. Commun., 2014, 50, 4331 RSC.
- (a) G. Choudhary and R. K. Peddinti, Green Chem., 2011, 13, 3290 RSC; (b) C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174 CrossRef CAS PubMed.
- For a C–H cyclization example with alkenes, see: (a) R. Shi, L. Lu, H. Zhang, B. Chen, Y. Sha, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 10582 CrossRef CAS PubMed; for intra-molecular cyclization via C–N bond cleavage, see: (b) D. Yue and R. C. Larock, Org. Lett., 2004, 6, 1037 CrossRef CAS PubMed; (c) B. Yao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2012, 51, 5170 CrossRef CAS PubMed; (d) X.-F. Xia, L.-L. Zhang, X.-R. Song, Y.-N. Niu, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2013, 49, 1410 RSC; (e) B. Yao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12992 CrossRef CAS PubMed; (f) J. Sheng, S. Li and J. Wu, Chem. Commun., 2014, 50, 578 RSC; (g) R. Shi, H. Niu, L. Lu and A. Lei, Chem. Commun., 2017, 53, 1908 RSC.
- (a) X. Zhao, D. Liu, H. Guo, Y. Liu and W. Zhang, J. Am. Chem. Soc., 2011, 133, 19354 CrossRef CAS PubMed; (b) Y. Kuninobu, M. Nishi and K. Takai, Chem. Commun., 2010, 46, 8860 RSC; (c) J. Shearer, C. X. Zhang, L. Q. Hatcher and K. D. Karlin, J. Am. Chem. Soc., 2003, 125, 12670 CrossRef CAS PubMed; (d) S. Ueno, N. Chatani and F. Kakiuchi, J. Am. Chem. Soc., 2007, 129, 6098 CrossRef CAS PubMed; (e) S. B. Blakey and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 6046 CrossRef CAS PubMed; (f) L.-G. Xie and Z.-X. Wang, Angew. Chem., Int. Ed., 2011, 50, 4901 CrossRef CAS PubMed; C–N bond cleavage of amines: (g) Y. Xie, J. Hu, Y. Wang, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 20613 CrossRef CAS PubMed; (h) M.-B. Li, Y. Wang and S.-K. Tian, Angew. Chem., Int. Ed., 2012, 51, 2968 CrossRef CAS PubMed.
- (a) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed; (b) P. T. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC; (c) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed; (d) J. Piera and J. E. Backvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS PubMed; (e) F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508 CrossRef CAS PubMed; (f) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932 CrossRef CAS PubMed.
- For examples of Rh-catalyzed aerobic oxidative annulations, see: (a) G. Zhang, L. Yang, Y. Wang, Y. Xie and H. Huang, J. Am. Chem. Soc., 2013, 135, 8850 CrossRef CAS PubMed; (b) Y. Lu, H.-W. Wang, J. Spangler, K. Chen, P.-P. Cui, Y. Zhao, W.-Y. Sun and J.-Q. Yu, Chem. Sci., 2015, 6, 1923 RSC; (c) Q. Jiang, C. Zhu, H. Zhao and W. Su, Chem.–Asian J., 2016, 11, 356 CrossRef CAS PubMed; (d) G. Zhang, H. Yu, G. Qin and H. Huang, Chem. Commun., 2014, 50, 4331 RSC; (e) C.-Z. Luo, P. Gandeepan, J. Jayakumar, K. Parthasarathy, Y.-W. Chang and C.-H. Cheng, Chem.–Eur. J., 2013, 19, 14181 CrossRef CAS PubMed; (f) J. Jayakumar and C.-H. Cheng, Chem.–Eur. J., 2016, 22, 1800 CrossRef CAS PubMed.
- (a) G. Claassen, E. Brin, C. Crogan-Grundy, M. T. Vaillancourt, H. Z. Zhang, S. X. Cai, J. Drewe, B. Tseng and S. Kasibhatla, Cancer Lett., 2009, 274, 243 CrossRef CAS PubMed; (b) R. M. Forbis and K. L. Rinehart, J. Am. Chem. Soc., 1973, 95, 5003 CrossRef CAS PubMed; (c) A. L. Hopkins, J. Ren, J. Milton, R. J. Hazen, J. H. Chan, D. I. Stuart and D. K. Stammers, J. Med. Chem., 2004, 47, 5912 CrossRef CAS PubMed; (d) A. Doleans-Jordheim, J.-B. Veron, O. Fendrich, E. Bergeron, A. Montagut-Romans, Y.-S. Wong, B. Furdui, J. Freney, C. Dumontet and A. Boumendjel, ChemMedChem, 2013, 8, 652 CrossRef CAS PubMed; (e) R. W. Carling, P. D. Leeson, K. W. Moore, J. D. Smith, C. R. Moyes, I. M. Mawer, S. Thomas, T. Chan and R. Baker, J. Med. Chem., 1993, 36, 3397 CrossRef CAS PubMed.
- (a) D. V. Kadnikov and R. C. Larock, J. Org. Chem., 2004, 69, 6772 CrossRef CAS PubMed; (b) T. Iwai, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2010, 132, 9602 CrossRef CAS PubMed; (c) R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408 CrossRef CAS PubMed; (d) J. Wu, S. Xiang, J. Zeng, M. Leow and X.-W. Liu, Org. Lett., 2015, 17, 222 CrossRef CAS PubMed; (e) R. Manikandan and M. Jeganmohan, Org. Lett., 2014, 16, 3568 CrossRef CAS PubMed; (f) B.-X. Tang, R.-J. Song, C.-Y. Wu, Z.-Q. Wang, Y. Liu, X.-C. Huang, Y.-X. Xie and J.-H. Li, Chem. Sci., 2011, 2, 2131 RSC; (g) P. J. Manley and M. T. Bilodeau, Org. Lett., 2004, 6, 2433 CrossRef CAS PubMed; (h) X.-F. Yang, X.-H. Hu and T.-P. Loh, Org. Lett., 2015, 17, 1481 CrossRef CAS PubMed.
- For reviews on carbonylation reactions, see: (a) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC; (b) X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447 CrossRef CAS; (c) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed; (d) Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788 CrossRef CAS PubMed.
- For examples of carbonylation reactions, see: (a) N. Chatani, T. Asaumi, T. Ikeda, S. Yorimitsu, Y. Ishii, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2000, 122, 12882 CrossRef CAS; (b) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 7316 CrossRef CAS PubMed; (c) X. Fang, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14089 CrossRef CAS PubMed; (d) K. Dong, X. Fang, R. Jackstell, G. Laurenczy, Y. Li and M. Beller, J. Am. Chem. Soc., 2015, 137, 6053 CrossRef CAS PubMed; (e) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed; (f) Z.-H. Guan, Z.-H. Ren, S. M. Spinella, S. Yu, Y.-M. Liang and X. Zhang, J. Am. Chem. Soc., 2009, 131, 729 CrossRef CAS PubMed; (g) C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. G. Tyler, M. R. Gagne, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830 CrossRef CAS PubMed; (h) J. S. Quesnel, A. Fabrikant and B. A. Arndtsen, Chem. Sci., 2016, 7, 295 RSC; (i) Z.-H. Guan, M. Chen and Z.-H. Ren, J. Am. Chem. Soc., 2012, 134, 17490 CrossRef CAS PubMed; (j) W. Li, Z. Duan, X. Zhang, H. Zhang, M. Wang, R. Jiang, H. Zeng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1893 CrossRef CAS PubMed; (k) W. Li, C. Liu, H. Zhang, K. Ye, G. Zhang, W. Zhang, Z. Duan, S. You and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 2443 CrossRef CAS PubMed; (l) J. Tjutrins and B. A. Arndtsen, Chem. Sci., 2017, 8, 1002 RSC.
- (a) J. DeRuiter, B. E. Swearingen, V. Wandrekar and C.A. Mayfield, J. Med. Chem., 1989, 32, 1033 CrossRef CAS PubMed; (b) C. Peifer, R. Urich, V. Schattel, M. Abadleh, M. Röttig, O. Kohlbacher and S. Laufer, Bioorg. Med. Chem. Lett., 2008, 18, 1431 CrossRef CAS PubMed.
- E. Sheiban and K. Wärnmark, Org. Biomol. Chem., 2012, 10, 2059 Search PubMed.
- T. Okuzumi, T. Yoshimura, E. Nakanishi, M. Ono and M. Murata, Novel Phenylalanine Derivatives, WO 03/053926 A1, July 3, 2003.
- Computational details and references are given in the ESI.†.
- (a) E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047 CrossRef CAS PubMed; (b) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed; (c) S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864 CrossRef CAS PubMed; (d) W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS; (e) J.-S. Tian and T.-P. Loh, Angew. Chem., Int. Ed., 2010, 49, 8417 CrossRef CAS PubMed; (f) S. Guo, B. Qian, Y. Xie, C. Xia and H. Huang, Org. Lett., 2011, 13, 522 CrossRef CAS PubMed; (g) S. Mahapatra, J. A. Halfen and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 11575 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/c7sc02181j |
| This journal is © The Royal Society of Chemistry 2017 |