
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired enantioselective synthesis of crinine-type alkaloids via iridium-catalyzed asymmetric hydrogenation of enones†
Xiao-Dong
Zuo
a,
Shu-Min
Guo
a,
Rui
Yang
a,
Jian-Hua
Xie
 *a and
Qi-Lin
Zhou
*ab
*a and
Qi-Lin
Zhou
*ab
aState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: jhxie@nankai.edu.cn; qlzhou@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China
First published on 3rd July 2017
Abstract
A bioinspired enantioselective synthesis of crinine-type alkaloids has been developed by iridium-catalyzed asymmetric hydrogenation of racemic cycloenones. The method features a biomimetic stereodivergent resolution of the substrates bearing a remote arylated quaternary stereocenter. Using this protocol, 24 crinine-type alkaloids and 8 analogues were synthesized in a concise and rapid way with high yield and high enantioselectivity.
Introduction
Amaryllidaceae alkaloids have long attracted the attention of synthetic chemists due to their significant bioactivities such as antitumor, antiviral, and anti-acetylcholinesterase activities, and the fascinating diversity of their structures. To date, more than 500 Amaryllidaceae alkaloids have been isolated from Amaryllidaceae plants.1 However, only one member of them, galanthamine, has been approved as a prescription drug for the treatment of Alzheimer’s disease.2 Because these alkaloids are structurally intricate, general methods for their synthesis are lacking, which has hindered the study of their medicinal chemistry.We recently became particularly interested in crinine-type alkaloids, a large subclass (more than 80 have been isolated) of the Amaryllidaceae alkaloid family, that possess an antipodal chiral 5,10b-ethanophenanthridine core skeleton with a benzylic all-carbon quaternary centre1c (Fig. 1). For example, the parent alkaloid of the subclass, (−)-crinine, was first isolated in 1955 from the bulbs of two unidentified Crinum species from South Africa;3 (+)-vittatine, which has the opposite configuration to that of (−)-crinine, is another crinine-type alkaloid.4 Importantly, the crinine-type alkaloids are considered to be biogenic precursors of several types of Amaryllidaceae alkaloids including tazettine-, haemanthamine-, and narciclasine-type alkaloids5 (Fig. 1). Thus, exploring efficient methods for the rapid synthesis of diverse crinine-type alkaloids is particularly important and desirable. However, although great efforts have been devoted to the development of synthetic methods to obtain crinine-type alkaloids, most of the reported approaches provided racemic products.6
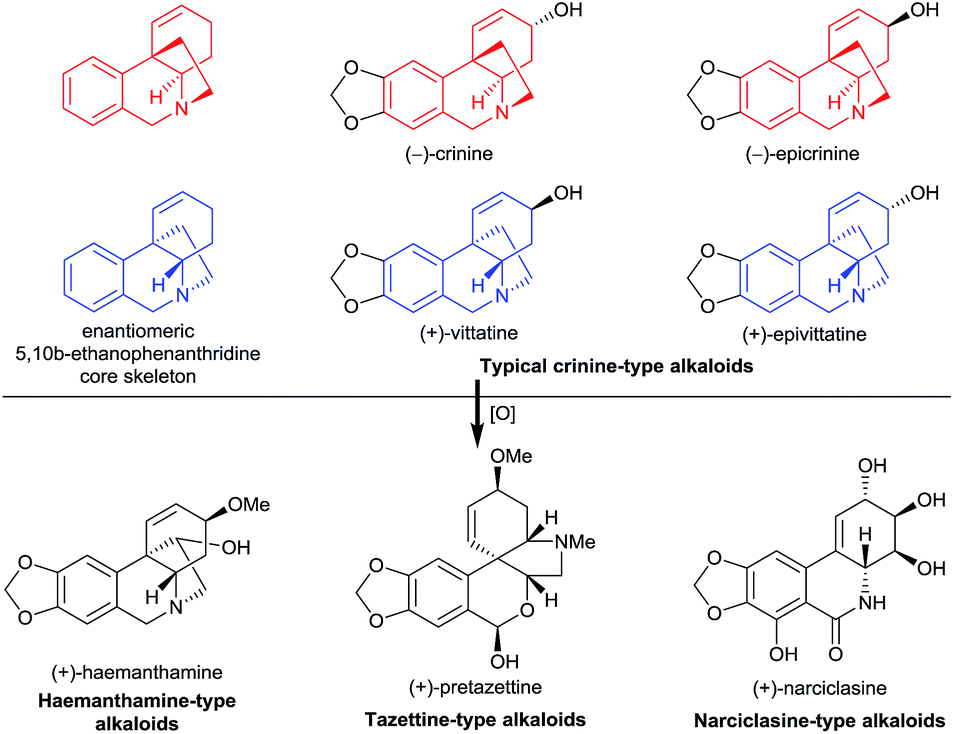 | ||
| Fig. 1 Representative crinine-type alkaloids and Amaryllidaceae alkaloids derived possibly from crinine-type alkaloids. | ||
We noticed that some Amaryllidaceae species can produce two similar alkaloids with enantiomeric skeletons. For example, both (−)-crinine and (+)-epivittatine have been isolated simultaneously from the bulbs of Nerine bowdenii, Boophane flava, and Crinum moorei (Scheme 1a).4b,9 Inspired by this natural phenomenon and the biosynthetic pathway of crinine-type alkaloids, we envisaged a new synthetic strategy that mimics the enzymatic reduction resolution of racemic oxocrinines 1 using synthetic chiral catalysts (Scheme 1b). Because racemic oxocrinines 1 can be prepared in only four steps in high yields by a biomimetic intramolecular phenolic oxidative coupling of O-methylnorbelladine derived from L-phenylalanine and L-tyrosine,6f this asymmetric catalytic stereodivergent resolution strategy10 will provide a concise and rapid approach to diverse crinine-type alkaloids and their analogues.
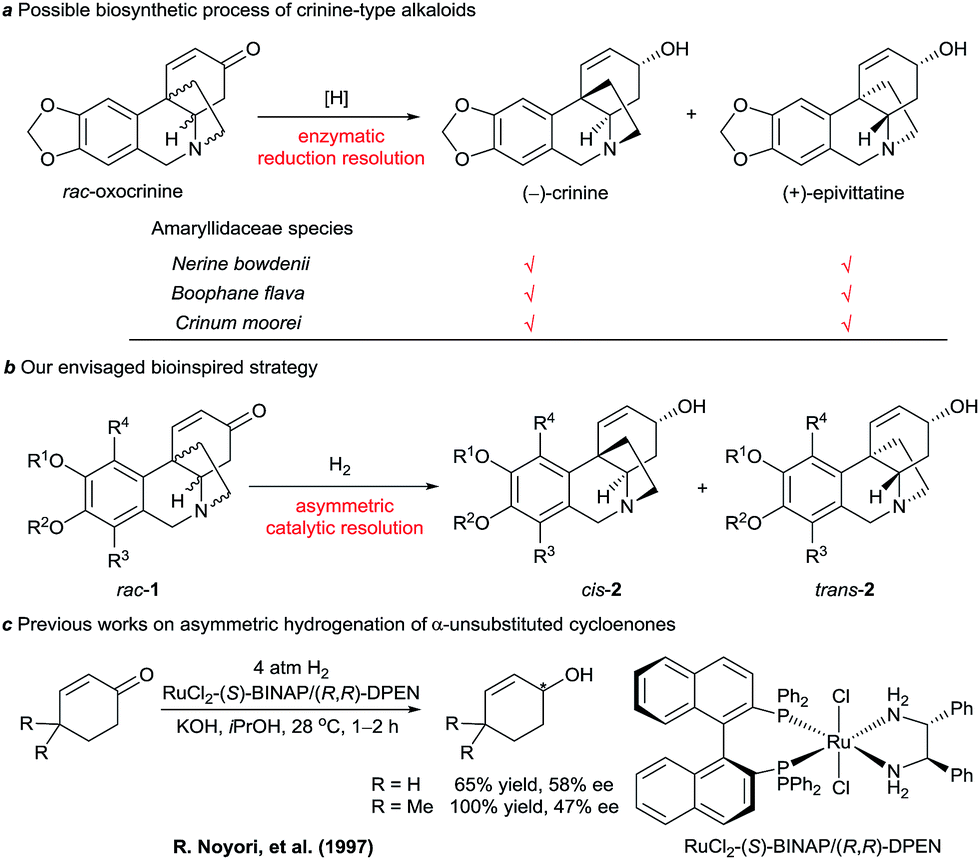 | ||
| Scheme 1 Possible biosynthetic process of crinine-type alkaloids and our envisaged bioinspired strategy. | ||
Results and discussion
Although asymmetric hydrogenation of the ketone group of α,β-unsaturated ketones is an efficient way for synthesising optically active allylic alcohols,11 the asymmetric hydrogenation of cyclohexenones with no substituent at the α-position is still a challenge (Scheme 1c).12 To find efficient chiral catalysts for the asymmetric hydrogenation of racemic oxocrinines 1, we investigated chiral spiro iridium catalysts Ir-SpiroPAP (3)13 developed in our laboratory. The racemic oxocrinines 1 were prepared in four steps with 55–66% yield from commercially available starting materials using Node’s biomimetic procedure6f (see the ESI†). Asymmetric hydrogenation of rac-1a (R1, R2 = –CH2–, R3, R4 = H) was firstly carried out using the catalyst (R)-3a. When the hydrogenation of rac-1a was performed under 1 atm of H2 pressure with KOtBu as the base in EtOH at room temperature, the reaction was completed within 0.8 h and the desired products (−)-cis-2a and (+)-trans-2a were obtained in 91% yield with a (−)-cis-2a/(+)-trans-2a ratio of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :88. However, the enantiomeric excess (ee) of the major product (+)-trans-2a was only 27% (Table 1, entry 1). A comparison of the various Ir-SpiroPAP catalysts showed that (R)-3d, containing P(3,5-di-tert-butylphenyl)2 groups and a 3-Me–pyridine moiety, is the best catalyst, which afforded the products (−)-cis-2a and (+)-trans-2a in 93% yield with 97% ee and 87% ee, respectively, in a ratio of 45:55 (entry 4). The effect of solvent was examined, and nPrOH gave comparable results to EtOH (entry 8). In addition to KOtBu, other bases such as KOH, K2CO3 and Et3N can also be used, although the reaction with Et3N needs a longer time for completion and the yield is lower (entry 11). Further study of the reaction temperature, base concentration, hydrogen pressure, and co-solvent established the optimal reaction conditions to be as follows: 0.1 mol% (R)-3d, [rac-1a] = 0.17 M, [KOtBu] = 0.008 M, 0 °C, 1 atm of H2, EtOH/DCM (5:2). Under these conditions, hydrogenation of rac-1a afforded two crinine-type alkaloids: (−)-cis-2a (crinine, 97% ee) and (+)-trans-2a (epivittatine, 93% ee) in 94% overall yield with a (−)-cis-2a/(+)-trans-2a ratio of 46:54 (entry 16). It is to be noted that we have tried the direct isolation of the mixture of (−)-cis-2a and (+)-trans-2a by chromatography on a Sephadex LH-20 column according to Codina’s protocol,14 but it was demonstrated to be difficult. Fortunately, we found that the mixture can be isolated by converting them into benzoyl esters, and after hydrolysis of the isolated benzoyl esters (−)-cis-2a and (+)-trans-2a can be obtained in pure form with high yield.
:88. However, the enantiomeric excess (ee) of the major product (+)-trans-2a was only 27% (Table 1, entry 1). A comparison of the various Ir-SpiroPAP catalysts showed that (R)-3d, containing P(3,5-di-tert-butylphenyl)2 groups and a 3-Me–pyridine moiety, is the best catalyst, which afforded the products (−)-cis-2a and (+)-trans-2a in 93% yield with 97% ee and 87% ee, respectively, in a ratio of 45:55 (entry 4). The effect of solvent was examined, and nPrOH gave comparable results to EtOH (entry 8). In addition to KOtBu, other bases such as KOH, K2CO3 and Et3N can also be used, although the reaction with Et3N needs a longer time for completion and the yield is lower (entry 11). Further study of the reaction temperature, base concentration, hydrogen pressure, and co-solvent established the optimal reaction conditions to be as follows: 0.1 mol% (R)-3d, [rac-1a] = 0.17 M, [KOtBu] = 0.008 M, 0 °C, 1 atm of H2, EtOH/DCM (5:2). Under these conditions, hydrogenation of rac-1a afforded two crinine-type alkaloids: (−)-cis-2a (crinine, 97% ee) and (+)-trans-2a (epivittatine, 93% ee) in 94% overall yield with a (−)-cis-2a/(+)-trans-2a ratio of 46:54 (entry 16). It is to be noted that we have tried the direct isolation of the mixture of (−)-cis-2a and (+)-trans-2a by chromatography on a Sephadex LH-20 column according to Codina’s protocol,14 but it was demonstrated to be difficult. Fortunately, we found that the mixture can be isolated by converting them into benzoyl esters, and after hydrolysis of the isolated benzoyl esters (−)-cis-2a and (+)-trans-2a can be obtained in pure form with high yield.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | (R)-3 | Sol. | Base | Time (h) | Yieldb (%) | cis/transc | eed (%) | |
| cis | trans | |||||||
|
a Reaction conditions: 1 mmol scale, [1a] = 0.17 M, 0.1 mol% of (R)-3, [base] = 0.017 M, solvent (6.0 mL), 1 atm of H2, room temperature (22–27 °C), 100% conversion.
b Isolated yield of the mixture of (−)-cis-2a and (+)-trans-2a.
c The ratio of cis to trans was determined by 1H NMR.
d The ee values of (−)-cis-2a and (+)-trans-2a were determined by chiral HPLC (Chiralcel OD-3 column) after converting them into benzoyl esters.
e Under 5 atm of H2.
f At 0 °C.
g At 0 °C, and [KOtBu] = 0.034 M.
h At 0 °C, and [KOtBu] = 0.008 M.
i At 0 °C, [KOtBu] = 0.008 M, and DCM as co-solvent (ethanol/DCM = 5:2).
|
||||||||
| 1 | (R)-3a | EtOH | KOtBu | 0.8 | 91 | 11:88 |
90 | 27 |
| 2 | (R)-3b | EtOH | KOtBu | 0.5 | 89 | 15:85 |
88 | 23 |
| 3 | (R)-3c | EtOH | KOtBu | 0.8 | 95 | 46:54 |
90 | 86 |
| 4 | (R)-3d | EtOH | KOtBu | 0.5 | 93 | 45:55 |
97 | 87 |
| 5 | (R)-3e | EtOH | KOtBu | 1.0 | 90 | 42:58 |
91 | 77 |
| 6 | (R)-3f | EtOH | KOtBu | 0.3 | 95 | 36:64 |
94 | 64 |
| 7 | (R)-3d | MeOH | KOtBu | 0.7 | 92 | 40:60 |
92 | 76 |
| 8 | (R)-3d | nPrOH | KOtBu | 0.8 | 91 | 43:57 |
95 | 90 |
| 9 | (R)-3d | EtOH | KOH | 0.3 | 90 | 46:54 |
95 | 87 |
| 10 | (R)-3a | EtOH | K2CO3 | 0.5 | 91 | 46:54 |
95 | 87 |
| 11 | (R)-3d | EtOH | Et3N | 19 | 79 | 38:62 |
90 | 90 |
| 12e | (R)-3d | EtOH | KOtBu | 0.3 | 90 | 44:56 |
99 | 85 |
| 13f | (R)-3d | EtOH | KOtBu | 3 | 93 | 46:54 |
98 | 88 |
| 14g | (R)-3d | EtOH | KOtBu | 7.5 | 91 | 46:54 |
94 | 88 |
| 15h | (R)-3d | EtOH | KOtBu | 9 | 89 | 46:54 |
99 | 90 |
| 16i | (R)-3d | EtOH | KOtBu | 8.5 | 94 | 46:54 |
97 | 93 |

Using this bioinspired asymmetric hydrogenation stereodivergent resolution method, a range of crinine-type alkaloids and analogues were synthesized from the corresponding racemic oxocrinines 1 (Table 2). In each reaction, two crinine-type alkaloids or analogues were obtained in high yield with high enantioselectivity. The absolute configurations of the products were determined by the configuration of the catalyst.
|
a Reaction conditions: 1 mmol scale, [1] = 0.17 M, 0.1 mol% of (R)-3d or (S)-3d, [KOtBu] = 0.008 M, EtOH/DCM (5:2, 6.0 mL), 1 atm H2, 0 °C, 100% conversion. The cis/trans ratio was determined by 1H NMR. The products were obtained by converting them into benzoyl esters, followed by chromatography on silica gel and hydrolysis of the resulting isolated products with NaOH aqueous solution. The ee values of the products were determined by chiral HPLC analysis of the corresponding benzoyl esters. All the yields are isolated yields.
|
|---|

|
The Ir-SpiroPAP-catalyzed asymmetric hydrogenation of oxocrinines 1 could be performed on a multigram scale (Scheme 2). For example, hydrogenation of substrates rac-1a and rac-1c on a 2 g scale produced (−)-cis-2a and (+)-trans-2a, and (−)-cis-2c and (+)-trans-2c in the presence of the catalyst (R)-3d and produced (+)-cis-2a and (−)-trans-2a, and (+)-cis-2c and (−)-trans-2c in the presence of the catalyst (S)-3d, with high yields and high enantioselectivities. After saturation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of alkaloids 2a over Pd/C, four dihydrocrinine-type alkaloids were obtained: (−)-dihydrocrinine,15 (+)-dihydrovittatine,16 (+)-dihydroepivittatine,17 and (−)-dihydroepicrinine.17 By debenzylation of alkaloids 2c with boron trichloride (BCl3), four naturally occurring crinine-type alkaloids were obtained: (+)-8-O-demethylmaritidine,18 (−)-8-O-demethylmaritidine,19 (+)-siculine,16 and (−)-siculine.20
C bonds of alkaloids 2a over Pd/C, four dihydrocrinine-type alkaloids were obtained: (−)-dihydrocrinine,15 (+)-dihydrovittatine,16 (+)-dihydroepivittatine,17 and (−)-dihydroepicrinine.17 By debenzylation of alkaloids 2c with boron trichloride (BCl3), four naturally occurring crinine-type alkaloids were obtained: (+)-8-O-demethylmaritidine,18 (−)-8-O-demethylmaritidine,19 (+)-siculine,16 and (−)-siculine.20
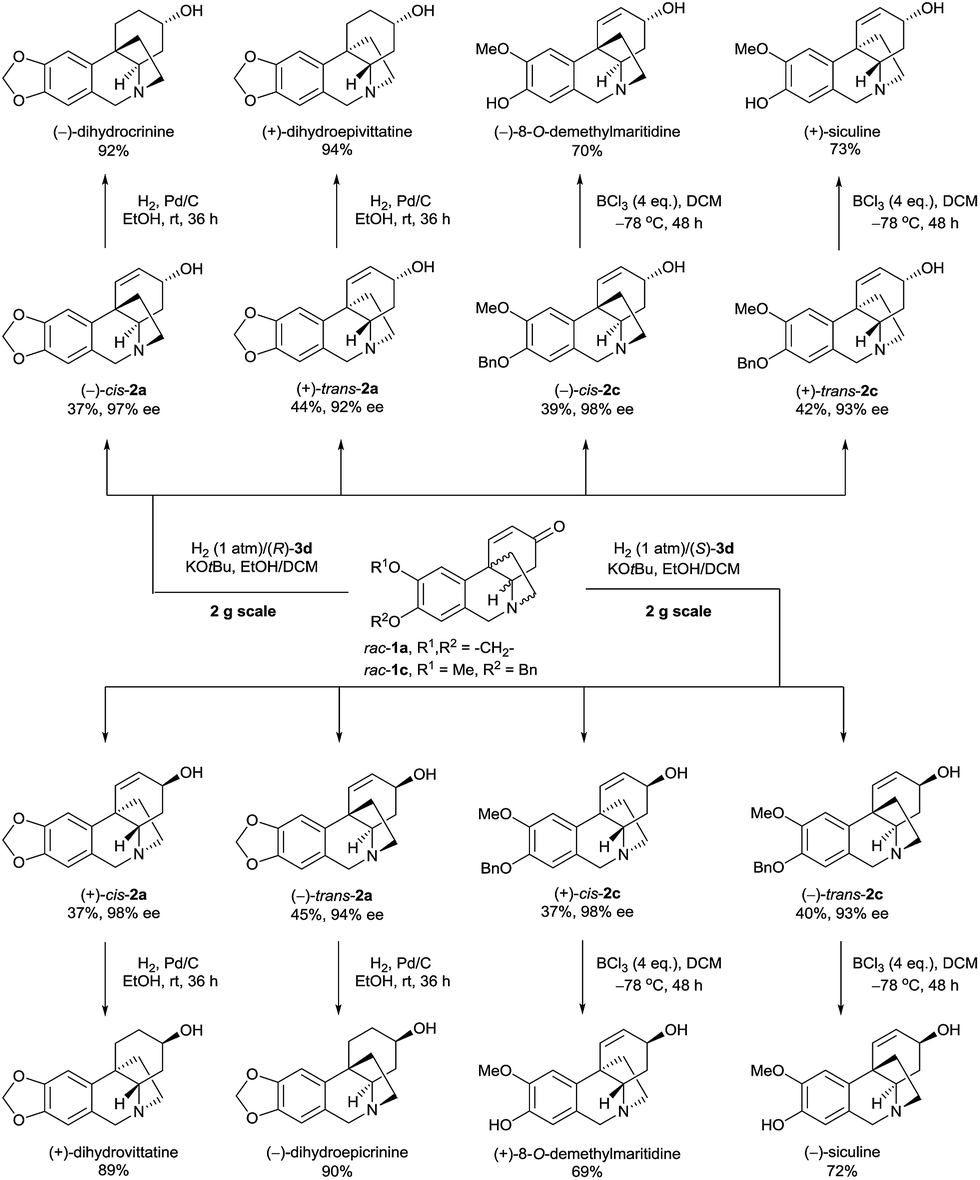 | ||
| Scheme 2 Asymmetric syntheses of 8-O-demethylmaritidines, siculines and dihydrocrinine-type alkaloids on the gram-scale. | ||
The methylation of the hydroxy group of the alkaloids 2 can offer O-methyl crinine-type alkaloids. When (−)-cis-2a and (−)-cis-2d were treated with TMSCH2N2 (trimethylsilyl diazomethane) in the presence of HBF4 in DCM, another two alkaloids, (−)-buphanisine and (−)-buphanidrine,21 were obtained in 62% and 68% yield, respectively (Scheme 3). According to Guillou’s procedure,6j,22 (−)-trans-2a was successfully oxidized with peroxymidic acid generated in situ from CCl3CN/H2O2 to the epoxide 4 in 68% yield in the presence of trifluoroacetic acid. A Mitsunobu reaction converted the epoxide 4 to the benzoate, followed by the removal of the benzoyl group with LiAlH4, yielding (−)-flexinine23 in 60% yield (2 steps). The (−)-flexinine was reacted with TMSCH2N2 in the presence of HBF4 in DCM to produce (−)-augustine in 69% yield. Thus, the enantioselective syntheses of (−)-buphanisine, (−)-buphanidrine, (−)-flexinine, and (−)-augustine were also achieved.
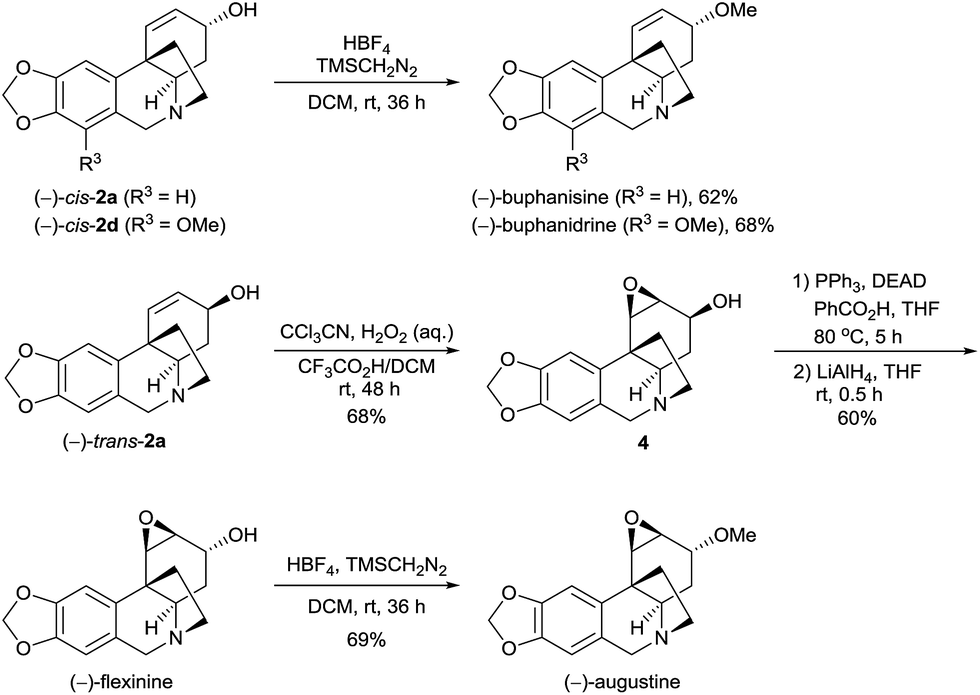 | ||
| Scheme 3 Asymmetric synthesis of O-methyl crinine-type alkaloids and analogues. | ||
Conclusions
In conclusion, we have developed a bioinspired strategy for the rapid enantioselective synthesis of crinine-type alkaloids. This strategy features an iridium-catalyzed asymmetric hydrogenation of racemic cycloenones with a remote arylated quaternary stereocenter via stereodivergent resolution. Using this new strategy, we synthesized a total of 24 crinine-type alkaloids and 8 analogues in 7.9–29.4% yields in only five or nine steps (seven or eleven steps including the esterification and hydrolysis in the separation of the hydrogenation products). Among these crinine-type alkaloids, seven have been previously synthesized with 8–20 steps in 0.6–13.2% yields (see also ESI†).7,8 Eleven were synthesized for the first time: (+)- and (−)-8-O-demethylmaritidine, (+)- and (−)-siculine, (−)-maritidine, (−)-epimaritidine, (+)-powelline, (+)-epipowelline, (−)-buphanidrine, (−)-flexinine and (−)-augustine. The concise, practical strategy reported here for the synthesis of crinine-type alkaloids and analogues is expected to be applicable for the rapid syntheses of other types of Amaryllidaceae alkaloids or even other types of natural products.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21325207, 21532003, and 21421062), and the “111” project (B06005) of the Ministry of Education of China for financial support.Notes and references
- (a) J. J. Nair, J. Bastida, C. Codina, F. Viladomat and J. V. Staden, Nat. Prod. Commun., 2013, 8, 1335 CAS; (b) Z. Jin and X.-H. Xu, in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, ed. K. G. Ramawat and J. M. Mérillon, Springer-Verlag, Berlin Heidelberg, 2013, p. 479 Search PubMed; (c) Y. Ding, D. Qu, K.-M. Zhang, X.-X. Cang, Z.-N. Kou, W. Xiao and J.-B. Zhu, J. Asian Nat. Prod. Res., 2016, 12, 1 CrossRef PubMed.
- P. Williams, A. Sorribas and M.-J. R. Howes, Nat. Prod. Rep., 2011, 28, 48 RSC.
- L. H. Masone, E. R. Puschett and W. C. Wildman, J. Am. Chem. Soc., 1955, 77, 1253 CrossRef.
- (a) H.-G. Boit, Chem. Ber., 1956, 89, 1129 CrossRef CAS; (b) A. I. Feistein and W. C. Wildman, J. Org. Chem., 1976, 41, 2447 CrossRef.
- M. B. Kilgore and T. M. Kutchan, Phytochem. Rev., 2016, 15, 317 CrossRef CAS PubMed.
- (a) H. Muxfeldt, R. S. Schneider and J. B. Mooberry, J. Am. Chem. Soc., 1966, 88, 3670 CrossRef CAS; (b) H. W. Whitlock and G. L. Smith, J. Am. Chem. Soc., 1967, 89, 3600 CrossRef CAS; (c) R. V. Stevens, L. E. DuPree Jr and P. L. Loewenstein, J. Org. Chem., 1972, 37, 977 CrossRef CAS; (d) G. E. Keck and R. R. Webb II, J. Am. Chem. Soc., 1981, 103, 3173 CrossRef CAS; (e) I. H. Sánchez, F. J. López, J. J. Soria, M. I. Larraza and H. J. Flores, J. Am. Chem. Soc., 1983, 105, 7640 CrossRef; (f) S. F. Martin and C. L. Campbell, Tetrahedron Lett., 1987, 28, 503 CrossRef CAS; (g) W. H. Pearson and F. E. Lovering, Tetrahedron Lett., 1994, 35, 9173 CrossRef CAS; (h) C. Bru, C. Thal and C. Guillou, Org. Lett., 2003, 5, 1845 CrossRef CAS PubMed; (i) S. Kodama, H. Takita, T. Kajimoto, K. Nishide and M. Node, Tetrahedron, 2004, 60, 4901 CrossRef CAS; (j) C. Bru and C. Guillou, Tetrahedron, 2006, 62, 9043 CrossRef CAS; (k) H. F. Anwar and T. V. Hansen, Synlett, 2008, 2681 CAS; (l) N. T. Tam and C.-G. Cho, Org. Lett., 2008, 10, 601 CrossRef CAS PubMed; (m) K. M. Bogle, D. J. Hirst and D. J. Dixon, Org. Lett., 2010, 12, 1252 CrossRef CAS PubMed; (n) K. M. Boglea, D. J. Hirst and D. J. Dixon, Tetrahedron, 2010, 66, 6399 CrossRef.
- (a) S.-I. Yamada, K. Tomioka and K. Koga, Tetrahedron Lett., 1976, 17, 57 CrossRef; (b) K. Tomioka, K. Koga and S.-I. Yamada, Chem. Pharm. Bull., 1977, 25, 2681 CrossRef CAS; (c) L. E. Overman and S. Sugai, Helv. Chim. Acta, 1985, 68, 745 CrossRef CAS; (d) Y. Kita, T. Takeda, M. Gyoten, H. Tohma, M. H. Zenk and J. Eichhorn, J. Org. Chem., 1996, 61, 5857 CrossRef CAS; (e) M. Bohno, H. Imase and N. Chida, Chem. Commun., 2004, 1086 RSC; (f) M. Bohno, K. Sugie, H. Imase, Y. N. Yusof, T. Oishib and N. Chida, Tetrahedron, 2007, 63, 6977 CrossRef CAS; (g) G. Rousseau, R. Lebeuf, K. Schenk, F. Castet, F. Robert and Y. Landais, Chem.–Eur. J., 2014, 20, 14771 CrossRef CAS PubMed.
- M.-X. Wei, C.-T. Wang, J.-Y. Du, H. Qu, P.-R. Yin, X. Bao, X.-Y. Ma, X.-H. Zhao, G.-B. Zhang and C.-A. Fan, Chem.–Asian J., 2013, 8, 1966 CrossRef CAS PubMed.
- (a) R. E. Lyle, E. A. Kielar, J. R. Crowder and W. C. Wildman, J. Am. Chem. Soc., 1960, 82, 2625 CrossRef; (b) F. Viladomat, J. Bastida, C. Codina, W. E. Campbell and S. Mathee, Phytochemistry, 1995, 40, 307 CrossRef CAS.
- For recent reviews on stereodivergent reactions of racemic mixtures, see: (a) H. B. Kagan, Tetrahedron, 2001, 57, 2449 CrossRef CAS; (b) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed; (c) L. C. Miller and R. Sarpong, Chem. Soc. Rev., 2011, 40, 4550 RSC ; For selected papers, see; (d) N. T. Reynolds and T. Rovis, Tetrahedron, 2005, 61, 6368 CrossRef CAS; (e) C. K. Jana and A. Studer, Angew. Chem., Int. Ed., 2007, 46, 6542 CrossRef CAS PubMed; (f) L. C. Miller, J. M. Ndungu and R. Sarpong, Angew. Chem., Int. Ed., 2009, 48, 2398 CrossRef CAS PubMed; (g) Y. Lian, L. C. Miller, S. Born, R. Sarpong and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 12422 CrossRef CAS PubMed; (h) S. Samanta, S. Perera and C.-G. Zhao, J. Org. Chem., 2010, 75, 1101 CrossRef CAS PubMed.
- For reviews of asymmetric hydrogenation of enones to chiral allylic alcohols see: (a) R. Noyori and T. Okuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS ; for selected recent papers, see:; (b) N. Arai, K. Azuma, N. Nii and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 7457 CrossRef CAS PubMed; (c) H. Shimizu, T. Nagano, N. Sayo, T. Saito, T. Ohshima and K. Mashima, Synlett, 2009, 3143 CrossRef CAS; (d) J.-Β. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538 CrossRef CAS PubMed; (e) Q.-Q. Zhang, J.-H. Xie, X.-H. Yang, J.-B. Xie and Q.-L. Zhou, Org. Lett., 2012, 14, 6158 CrossRef CAS PubMed; (f) X. Chen, H. Zhou, K. Zhang, J. Li and H. Huang, Org. Lett., 2014, 16, 3912 CrossRef CAS PubMed.
- The existence of a substituent such as a methyl group at the α-position of the cyclohexenones is crucial for obtaining high enantioselectivity, see: (a) T. Ohkuma, H. Ooka, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 10417 CrossRef CAS; (b) T. Ohkuma, H. Ikehira, T. Ikariya and R. Noyori, Synlett, 1997, 467 CrossRef CAS; (c) T. Ohkuma, T. Hattori, H. Ooka, T. Inoue and R. Noyori, Org. Lett., 2004, 6, 2681 CrossRef CAS PubMed.
- (a) J.-H. Xie, X.-Y. Liu, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 7329 CrossRef CAS PubMed; (b) J.-H. Xie, X.-Y. Liu, X.-H. Yang, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 201 CrossRef CAS PubMed.
- Codina, et al. reported that (−)-crinine and (+)-epivittatine can be isolated by chromatography on a Sephadex LH-20 column. See ref. 9b.
- W. C. Wildman, J. Am. Chem. Soc., 1958, 80, 2567 CrossRef CAS.
- C.-K. Chen, F.-H. Lin, L.-H. Tseng, C.-L. Jiang and S.-S. Lee, J. Nat. Prod., 2011, 74, 411 CrossRef CAS PubMed.
- H. Irie, S. Uyeo and A. Yoshitake, J. Chem. Soc. C, 1968, 1802 RSC.
- M. Kihara, T. Koike, Y. Imakura, K. Kida, T. Shingu and S. Kobayashi, Chem. Pharm. Bull., 1987, 35, 1070 CrossRef CAS.
- A. H. Abou-Donia, M. E. Amer, F. A. Darwish, F. F. Kassem, H. M. Hammoda, M. S. Abdel-Kader, B.-N. Zhou and D. G. Kingston, Planta Med., 2002, 68, 379 CrossRef CAS PubMed.
- V. Pabuççuoğlu, P. Richomme, T. Gözler, B. Kivçak, A. J. Freyer and M. Shamma, J. Nat. Prod., 1989, 52, 785 CrossRef.
- A. Machocho, S. C. Chhabra, F. Viladomat, C. Codina and J. Bastida, Phytochemistry, 1999, 51, 1185 CrossRef CAS.
- (a) K. Likhitwitayawuid, C. K. Angerhofer, H. Chai, J. M. Pezzuto, G. A. Cordell and N. Ruangrungsi, J. Nat. Prod., 1991, 56, 1331 CrossRef; (b) L. H. Pham, W. Dopke, J. Wagner and C. Mugge, Phytochemistry, 1998, 48, 371 CrossRef CAS.
- (a) A. A. Ali, H. M. El Sayed, O. M. Abdallah and W. Steglish, Phytochemistry, 1986, 25, 2399 CrossRef CAS; (b) A. K. Machocho, J. Bastida, C. Codina, F. Viladomat, R. Brun and S. C. Chhabra, Phytochemistry, 2004, 65, 3143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02112g |
| This journal is © The Royal Society of Chemistry 2017 |