
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple and traceless solid phase method simplifies the assembly of large peptides and the access to challenging proteins†
N.
Ollivier
,
R.
Desmet
,
H.
Drobecq
,
A.
Blanpain
,
E.
Boll
,
B.
Leclercq
,
A.
Mougel
,
J.
Vicogne
 and
O.
Melnyk
*
and
O.
Melnyk
*
UMR CNRS 8161 CNRS, Université de Lille, Institut Pasteur de Lille, 1 rue du Pr Calmette, 59021 Lille Cedex, France. E-mail: oleg.melnyk@ibl.cnrs.fr
First published on 30th May 2017
Abstract
Chemical protein synthesis gives access to well-defined native or modified proteins that are useful for studying protein structure and function. The majority of proteins synthesized up to now have been produced using native chemical ligation (NCL) in solution. Although there are significant advantages to assembling large peptides or proteins by solid phase ligation, reports of such approaches are rare. We report a novel solid phase method for protein synthesis which relies on the chemistry of the acetoacetyl group and ketoxime ligation for the attachment of the peptide to the solid support, and on a tandem transoximation/rearrangement process for the detachment of the target protein. Importantly, we show that the combination of solid phase and solution ligation techniques facilitates the production of a challenging and biologically active protein made of 180 amino acids. We show also that the solid phase method enables the purification of complex peptide segments through a chemoselective solid phase capture/release approach.
A Introduction
Chemical protein synthesis1 is an established method for accessing well-defined native or modified proteins that are used for studying protein structure and function. In the future, chemical synthesis might be a popular alternative to living systems for protein engineering and optimization through the production of protein libraries. Chemical synthesis is particularly powerful for accessing small proteins (<150 amino acids).2 Analysis of domain length distribution in 3D-structure databases reveals that the highest frequency is found for domains composed of ∼80–120 amino acids.3 Thus, synthetic methods that give simplified access to these protein domains potentially meet a lot of the needs of academic and industrial laboratories involved in the study of protein function.Native chemical ligation (NCL)4 of peptide thioesters with cysteinyl (Cys) peptides is the most popular chemoselective amide bond forming reaction for assembling proteins from unprotected peptide segments. The majority of proteins synthesized up to now have been produced using solution phase methods. In recent years, there has been keen interest in simplifying protein assembly while minimizing isolation and purification steps. A dynamic area of research is the conception of one-pot peptide segment assembly schemes, which rely on the development of novel Cys protection strategies,5–13 latent thioester or selenoester surrogates,14–16 or kinetically controlled ligation methods17–21 (and combinations thereof). An alternative approach is to perform stepwise assembly of peptide segments on a water-compatible solid support.22–27 Solid phase synthesis benefits from several advantages over solution phase methods, which have made solid phase peptide synthesis (SPPS28) successful: it avoids the isolation of poorly soluble intermediates, allows forcing of the coupling conditions and importantly replaces intermediate purifications by simple washing steps. Assembly in the N-to-C direction offers an important advantage over the alternative C-to-N direction, which is self-purification of the incoming peptide segments produced by SPPS (Scheme 1). The peptide segments II produced by SPPS can be contaminated by shorter acetylated peptides III even after the application of a purification procedure (Scheme 1A). In the N-to-C direction only a full length peptide segment equipped with the appropriate N-terminal functionality, i.e. a functional linker (FL) for segment 1 or Cys residue for the other segments, can be incorporated in the growing polypeptide chain (Scheme 1B). Acetylated and unreactive peptide impurities are therefore eliminated after each elongation step during the washing steps and the accumulation of truncated polypeptide chains on the solid phase is avoided. The concept of purification by selective capture-and-release has been illustrated by several groups24,29–43 and recently applied to the solid phase synthesis of MUC1 polypeptides by combining His6 tag/Ni-NTA and hydrazone capture methods.43
 | ||
| Scheme 1 Protein synthesis by assembly of unprotected peptide segments in the N-to-C direction using NCL. (A) Synthesis of the peptide segments by SPPS; (B) general assembly strategy. | ||
Although there are significant advantages of assembling proteins by solid phase N-to-C elongation, studies that report such approaches can be counted on the fingers of one hand (Scheme 1, Table 1). The first reason is the paucity of chemical systems that can act as latent thioesters (LT) during NCL, while being easily activated in a subsequent step (V → VI → VII in Scheme 1B). The second reason, which is the focus of this report, is the challenge represented by designing appropriate linkers that (i) can be installed on the first peptide segment using conventional Fmoc-SPPS and simple reagents (Scheme 1A), (ii) allow efficient and chemoselective attachment of the first segment while preserving its C-terminal functionality (IV → V, Scheme 1B), (iii) are stable during the elongation procedure (V → VIII, Scheme 1B) and (iv) can be cleaved under mild conditions (VIII → IX, Scheme 1B). Methods developed so far rely on the use of linkers that require multistep procedures for their synthesis.32,40,44 However, the most striking feature of these approaches is the strong basic (pH 13, Table 1, entries 1 & 2)32,40 or nucleophilic (1 M hydroxylamine, pH 7–8.5, Table 1, entry 3)44 conditions used for the final cleavage step.
| Entry | Functional linker (FL) | Attachment method for peptide segment 1 | Latent thioester (LT) | Cleavage | Ref | |
|---|---|---|---|---|---|---|
| Structure | Activation method | |||||
| a The method was used for the synthesis of large protein mimetics through CuAAC ligation. | ||||||
| 1 |

|
Oximation pH 4 |

|
Alkylation with bromoacetic acid, pH 4.6 | β-Elimination, aqueous base, pH 13 | 22 |
| 2 |

|
CuAAC pH 7 or SPAAC pH 2 |

|
Reduction (TCEP) and exchange by 3-mercaptopropionic acid, pH 4 | See entry 1 | 40 |
| 3 |

|
CuAAC | Notea | Transimination, 1 M H2NOH, pH 7–8.5 | 44 | |
| 4 (This work) |
|
Oximation pH 3–4 |

|
See entry 2 | Transoximation 0.025 M H2NOH, 3 M aniline, pH 3 | |
We sought to develop a simple and mild traceless linker methodology with the aim of making the solid phase synthesis of proteins more popular. In addition to abiding by the above specifications, an important objective was to avoid multistep synthetic work for linker preparation. The use of an affordable and commercially available reagent would be ideal. Also, the products resulting from linker decomposition must be benign. Thus another significant goal was to avoid the formation of harmful species during cleavage, such as 2-(alkylsulfonyl)ethyloxycarbonyl linkers (Table 1, entries 1–2) that decompose into vinylsulfones.32,40
We describe here that the acetoacetyloxime linker shown in Scheme 2 meets all these criteria. Peptides are modified after Fmoc-SPPS by the commercially available acetoacetyl N-succinimidyl ester (AcA-OSu) and are attached to the water-compatible solid support through ketoxime bond formation. Traceless cleavage is triggered by transoximation with hydroxylamine at a mildly acidic pH. The resulting acetoacetyloxime (AcAO) undergoes spontaneous cyclization that yields harmless and hydrophilic 3-methyl-5-oxo–iso-oxazoline and the free peptide, which is easily separated. Importantly, we show that the method gives access to large polypeptides and in particular large Cys peptide segments, and thereby considerably facilitates the synthesis of challenging proteins. The utility of the method is illustrated by the total synthesis of a 180 amino acid biologically active protein derived from hepatocyte growth factor (HGF).
 | ||
| Scheme 2 Protein synthesis by assembly of unprotected peptide segments in the N-to-C direction using AcAO linker and NCL. (A) Synthesis of the AcA peptide segment by SPPS; (B) general assembly strategy. | ||
B Results and discussion
Synthesis of AcA peptides
In order to develop the strategy described in Scheme 2, we needed to prepare peptide segments featuring a C-terminal bis(2-sulfanylethyl)amido (SEA) group in the form of the cyclic disulfide SEAoff and an acetoacetyl group at the N-terminus. The SEAoff group serves as a latent thioester surrogate for chain elongation in the N-to-C direction.14,25 These bifunctional peptides were produced by two different methods, as shown in Scheme 3.13,16 Succinimidyl acetoacetate (AcA-OSu, commercially available) can be used for modifying the peptidyl resin after Fmoc-SPPS, starting from SEA PS resin 1 (Scheme 3A, method A). The peptide cleavage and deprotection in TFA is carried out in the absence of triisopropylsilane (TIS), and any standard thiol scavenger is replaced by 3,4-difluorothiophenol to preserve the AcA moiety from degradation during this step.13 However, using these conditions we observed partial reduction of the Cys(StBu) disulfide that protects the Cys thiol during the oxidation of the SEAon dithiol into the SEAoff cyclic disulfide. Therefore, when the peptide features at least one Cys residue in its sequence, an interesting alternative is to graft the AcA group in solution by NCL using AcA thioester 6, as shown in Scheme 3B (method B).16 This method was used for preparing AcA peptide 7, whose sequence corresponds to amino acids 128–148 from hepatocyte growth factor (HGF).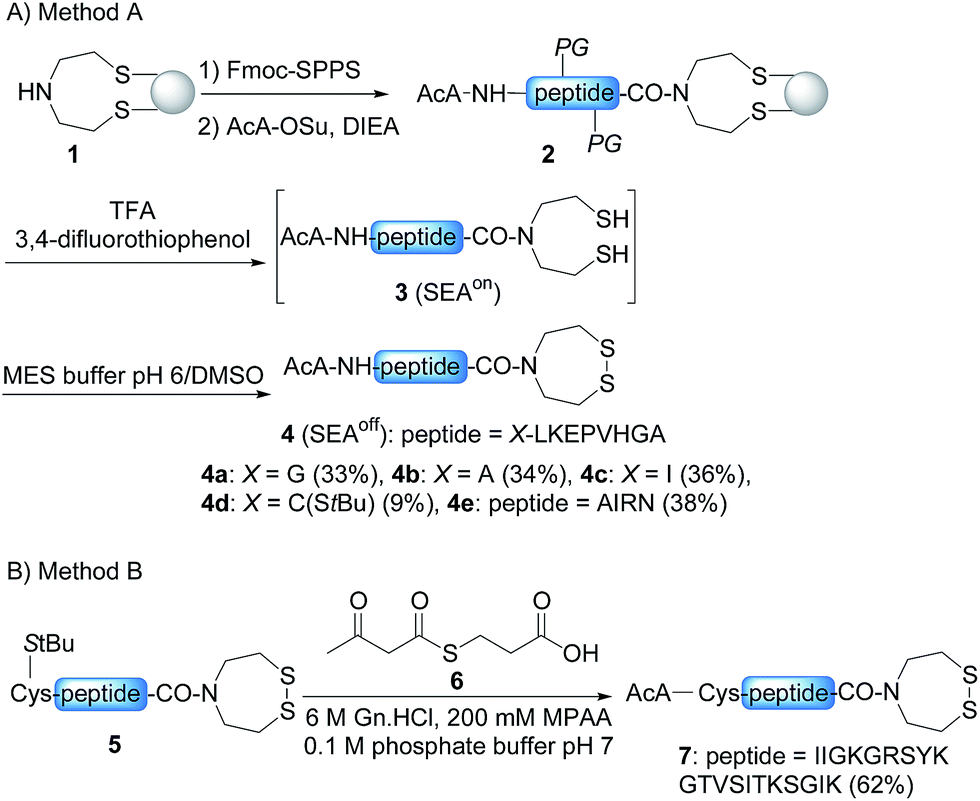 | ||
| Scheme 3 Solid phase (A) and solution (B) synthesis of AcA peptides. | ||
Chemoselective ketoxime ligation
Scheme 2 shows that the attachment of the first segment on the water-compatible solid support relies on the chemoselective formation of a ketoxime bond involving the AcA group and a solid supported hydroxylamine. We chose the aminooxyacetyl functionality that has been extensively used for conjugate synthesis using oxime ligation.45 The reaction of β-ketoacid derivatives with hydroxylamine has been mainly used in the past as a way to access 5-oxo-iso-oxazolines.46 Since the AcA group has not been used so far for preparing ketoxime-linked conjugates, we first examined the reactivity of model AcA peptide 8 (AcA-ALKEPVHGA-NH2, prepared from Rink resin in a manner similar to that described in Scheme 3A, see ESI†) towards ketoxime formation in 6 M Gn·HCl at pH 4 (Scheme 4). | ||
| Scheme 4 Synthesis of acetoacetyloxime (AcAO) peptides in solution. | ||
The reaction of AcA peptide 8 with aminooxyacetic acid (1 equiv.) proceeded chemoselectively and efficiently in a few hours. The resulting AcAO peptide 9 was purified by HPLC (71%) and thoroughly analysed by 1H and 13C NMR (see ESI†). The NMR spectra for AcAO peptide 9 show the presence of two oxime isomers in a ∼1/1 ratio and a methylene group (13C δ 39.5, 44.4 ppm) linked to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (13C δ 159 ppm) and CONH (13C 173.6 δ ppm) groups, in accordance with the proposed structure for the AcAO moiety. The conditions for ketoxime bond formation proved equally useful for accessing AcAO-peptides 10a–d, which are equipped with a SEAoff latent thioester at the C-terminus.
N (13C δ 159 ppm) and CONH (13C 173.6 δ ppm) groups, in accordance with the proposed structure for the AcAO moiety. The conditions for ketoxime bond formation proved equally useful for accessing AcAO-peptides 10a–d, which are equipped with a SEAoff latent thioester at the C-terminus.
Having demonstrated the chemoselective formation of an acetoacetyloxime bond in solution we next examined the reaction of AcA peptides 4b and 7 with the water-compatible solid support 11 (Fig. 1), which was produced by coupling Fmoc-aminooxyacetic acid to Rink PEGA resin, followed by Fmoc removal using piperidine in DMF. The immobilization of peptide 4b and formation of peptidyl resin 12b proceeded to completion in about 10 h at pH 4 and 37 °C, as seen by HPLC analysis of the supernatant in the presence of 2-hydroxy 5-methoxybenzoic acid, which was used as an internal reference (Fig. 1). The cleavage of peptidyl resin 12b in TFA formed ketoxime peptide 14b (Scheme 5), thereby demonstrating the successful formation of a ketoxime bond in the solid phase.
 | ||
| Fig. 1 Immobilization of AcA peptides 4b and 7. Conditions: 0.1 M sodium acetate buffer, pH 4.6, peptide 4b (38 mM) or peptide 7 (11.9 mM), 1.5 equiv. PEGA resin 11 (0.098 mmol g−1), 37 °C. 2-Hydroxy 5-methoxybenzoic acid was used as an internal reference for quantification of the peptides in solution by HPLC (UV detection at 215 nm). | ||
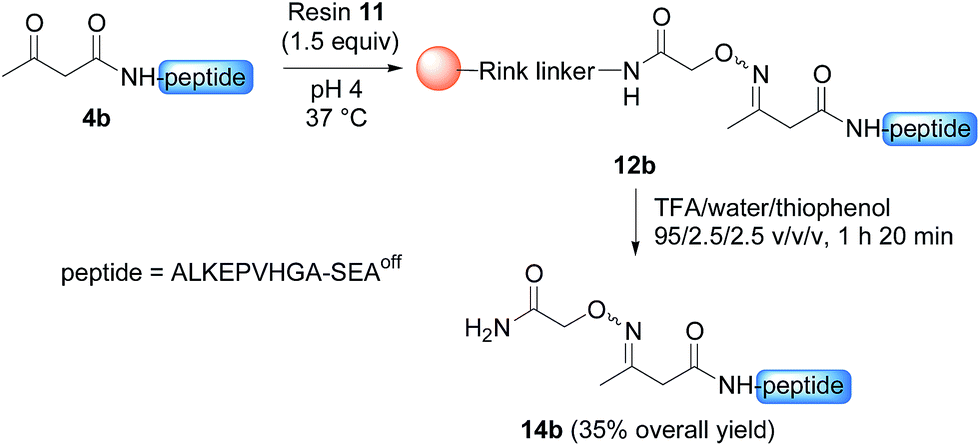 | ||
| Scheme 5 Synthesis of peptide 14b. | ||
Interestingly, the process described in Scheme 5 enabled the purification of a highly hydrophobic peptide segment derived from AS-48 antimicrobial cyclic protein (peptide 3f, Schemes S1 & S2†). The segment was equipped with a protected N-terminal Cys residue, three internal O-acyl isodipeptide units to favour solubility and a C-terminal thioester group. All these functionalities were stable in the acidic conditions employed for the solid phase capture/release procedure.
The immobilization of AcA peptide 7 proceeded successfully too, albeit with a slower rate due to the lower concentration of the AcA peptide in this experiment (Fig. 1). Note that only 1.5 equivalents of the hydroxylamine PEGA resin 11 were used for the immobilization step.
Optimization of the tandem transoximation/rearrangement process
Stability studies in solution using peptide 10b proved the high stability of the AcAO bond in the conditions used for solid phase protein synthesis using SEAoff latent thioester chemistry (see later). We therefore concentrated our efforts on the optimization of the cleavage step which relies on a tandem transoximation/rearrangement sequence (Fig. 2).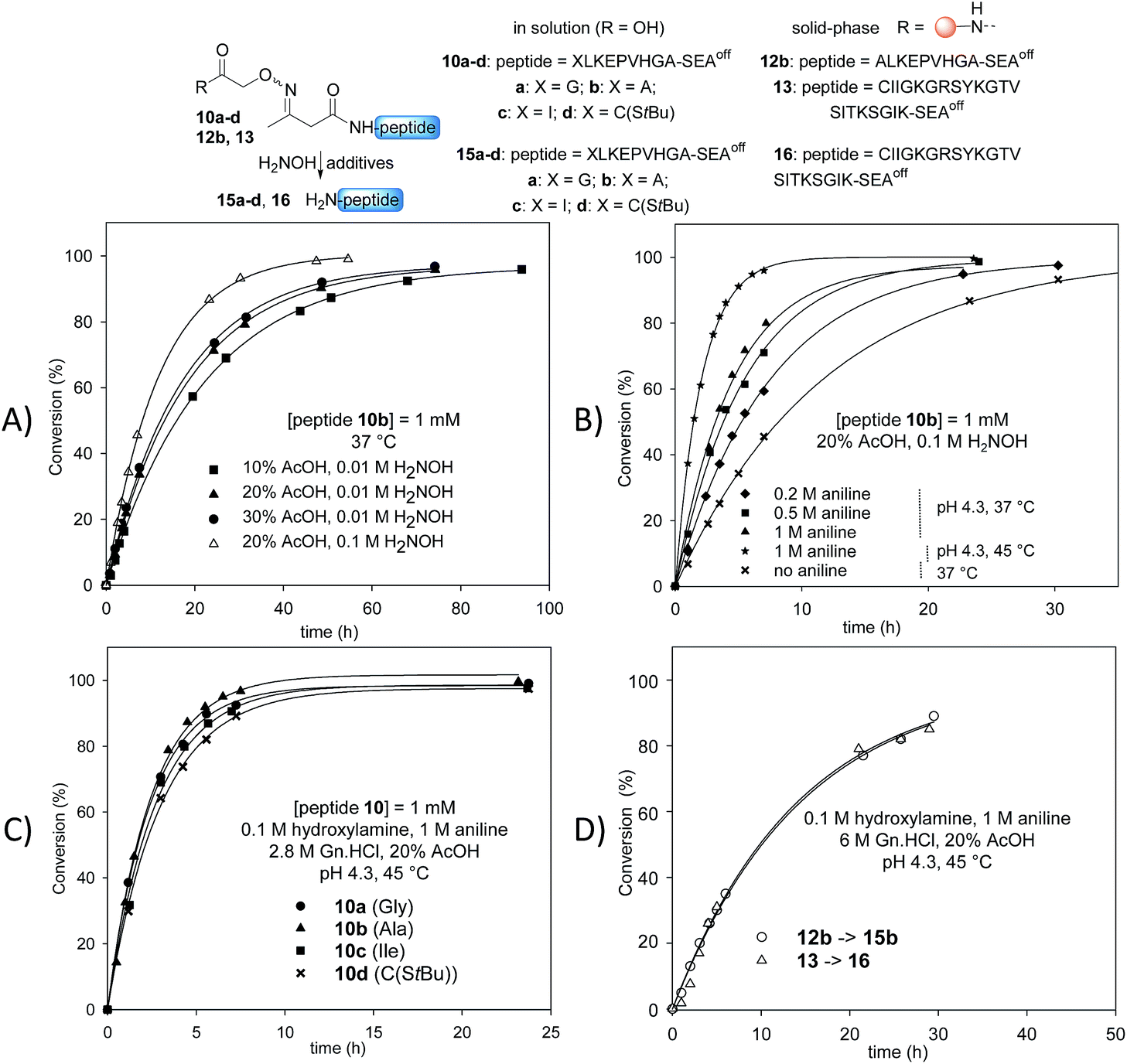 | ||
| Fig. 2 Optimization of the tandem transoximation/rearrangement sequence. (A) Effect of AcOH and hydroxylamine concentration. (B) Effect of aniline catalysis and temperature. (C) Effect of N-terminal residue. (D) Cleavage of peptidyl resins 12b and 13. | ||
Previous studies showed that the removal of the AcA group in the presence of hydroxylamine is particularly efficient in aqueous acetic acid.13,47 Since the transoximation reaction is also subject to acid-catalysis,48 we first examined the cleavage of model AcAO peptide 10b in this solvent mixture (Fig. 2A). Using 0.01 M hydroxylamine at 37 °C, the rate of the cleavage reaction increased by increasing the proportion of acetic acid from 10% to 20%. A further increase to 30% had only a minor effect on the cleavage rate, thus the concentration of acetic acid was fixed at 20% for the rest of this study. Raising the concentration of hydroxylamine from 0.01 M to 0.1 M in 20% aqueous acetic acid favourably impacted the rate of the cleavage reaction (Fig. 2A). Using 0.1 M hydroxylamine in 20% aqueous AcOH, we next examined the effect of aniline on the cleavage rate (Fig. 2B). Indeed, aniline is known to catalyze hydrazone and oxime formation and exchange.49,50 Therefore, we hoped that the addition of aniline would accelerate the oxime exchange step of the process described in Scheme 2 (XIII → XIV), which is rate limiting, and thus increase the global cleavage rate. Interestingly, the addition of aniline from 0.2 M to 1 M resulted in a significant increase of the cleavage rate (see note ‡). The pH of the hydroxylamine/AcOH/aniline mixture had no effect on the cleavage rate in the range of 2.3–4.3 (see Fig. S39†). Thus we selected the mildest conditions, i.e. 1 M aniline at pH 4.3 to further study the effect of the temperature (Fig. 2B). Slightly raising the temperature from 37 °C to 45 °C, using 20% AcOH, 0.1 M H2NOH and 1 M aniline, had a significant impact on the rate of the cleavage reaction, which proceeded about 6 times faster than in the absence of aniline at 37 °C. Guanidine hydrochloride (Gn·HCl) had no influence (data not shown).
Importantly, the nature of the N-terminal residue had no significant effect on the rate of cleavage (Fig. 2C), showing the generality of the method. For example, the cleavage of AcAO peptide 10c with an N-terminal Ile residue proceeded at a similar rate to the cleavage of peptide 10a equipped with an N-terminal Gly. The best conditions at this stage (20% AcOH, 0.1 M H2NOH and 1 M aniline, pH 4.3, 45 °C, called method A) were further used successfully for cleaving peptidyl resins 12b and 13 (Fig. 2D). Note that peptides 15b and 16 were released in solution at about the same rate. Since the rate of cleavage is not affected by the N-terminal residue to a significant extent (Ala for 15b, Cys for 16), it is probably imposed by the concentration of the peptide within the solid support, which is the same for both samples. Pleasingly, peptide 15b was isolated by HPLC in 82% overall yield (4b → 12b → 15b).
Solid phase synthesis of K1 domains from HGF
Having established suitable conditions for the chemoselective immobilization of AcA peptides as well as for the cleavage of the AcAO linker, the stage was set for the solid phase assembly of our target proteins (Table 2). These are variants of the HGF Kringle 1 (K1) domain, which contains a high affinity binding site for HGF Receptor Tyrosine Kinase (RTK), also called MET.51 The first segment equipped with the AcA group was immobilized on the hydroxylamine solid support as described before. The peptide chain was elongated in the N-to-C direction by activating the transformation of the SEAoff group into a thioester with an excess of 3-mercaptopropionic acid (MPA) and then performing an NCL reaction in the presence of 4-mercaptophenylacetic acid (MPAA) as described previously.14,25 Finally, the peptides were released in solution using method A. This procedure successfully formed K1 domains 17 and 18 (entries 1 and 2, Table 2) with an overall yield of 23–28%, corresponding to an average yield per step of ∼80%. Note that peptide 17 was released in solution at a rate similar to the rate observed for the detachment of peptides 15b and 16 (compare Fig. 2D and Fig. S47†). However, careful inspection of the MALDI-TOF spectra for K1 polypeptides 17 and 18 suggested the presence of a contaminant with +16 mass units (u) relative to the target polypeptide. Nevertheless, peptide 18 was further folded using GSH/GSSG in sodium phosphate at pH 7.4/glycerol (9/1 v/v) to give K1 protein 19, which was assayed for its capacity to activate the MET receptor in a cell-based assay (see ESI†). Pleasingly, the folded K1 domain 19 displayed the expected agonistic activity by activating the MET receptor. Moreover, it was as active as an analogue produced by another method (see Fig. S59 in the ESI†).14|
|
|||||
|---|---|---|---|---|---|
| Entry | Polypeptideb | Peptide (# of AA) | Cleavage methodc | % +16 u by-productd | % Yield (average yield per step)e |
| a Elongation cycle: (1) SEAoff → MPA thioester: 6 M Gn·HCl, TCEP, 5% vol HSCH2CH2CO2H, pH 4.0, 37 °C, 24 h; (2) NCL: 6 M Gn·HCl, MPAA, pH 7.2, 37 °C, 24 h (see ref. 25). b The formed junctions are underlined. The residue modified by the AcA group is indicated in bold. c Method A: 20% AcOH, 0.1 M H2NOH and 1 M aniline, pH 4.3, 45 °C. Method B: 20% AcOH, 0.025 M H2NOH and 3 M aniline, pH 3.0, 45 °C. d The proportion of the by-product was estimated by high resolution ESI MS. e Overall yield starting from AcA segment 1. Isolated by HPLC. | |||||
| 1 |
CIIGKGRSYKGTVSITKSGI QPWSSMIPHEHSFLPSSYRGKDLQEN QPWSSMIPHEHSFLPSSYRGKDLQEN RNPRGEEGGPWCFTSNPEVRYEVCDIPQCSEVK(biotin)-NH2 RNPRGEEGGPWCFTSNPEVRYEVCDIPQCSEVK(biotin)-NH2 |
17 (83) | A | 40 | 28 (81) |
| 2 |
AIR IIGKGRSYKGTVSITKSGI IIGKGRSYKGTVSITKSGI QPWSSMIPHEHSFLPSSYRGKDLQEN QPWSSMIPHEHSFLPSSYRGKDLQEN RNPRGEEGGPWCFTSNPEVRYEVCDIPQCSEV-NH2 RNPRGEEGGPWCFTSNPEVRYEVCDIPQCSEV-NH2 |
18 (87) | A | 45 | 23 (83) |
| 3 | See entry 1 | 17 (83) | B | < Detection limit | 21 (73) |

To clarify the origin of the contaminant in the K1 polypeptides, we analysed polypeptides 17 and 18 and protein 19 by high resolution mass spectrometry (HR MS). Furthermore, K1 polypeptide 18 was subjected to proteomic analysis after reduction with DTT, alkylation with iodoacetamide and trypsin digestion. HR MS spectra confirmed the presence of a byproduct with +16 u relative to the target peptide in all three peptides. The proportion was estimated to be ∼40% on the basis of the HR MS spectra (Fig. 3A) but was less (∼20%) based on MALDI-TOF analysis. The proteomic analysis of polypeptide 18 ruled out the possibility of Met oxidation. However, a precise site of modification could not be identified.
 | ||
| Fig. 3 HR MS and MALDI-TOF analysis of peptide 17. (A) Method A; (B) method B. | ||
Hydroxylamines are popular reagents in the field of protein chemical synthesis.5,44 However, hydroxylamines are strong nucleophiles that can react with carboxylic acid derivatives such as esters52 and thioesters.53 Much less discussed is the reaction of hydroxylamines with amides. Nevertheless, the capacity of hydroxylamine to react with primary amides54, and in particular Asn and Gln side-chains, has been mentioned in the past.55,56 For example, the formation of hydroxamate derivatives of Asn and Gln residues has been observed by Canova-Davis and coworkers during the hydroxylamine cleavage of a fusion protein of human insulin-like growth factor I (IGF-I).55 Since K1 polypeptides contain 7 or 8 primary amide groups, we suspected that the origin of the +16 u side-products was the formation of hydroxamates by the reaction of hydroxylamine with Asn and Gln side-chain carboxamides (see note §). For a proportion of the +16 u side-product of 20–40% and if we assume a random attack of Asn/Gln residues, the proportion of hydroxamate impurity per Asn/Gln residue is ∼2.5–5%. This may explain why the modifications could not be mapped by proteomic analysis.
Faced with this problem, we optimized the cleavage protocol further to minimize hydroxamate side-product formation (see note ¶). To make a long story short, decreasing the pH from 4.3 to 3.0 and the concentration of hydroxylamine from 0.1 M to 0.025 M was found to significantly decrease hydroxamate formation, while raising the concentration of aniline from 1 M to 3 M allowed the rate of cleavage to be maintained close to that under the initial conditions (see Fig. S44 in the ESI†). Under these new conditions (method B), peptide 15b was isolated by HPLC with a 64% overall yield (4b → 12b → 15b). To our delight, peptide 17 was resynthesized using this novel protocol with a similar overall yield (entry 3, Table 1) and importantly without being contaminated by +16 u impurities (Fig. 3B). The HPLC profiles for the crude and purified protein 17 can be found in Fig. S51 in the ESI.†
Total synthesis of a biotinylated NK1 domain from HGF by a mixed solid phase/liquid phase approach
The possibility of accessing large peptide segments such as K1 polypeptide 17 in high purity and good yield facilitates the total synthesis of challenging proteins. To illustrate this point we assembled biotinylated NK1 protein NK1-B, which is made of 180 amino acids. NK1 is a natural variant of HGF, a growth factor that triggers strong phenotypes in epithelial cells such as proliferation, migration, survival or morphogenesis through MET RTK activation. Strong agonists of the MET receptor have potential applications in regenerative medicine. Indeed, HGF is poorly active in vivo due to limited diffusion within tissue and rapid degradation by proteolytic enzymes. NK1 is a good agonist, although it is significantly less potent than HGF. Nevertheless and in contrast to HGF, NK1 analogues are promising candidates for in vivo applications such as organ regeneration. Therefore, a method that can give access to NK1 derivatives is of utmost interest for protein optimization. Synthetic NK1-B was produced by combining peptide segments 20, 21 and K1 polypeptide 17 (1 equiv. of each) by a sequential one-pot NCL/SEA ligation process (Scheme 6). The efficiency of the process is illustrated by Fig. 4A, which corresponds to the LC-MS analysis of the crude one-pot mixture. To our delight, NK1-B polypeptide was isolated with a 35% yield after HPLC purification (Fig. 4B). The global yield of the synthesis, which includes the solid phase synthesis of K1 domain 17, was 7.4%. | ||
| Scheme 6 Total synthesis of a biotinylated HGF NK1 domain (180 amino acids). | ||
 | ||
| Fig. 4 Total synthesis of NK1-B polypeptide. (A) HPLC of the crude mixture after one-pot assembly. (B and C) LC-MS of purified NK1-B. We noticed that multicharged ions tend to decompose upon ESI-MS analysis. (D) NK1-B was resolved on a NuPAGE (12–4% gradient) polyacrylamide gel. Proteins were fixed in methanol/acetic acid solution (40%/8%) and stained overnight with Coomassie Blue (Brilliant blue R 250). MW: molecular weight ladder. (E) Western blot of MCF10A cells signaling analysis upon NK1-B stimulation. Cells were treated for 10 min with 100 pM mature HGF/SF (HGF) or 50–100 nM NK1-B. Cell lysates were then analyzed by SDS-PAGE using specific total or phospho MET, ERK or AKT antibodies. Ctrl: vehicle, MW: molecular weight. | ||
Recently, we produced NK1-B polypeptide by another route which combined latent thioester and selenoester chemistries. NK1-B was assembled by performing several one-pot reactions, thereby saving several intermediate purification steps, with a global yield of 6.7%. Although this assembly strategy was highly efficient, the difficulty in accessing some of the peptide segments limited the scale of the synthesis and thus the quantity of the protein produced at the end (∼140 μg). In the present work, the overall yield was only slightly improved (7.4%) but the quantity of NK1-B polypeptide obtained (1.8 mg) was 10-fold higher. This is due to the simplicity of the mixed solid phase/solution phase approach and the use of peptide segments that are easier to produce.
The folding of NK1-B was expected to be challenging due to the presence of five disulfide bonds in the protein (two in the N domain and three in the K1 domain) and a cis peptide bond to proline 157 in the K1 domain. In a preliminary experiment, the NK1-B linear precursor was folded using GSH/GSSG in sodium phosphate at pH 7.4/glycerol (9/1 by volume) at 4 °C. LC-MS monitoring of the folding mixture showed the formation of a −10 u product along with misfolded aggregates which precipitated and resulted in a low yield after dialysis. Nevertheless, the dialyzed NK1-B protein showed a single band by SDS-PAGE with Coomassie blue staining (Fig. 4D), and to our delight was found to activate the MET receptor through its phosphorylation (P-MET) and downstream signalling pathways (phosphorylated ERK and AKT, P-ERK and P-AKT, respectively) at low concentrations (50–100 nM) in two different cell types, MCF10A cells (Fig. 4E) and HeLa cells (see ESI†). These results are highly encouraging and more work is now in progress to improve the folding step and exploit the C-terminal biotin group for fundamental studies that aim to clarify the mechanism of action of this protein.
C Conclusions
The acetoacetyloxime linker methodology reported here can be set up easily since the reagents are commercially available and affordable. The cleavage protocol has been thoroughly optimized to minimize the side-reaction of hydroxylamine with Asn or Gln side chain carboxamides. The tandem transoximation/rearrangement sequence in the presence of hydroxylamine results in the cleavage and self-immolation of the linker. The method is insensitive to the nature of the N-terminal residue and was validated by the synthesis of a large Cys peptide, which was subsequently used to assemble NK1-B protein in a one-pot process. By using this mixed solid phase/solution assembly strategy, the global yield was significantly improved in comparison with a previous solution approach. We believe that combining solid phase and solution assembly strategies and the powerfulness of NCL and related ligations will considerably facilitate the synthesis of challenging proteins. We show also that the acetoacetyloxime linker enables the purification of complex peptide segments equipped with all the functionalities required for further ligation work.Acknowledgements
We thank the CSB platform for technical help and ANR for financial support (CyProt, ANR-15-CE07-0020).Notes and references
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC.
- S. J. Wheelan, A. Marchler-Bauer and S. H. Bryant, Bioinformatics, 2000, 16, 613–618 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- D. Bang and S. B. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS PubMed.
- S. Ueda, M. Fujita, H. Tamamura, N. Fujii and A. Otaka, ChemBioChem, 2005, 6, 1983–1986 CrossRef CAS PubMed.
- J. Li, Y. Li, Q. He, Y. Li, H. Li and L. Liu, Org. Biomol. Chem., 2014, 12, 5435–5441 CAS.
- M. Pan, Y. He, M. Wen, F. Wu, D. Sun, S. Li, L. Zhang, Y. Li and C. Tian, Chem. Commun., 2014, 50, 5837–5839 RSC.
- S. Tang, Y.-Y. Si, Z.-P. Wang, K.-R. Mei, X. Chen, J.-Y. Cheng, J.-S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2015, 19, 5713–5717 CrossRef PubMed.
- K. Aihara, K. Yamaoka, N. Naruse, T. Inokuma, A. Shigenaga and A. Otaka, Org. Lett., 2016, 18, 596–599 CrossRef CAS PubMed.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
- E. Boll, J. P. Ebran, H. Drobecq, O. El-Mahdi, L. Raibaut, N. Ollivier and O. Melnyk, Org. Lett., 2015, 17, 130–133 CrossRef CAS PubMed.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El-Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed.
- L. Raibaut, H. Drobecq and O. Melnyk, Org. Lett., 2015, 17, 3636–3639 CrossRef CAS PubMed.
- L. Raibaut, M. Cargoet, N. Ollivier, Y. M. Chang, H. Drobecq, E. Boll, R. Desmet, J.-C. M. Monbaliu and O. Melnyk, Chem. Sci., 2016, 7, 2657–2665 RSC.
- D. Bang, B. L. Pentelute and S. B. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed.
- H. Ding, A. Shigenaga, K. Sato, K. Morishita and A. Otaka, Org. Lett., 2011, 13, 5588–5591 CrossRef CAS PubMed.
- R. Yang, W. Hou, X. Zhang and C.-F. Liu, Org. Lett., 2012, 14, 374–377 CrossRef CAS PubMed.
- T. Kawakami and S. Aimoto, Tetrahedron Lett., 2007, 48, 1903–1905 CrossRef CAS.
- K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, Y. Sumikawa, K. Sakamoto and A. Otaka, ChemBioChem, 2011, 12, 1840–1844 CrossRef CAS PubMed.
- L. E. Canne, P. Botti, R. J. Simon, Y. Chen, E. A. Dennis and S. B. H. Kent, J. Am. Chem. Soc., 1999, 121, 8720–8727 CrossRef CAS.
- A. Brik, E. Keinan and P. E. Dawson, J. Org. Chem., 2000, 65, 3829–3835 CrossRef CAS PubMed.
- E. C. Johnson, T. Durek and S. B. Kent, Angew. Chem., Int. Ed., 2006, 45, 3283–3287 CrossRef CAS PubMed.
- L. Raibaut, H. Adihou, R. Desmet, A. F. Delmas, V. Aucagne and O. Melnyk, Chem. Sci., 2013, 4, 4061–4066 RSC.
- M. Jbara, M. Seenaiah and A. Brik, Chem. Commun., 2014, 50, 12534–12537 RSC.
- L. Raibaut, O. El Mahdi and O. Melnyk, Top. Curr. Chem., 2015, 363, 103–154 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- D. E. Krieger, B. W. Erickson and R. B. Merrifield, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3160–3164 CrossRef CAS.
- S. Funakoshi, H. Fukuda and N. Fujii, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6981–6985 CrossRef CAS.
- I. Sucholeiki and P. T. Lansbury, J. Org. Chem., 1993, 58, 1318–1324 CrossRef CAS.
- L. E. Canne, R. L. Winston and S. B. H. Kent, Tetrahedron Lett., 1997, 38, 3361–3364 CrossRef CAS.
- M. Villain, J. Vizzavona and K. Rose, Chem. Biol., 2001, 8, 673–679 CrossRef CAS PubMed.
- J. Vizzavona, M. Villain and K. Rose, Tetrahedron Lett., 2002, 43, 8693–8696 CrossRef CAS.
- M. Davies and M. Bradley, Angew. Chem., Int. Ed., 1997, 36, 1097–1099 CrossRef CAS.
- M. Davies and M. Bradley, Tetrahedron, 1999, 55, 4733–4746 CrossRef CAS.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2007, 46, 4577–4580 CrossRef CAS PubMed.
- T. Hara, A. Tainosho, K. Nakamura, T. Sato, T. Kawakami and S. Aimoto, J. Pept. Sci., 2009, 15, 369–376 CrossRef CAS PubMed.
- F. Mende, M. Beisswenger and O. Seitz, J. Am. Chem. Soc., 2010, 132, 11110–11118 CrossRef CAS PubMed.
- V. Aucagne, I. E. Valverde, P. Marceau, M. Galibert, N. Dendane and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 11320–11324 CrossRef CAS PubMed.
- M. Zhang, D. Pokharel and S. Fang, Org. Lett., 2014, 16, 1290–1293 CrossRef CAS PubMed.
- I. E. Decostaire, D. Lelievre, V. Aucagne and A. F. Delmas, Org. Biomol. Chem., 2014, 12, 5536–5543 CAS.
- S. F. Loibl, Z. Harpaz, R. Zitterbart and O. Seitz, Chem. Sci., 2016, 7, 6753–6759 RSC.
- M. Galibert, V. Piller, F. Piller, V. Aucagne and A. F. Delmas, Chem. Sci., 2015, 6, 3617–3623 RSC.
- K. Rose, J. Am. Chem. Soc., 1994, 116, 30–33 CrossRef CAS.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1891, 24, 495–506 CrossRef.
- C. Di Bello, F. Filira, V. Giormani and F. D’Angeli, J. Chem. Soc. C, 1969, 350–352 RSC.
- M. S. Newman and W. B. Lutz, J. Am. Chem. Soc., 1956, 78, 2469–2473 CrossRef CAS.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- C. Simonneau, L. Berenice, A. Mougel, E. Adriaenssens, C. Paquet, L. Raibaut, N. Ollivier, H. Drobecq, J. Marcoux, S. Cianferani, D. Tulasne, H. de Jonge, O. Melnyk and J. Vicogne, Chem. Sci., 2015, 6, 2110–2121 RSC.
- S. Hestrin, J. Biol. Chem., 1949, 180, 249–261 CAS.
- L. H. Noda, S. A. Kuby and H. A. Lardy, J. Am. Chem. Soc., 1953, 75, 913–917 CrossRef CAS.
- C. L. Allen, B. N. Atkinson and J. M. Williams, Angew. Chem., Int. Ed., 2012, 51, 1383–1386 CrossRef CAS PubMed.
- E. Canova-Davis, M. Eng, V. Mukku, D. H. Reifsnyder, C. V. Olson and V. T. Ling, Biochem. J., 1992, 285(1), 207–213 CrossRef CAS PubMed.
- M. Antorini, U. Breme, P. Caccia, C. Grassi, S. Lebrun, G. Orsini, G. Taylor, B. Valsasina, E. Marengo, R. Todeschini, C. Andersson, P. Gellerfors and J. G. Gustafsson, Protein Expression Purif., 1997, 11, 135–147 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic protocols and characterization for all compounds. See DOI: 10.1039/c7sc01912b |
| ‡ Since aniline catalyzes the transoximation of acetoacetoximes, it can probably catalyze the attachment of the first segment to the solid support (XI → XII in Scheme 2). |
| § In Canova-Davis’s work, the chemical heterogeneity induced by hydroxylamine treatment did not affect the biological activity of IGF-I. Similarly, the biological activity of K1 domain 19 was not affected by the presence of the +16 u side-products. |
| ¶ Unfortunately, the use of acetamide as a cosolvent and competitor for primary amides favoured hydroxamate formation. |
| This journal is © The Royal Society of Chemistry 2017 |