DOI:
10.1039/C7SC01859B
(Edge Article)
Chem. Sci., 2017,
8, 6247-6256
Efficient syntheses of (−)-crinine and (−)-aspidospermidine, and the formal synthesis of (−)-minfiensine by enantioselective intramolecular dearomative cyclization†
Received
26th April 2017
, Accepted 30th June 2017
First published on 3rd July 2017
Abstract
Polycyclic alkaloids bearing all-carbon quaternary centers possess a diversity of biological activities and are challenging targets in natural product synthesis. The development of a general and asymmetric catalytic method applicable to the efficient syntheses of a series of complex polycyclic alkaloids remains highly desirable in synthetic chemistry. Herein we describe an efficient palladium-catalyzed enantioselective dearomative cyclization which is capable of synthesizing two important classes of tricyclic nitrogen-containing skeleton, chiral dihydrophenanthridinone and dihydrocarbazolone derivatives bearing all-carbon quaternary centers, in excellent yields and enantioselectivities. The P-chiral monophosphorus ligand AntPhos is crucial for the reactivity and enantioselectivity, and the choice of the N-phosphoramide protecting group is essential for the desired chemoselectivity. This method has enabled the enantioselective total syntheses of three distinctive and challenging biologically important polycyclic alkaloids, specifically a concise and gram-scale synthesis of (−)-crinine, an efficient synthesis of indole alkaloid (−)-aspidospermidine and a formal enantioselective synthesis of (−)-minfiensine.
Introduction
Numerous biologically important natural products are polycyclic alkaloids bearing one or more all-carbon quaternary center. As a representative alkaloid of the Amaryllidaceae family with significant biological activity, crinine (1) is characterized by a 5,10b-ethanophenathridine skeleton bearing an all-carbon quaternary center (Fig. 1a).1 Aspidospermidine (2) is a representative pentacyclic indole alkaloid of over 250 members of Aspidosperma alkaloids that exhibit significant respiratory stimulation and antibiotic activities.2 Minfiensine (3) is an important member of the Strychnos alkaloids possessing potent anticancer activity. Many structurally related alkaloids such as haemanthamine, strychnine and strictamine exhibit a variety of biological properties including potent anticancer, antimalarial and anti-inflammatory activities.3 Despite their biological importance, the efficient preparation of these natural products is a significant challenge in synthetic chemistry. A general, efficient and asymmetric catalytic method for the facile preparation of all of these polycyclic alkaloids remains highly desirable for the discovery of new therapeutic agents and drugs.
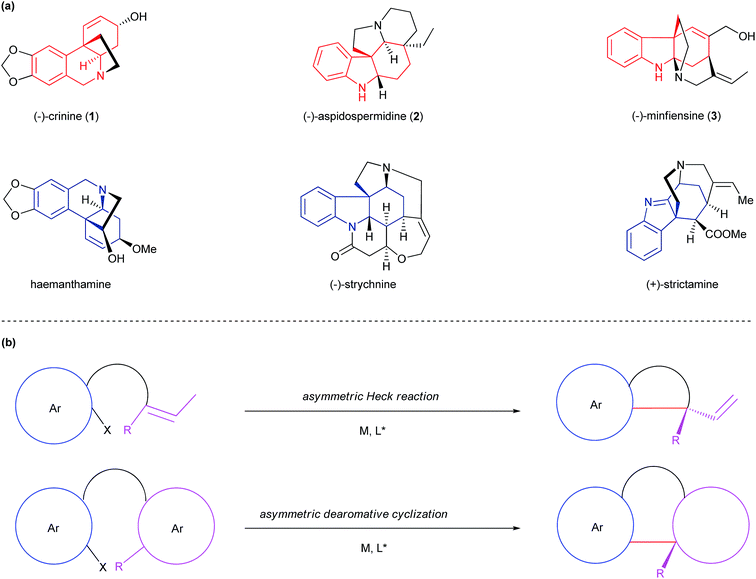 |
| | Fig. 1 (a) Selected polycyclic alkaloids bearing all-carbon quaternary centers. (b) Asymmetric Heck reaction vs. asymmetric dearomative cyclization. | |
The development of a general and efficient asymmetric catalytic method for the concise syntheses of polycyclic natural products has become an important subject in organic chemistry. As a result, several elegant catalytic methods for the enantioselective construction of a polycyclic framework possessing all-carbon quaternary centers have been developed.4 Among them, the asymmetric intramolecular Heck reaction has become one of the most important methods (Fig. 1b).5 Despite its synthetic versatility, the asymmetric Heck cyclization employs an olefinic starting material which often requires multiple synthetic steps to prepare. In addition, the transformation of the olefinic product to a target molecule is not always straightforward. An alternative method is an enantioselective intramolecular dearomative cyclization,6 which usually employs a more accessible substrate with an aryl moiety and leads to a multi-cyclic skeleton bearing an all-carbon quaternary center. Because of the closer resemblance of the cyclic product to a variety of chiral natural products, this method offers advantages over the Heck reaction under certain circumstances for the asymmetric synthesis of polycyclic natural products.
We have previously developed an asymmetric palladium-catalyzed dearomative cyclization for the construction of chiral phenanthrenone and spiroenone derivatives bearing all-carbon quaternary centers and applied this method to terpene, steroid and polyketide syntheses.7 To accomplish the efficient syntheses of the challenging polycyclic skeletons possessed by crinine, aspidospermidine and minfiensine, a highly enantioselective palladium-catalyzed dearomative cyclization is reported in this paper. By employing this method, two important classes of tricyclic nitrogen-containing skeleton, 6,10b-dihydrophenanthridin-3(5H)-one and dihydrocarbazolone derivatives bearing all-carbon quaternary centers, are efficiently constructed with excellent enantioselectivities. The employment of a P-chiral monophosphorus ligand AntPhos and the use of a bulky phosphoramide protecting group at nitrogen are critical for the excellent reactivity, chemoselectivity and enantioselectivity of the dearomative cyclization. The advanced cyclization products have enabled us to accomplish the concise and gram-scale synthesis of (−)-crinine, offering a practical synthetic route to a series of crinine-type alkaloids. Although the two complex chiral natural products (−)-aspidospermidine and (−)-minfiensine belong to a different indoline alkaloid family, their distinctive structures can be derived from a common chiral dihydrocarbazolone intermediate which can be efficiently prepared by the enantioselective dearomative cyclization. This strategy has allowed us to accomplish for the first time the efficient synthesis of (−)-aspidospermidine as well as the formal enantioselective synthesis of (−)-minfiensine using the same asymmetric catalytic method. Herein we report our results.
Results and discussion
Retrosynthetic analysis of crinine
The synthesis of crinine has gained significant interest, resulting in many elegant synthetic strategies for the construction of the 5,10b-ethanophenathridine skeleton bearing an all-carbon quaternary center.8 Surprisingly, few asymmetric syntheses of crinine or vittatine have been reported.9 Early work by Overman described a beautiful synthesis of crinine by employing a chiral auxiliary.9c Chida completed the asymmetric synthesis of vittatine, the antipode of crinine, by utilizing a chiral pool strategy.9a A notable enantioselective synthesis of vittatine was developed by Fan using an organocatalytic Michael addition of α-cyanoketones to acrylates with up to 85% ee through a 15-step sequence.9b Despite these reported synthetic efforts, a concise and highly enantioselective synthesis of 1 remains highly desirable.
It is regarded that the biogenetic synthesis of crinine (1) originates from norbelladine 4 through an intramolecular oxidative dearomative coupling followed by a facile intramolecular aza-Michael addition of 5 (Fig. 2).10 However, the asymmetric biomimetic synthesis of crinine remains to be achieved mainly due to two limitations in this pathway: (1) the oxidative dearomative coupling is nonselective and controlling its chemoselectivity is extremely difficult; (2) the intramolecular Michael addition is too facile to develop an enantioselective version, which indicates why both crinine and its antipode vittatine exist in nature. Inspired by the brevity as well as the limitation of this biogenetic pathway, we envisioned that a concise and enantioselective synthesis of crinine could be achieved by employing a transition-metal catalyzed intramolecular dearomative coupling strategy. The advantage of this approach is that it adopts a dearomative coupling reaction bearing resemblance to the biogenetic pathway. More importantly, the palladium-catalyzed intramolecular dearomative coupling could offer excellent chemo- and enantioselectivity that the biogenetic pathway lacks. Thus, crinine could be prepared by the ring closure of structure I, which could be synthesized from a chiral 6,10b-dihydrophenanthridin-3(5H)-one II by selective reductions. The key transformation is to construct structure II bearing a chiral all-carbon quaternary center from aryl bromide III through enantioselective palladium-catalyzed intramolecular dearomative coupling. We proposed that a chiral monophosphorus ligand developed in our laboratory could provide excellent reactivity, chemoselectivity and enantioselectivity for this challenging reaction.11 Bromide III could be synthesized from the readily available aryl aldehyde 6 and aniline 7 by a reductive amination process.
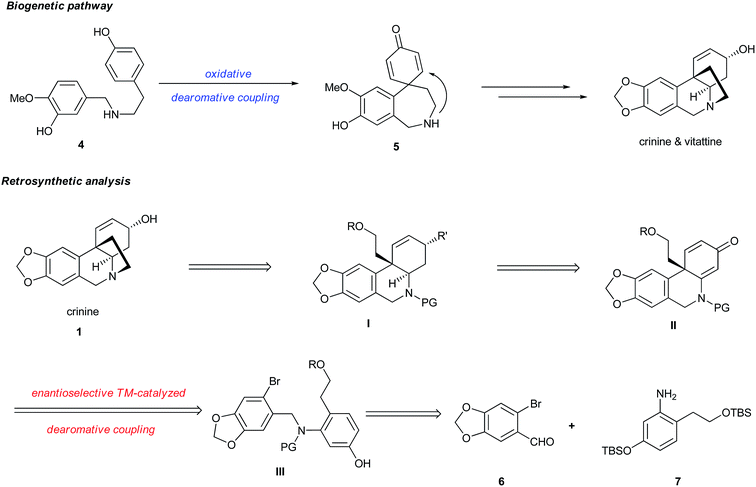 |
| | Fig. 2 Retrosynthetic analysis of crinine (1). | |
Methodology development
Based on the retrosynthetic analysis of crinine, we considered that its asymmetric synthesis could be efficiently accomplished if dihydrophenanthridine 5a could be constructed by an efficient enantioselective dearomative cyclization of aniline 4 (Fig. 3). Although we previously reported the synthesis of chiral phenathrenone compounds,7b the preparation of dihydrophenanthridine 5a from aniline 4 could be challenging due to the conformational change caused by the nitrogen atom in the skeleton. Pathway a during the nucleophilic substitution of the palladium species I would lead to the formation of the desired chiral product 5a after reductive elimination, while pathway b would provide the undesired non-chiral product 5b. The choice between pathway a and b could be largely affected by the conformation of the palladium complex I.
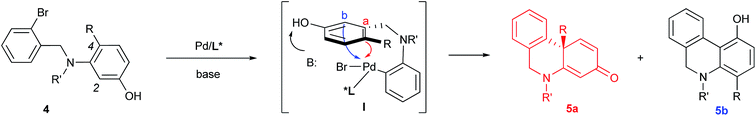 |
| | Fig. 3 Chemoselectivity in asymmetric dearomative cyclization. | |
We reasoned that the conformation of this palladium species could be adjusted by the N-R′ protecting group, which could have a significant effect on the chemoselectivity of the transformation. Thus, a series of nitrogen-containing substrates 8a–i with various R protecting groups were prepared for the study (Table 1). The reactions were performed in toluene at 90 °C for 16 hours with K2CO3 as the base and a palladium catalyst loading of 2 mol% using (S)-L1 (AntPhos) as the ligand (entries 1–9). The cyclization did not occur when free amine 8a (R = H) was directly employed (entry 1). Surprisingly, a Piv-protected substrate 8b proceeded to form solely the undesired cyclization product 10b (entry 2). In order to alter the chemoselectivity, substrates with bulky sulfonyl protecting groups were tested (entries 3–8). Encouragingly, the Ms-protected substrate 8c provided the desired cyclization product 9c in 16% yield with 95% ee (entry 3). A Ts-protected substrate 8d provided a significantly higher yield of 9d (61%, entry 4). However, no better results were obtained from substrates with a Tris- or Nos-protecting group (entries 5–6). A slight improvement (yields > 70%) was observed when Tf- or Me2NSO2-protecting groups were employed (entries 7–8). Finally, when substrate 8i with a bulky phosphoramide protecting group (Me2N)2P(O)- was subjected to the cyclization, the desired chiral dihydrophenanthridin-3(5H)-one product 9i was isolated in 96% yield with 96% ee (entry 9). It is important to note that the ligand structure plays a significant role in the reactivity and selectivity of this reaction. (S)-L1 (AntPhos) is responsible for the excellent reactivity, chemoselectivity and enantioselectivity, since the other related monophosphorus ligands L2, L3, L4 and L5 provided either diminished chemoselectivities, yields or ees (entries 10–13).
Table 1 Asymmetric dearomative cyclization of 8a–i
The effects of the N-R protecting groups on the chemoselectivity observed in our experiments were in accordance with the DFT calculations of the optimized structures of substrates 8b and 8i (Fig. 4). The C-2′ position is in closer proximity to the C-4 position in substrate 8i (3.526 Å) than to that in substrate 8b (3.910 Å). In addition, according to the NBO analysis, the charge on the C-4 position in 8i is −0.071, which is more negative than that in 8b (−0.050) and that on the C-2 position in 8i (−0.017), indicating the higher nucleophilicity of the C-4 position in substrate 8i which participates smoothly in the intramolecular dearomative cyclization.
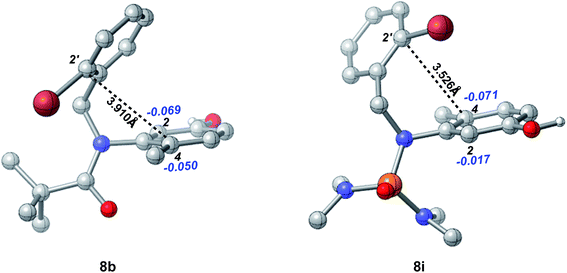 |
| | Fig. 4 The optimized structures of substrates 8b and 8i at the B3LYP/6-31+G(d,p) level (the black bold numbers are the distances between C-2′ and C-4. The blue italic numbers are the NPA charges on the carbon atoms and their attached hydrogen atoms. Hydrogens (except those in the OH groups) have been omitted for clarity). | |
Synthesis of (−)-crinine
With the successful development of an efficient enantioselective dearomative cyclization, we turned our attention to complete the synthesis of crinine (Fig. 5). Thus, the reductive amination between 6-bromopiperonal 6 and readily accessible aniline 7 using NaBH3CN/HOAc provided secondary amine 11 in 92% yield. This was followed by the installation of the phosphoramide group at the nitrogen with LiHMDS/ClP(NMe2)2/H2O2 and subsequent treatment with KF/tetraethylene glycol to selectively deprotect the TBS aryl ether.12 The next step was the key intramolecular dearomative cyclization of bromo phenol 12. Gratifyingly, the cyclization proceeded smoothly in the presence of 1 mol% [Pd(cinnamyl)Cl]2 and 2 mol% (S)-L1 with potassium carbonate as the base to form compound 13 bearing an all-carbon quaternary center in 96% yield with 94% ee. This result further demonstrated the generality and excellent functional group compatibility of the enantioselective dearomative cross-coupling. Treatment of 13 with DIBAL-H at −78 °C selectively reduced the enamide double bond, which was followed by a Luche reduction and treatment with TBAF to stereospecifically give the allylic alcohol 14, whose absolute structure and relative stereochemistry were confirmed by X-ray crystallographic analysis.13 The final tetrahydropyrrole ring formation required the deprotection of the phosphoramide moiety, as well as the activation of the primary alcohol in 14. This was accomplished effectively in a single step by the treatment of 14 with triphosgene/Et3N to afford the cyclization product 15 in 93% yield. It is noteworthy that the allylic alcohol moiety in 14 was stereospecifically transformed into the allylic chloride functionality in 15. The final task was the transformation of the allylic chloride to an allylic alcohol with the retention of its stereochemistry to give the final product crinine. A number of reaction conditions such as using H2O, H2O/AcOH and AgOAc/AcOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 were studied and they all provided mixtures of stereoisomers along with a diene side-product. We were delighted that the employment of [Pd(cinnamyl)Cl]2, PPh3 and AgOAc15 stereoselectively afforded an allylic acetate with the desired stereochemistry, which, after basic hydrolysis, led to (−)-crinine (1) in 90% yield and a 35% overall yield from 6-bromopiperonal 6. Over 1 g of (−)-crinine was successfully prepared, demonstrating the practicality of this synthetic route. The work constitutes the most efficient enantioselective synthesis of (−)-crinine to date.
14 were studied and they all provided mixtures of stereoisomers along with a diene side-product. We were delighted that the employment of [Pd(cinnamyl)Cl]2, PPh3 and AgOAc15 stereoselectively afforded an allylic acetate with the desired stereochemistry, which, after basic hydrolysis, led to (−)-crinine (1) in 90% yield and a 35% overall yield from 6-bromopiperonal 6. Over 1 g of (−)-crinine was successfully prepared, demonstrating the practicality of this synthetic route. The work constitutes the most efficient enantioselective synthesis of (−)-crinine to date.
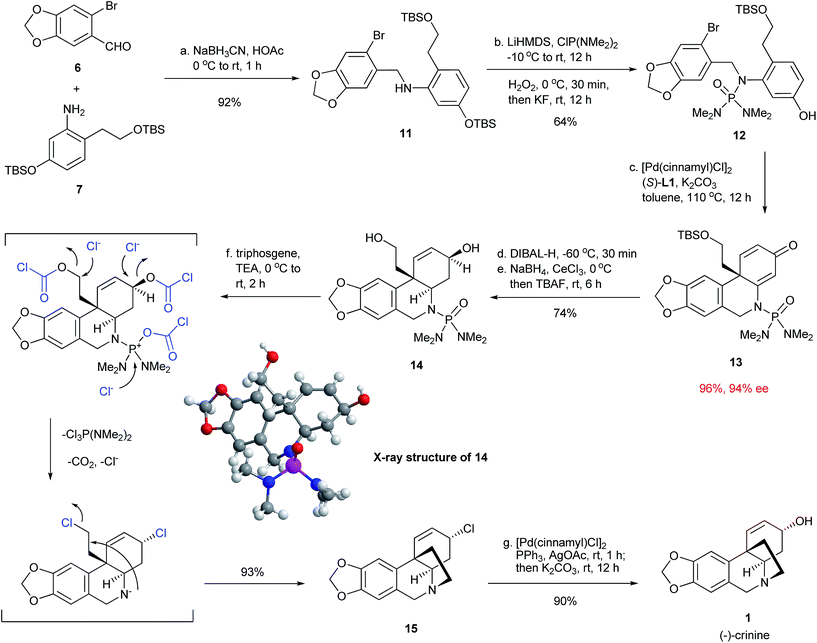 |
| | Fig. 5 Enantioselective synthesis of (−)-crinine. | |
The chiral allylic chloride 15 can also be used for the synthesis of other crinine-type alkaloids (Fig. 6). Methanolysis of 15 led to the formation of two natural products, buphanisine (16)8h,9b and epibuphanisine (17).16 Alternatively, the reduction of 15 with LiEt3BH followed by dihydroxylation led to the formation of amabiline (18)8c,17 in 50% overall yield. Thus, a series of crinine-type alkaloids were conveniently synthesized using this synthetic route.
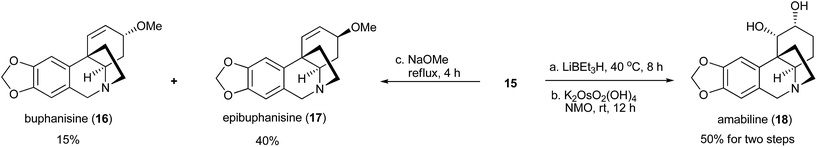 |
| | Fig. 6 Preparation of buphanisine, amabiline and epibuphanisine. | |
Retrosynthetic analysis of aspidospermidine and minfiensine
The unique structures of aspidospermidine and minfiensine have attracted considerable synthetic efforts. Although a number of beautiful total syntheses have been reported, the asymmetric syntheses of aspidospermidine18 and minfiensine19 using enantioselective catalytic methods remain scarce and highly desirable. Despite having different biological origins, both aspidospermidine and minfiensine share a common chiral hydrocarbazole skeleton bearing an all-carbon quaternary stereocenter. We envisioned that a dearomative cyclization of bromo phenol IV could lead to the formation of dihydrocarbazolone III bearing an all-carbon quaternary center, which could be followed by two ring closures via structures II and I to form (−)-aspidospermidine (2) in a concise manner (Fig. 7). The advanced intermediate VI in the minfiensine synthesis could be readily afforded from the dearomative cyclization of product IIIvia intermediate V. The bromo phenol substrate IV could be readily synthesized from the Buchwald–Hartwig amination of 1,2-dibromobenzene with aniline VII.
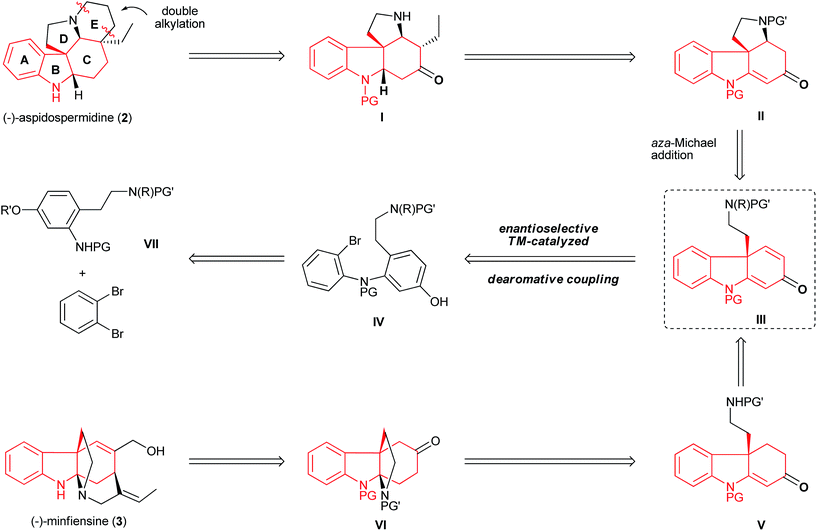 |
| | Fig. 7 Retrosynthetic analysis of aspidospermidine (2) and minfiensine (3). | |
Total synthesis of (−)-aspidospermidine
The total synthesis of (−)-aspidospermidine commenced from the construction of diaryl amine structure IV in Fig. 7 for the asymmetric dearomative cyclization. Thus, the installation of the N-Boc and N-Bn protecting group on the commercially available material 19 formed compound 20, which was hydrogenated over RANEY® nickel to provide aniline 21 in 58% yield over three steps (Fig. 8). A Pd-catalyzed Buchwald–Hartwig amination of 1,2-dibromobenzene with 21 furnished bromo aniline 22 in 70% yield. The installation of the N-P(O)(NMe2)2 group using LiHMDS/P(NMe2)2Cl/H2O2 followed by the deprotection of the MeO group using NaSEt as the reagent afforded the cyclization substrate 23 in 58% yield, which was subjected to the Pd-catalyzed dearomative cyclization. To our delight, the chiral carbazolone 24 possessing an all-carbon quaternary stereocenter was successfully afforded in 63% yield and 90% ee with the Pd-(S)-AntPhos catalyst. The N-phosphoramide protecting group in 23 again proved to be highly important for the success of the dearomative cyclization.20 Treatment of 24 with TMSOTf provided tetracyclic compound 25 in 76% yield through a Boc deprotection and an intramolecular aza-Michael addition. This was followed by the stereoselective installation of the ethyl group at the α-position of the carbonyl group using EtI/LDA to give 26 as a single diastereoisomer in 90% yield. A homogeneous hydrogenation with the Rh-(S,S)-MeO-BIBOP catalyst21 was employed and the reduction of the double bond in 26 took place exclusively at the Re face to afford 27 in 72% yield. After debenzylation of 27 by hydrogenolysis using PdCl2 as the catalyst, the construction of the E ring in 28 was accomplished through a double-alkylation protocol using I(CH2)3I/DIPEA/tBuOK.22 A Wolff–Kishner–Huang Minlon reduction and subsequent acidic hydrolysis successfully delivered (−)-aspidospermidine in 7 steps and 10% overall yield from the key chiral intermediate 24.
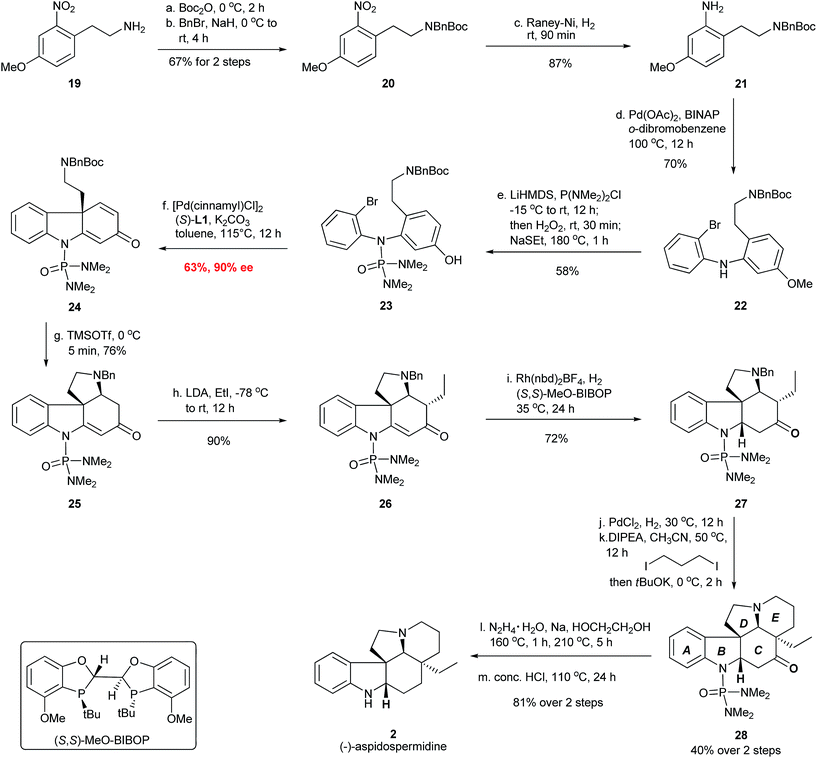 |
| | Fig. 8 Enantioselective synthesis of (−)-aspidospermidine (2). | |
Formal synthesis of (−)-minfiensine
We envisioned that the enantioenriched dienone 24 could be used for the synthesis of (−)-minfiensine. Thus, the transformation of 24 to the reported key chiral intermediate 32 in the minfiensine synthesis19b was conducted (Fig. 9). In order to avoid the intramolecular reaction described in Fig. 8, compound 24 was subjected to hydrogenation over PdCl2 to reduce the less-hindered carbon–carbon double bond. The concomitant removal of the N-benzyl group led to the formation of compound 29 in 77% yield, whose absolute configuration was confirmed by its X-ray crystal structure.23 Next, the cyclization of enone 29via an intramolecular aza-Michael addition was attempted under various acidic or basic conditions and all attempts led to the formation of complex mixtures. Interestingly, the installation of a methylene group at the α position of the carbonyl group using Triton B/paraformaldehyde followed by a Luche reduction cleanly formed the tetracyclic diene 30 in 84% yield. Exchange of the phosphoramide protecting group in 30 to form the methyl carbamate 31 was successfully accomplished by acidic hydrolysis followed by sequential treatment with Boc2O and methyl chloroformate. Finally, the Lemieux–Johnson oxidation followed by hydrogenation afforded the chiral ketone 32, a key advanced intermediate for the synthesis of (−)-minfiensine.19b Thus, we have accomplished the formal synthesis of (−)-minfiensine using the enantioselective dearomative cyclization protocol.
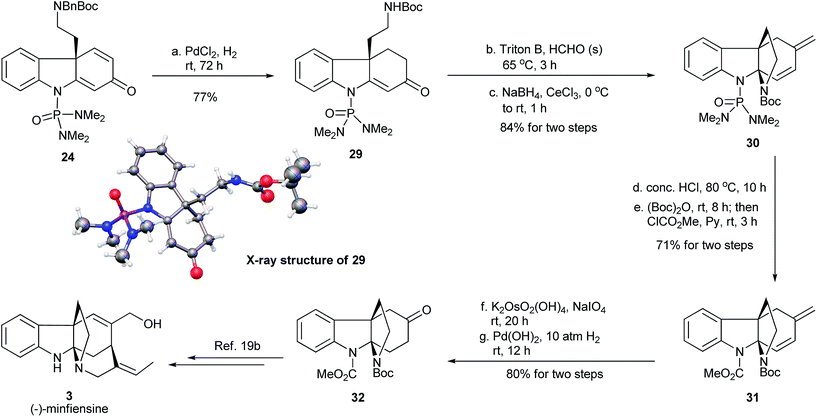 |
| | Fig. 9 Formal synthesis of (−)-minfiensine. | |
Conclusions
We have established a highly efficient and enantioselective Pd-catalyzed intramolecular dearomative cyclization for the synthesis of two important classes of tricyclic nitrogen-containing skeleton, dihydrophenanthridinone and dihydrocarbazolone derivatives bearing an all-carbon quaternary center. It has been demonstrated that the choice of an N-phosphoramide protecting group is essential for the excellent chemoselectivity and the P-chiral monophosphorus ligand AntPhos is crucial for the high reactivity and enantioselectivity. This synthetic method has enabled the enantioselective total syntheses of three distinctive and challenging biologically important polycyclic alkaloids, specifically a concise and gram-scale synthesis of (−)-crinine, an efficient synthesis of indole alkaloid (−)-aspidospermidine and a formal enantioselective synthesis of (−)-minfiensine. This enantioselective dearomative cyclization is expected to be a powerful asymmetric catalytic method for the efficient and scalable syntheses of a number of biologically important natural products, which will certainly facilitate the research and discovery of new therapeutic agents and drugs.
Acknowledgements
We are grateful for the financial support from the Strategic Priority Research Program of the Chinese Academy of Sciences XDB20000000, CAS (QYZDY-SSW-SLH029), NSFC-21432007, 21572246, and the K. C. Wong Educational Foundation. X-ray crystallographic support from Professor Jie Wu at Fudan University is greatly appreciated.
Notes and references
-
(a) Z. Jin, Nat. Prod. Rep., 2016, 33, 1318 RSC;
(b) M. M. He, C. R. Qu, O. D. Gao, X. M. Hu and X. C. Hong, RSC Adv., 2015, 5, 16562 RSC.
-
J. E. Saxton, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, Sandiego, 1998, vol. 50, pp. 343–376 Search PubMed.
-
(a) A. Ramirez and S. Garcia-Rubio, Curr. Med. Chem., 2003, 10, 1891 CrossRef CAS PubMed;
(b) R. Eckermann and T. Gaich, Synthesis, 2013, 45, 2813 CrossRef CAS.
- For recent reviews, see:
(a) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed;
(b) R. Long, J. Huang, J. X. Gong and Z. Yang, Nat. Prod. Rep., 2015, 32, 1584 RSC;
(c) T. T. Ling and F. Rivas, Tetrahedron, 2016, 72, 6729 CrossRef CAS.
- For recent reviews, see:
(a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed;
(b) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS.
-
(a) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC;
(b) W. Fu and W. Tang, ACS Catal., 2016, 6, 4814 CrossRef CAS;
(c) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed;
(d) J. García-Fortanet, F. Kessler and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 6676 CrossRef PubMed;
(e) S. Rousseaux, J. García-Fortanet, M. A. Del Aguila Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282 CrossRef CAS PubMed.
-
(a) G. Zhao, G. Xu, C. Qian and W. Tang, J. Am. Chem. Soc., 2017, 139, 3360 CrossRef CAS PubMed;
(b) K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033 CrossRef CAS PubMed.
- For racemic syntheses of crinine, see:
(a) H. Muxfeldt, R. S. Schneider and J. B. Mooberry, J. Am. Chem. Soc., 1966, 88, 3670 CrossRef CAS;
(b) L. E. Overman and L. T. Mendelson, J. Am. Chem. Soc., 1981, 103, 5579 CrossRef CAS;
(c) W. H. Pearson and F. E. Lovering, J. Org. Chem., 1998, 63, 3607 CrossRef CAS;
(d) N. T. Tam and C.-G. Cho, Org. Lett., 2008, 10, 601 CrossRef CAS PubMed;
(e) C. Bru and C. Guillou, Tetrahedron, 2006, 62, 9043 CrossRef CAS;
(f) J.-D. Liu, S.-H. Wang, F.-M. Zhang, Y.-Q. Tu and Y.-Q. Zhang, Synlett, 2009, 18, 3040 Search PubMed;
(g) D. A. Candito, D. Dobrovolsky and M. Lautens, J. Am. Chem. Soc., 2012, 134, 15572 CrossRef CAS PubMed;
(h) S. F. Martin and C. L. Campbell, J. Org. Chem., 1988, 53, 3184 CrossRef CAS;
(i) L. E. Overman and E. J. Jacobsen, Tetrahedron Lett., 1982, 23, 2741 CrossRef CAS;
(j) H. W. Whitlock and G. L. Smith, J. Am. Chem. Soc., 1967, 89, 3600 CrossRef CAS;
(k) G. Pandey and G. S. R. Gadre, Pure Appl. Chem., 2012, 84, 1597 CrossRef CAS;
(l) L. Yang, X. Wang, Z. Pan, M. Zhou, W. Chen and X. Yang, Synlett, 2011, 2, 207 CrossRef.
- For asymmetric syntheses of crinine or its antipode, see:
(a) M. Bohno, H. Imase and N. Chida, Chem. Commun., 2004, 1086 RSC;
(b) M.-X. Wei, C.-T. Wang, J.-Y. Du, H. Qu, P.-R. Yin, X. Bao, X.-Y. Ma, X.-H. Zhao, G.-B. Zhang and C.-A. Fan, Chem.–Asian J., 2013, 8, 1966 CrossRef CAS PubMed;
(c) L. E. Overman and S. Sugai, Helv. Chim. Acta, 1985, 68, 745 CrossRef CAS.
-
J. C. Cedrón, M. Del Arco-Aguilar, A. Estévez-Braun, Á. G. Ravelo, in The Alkaloids, ed. G. A. Cordell, Elsevier Scientific Publishing, Amsterdam, 2010, vol. 68, pp. 1–37 Search PubMed.
-
(a) M. Nie, W. Fu, Z. Cao and W. Tang, Org. Chem. Front., 2015, 2, 1322 RSC;
(b) Z. Cao, K. Du, J. Liu and W. Tang, Tetrahedron, 2016, 72, 1782 CrossRef CAS;
(c) W. Tang, N. D. Patel, G. Xu, X. Xu, J. Savoie, S. Ma, M.-H. Hao, S. Keshipeddy, A. G. Capacci, X. Wei, Y. Zhang, J. J. Gao, W. Li, S. Rodriguez, B. Z. Lu, N. K. Yee and C. H. Senanayake, Org. Lett., 2012, 14, 2258 CrossRef CAS PubMed;
(d) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed;
(e) W. Fu, M. Nie, A. Wang, Z. Cao and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 2520 CrossRef CAS PubMed;
(f) N. Hu, K. Li, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2016, 55, 5044 CrossRef CAS PubMed.
- H. Yan, J. S. Oh and C. E. Song, Org. Biomol. Chem., 2011, 9, 8119 CAS.
- The CIF data for the crystal structure of 14 have been deposited with the CCDC and have been given the deposition number CCDC 1444649.†.
- N. Ma, Y. Yao, B.-X. Zhao, Y. Wang, W.-C. Ye and S. Jiang, Chem. Commun., 2014, 50, 9284 RSC.
- For a transformation of allylic chloride to allylic fluoride, see: M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402 CrossRef CAS PubMed.
- F. Viladomat, J. Bastida, C. Codina, W. E. Campbell and S. Mathee, Phytochemistry, 1995, 40, 307 CrossRef CAS.
- A. D. Findlay and M. G. Banwell, Org. Lett., 2009, 11, 3160 CrossRef CAS PubMed.
- For asymmetric syntheses of aspidospermidine, see:
(a) Z. Li, S. Zhang, S. Wu, X. Shen, L. Zou, F. Wang, X. Li, F. Peng, H. Zhang and Z. Shao, Angew. Chem., Int. Ed., 2013, 52, 4117 CrossRef CAS PubMed;
(b) S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183 CrossRef CAS PubMed;
(c) S. Z. Zhao and R. B. Andrade, J. Am. Chem. Soc., 2013, 135, 13334 CrossRef CAS PubMed;
(d) G. Pandey, S. K. Burugu and P. Singh, Org. Lett., 2016, 18, 1558 CrossRef CAS PubMed;
(e) S. Zhao and R. B. Andrade, J. Org. Chem., 2017, 82, 521 CrossRef CAS PubMed.
- For asymmetric syntheses of minfiensine, see:
(a) A. B. Dounay, L. E. Overman and A. D. Wrobleski, J. Am. Chem. Soc., 2005, 127, 10186 CrossRef CAS PubMed;
(b) A. B. Dounay, P. G. Humphreys, L. E. Overman and A. D. Wrobleski, J. Am. Chem. Soc., 2008, 130, 5368 CrossRef CAS PubMed;
(c) S. B. Jones, B. Simmons and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 13606 CrossRef CAS PubMed;
(d) Z.-X. Zhang, S.-C. Chen and L. Jiao, Angew. Chem., Int. Ed., 2016, 55, 8090 CrossRef CAS PubMed.
- Model reactions were conducted using diarylamine substrates with N-P(O)Ph2 and N-P(O)(NMe2)2 protecting groups. Please see ESI† for the details.
-
(a) W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176 CrossRef CAS PubMed;
(b) W. Li, S. Rodriguez, A. Duran, X. Sun, W. Tang, A. Premasiri, J. Wang, K. Sidhu, N. D. Patel, J. Savoie, B. Qu, H. Lee, N. Haddad, J. C. Lorenz, L. Nummy, A. Hossain, N. Yee, B. Lu and C. H. Senanayake, Org. Process Res. Dev., 2013, 17, 1061 CrossRef CAS.
- B. N. Laforteza, M. Pickworth and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2013, 52, 11269 CrossRef CAS PubMed.
- The CIF data for the crystal structure of 29 have been deposited with the CCDC and have been given the deposition number CCDC 1537084.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1444649 and 1537084. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01859b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Wenjun
Tang
b and
Wenjun
Tang



![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
:






