
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bonding vs. halogen bonding: the solvent decides†
Craig. C.
Robertson
 a,
James S.
Wright
a,
Elliot J.
Carrington‡
a,
Robin N.
Perutz
*b,
Christopher A.
Hunter
*c and
Lee
Brammer
*a
a,
James S.
Wright
a,
Elliot J.
Carrington‡
a,
Robin N.
Perutz
*b,
Christopher A.
Hunter
*c and
Lee
Brammer
*a
aDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: lee.brammer@sheffield.ac.uk
bDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: robin.perutz@york.ac.uk
cDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: ch664@cam.ac.uk
First published on 1st June 2017
Abstract
Control of intermolecular interactions is integral to harnessing self-assembly in nature. Here we demonstrate that control of the competition between hydrogen bonds and halogen bonds, the two most highly studied directional intermolecular interactions, can be exerted by choice of solvent (polarity) to direct the self-assembly of co-crystals. Competitive co-crystal formation has been investigated for three pairs of hydrogen bond and halogen bond donors, which can compete for a common acceptor group. These competitions have been examined in seven different solvents. Product formation has been determined and phase purity has been examined by analysis of powder X-ray diffraction patterns. Formation of hydrogen-bonded co-crystals is favoured from less polar solvents and halogen-bonded co-crystals from more polar solvents. The solvent polarity at which the crystal formation switches from hydrogen-bond to halogen-bond dominance depends on the relative strengths of the interactions, but is not a function of the solution-phase interactions alone. The results clearly establish that an appreciation of solvent effects is critical to obtain control of the intermolecular interactions.
Introduction
Inspiration from molecular recognition and self-assembly processes in nature has led to the exploration of self-assembly in chemistry with a view to exerting synthetic control. Such efforts are central to prominent research fields such as supramolecular chemistry1,2 and crystal engineering,3,4 and are of increasing impact in areas such as materials chemistry5 and catalysis.6 Control of self-assembly processes remains highly challenging as a consequence of the relatively weak interactions involved. Most prominent of these interactions, both in naturally occurring systems and in synthetic assemblies, are hydrogen bonds (HBs), which are among the strongest and most directional of intermolecular interactions. Another class of important, directional intermolecular interactions are halogen bonds (XBs). Halogen bonds have come to prominence over the past 15 years,7–10 and have been exploited in the fields of crystal engineering,11,12 soft matter,13 protein–ligand interactions,14 anion recognition and transport,15,16 catalysis17 and materials chemistry.18,19 Halogen bonds involve the interaction of a covalently-bound halogen atom, usually iodine, with an electron-rich region of a neighbouring atom or molecule. The halogen adopts a Lewis acidic role, exemplified by the region of positive electrostatic potential on the surface of the halogen, which lies trans to its σ-bond and is usually referred to as the σ-hole.20 In this article we clearly demonstrate for the first time, using co-crystal formation as an exemplar, that appreciation of solvent properties is essential to control the outcome of direct competition between formation of hydrogen bonds and halogen bonds in self-assembly.Understanding the role of specific intermolecular interactions in directing crystal growth is of particular interest in designing crystalline forms of commodity chemicals, such as pharmaceuticals,21,22 agrochemicals,23 pigments24 and energetic materials,25,26 whose physical properties depend on their solid form. Such forms include not only single phases and their polymorphs, but also solvates and, increasingly, co-crystals, which allow the inclusion of a variety of co-crystal former molecules to enable tuning of physical properties through changes in crystal structure. A number of approaches have been taken to understand the formation of co-crystals and enable development of new materials.27 Computational approaches include crystal structure prediction (CSP), which is based on calculation of lattice enthalpies for putative crystal structures.28,29 Alternatively, interaction propensity calculations can be used to predict co-crystal formation based on likelihood of formation of intermolecular interactions,30 principally hydrogen bonds, between available functional groups, and are based on knowledge from the large number of known crystal structures present in the Cambridge Structural Database.31 Experimental approaches may involve screening of a number of potential co-formers (molecules used to form co-crystals with a desired compound). Co-formers are selected based upon their likely formation of intermolecular interactions and screened in a series of co-crystallization experiments.32,33 Absent from most approaches is a specific consideration of the role of solvent in crystal structure formation although solvent effects on directing the crystallization of different polymorphs of molecular crystals have been observed.34 One reason for this is the complexity of accounting for the kinetic and thermodynamic role of solvent in the methodology used, but another reason is a lack of understanding of its importance. The potential influence of solvent on solution-phase self-assembly, however, extends far beyond crystallization processes and improved control of molecular self-assembly remains a goal for supramolecular chemistry if we are to harness intermolecular interactions in the manner in which nature has evolved to do so.
The effect of solvent on hydrogen bonding in solution has been studied. Notably, the hydrogen bonding scale developed by Hunter quantitatively relates empirical hydrogen bond donor strength (α) and hydrogen bond acceptor strength (β) parameters to the free energy of interaction for the hydrogen bond in solution.35 This model explicitly accounts for the role of the solvent by taking into account its hydrogen bond donor and acceptor strengths (αs, βs). Recently we have demonstrated that the strongly-bound halogen-bonded complex iodine·thiourea is stable in a wide range of solvent environments in contrast to hydrogen-bonding interactions, which may be strong in nonpolar solvents, but weak in polar solvents.36 These results led us to consider more broadly the role of solvent in directing the competition between intermolecular interactions in the formation of crystals, and specifically co-crystals as a prominent exemplar of self-assembly. We chose the competition between hydrogen bonding and halogen bonding, the two most widely studied intermolecular interactions, for this investigation. The competition between 1,4-diiodotetrafluorobenzene (1) and hydroquinone (2a) to form co-crystals with 1,2-bis(4-pyridyl)ethane (3) via halogen bonding or hydrogen bonding, respectively, was examined by Metrangolo, Resnati and coworkers.37 Their report, showing that the halogen-bonded co-crystals 1·3 formed exclusively, in preference to the hydrogen-bonded co-crystals 2a·3, when 1, 2a and 3 are dissolved in equimolar quantities in acetone, provided a starting point for our investigations.
Here we report a series of co-crystal formation experiments using halogen-bond donor 1 in competition with either hydrogen-bond donor 2a or its monofluoro- or tetrafluoro-substituted analogues, 2b and 2c, respectively (Scheme 1). These studies were conducted in seven different solvents with 3 as the acceptor, forming a co-crystal in each case. Thus, we have examined hydrogen bond vs. halogen bond pairings of different relative interaction strengths and the role of solvent polarity in controlling the competition. In all cases the hydrogen-bonded co-crystals (2·3) are formed selectively in the least polar solvent, toluene, but formation of the halogen-bonded co-crystal (1·3) ultimately dominates as solvent polarity increases. These results are discussed in the context of solution-phase NMR titrations that determine hydrogen bond and halogen bond strengths, and demonstrate the critical role of solvent in tuning the competition between intermolecular interactions and controlling the form of the co-crystal product. The crossover point (in solvent polarity) between formation of the two crystalline products correlates with the relative strength of the halogen bonds and hydrogen bonds formed.
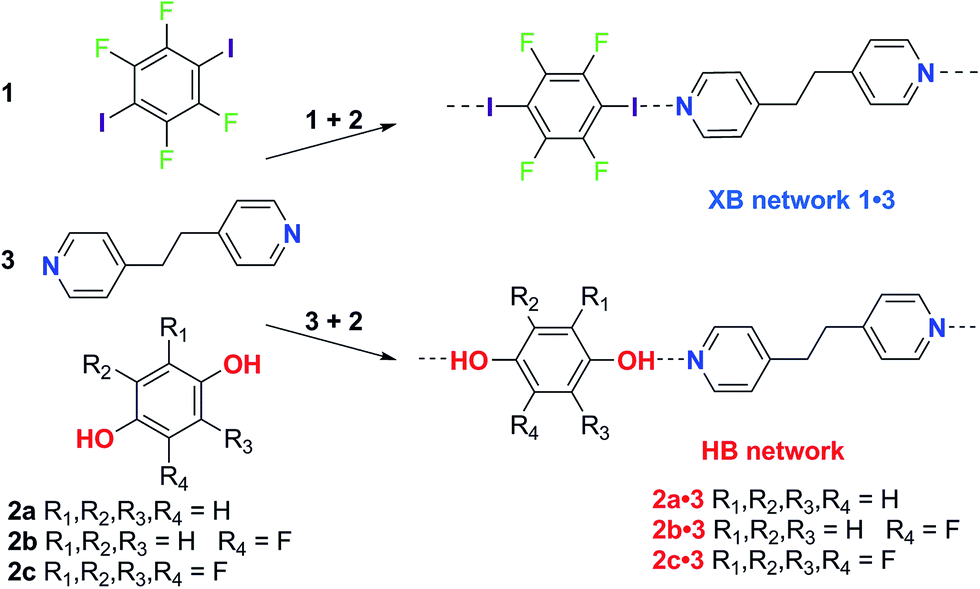 | ||
| Scheme 1 Synthesis of hydrogen-bonded (HB) and halogen-bonded (XB) co-crystals. Co-crystallization of XB donor 1 or HB donor 2 with 1,2-bis(4-pyridyl)ethane 3 to form the XB network 1·3 or HB network as 2·3, respectively. | ||
Results and discussion
Single crystals of the co-crystals 1·3 (XB network), 2a·3 (HB network), 2b·3 (HB) and 2c·3 (HB) were prepared separately by co-crystallization of each pair of components in acetone. The crystal structures of the four co-crystals establish that each comprises 1D networks in which the two molecular components alternate and are linked via either C–I⋯N halogen bonds (1·3) or O–H⋯N hydrogen bonds (2a·3, 2b·3 and 2c·3). In each case the 1D networks adopt stacking arrangements of the aromatic rings in the two molecular components. Structure 1·3 is arranged in offset homomolecular stacks (Fig. 1a), structures 2a·3 and 2b·3 are isostructural and exhibit separate homomolecular and heteromolecular stacks (Fig. 1b and c), and 2c·3 exhibits stacks containing both homomolecular and heteromolecular interactions (Fig. 1d).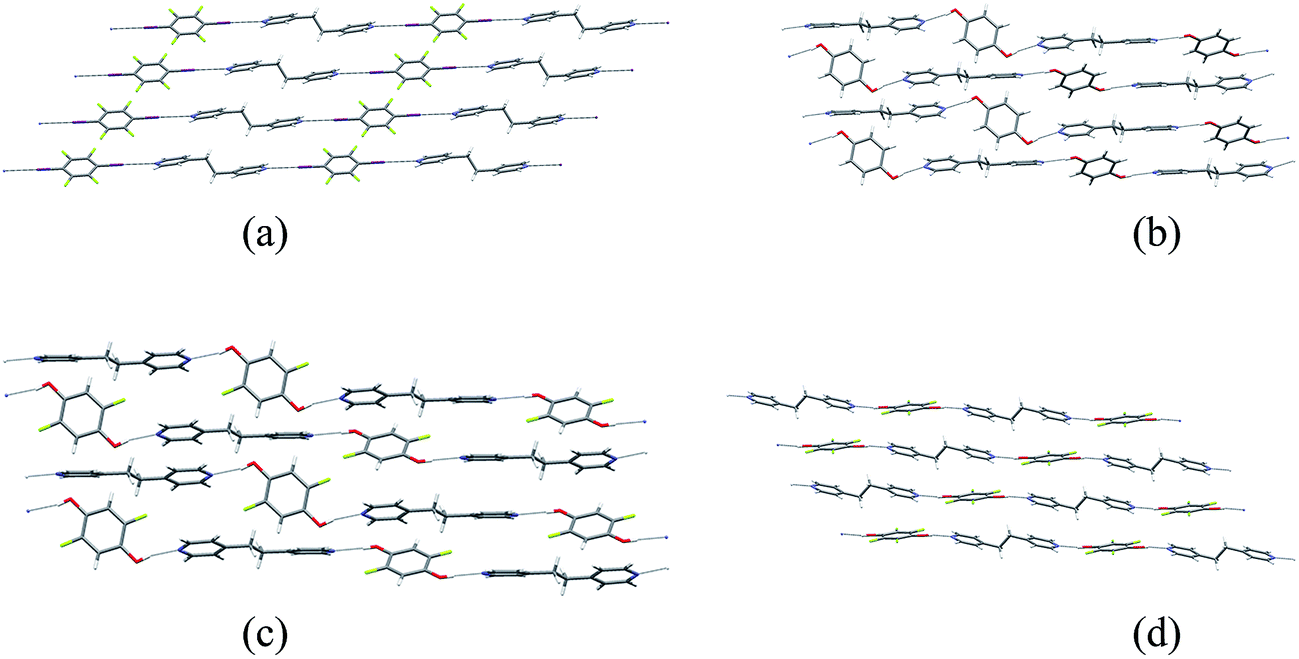 | ||
Fig. 1 Crystal structures of co-crystals: (a) XB network 1·3, (b) HB network 2a·3, (c) HB network 2b·3, (d) HB network 2c·3. The molecule of 2b (in (c)) exhibits 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 F/H disorder at the 2- and 5-positions; disordered H atom sites are not shown. The crystal structures of 1·3 and 2a·3 have been previously reported37 and are consistent with the structures determined herein. :50 F/H disorder at the 2- and 5-positions; disordered H atom sites are not shown. The crystal structures of 1·3 and 2a·3 have been previously reported37 and are consistent with the structures determined herein. | ||
Having established the crystal structures of the possible co-crystals, bulk syntheses of each co-crystal were undertaken by co-crystallization of each pair of components from acetone. In each case Pawley fitting38 of the resultant powder diffraction pattern established that the co-crystals form as the only crystalline product (see Section 9 of ESI†). Powder diffraction further established that the patterns for the hydrogen-bonded co-crystals 2·3 could be readily distinguished from those of the halogen-bonded co-crystal 1·3, and that all co-crystals could be readily distinguished from crystals of the single molecular components (1, 2 and 3).
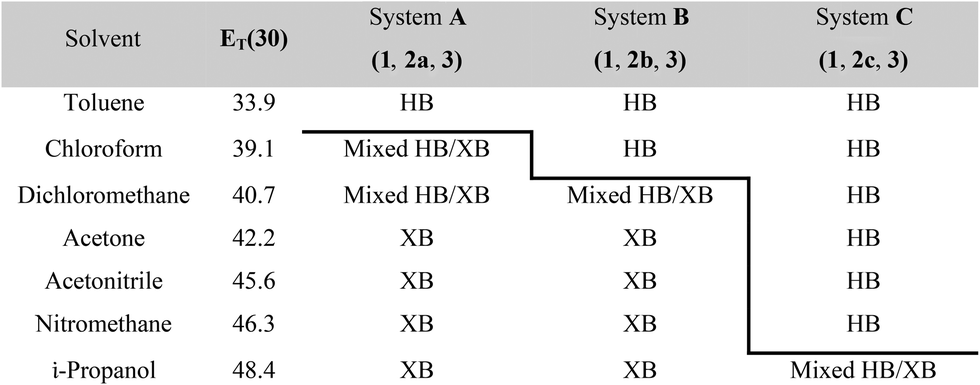
Three competitive co-crystallization systems (A, B and C) were then investigated in which a halogen bond donor directly competes with a hydrogen bond donor to form co-crystals with a common acceptor molecule. 1,2-Bis(4-pyridyl)ethane 3 was used as the ditopic acceptor of hydrogen bonds or halogen bonds throughout all experiments. 1,4-Diiodotetrafluorobenzene 1 served as the ditopic XB donor and the strength of the competing HB donor was adjusted by using either hydroquinone 2a (the weakest HB donor, used in system A), fluorohydroquinone 2b (intermediate strength, system B) or tetrafluorohydroquinone 2c (strongest HB donor, system C). The ranking of the hydrogen bond donor strengths was confirmed by solution-phase NMR spectroscopic titrations and is discussed in more detail below. An equimolar solution of 1, 3, and either 2a, 2b or 2c was prepared in each of seven solvents and the solvent was allowed to evaporate slowly for 24 h at room temperature. The solid formed was isolated by filtration, dried and ground before characterization by powder X-ray diffraction. Where the powder pattern could be indexed as a single phase, the phase purity of the product was established by Pawley refinement and where a mixture of products was identified upon indexing, mixed-phase Rietveld refinement39 was conducted to establish the relative proportions of the products (see Sections 10–12 in ESI†).
The results from system A (1, 2a and 3) confirm the previously reported formation of the XB co-crystal 1·3 in acetone and clearly establish by PXRD that it is the sole product under the experimental conditions used (Fig. 2). The experiments in the other six solvents, however, establish the pivotal role of the solvent in determining the outcome of the competition between halogen bonding and hydrogen bonding in the self-assembly process that leads to co-crystal formation. The XB co-crystal also forms exclusively in the three solvents more polar than acetone, but decreasing the solvent polarity increases the likelihood of formation of the HB network 2a·3, such that mixtures of the two co-crystals are formed in CHCl3 and CH2Cl2, but the HB network is formed exclusively in toluene (Table 1, Fig. 3).
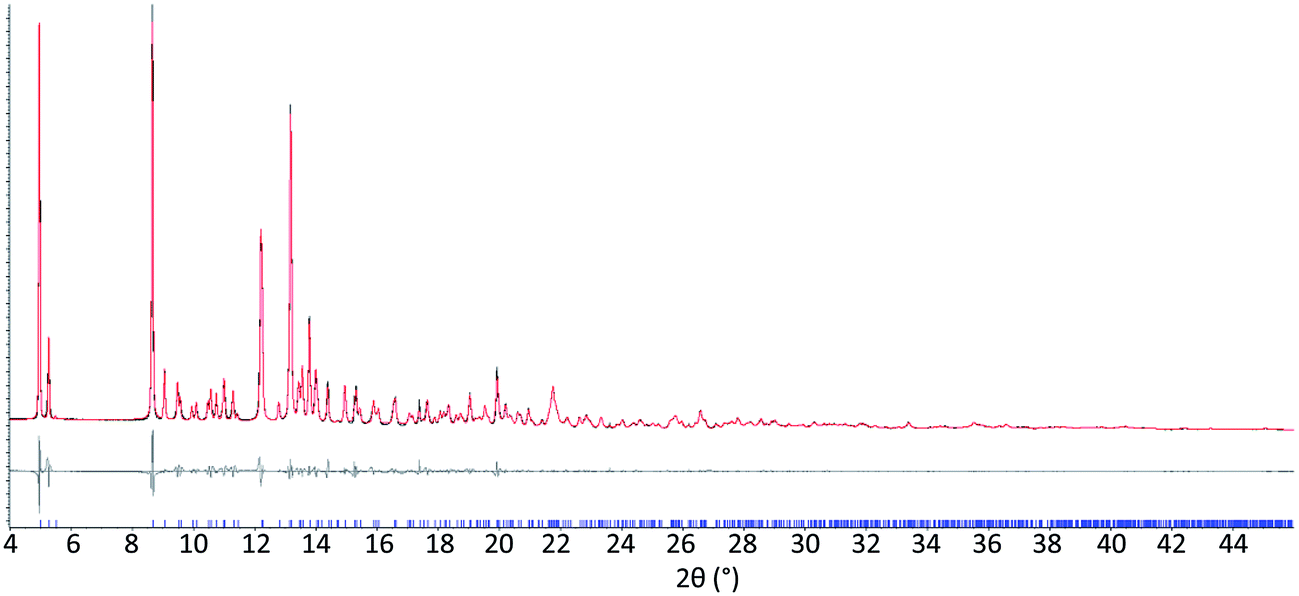 | ||
| Fig. 2 Exclusive formation of halogen-bonded co-crystal 1·3 obtained from system A competitive co-crystallization in acetone. Observed (black) and calculated (red) powder X-ray diffraction profiles and difference plot [Iobs − Icalc] (grey) of the Pawley refinement showing the fit for co-crystal 1·3 as a single phase obtained from system A competitive co-crystallization in acetone (synchrotron radiation λ = 0.82582(1) Å, 4.0 ≤ 2θ ≤ 46.0°, dmin = 1.06 Å; Rwp 0.0511, Rwp′ = 0.1143). | ||
| a Solvent polarity lacks a formal quantitative IUPAC definition and is rather a loosely defined term that encompasses a number of solvent properties. We have used the Reichardt ET(30) parameter,40 which is one of the most commonly used quantitative representations of the solvent properties associated with the concept of solvent polarity. HB refers to hydrogen-bonded co-crystal 2a·3 (system A), 2b·3 (system B) or 2c·3 (system C); XB refers to halogen-bonded co-crystal 1·3. |
|---|
|
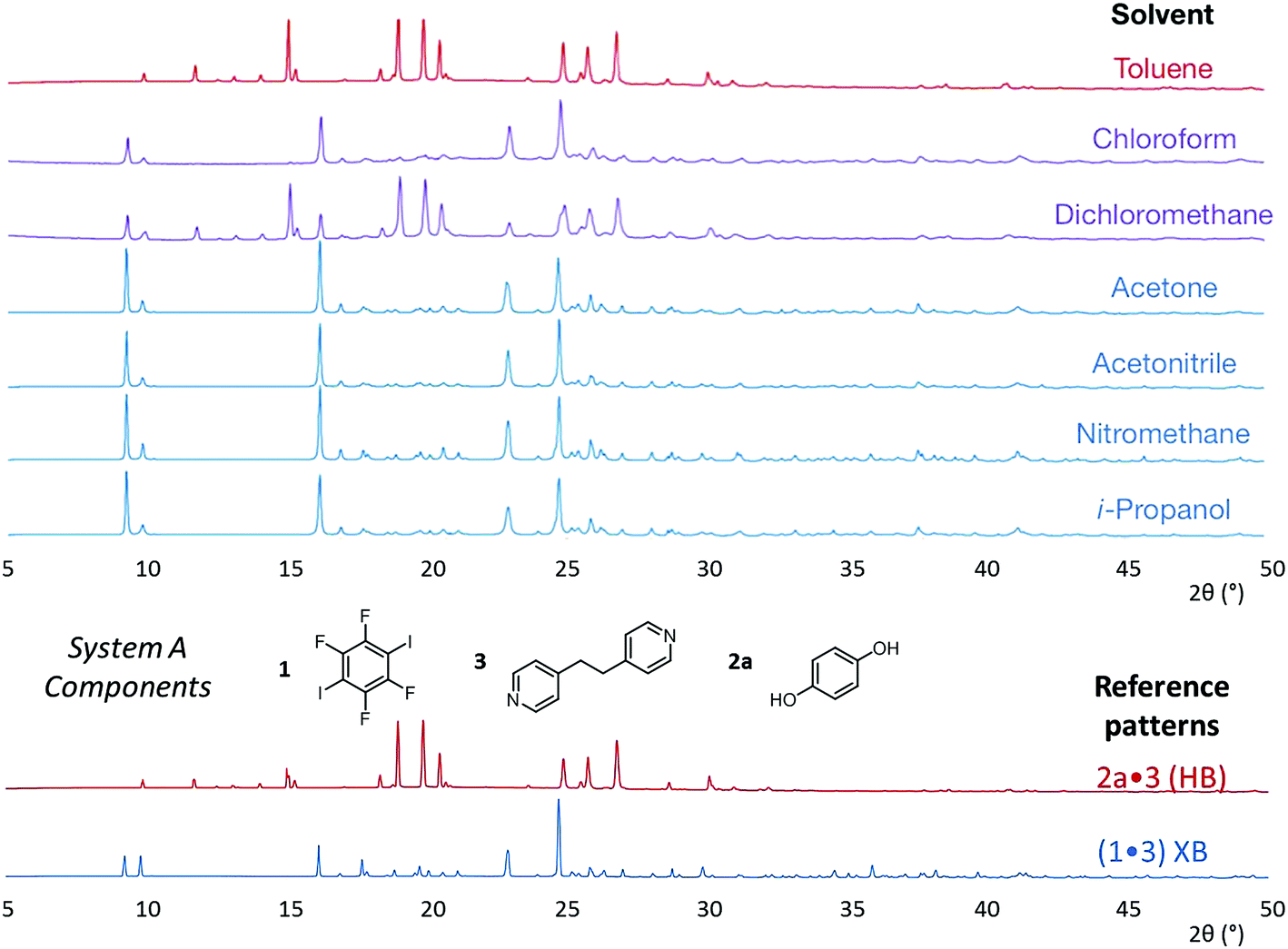 | ||
| Fig. 3 Powder X-ray diffraction patterns for products of system A competitive co-crystallizations from 7 solvents. Patterns shown in red when only HB co-crystals were formed, blue when only XB co-crystals were formed and purple when mixtures of HB and XB co-crystals were detected. All patterns are shown in the range 17.7 Å > d > 1.82 Å. For clarity of presentation, the scales are adjusted to allow representation as 2θ (°) appropriate for Cu-Kα radiation. Fits to patterns are provided in Section 10 of ESI.† | ||
The outcomes from systems B and C establish the generality of the findings (Table 1). Thus, increasing the HB donor strength by changing from 2a to 2b results in formation of the HB co-crystal (now 2b·3) becoming more probable in increasingly polar solvents. Thus, formation of 2b·3 occurs almost exclusively (HB ≥ 95%) in all solvents less polar than acetone, whereas the XB co-crystals 1·3 form exclusively in acetone and the more polar solvents. Further increase in the HB donor strength by using 2c leads to formation only of the HB co-crystal (2c·3) in all solvents except i-propanol, the most polar solvent used, in which a mixture of the HB and XB co-crystals results, with former dominating.
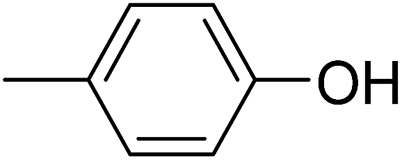
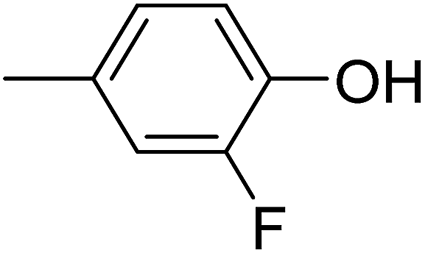
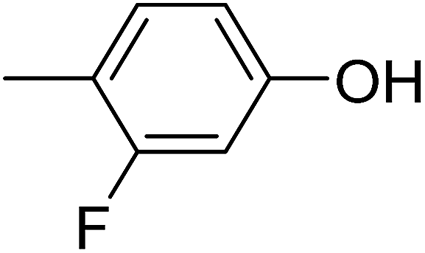
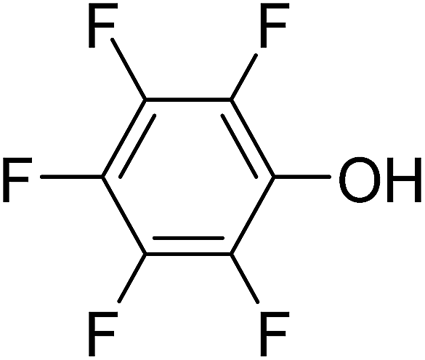
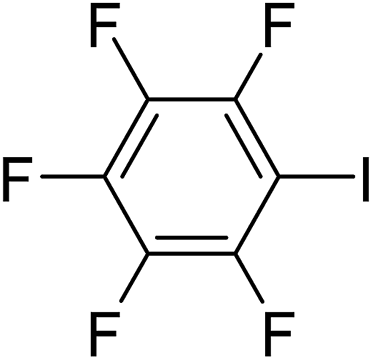
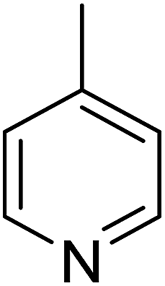
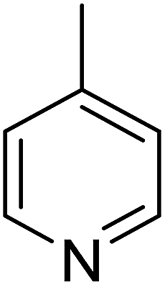
In parallel to the diffraction studies of co-crystal formation, the strengths of the hydrogen bonding and halogen bonding interactions present in the co-crystals were examined in solution by NMR spectroscopic titration. In order to limit the solution-phase studies to 1:1 binding and to ensure solubility at the concentrations needed, monotopic analogues of 1–3 were used for the titrations (C6F5I for 1; p-cresol for 2a, 2-fluoro-p-cresol and 3-fluoro-p-cresol for 2b, C6F5OH for 2c, and 4-picoline for 3). Titrations were carried out in three of the seven solvents used for the co-crystallization experiments, toluene, chloroform and acetonitrile and the changes in 1H and/or 19F chemical shifts were fitted to 1:1 binding isotherms, enabling association constants for the hydrogen bonding or halogen bonding interaction to be determined (Table 2). The titrations establish that monofluorination of the phenol leads to a small increase in association constant and perfluorination leads to a more dramatic increase, for titrations in a given solvent, confirming the increase in hydrogen bond donor capability from 2a to 2b to 2c, with the greater change being from 2b to 2c. Association constants for all hydrogen bonds decrease with increasing solvent polarity from toluene to chloroform to acetonitrile. It is also noted that the association constants for the halogen-bonded complexes are effectively too small to measure in all three solvents. These association constants are smaller than those of all hydrogen bonds in toluene and chloroform and any change in halogen bond strength upon increasing solvent polarity cannot be discerned.
| Solvent | Toluene | Chloroform | Acetonitrile |
|---|---|---|---|
| Guest |
|
|
|
| Host | |||
| a Errors determined by 2 × standard deviation of multiple repeat titrations. | |||
|
31 ± 1 | 19 ± 2 | <1 |
|
40 ± 1 | 20 ± 1 | <1 |
|
62 ± 5 | 52 ± 2 | <1 |
|
1300 ± 50 | 850 ± 60 | 19 ± 1 |
|
1 ± 1 | <1 | <1 |
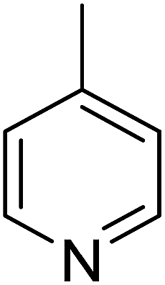
The solution-phase interactions require the formation of the HB or XB and the desolvation of the interacting functional groups. Co-crystal formation also requires formation of the HB or XB, but involves desolvation of the entire molecule and formation of additional intermolecular interactions, such as stacking of the aromatic rings and C–H⋯O hydrogen bonds. Kinetics effects may also play a role in the co-crystal competition experiment, whereas the solution-phase measurements are made at thermodynamic equilibrium. Thus the solution-phase bimolecular association and the co-crystal formation have important common features, but are not identical. Our measurements show the importance of the solvent in determining the strength of interaction in solution and the type of co-crystal formed. They show that solvent polarity has a major effect on the outcome of co-crystallization experiments. More polar solvents weaken HB interactions in solution and favour XB co-crystals.
The boundary between HB and XB co-crystal formation shifts to higher polarity solvents as the HB interaction energy increases, and the solution-phase HB and XB association constants are consistent with this outcome. Thus, in toluene solution the association constants for hydrogen bonding are maximised and are stronger than halogen bonding for all three HB donors, which is consistent with the fact that only HB co-crystals are observed from this solvent. The solution-phase association constants are much lower in acetonitrile, so that only the stability of the hydrogen-bonded complex with the perfluorinated phenol could be measured. This result is again in agreement with the observation of HB co-crystals for the competition experiment with perfluorinated hydroquinone 2c in acetonitrile. In chloroform solution, hydrogen bonding is stronger than halogen bonding for all three HB donors, and HB co-crystals are formed for all three HB donors in this solvent. The solution-phase association constants are consistent with the outcome of the co-crystal competition experiment in chloroform, but in the case of hydroquinone some XB co-crystals are also observed. The consistency between solution-phase measurements and co-crystal formation indicates that the HB/XB are the dominant intermolecular interactions in determining the outcome of the co-crystal competition experiment. The only discrepancy is the system A result in chloroform, which shows that the other factors do have some influence on the crystallization outcome.
Previous studies of competition between hydrogen bonding and halogen bonding in co-crystal formation37,41–43 have focused on tuning halogen bond strength, which is often empirically estimated or qualitatively ranked rather than quantified. In all prior studies the role of solvent has not been taken into account. In most cases a single crystallization solvent has been used exclusively, or at least predominantly, this solvent often being an alcohol or acetone. In the very few studies that consider the role of solvent, focus is restricted to solution-phase binding15,36,44,45 and does not consider the impact on controlling crystallization, but supports the potentially important role for halogen bonding in more polar solvents.
Conclusions
We have demonstrated the critical role of solvent in directing self-assembly by examining a system in which either hydrogen bonding or halogen bonding may play a prominent role and showing that either hydrogen-bonded co-crystals or halogen-bonded co-crystals can be selected exclusively by appropriate choice of solvent. The role of solvent choice has been evaluated experimentally in seven solvents using molecular components that are representative of commonly used molecular building blocks in supramolecular systems. All products have been identified and their phase purity or composition determined by analysis of powder X-ray diffraction data. Specifically, hydrogen-bonded co-crystals form in the least polar solvents and halogen-bonded co-crystals in the more polar solvents. The solvent polarity at which the preference switches from hydrogen bonding to halogen bonding depends upon the relative strength of the two types of interaction. The influence of solvent is all the more striking because it is not immediately evident that halogen bonds should be favoured in the more polar solvents.The implications of the results are not restricted to the development of co-crystals, which are seen as a practical option for tuning the physical properties of the solid forms of the active ingredients of pharmaceuticals, agrochemicals, high-energy-density materials (e.g. explosives & propellants), and other molecular crystalline materials. The results point to the wider importance of understanding solvation in the context of numerous physical and chemical phenomena,46 most directly those involving self-assembly and molecular recognition, but encompassing areas such as materials chemistry, catalysis and ion transport and recognition. We believe the results will stimulate examination of solvent-control of intermolecular interactions in many of these areas of application.
Experimental section
1H and 19F NMR spectroscopic titrations
1H and 19F NMR spectra were recorded on a Bruker Avance II 400 spectrometer at 400.1 MHz and 374.9 Hz respectively at 298 K, using the residual proton signal of the deuterated solvent or a capillary insert with D2O as the reference. For 1H NMR spectra, the chemical shifts (δH) are assigned relative to tetramethylsilane (TMS) at δH = 0. For 19F NMR spectra, the chemical shifts (δF) are assigned relative to CFCl3 at δF 0. Analyses of NMR spectra were carried out using Topspin version 3.2 or iNMR version 5.2.1. Measurement of mass of solids was carried out on a Precisa 125A balance. Measurement of volumes of liquids was carried out using Eppendorf Multipette XStream electronic pipettors. In a typical titration 10 Norell S-400 NMR tubes, each with different concentrations of host and guest as measured with a programmed Multipette XStream, were used to record NMR spectra using the automated sample recording system (BACS). The concentration of guest (4-picoline) was chosen to obtain a binding isotherm of >50% saturation in each titration. Titrations were repeated at least twice for reproducibility and estimation of errors. Binding constants, Ka, were determined by non-linear least-squares fitting the observed and calculated chemical shifts to a 1:1 binding isotherm using a macro-based Microsoft Excel fitting program written by Christopher A. Hunter (University of Cambridge). Where possible fits were conducted using multiple 1H or 19F signals for each titration. For further details see ESI section 4.†
Single crystal X-ray diffraction studies of co-crystals
Intensity data for 2b·3 and 2c·3 were collected at 100 K on a either a Bruker SMART APEX-II CCD diffractometer operating with a Mo-Kα sealed-tube X-ray source or Bruker D8 VENTURE diffractometer equipped with the PHOTON 100 CMOS detector, using a Cu-Kα microfocus X-ray source. A summary of data collection and structure refinement parameters is provided in the ESI (Table S3†). Data were corrected for absorption using empirical methods (SADABS) based upon symmetry equivalent reflections combined with measurements at different azimuthal angles.47 The crystal structures were solved and refined against all F2 values using SHELXL48 accessed via the Olex2 program.49 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions with idealized geometries and then refined by employing a riding model and isotropic displacement parameters. Crystal structures of 1·3 and 2a·3 have been previously reported and are therefore not reported in full here.37Powder X-ray diffraction studies
Microcrystalline powder samples were loaded into 0.7 mm borosilicate capillaries. X-ray diffraction data were collected using either synchrotron radiation at beamline I11 at Diamond Light Source50 or at the University of Sheffield using Cu-Kα radiation on a Bruker D8 ADVANCE X-ray powder diffractometer equipped with focusing Göbel mirror optics and a high-resolution energy-dispersive Lynxeye XE detector. Full details of data collections are provided in ESI.† Powder pattern indexing and fitting was carried out using the TOPAS program.51,52 Where the powder pattern could be indexed as a single phase, the phase purity of the material was established by Pawley refinement38 and where a mixture of products was identified upon indexing, mixed-phase Rietveld refinement39 was conducted to establish the relative proportions of the products. Comparison with powder patterns calculated from single crystal structures was used to provide a preliminary qualitative assessment of experimental powder patterns prior to quantitative fitting. Full details of fitting of powder patterns are provided in Sections 8–12 of the ESI.†Acknowledgements
This work was supported by the EPSRC (EP/J012955/1 and EP/J012998/1) and by the University of Sheffield. We are grateful to Diamond Light Source for provision of beam time at beamline I11.Notes and references
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley, Weinheim, 2012 Search PubMed.
- G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: a Textbook, World Scientific Publishing Co., Singapore, 2011 Search PubMed.
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- S. Das, G. W. Brudvig and R. H. Crabtree, Chem. Commun., 2008, 413–424 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem.–Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2012, 5, 42–47 CrossRef PubMed.
- M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- A. Vargas Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef PubMed.
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- T. Shirman, R. Kaminker, D. Freeman and M. E. van der Boom, ACS Nano, 2011, 5, 6553–6563 CrossRef CAS PubMed.
- P. Politzer, J. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zawarotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS PubMed.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- B. S. Sekhon, Int. J. Agrochem. Plant Prot., 2014, 2, 44–47 Search PubMed.
- D.-K. Bučar, S. Filip, M. Arhangelskis, G. O. Lloyd and W. Jones, CrystEngComm, 2013, 15, 6289–6291 RSC.
- O. Bolton, L. R. Simke, P. F. Pagoria and A. J. Matzger, Cryst. Growth Des., 2012, 12, 4311–4314 CAS.
- C. B. Aakeröy, T. K. Wijethunga and J. Desper, Chem.–Eur. J., 2015, 21, 11029–11037 CrossRef PubMed.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS.
- M. Habgood, M. A. Deij, J. Mazurek, S. L. Price and J. H. ter Horst, Cryst. Growth Des., 2010, 10, 903–912 CAS.
- H. C. S. Chan, J. Kendrick, M. A. Neumann and F. J. J. Leusen, CrystEngComm, 2013, 15, 3799–3807 RSC.
- P. A. Wood, N. Feeder, M. Furlow, P. T. A. Galek, C. R. Groom and E. Pidcock, CrystEngComm, 2014, 16, 5839–5848 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- L. Zhao, V. Raval, N. E. B. Briggs, R. M. Bhardwaj, T. McGlone, I. D. H. Oswald and A. J. Florence, CrystEngComm, 2014, 16, 5769–5780 RSC.
- M. D. Eddleston, B. Patel, G. M. Day and W. Jones, Cryst. Growth Des., 2013, 13, 4599–4606 CAS.
- C.-Q. Wan, A.-M. Li, S. A. Al-Thabaiti, E.-S. H. El-Mosslamy and T. C. W. Mak, CrystEngComm, 2014, 16, 8960–8968 RSC.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC.
- E. Corradi, S. V. Meille, M. T. Messina, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2000, 39, 1782–1786 CrossRef CAS PubMed.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- C. B. Aakeröy, C. L. Spartz, S. Dembowski, S. Dwyre and J. Desper, IUCrJ, 2015, 2, 498–510 Search PubMed.
- G. Mínguez Espallargas, F. Zordan, L. Arroyo Marín, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Chem.–Eur. J., 2009, 15, 7554–7568 CrossRef PubMed.
- T. Shirman, M. Boterashvili, M. Orbach, D. Freeman, L. J. W. Shimon, M. Lahav and M. E. van der Boom, Cryst. Growth Des., 2015, 15, 4756–4759 CAS.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed.
- Q.-Z. Li, B. Jing, R. Li, Z.-B. Liu, W.-Z. Li, F. Luan, J.-B. Cheng, B.-A. Gong and J.-Z. Sun, Phys. Chem. Chem. Phys., 2011, 13, 2266–2271 RSC.
- C. A. Hunter, Chem. Sci., 2013, 4, 1687–1700 RSC.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Rev. Sci. Intrum., 2009, 80, 075107 CrossRef CAS PubMed.
- A. A. Coelho, TOPAS Academic, version 4.1, 2007, see http://www.topas-academic.net Search PubMed.
- A. A. Coelho, J. S. O. Evans, I. R. Evans, A. Kern and S. Parsons, Powder Diffr., 2011, 26, S22–S25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full details of all experimental procedures and analysis of NMR spectroscopic data, powder X-ray data and single-crystal X-ray diffraction data. CCDC 1478819 and 1478820, for compounds 2b·3 and 2c·3 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01801k |
| ‡ Current address: Department of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. |
| This journal is © The Royal Society of Chemistry 2017 |