
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective C–O bond formation via a photocatalytic radical coupling strategy: access to perfluoroalkoxylated (ORF) arenes and heteroarenes†
Johnny W.
Lee
 ab,
Dominique N.
Spiegowski
ab and
Ming-Yu
Ngai
*ab
ab,
Dominique N.
Spiegowski
ab and
Ming-Yu
Ngai
*ab
aDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA. E-mail: ming-yu.ngai@stonybrook.edu
bInstitute of Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, New York 11794-3400, USA
First published on 5th June 2017
Abstract
Development of an efficient process that employs commercially available and cost effective reagents for the synthesis of perfluoroalkoxylated aromatic compounds (Ar–ORF) remains a daunting challenge in organic synthesis. Herein, we report the first catalytic protocol using readily available perfluoroalkyl iodides (RFI) and N-(hetero)aryl-N-hydroxylamides to access a wide range of perfluoroalkoxylated (hetero)arenes. Mild reaction conditions allow for selective O–RF bond formation over a broad substrate scope and are tolerant of a wide variety of functional groups. Mechanistic studies suggest the formation and recombination of persistent N-hydroxyl radicals and transient RF radicals under photocatalytic reaction conditions to generate N–ORF compounds that rearrange to afford the desired products.
Introduction
Molecules containing a perfluoroalkoxy group (ORF) have emerged as an important class of compounds in the fields of pharmaceutical, agrochemical, and materials science because incorporation of an ORF group into organic compounds often improves thermal, chemical and metabolic stability, lipophilicity, and bioavailability of parent molecules.1–10 While much progress has been made for late stage fluorination,11,12 perfluoroalkylation,13–15 and perfluoroalkylthiolation16–19 of (hetero)arenes, the facile synthesis of perfluoroalkoxylated (hetero)aromatic compounds remains an unmet challenge in synthetic organic chemistry.9,20–25 Unlike their analogous alkoxy groups, formation of an O–RF bond (e.g. RF = CF3) via direct SN2 type displacement is unfavorable due to (i) strong electron repulsion between fluorine atoms and incoming nucleophiles and (ii) the formation of an energetically adverse CF3 carbocation transition state (TS) structure (Fig. 1a).2,26 Umemoto et al. addressed this issue with an elegant electrophilic O–RF bond formation strategy via radical intermediates,20 yet the non-selective formation of O- and C-perfluoroalkylated products limited its synthetic utility. Although new strategies for the synthesis of perfluoroalkoxylated (hetero)arenes have emerged over the past few years,23,27–32 a general and mild catalytic process has yet to be developed. As a result, the full potential of perfluoroalkoxylated (hetero)aromatic compounds has not been fully exploited across a broad spectrum of technological applications. | ||
| Fig. 1 Photocatalytic radical coupling for the synthesis of perfluoroalkoxylated (hetero)arenes. | ||
To address this challenge, we recently developed trifluoromethoxylation reactions of aromatic compounds using N-(hetero)aryl-N-hydroxylamides and Togni reagents under mild reaction conditions.33,34 Our operationally simple and scalable protocols provide access to a diverse array of trifluoromethoxylated (hetero)aromatics with complex molecular architectures. Nevertheless, the high cost and multi-step synthesis of Togni reagents (e.g. Togni reagent I costs $55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 980 mol−1)35 might hinder their synthetic application. Furthermore, preparation of other O-perfluoroalkylated analogues requires the use of unique hypervalent iodine(III) perfluorinating reagents, which are commercially unavailable, synthetically-inaccessible, and thermally unstable. In order to develop a general method to access perfluoroalkoxylated (hetero)arenes, we turned our attention to RF–I reagents (RF = perfluoroalkyl) that are commercially available and cost efficient (e.g. CF3I costs $83 mol−1).35 Based on our prior mechanistic studies,36 selective O–RF bond formation is feasible if N-hydroxyl and RF radicals are generated simultaneously.37 Although direct single electron transfer (SET) from N-(hetero)aryl-N-hydroxylamides to RF–I is kinetically and thermodynamically unfavorable, we hypothesize that such a SET process could be facilitated by using an appropriate photoredox catalyst.15,38,39 Herein, we describe our efforts to develop the first photocatalytic radical coupling reaction of N-(hetero)aryl-N-hydroxylamides with RF–I to form N–ORF compounds, which then undergo ORF-migration to afford a wide variety of perfluoroalkoxylated (hetero)arenes (Fig. 1b).
980 mol−1)35 might hinder their synthetic application. Furthermore, preparation of other O-perfluoroalkylated analogues requires the use of unique hypervalent iodine(III) perfluorinating reagents, which are commercially unavailable, synthetically-inaccessible, and thermally unstable. In order to develop a general method to access perfluoroalkoxylated (hetero)arenes, we turned our attention to RF–I reagents (RF = perfluoroalkyl) that are commercially available and cost efficient (e.g. CF3I costs $83 mol−1).35 Based on our prior mechanistic studies,36 selective O–RF bond formation is feasible if N-hydroxyl and RF radicals are generated simultaneously.37 Although direct single electron transfer (SET) from N-(hetero)aryl-N-hydroxylamides to RF–I is kinetically and thermodynamically unfavorable, we hypothesize that such a SET process could be facilitated by using an appropriate photoredox catalyst.15,38,39 Herein, we describe our efforts to develop the first photocatalytic radical coupling reaction of N-(hetero)aryl-N-hydroxylamides with RF–I to form N–ORF compounds, which then undergo ORF-migration to afford a wide variety of perfluoroalkoxylated (hetero)arenes (Fig. 1b).
Results and discussion
To examine the feasibility of our hypothesis, we started our investigation using N-(p-tert-butylphenyl)-N-hydroxylamide (1a) and perfluoroisopropyl iodide (2a) as model substrates. Pleasingly, after exposure of 1a (1.00 equiv.) and 2a (8.00 equiv.) to visible light irradiation [3 W blue light-emitting diodes (LEDs)] in the presence of a ruthenium photoredox catalyst [Ru(bpy)3(PF6)2, (0.500 mol%)] and potassium carbonate (3.00 equiv.) in acetonitrile (0.100 M) at 23 °C for 12 hours, we obtained the desired product 3a in 38% yield (Table 1, entry 1). Exploration of photoredox catalysts, solvents, bases, concentrations, reactant stoichiometry and catalyst loading did not improve the product yield (entries 2–5). A breakthrough in optimization came when we lowered the reaction temperature to 0 °C, at which an 80% yield of the desired product 3a was obtained (entry 6). It is noteworthy that we did not observe addition of RF radicals directly to arenes even though such a reaction has been developed under photoredox-catalyzed reaction conditions.15,40 Apparently, this is due to the persistent radical effect that coupling of O- and RF-radicals is more favorable than the addition of RF radicals to arenes.41,42 Finally, control experiments showed that a photoredox catalyst, a base, light, and an oxygen-free atmosphere are critical for the success of the perfluoroalkylation reaction (entries 7–10).|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Base | Temp (°C) | Yielda (%) |
| a Reaction conditions: 1a (1.00 equiv.), 2a (8.00 equiv.), photocatalyst (0.500 mol%) and base (3.00 equiv.) in MeCN (0.100 M) for 12 h. Yields were determined by 19F NMR using trifluorotoluene as the internal standard. b No light. c Exposed to air. | ||||
| 1 | Ru(bpy)3(PF6)2 | K2CO3 | 23 | 38 |
| 2 | Rhodamine 6-G | K2CO3 | 23 | <5 |
| 3 | fac-Ir(ppy)3 | K2CO3 | 23 | 17 |
| 4 | Ru(bpy)3(PF6)2 | K3PO4 | 23 | 12 |
| 5 | Ru(bpy)3(PF6)2 | 2,6-Lutidine | 23 | 12 |
| 6 | Ru(bpy)3(PF6)2 | K2CO3 | 0 | 80 |
| 7 | — | K2CO3 | 0 | <5 |
| 8 | Ru(bpy)3(PF6)2 | — | 0 | <5 |
| 9 | Ru(bpy)3(PF6)2 | K2CO3 | 0 | <5b |
| 10 | Ru(bpy)3(PF6)2 | K2CO3 | 0 | <5c |

With the optimized reaction conditions in hand, we explored the scope of the perfluoroisopropylation reaction with respect to N-(hetero)aryl-N-hydroxylamides (1a–1t) (Table 2).43 The optimized reaction conditions were compatible with both aromatic and heteroaromatic hydroxylamides bearing a wide variety of functional groups and molecular scaffolds. For example, substrates with benzylic hydrogens, which are often prone to hydrogen atom abstraction in the presence of radical species, are tolerated (3b–3e and 3q–3s). Presumably, the rate of O- and RF-radical coupling is faster than that of benzylic hydrogen atom abstraction. These results further demonstrate the chemoselectivity of our protocol. In addition, halogen functionalities (3f–3i, 3n, 3o and 3q) remained intact after the reaction, providing easy handles for further synthetic elaborations. Substrates containing polyfluoromethyl ethers were also viable and afforded good yields of the desired products (3j and 3k). Moreover, products derived from the heterocyclic N-hydroxylamides such as benzofuran (3l) and benzothiophene (3m) were formed smoothly with high levels of regioselectivity. Other functional groups such as esters (3e and 3m), ketones (3l and 3s), ethers (3q–3t), carbamates (3e), 1,2,4-oxadiazoles (3n), oxindoles (3o), pyrazoles (3p), pyridines (3p–3t) and ketals (3t) were susceptible to ORF addition as well. Importantly, more complex N-pyridinyl-N-hydroxylamides derived from estrone and diacetone-D-glucose could be effectively converted to their perfluoroisopropylated analogs (3s and 3t), demonstrating that this method can be used in the preparation of pharmaceutically relevant compounds. Notably, none of the perfluoroisopropylated arenes and pyridines reported herein have been prepared prior to this study.
| a Reaction conditions: 1 (1.00 equiv.), 2a (8.00 equiv.), Ru(bpy)3(PF6) (0.500 mol%), K2CO3 (3.00 equiv.) in MeCN (0.100 M) at 0 °C. Cited yields are for isolated material. b −40 °C. c Following perfluoroalkylation, the reaction was heated to 40 °C. d Following O-perfluoroalkylation, the reaction was filtered, concentrated and the residue was dissolved in MeCN and heated to 80 °C. e Following O-perfluoroalkylation, the reaction was filtered, concentrated and the residue was dissolved in MeNO2 and heated to 120 °C. See ESI for further experimental details.† |
|---|

|
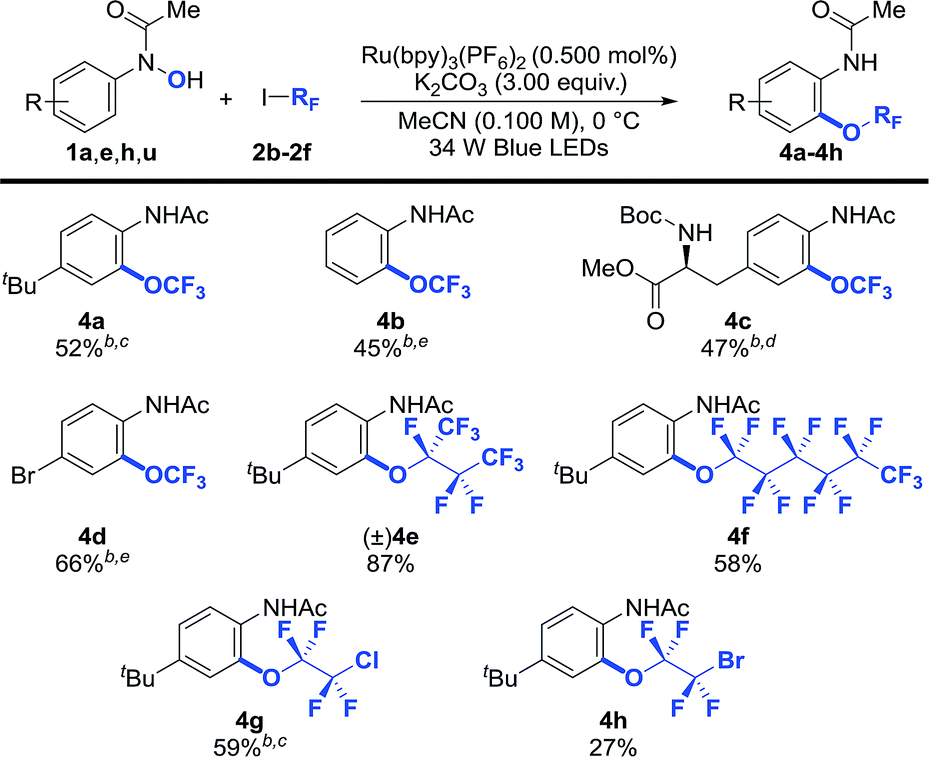
Trifluoromethoxy aryl ethers (Ar–OCF3) are constituents of several pharmaceutically active compounds, agrochemicals, and functional materials.3,5,6,8,9 As a result, significant effort has recently been directed towards uncovering general and practical protocols for their preparation,6,24,25 yet methods that use commercially available CF3I for their preparation have not been developed. We were pleased to see that our photocatalytic protocol can also be used for the synthesis of trifluoromethoxylated arenes (4a–4d) from CF3I (Table 3). In general, O-trifluoromethylation required a longer reaction time (48 h vs. 12–24 h for heptafluoroalkoxylation), possibly due to the lower reduction potential of CF3I (Ered1/2 = −1.52 V vs. SCE)44 in comparison with (CF3)2CFI (Ered1/2 = −0.66 V vs. SCE),44 which required an over-potential of 0.19 V for the reduction of CF3I to generate the CF3 radical using Ru(bpy)3+ (Ered1/2 = −1.33 V vs. SCE).45 In addition, other perfluoroalkyl iodides such as 1,1,1,2,2,3,4,4,4-nonafluoro-3-iodobutane and n-perfluorohexyliodide coupled smoothly to afford the desired products (4e and 4f) in synthetically useful yields. Importantly, our reaction is applicable to polyfluoroalkyl iodides such as 1-chloro-2-iodo-tetrafluoroethane and 1-bromo-2-iodotetrafluoroethane, albeit that 4h was obtained in a lower yield. This may be due to the instability of the 1-bromotetrafluoroethoxide species generated during the ORF-migration process. It is worth noting that the anilide moiety of the products could serve as a versatile handle for further synthetic functionalizations.34
| a Reaction conditions: 1 (1.00 equiv.), 2 (8.00 equiv.), Ru(bpy)3(PF6) (0.500 mol%), K2CO3 (3.00 equiv.) in MeCN (0.100 M) at 0 °C. Cited yields are for isolated material. b −40 °C. c Following O-perfluoroalkylation, the reaction was heated to 40 °C. d Following O-perfluoroalkylation, the reaction was filtered, concentrated and the residue was dissolved in MeCN and heated to 40 °C. e Following O-perfluoroalkylation, the reaction was filtered, concentrated and the residue was dissolved in MeCN and heated to 80 °C. See the ESI for further experimental details.† |
|---|
|
In order to get an insight into the mechanism of the photocatalytic reaction, we performed a series of Stern–Volmer quenching experiments (Fig. 2a). While deprotonated N-phenyl-N-hydroxylamide (Ia, Ered1/2 = 0.62 V vs. SCE)46 efficiently quenched *Ru(bpy)32+ in MeCN with a quenching constant of kq = 7.84 × 109 M−1 s−1, N-phenyl-N-hydroxylamide (1u) and perfluoroisopropyl iodide (2a) quenched the photoexcited photocatalyst (*Ru(bpy)32+) only to a minor extent. We also observed that the ORF migration is slower with more electron deficient aromatics, which is consistent with our previous observations and suggests an ionic ORF-migration pathway.36 Based on these results, a detailed description of our proposed photocatalytic cycle for selective O–RF bond formation and the consequent ORF-migration is outlined in Fig. 2b. Irradiation of Ru(bpy)32+ with visible light produces a long-lived (1.10 μs) photoexcited state, *Ru(bpy)32+,45 which engages in a SET with Ia to give N-hydroxyl radical (Ib) and a strong reductant Ru(bpy)3+ (Ered1/2 = −1.33 V vs. SCE).45 A single electron reduction of perfluoroalkyl iodide (RFI) with Ru(bpy)3+ forms a perfluoroalkyl radical (˙RF) and regenerates Ru(bpy)32+. Subsequent radical–radical coupling between Ib and ˙RF affords O-perfluoroalkylated N-phenyl-N-hydroxylamide Ic, which undergoes heterolytic N–ORF bond cleavage47,48 followed by recombination of the resulting short-lived ion pair (Id) and then tautomerization to yield the final perfluoroalkoxylated arene product.36
 | ||
| Fig. 2 a) Stern–Volmer quenching experiments. (b) The proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed the first photocatalytic protocol for the synthesis of structurally diverse perfluoroalkoxylated (hetero)arenes. The key to the success of our approach is the ability to concomitantly generate persistent and transient radicals under photoredox-catalyzed reaction conditions, which provide direct access to the challenging O–RF bond formation. Our approach is one of the mildest and most general perfluoroalkoxylations of (hetero)arenes reported to date.9,20–25 It features a broad substrate scope and high functional group compatibility. In addition, the use of commercially available RFI reagents and the excellent chemoselectivity of this reaction represents a considerable advance in the construction of the O–RF bond and should have a significant impact on the approach towards the synthesis of perfluoroalkoxylated aromatic building blocks. The success of this method not only provides access to unexplored chemical spaces to aid the discovery and development of novel drugs, agrochemicals, and functional materials, but also establishes a solid framework for further development of the photocatalytic radical coupling strategy using N-(hetero)aryl-N-hydroxylamides.Acknowledgements
This work was partially supported by the National Institute of General Medical Sciences (R35GM119652) and start-up funds from Stony Brook University (SBU). J. W. L. is grateful for the Chemistry Graduate Fellowship from the Department of Chemistry at SBU. We thank James Herbort, an NSF REU student (CHE-1358959), and Katarzyna N. Lee for the preparation of some N-(hetero)aryl-N-hydroxylamides. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- G. Siegemund, W. Schwertfeger, A. Feiring, B. Smart, F. Behr, H. Vogel and B. McKusick, in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a11_349.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827–856 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- P. Jeschke, E. Baston and F. R. Leroux, Mini-Rev. Med. Chem., 2007, 7, 1027–1034 CrossRef CAS PubMed.
- F. R. Leroux, B. Manteau, J. P. Vors and S. Pazenok, Beilstein J. Org. Chem., 2008, 4, 13 Search PubMed.
- A. Tressaud and G. N. Haufe, Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals, Elsevier Science, Amsterdam, 1st edn, 2008 Search PubMed.
- B. Manteau, S. Pazenok, J. P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140–158 CrossRef CAS.
- G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941–951 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956–960 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415 CrossRef CAS PubMed.
- M. G. Mormino, P. S. Fier and J. F. Hartwig, Org. Lett., 2014, 16, 1744–1747 CrossRef CAS PubMed.
- M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921–4925 CrossRef CAS PubMed.
- G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312–7314 CrossRef CAS PubMed.
- C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183–185 CrossRef CAS PubMed.
- X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457–3460 CrossRef CAS PubMed.
- R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 1650–1653 CrossRef CAS PubMed.
- T. Umemoto and O. Miyano, Bull. Chem. Soc. Jpn., 1984, 57, 3361–3362 CrossRef CAS.
- M.-L. Fu, J.-B. Liu, X.-H. Xu and F.-L. Qing, J. Org. Chem., 2017, 82, 3702–3709 CAS.
- K. E. Peterman and W. Dmowski, Org. Prep. Proced. Int., 1991, 23, 760–762 CrossRef CAS.
- T. M. Sokolenko, Y. A. Davydova and Y. L. Yagupolskii, J. Fluorine Chem., 2012, 136, 20–25 CrossRef CAS.
- K. N. Lee, J. W. Lee and M.-Y. Ngai, Synlett, 2016, 27, 313–319 CAS.
- A. Tlili, F. Toulgoat and T. Billard, Angew. Chem., Int. Ed., 2016, 55, 11726–11735 CrossRef CAS PubMed.
- T. Umemoto, K. Adachi and S. Ishihara, J. Org. Chem., 2007, 72, 6905–6917 CrossRef CAS PubMed.
- C. Huang, T. Liang, S. Harada, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 13308–13310 CrossRef CAS PubMed.
- F. Venturini, W. Navarrini, A. Famulari, M. Sansotera, P. Dardani and V. Tortelli, J. Fluorine Chem., 2012, 140, 43–48 CrossRef CAS.
- T. Khotavivattana, S. Verhoog, M. Tredwell, L. Pfeifer, S. Calderwood, K. Wheelhouse, T. Lee Collier and V. Gouverneur, Angew. Chem., Int. Ed., 2015, 54, 9991–9995 CrossRef CAS PubMed.
- J. B. Liu, C. Chen, L. Chu, Z. H. Chen, X. H. Xu and F. L. Qing, Angew. Chem., Int. Ed., 2015, 54, 11839–11842 CrossRef CAS PubMed.
- Q. W. Zhang, A. T. Brusoe, V. Mascitti, K. D. Hesp, D. C. Blakemore, J. T. Kohrt and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 9758–9762 CrossRef CAS PubMed.
- M. Zhou, C. F. Ni, Z. B. He and J. B. Hu, Org. Lett., 2016, 18, 3754–3757 CrossRef CAS PubMed.
- K. N. Hojczyk, P. J. Feng, C. B. Zhan and M. Y. Ngai, Angew. Chem., Int. Ed., 2014, 53, 14559–14563 CrossRef CAS PubMed.
- P. Feng, K. N. Lee, J. W. Lee, C. Zhan and M.-Y. Ngai, Chem. Sci., 2016, 7, 424–429 RSC.
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed.
- K. N. Lee, Z. Lei, C. A. Morales-Rivera, P. Liu and M. Y. Ngai, Org. Biomol. Chem., 2016, 14, 5599–5605 CAS.
- V. Matoušek, E. Pietrasiak, L. Sigrist, B. Czarniecki and A. Togni, Eur. J. Org. Chem., 2014, 2014, 3087–3092 CrossRef.
- D. A. Nagib, M. E. Scott and D. W. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed.
- A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS.
- A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC.
- For some substrates, reaction was run at −40 °C, and filtration and/or heating were required for the ORF-migration step. See the ESI† for detailed reaction conditions.
- V. G. Koshechko and L. A. Kiprianova, Theor. Exp. Chem., 1999, 35, 18–36 CrossRef CAS.
- A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- F. Xu, J. J. Kulys, K. Duke, K. C. Li, K. Krikstopaitis, H. J. W. Deussen, E. Abbate, V. Galinyte and P. Schneider, Appl. Environ. Microbiol., 2000, 66, 2052–2056 CrossRef CAS PubMed.
- A. Porzelle, A. W. J. Cooper, M. D. Woodrow and N. C. O. Tomkinson, Synlett, 2010, 2471–2473 CAS.
- A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01684k |
| This journal is © The Royal Society of Chemistry 2017 |